
The Herpes Simplex Virus Protein pUL31 Escorts Nucleocapsids to Sites of Nuclear Egress, a Process Coordinated by Its N-Terminal Domain
Herpesviral capsid assembly is initiated in the host nucleus. Due to size constraints, newly formed nucleocapsids are unable to leave the nucleus through the nuclear pore complex. Instead herpesviruses apply an evolutionarily conserved mechanism for nuclear export of capsids called nuclear egress. This process is initiated by docking of capsids at the inner nuclear membrane, budding of enveloped capsids into the perinuclear space followed by de-envelopment and release of capsids to the cytoplasm where further maturation occurs. Two viral proteins conserved throughout the herpesvirus family, the membrane protein pUL34 and the phosphoprotein pUL31 form the nuclear egress complex that is critical for primary envelopment. We show here that pUL31 and pUL34 enter the nucleus independently of each other. pUL31 is targeted to the nucleoplasm where it binds to nucleocapsids via the conserved C-terminal domain, while its N-terminal domain is important for capsid translocation to the nuclear envelope and for a coordinated interaction with pUL34. Our data suggest a mechanism that is apparently conserved among all herpesviruses with pUL31 escorting nucleocapsids to the nuclear envelope in order to couple capsid maturation with primary envelopment.
Published in the journal:
. PLoS Pathog 11(6): e32767. doi:10.1371/journal.ppat.1004957
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004957
Summary
Herpesviral capsid assembly is initiated in the host nucleus. Due to size constraints, newly formed nucleocapsids are unable to leave the nucleus through the nuclear pore complex. Instead herpesviruses apply an evolutionarily conserved mechanism for nuclear export of capsids called nuclear egress. This process is initiated by docking of capsids at the inner nuclear membrane, budding of enveloped capsids into the perinuclear space followed by de-envelopment and release of capsids to the cytoplasm where further maturation occurs. Two viral proteins conserved throughout the herpesvirus family, the membrane protein pUL34 and the phosphoprotein pUL31 form the nuclear egress complex that is critical for primary envelopment. We show here that pUL31 and pUL34 enter the nucleus independently of each other. pUL31 is targeted to the nucleoplasm where it binds to nucleocapsids via the conserved C-terminal domain, while its N-terminal domain is important for capsid translocation to the nuclear envelope and for a coordinated interaction with pUL34. Our data suggest a mechanism that is apparently conserved among all herpesviruses with pUL31 escorting nucleocapsids to the nuclear envelope in order to couple capsid maturation with primary envelopment.
Introduction
Morphogenesis of herpesviral capsids is an intricate process initiated in the infected nucleus [1]. A fragile procapsid is formed and packaged with one copy of the viral genome that is generated by cleavage of replicated concatameric DNA molecules. During this process, the rather spherical procapsids change their conformation and mature into the icosahedral and more stable C capsids. These accumulate in large numbers in capsid assembly sites and in the nucleoplasm. Over time, the infected nuclei are enlarged, concurrently the capsids get dispersed, the host chromatin is marginalized, and the nuclear lamina is partially disintegrated [2–5]. How mature capsids are released from sites of assembly, and how they translocate from there to the nuclear envelope is not completely understood, and their mode of transport to the nuclear periphery is discussed controversially [5–9].
With a diameter of 125 nm, herpesviral nucleocapsids exceed the nuclear pore diameter forcing them to take a different route out of the nucleus. Nuclear egress involves primary envelopment of capsids at the inner nuclear membrane (INM) resulting in a transiently enveloped perinuclear particle followed by de-envelopment at the outer nuclear membrane (ONM) and release of capsids to the cytoplasm [10,11]. Nuclear egress of all herpesviruses is mediated by a group of conserved viral proteins. In Herpes simplex virus type 1 (HSV-1), pUL31, a nucleo-phosphoprotein [12], and pUL34, a type II membrane protein [13], are recruited to the INM where they form the nuclear egress complex (NEC; [13,14]). Both proteins are required for nuclear egress of capsids out of the nucleus since deletion of either NEC component leads to their nuclear retention concomitant with a defect in viral propagation [15,16]. Moreover, the NEC recruits several viral and cellular kinases to partially disintegrate the major host barriers, namely the chromatin and the nuclear lamina, and to provide access of capsids to the INM [17–21]. Current data on pUL34 and pUL31 interaction(s) support a temporally regulated and orchestrated sequence of events at the INM, e.g. docking of capsids at the nucleoplasmic face, initiation of membrane curvature, wrapping of capsids by the INM, completion of budding by membrane scission and release of enveloped capsids into the perinuclear space [22–27]. In vivo, co-expression of the two NEC proteins in absence of any other viral protein is sufficient to form and accumulate empty vesicles in the perinuclear space [28,29]. Recently, insights into the membrane-associated NEC activity have been obtained by in vitro systems [30,31]. Recombinant HSV-1 pUL31 and pUL34 form ordered coats on artificial membranes and can induce membrane curving, invaginations, and vesicle formation. Thus, the NEC represents the minimal virus-encoded membrane-budding machinery with an intrinsic activity to drive membrane budding and scission of vesicles [30,31]. During infection, the situation is more complex due to the presence of other viral and cellular factors and their spatio-temporal regulation. Among them are the nonessential HSV-1 protein kinase pUS3 [32], the viral proteins pUL47 [33] and ICP22/pUS1 [34] as well as numerous host factors [33]. In addition to its well documented role in primary envelopment of nucleocapsids [18,25,26,35], pUL31 may assist in viral genome cleavage/packaging [15,36–41] and thus link capsid maturation to nuclear egress. Several studies have reported a preferred nuclear egress of C capsids over A or B capsids ([10,11]; and references therein); however, the molecular mechanism of these sorting events is poorly understood. The minor capsid proteins pUL25 and pUL17 that physically interact with pUL31 [38,39,41,42] are candidates to contribute to this quality control of nuclear capsid egress [10,11,27,43,44].
Orthologous pUL31 proteins share several features. The larger C-terminal domain can be divided into four conserved regions CR1 to 4 [45,46] with CR1 of all pUL31 orthologs containing a binding site for the respective pUL34 ortholog (Fig 1A; [45–47], and references therein); however, additional binding sites are likely to exist in their C-terminal domain [25,26,48–50]. In contrast, the smaller N-terminal domains are variable and enriched in basic residues clustered in several patches (red in Fig 1B). Furthermore, a putative classical bipartite nuclear localization signal (NLS; [45,50–52]) has been identified by in silico analysis (Fig 1A, grey in Fig 1B). To characterize the functions of the N-terminal domain of pUL31 reported to be phosphorylated by the US3 protein kinase [18], we generated a series of HSV-1 mutants with a particular focus on the basic patches (Fig 1C). We identified a classical bipartite NLS embedded in the N-terminal domain that was however not required for nuclear import of pUL31 during HSV-1 infection. Furthermore, we show here that pUL31 and pUL34 entered the nucleus independently of each other via separate routes. pUL31 lacking the N-terminal domain associated with capsids in the nucleoplasm but was unable to support nuclear egress and viral replication. A considerable amount of pUL31ΔN was retained in the cytoplasm if co-expressed with pUL34 suggesting that these proteins had prematurely interacted, and that the N-terminal domain of pUL31 controls the interaction with pUL34. Interestingly, while the C-terminal domain of pUL31 was sufficient to interact with nucleocapsids, the N-terminal domain was required for translocation of capsids from the nucleoplasm to the nuclear envelope and for viral propagation. Together, our data suggest a highly regulated sequence of events during nuclear egress: pUL31 is initially targeted to nuclear sites of capsid assembly and then escorts the nucleocapsids to the nuclear envelope for primary envelopment, a process coordinated by the N-terminal domain of pUL31.

Materials and Methods
Cells, viruses, and general methods
Hep2 (ATCC-No. CCL-23), HeLa (ATCC-No. CCL-2) and Vero cells (ATCC-No. CCL-81) were cultured as described previously [53]. HSV1(17+)Lox was used for all experiments [54,55]. The HSV-1 strain 17+ (kindly provided by D. J. McGeoch) and pHSV1(17+)Lox [54–56] were used for PCR amplification. HSV-1 propagation, titration and kinetics were done as described previously [13,53]. Plasmid transfection was performed using Effectene Transfection Reagent (Qiagen), while BAC transfection was done using Lipofectamine 2000 (Invitrogen). The yeast 2-hybrid method (Y2H; [53,57]), the NEX-TRAP assay [58] and the LUMIER assay [59,60] were described previously.
Plasmids
Cloning was performed by classical restriction or Gateway recombination according to the manufacturer’s protocol (Gateway, Invitrogen). Single base pair exchanges were introduced using the QuikChange Site-directed Mutagenesis Kit (Stratagene) and verified by sequencing. Constructs encoding pUL31 or mutants thereof were cloned into pCR3-N-myc destination vectors. Constructs encoding maltose-binding protein (MBP)-UL34 were cloned into the pCR3-MBP destination vector similar to the plasmid encoding Strep-pUL34 described previously [13]. The plasmids encoding bp1 (basic patch 1), bp2 or bp1bp2 were cloned using the plasmid EYFP (Clontech). Primers used to generate plasmids encoding EYFP-UL31-bp1bp2,-bp1,-bp2, and pUL31-mp1 (mutant patch 1),-mp2,-mp1mp2,-hbpmp1mp2, pSV40NLS-UL31-mp1mp2, pUL31ΔN, and pSV40NLS-UL31ΔN (Table 1) are described in Table 2. The plasmid EYFP-Nuc (Clontech) was used as control. Plasmids used for the yeast 2-hybrid (Y2H) and the LUMIER assays [59] and the plasmid EYFP-FRB-pUL31 [58] have been described before.


BAC mutagenesis
The HSV-1 UL31 mutants were generated using pHSV1(17+)Lox [54–56] and a modified galK positive counterselection scheme essentially as described ([13] and Striebinger et al., in revision). First, the non-overlapping coding region of UL31 (Nucleotides 9 to 865) was replaced by a galK-kan cassette, which had been amplified using the pGPS-galK-kan plasmid and primers equipped with 50bp homologies flanking the UL31 locus (Table 3: H5-UL31/galk and H3-UL31/galk). In a second step, the galK-kan cassette was substituted with a UL31 region encoding either wild type (wt) pUL31, pUL31ΔN, pSV40NLS-UL31ΔN, pUL31-mp1 (mutant patch 1), pUL31-mp2, pUL31-mp1mp2, or pUL31-hyperbasic patch 1 (hbp1)mp1mp2 (Fig 1C; Tables 1, 3 and 4). To rescue the ΔUL31/galk intermediate, the galK-kan cassette was replaced by the wt UL31 sequence. To reverse the pHSV1(17+)Lox-UL31-mp1mp2 to a pUL31 wt sequence, a two-step recombination process was applied resulting in pHSV1(17+)Lox-UL31-mp1mp2 revertant (rev). For PCR amplification, mutant plasmids that had been generated by site-directed mutagenesis using specific primers (Table 2) were used as templates. Details of BAC mutants are presented in Tables 1 and 4. Direct insertion of the SV40NLS coding sequence at the 5´end of UL31 would have perturbed the 3´coding sequence of UL32. To leave the UL32 coding sequence intact, a BAC was generated in which galK-kan was inserted into the UL31 locus while the original start site of UL31 was inactivated without changing the amino acid sequence of pUL32. Upon insertion of the coding sequence of pSV40NLS-UL31-mp1mp2, the overlapping 8 bp of UL31 and UL32 were duplicated. To reverse the pHSV1(17+)Lox-SV40NLS-UL31-mp1mp2 to a UL31 wt sequence, a two-step recombination process was applied resulting in pHSV1(17+)Lox-SV40NLS-UL31-mp1mp2 revertant (rev). This revertant still carries the 8 bp duplication of the 5´ UL31 region as well the mutated original start codon of UL31 (Tables 3–4). All BAC sequences were validated by sequencing of the DNA regions targeted by mutagenesis and by restriction pattern analysis of the entire BAC backbone. The pHSV1(17+)Lox strains were reconstituted by transfecting BAC DNA into Vero cells using Lipofectamine 2000 according to the manufacturer´s instructions (Invitrogen).


Immunofluorescence microscopy
Hep2, HeLa or Vero cells grown on coverslips, either transfected or infected, were fixed with 2% formaldehyde/PBS (15 min, room temperature) and permeabilized with 0.5% Triton X-100 (5 min, 4°C). Binding of antibodies to the HSV-1 Fc-receptor like proteins gE/gI was blocked with human blood sera of HSV-1 negative individuals/PBS for at least 3 h at room temperature [13]. Mouse monoclonal antibodies anti-myc (clone 9E10; kindly provided by J. von Einem), anti-MBP (NEB), anti-ICP0 (Santa Cruz), anti-ICP8 (kindly provided by R. Heilbronn) and anti-VP5 (clone 8F5; kindly provided by J. Brown) as well as rabbit anti-pUL31 and anti-pUL32 antibodies (kindly provided by B. Roizman and J. Baines [49]), anti-gM antibodies (kindly provided by T. Mettenleiter), and anti-pUL34 antibodies [13,53] were used. Anti-mouse and anti-rabbit fluorescently labelled secondary antibodies were from Invitrogen. Cells were examined using a confocal laser scanning microscope (LSM710; Zeiss, Oberkochen, Germany, or TCS SP5; Leica, Mannheim, Germany). Pictures were processed using Adobe Photoshop (Adobe) and Zen-Lite (Zeiss, Oberkochen, Germany). Fluorescence was measured along a 1 pixel thick and 6 μm long line using the "plot profile" tool of the software ImageJ (version 1.48K) on 8 bit images (Zeiss LSM710) taken with a 63x objective, NA 1.4, a pinhole aperture of 1 Airy unit, and a pixel size of 78 x 78 nm.
Amylose-affinity purification
To evaluate complex formation between pUL31 and pUL34, 3.5 x 106 HeLa cells were transfected with single plasmids encoding MBP-pUL34, myc-pUL31 or myc-pUL31ΔN, or a combination of a plasmid encoding MBP-pUL34 with one either encoding myc-pUL31 or myc-pUL31ΔN using Effectene according to the manufacturer´s protocol (Qiagen). Twenty-four hours post transfection (hpt), the cells were washed with ice-cold PBS, incubated for 20 min with ice-cold lysis buffer (20 mM Tris-HCl pH8, 150 mM NaCl, 10% (v/v) glycerol, 0.5% (v/v) Triton X-100, 2 mM EDTA, with complete Protease-Inhibitor Cocktail (Roche)). The lysates were pre-cleared by centrifugation (4°C, 12 000 rpm, 10 min) and incubation with Protein A Sepharose beads (GE Healthcare) for 10 min at 4°C. Following centrifugation (4°C, 5300 rpm, 10 min), the lysates were incubated with prewashed Amylose Resin (NEB). After incubation for 1 hour at 4°C on a rotating wheel, the supernatant was removed, and the beads were washed 3x using ice-cold lysis buffer. Proteins were released from the resin by incubation with 4x Lämmli buffer (room temperature, 15 min) and analyzed by SDS-PAGE followed by Western blotting using anti-MBP antibodies and anti-myc antibodies and peroxidase-conjugated secondary antibodies.
Electron microscopy
Vero cells were seeded onto coverslips 1 day prior to infection. The cells were pre-cooled for 20 min on ice, and incubated with HSV-1 at 1 pfu/cell in CO2-independent medium containing 0.1% (w/v) BSA for 2 h on ice on a rocking platform as described previously [54,61]. The cells were then shifted to regular growth medium at 37°C and 5% CO2 for 1 h. Non-internalized virus was inactivated by a short acid wash for 3 min (40 mM citrate, 135 mM NaCl, 10 mM KCl, pH 3), and the cells were transferred back to regular growth medium. After another 12 h, the cells were fixed with 2% (w/v) glutaraldehyde in 130 mM cacodylate buffer at pH 7.4 containing 2 mM CaCl2 and 10 mM MgCl2 for 1 h at room temperature. Subsequently the cells were washed and postfixed for 1 h with 1% (w/v) OsO4 in 165 mM cacodylate buffer at pH 7.4 containing 1.5% (w/v) K3[Fe(CN)6], followed by 0.5% (w/v) uranyl acetate in 50% (v/v) ethanol overnight. The cells were embedded in Epon, and 50 nm ultrathin sections were cut parallel to the substrate. Images were taken with an Eagle 4k camera at a Tecnai G2 electron microscope at 200 kV (FEI, Eindhoven, The Netherlands). For quantitation, images were taken at low magnification (6000x) and merged (Adobe Photoshop) to cover the whole cell area. The capsids in the nucleus and in the cytoplasm were counted and the areas of the nucleus and the cytoplasm were measured (ImageJ). Capsid numbers were calculated per area in mm2.
Results
The N-terminal domain of pUL31 contains a classical bipartite NLS
Bioinformatic analysis revealed two patches of positively charged residues composed of RRRSR (basic patch 1; bp1) and RRASRK (basic patch 2; bp2) separated by a linker region within the first 42 residues of HSV-1 pUL31 which resemble a classical bipartite NLS (http://www.expasy.org/; Fig 1A and 1B; [45,50–52]). To be classified as an NLS, a given sequence has to target an unrelated cytoplasmic protein to the nucleus. In addition, it should mediate physical interaction with transport factors of the importin α/β family [62], and its mutagenesis should result in a cytoplasmic localization while re-addition should restore the nuclear residence [62]. EYFP-pUL31-bp1bp2 comprising only residues 21 to 42 of pUL31 (grey in Fig 1B) fused to EYFP was as efficiently targeted to the nucleus as EYFP-SV40NLS (Fig 2A). Both bp1 and bp2 of pUL31 were able to individually target EYFP to the nucleus although less efficiently than the combination of both (Fig 2A) while EYFP alone was located to both cytoplasm and nucleus. Yeast 2-hybrid (Y2H; Fig 2B) and LUMIER experiments (S1A Fig) furthermore demonstrated a physical interaction of pUL31 with transport factors of the importin α family [62]. While pUL31, pUL31-mp1 (mutant patch 1; Fig 1C) as well as pUL31-mp2 (mutant patch 2; Fig 1C) interacted with importins (Fig 2B), pUL31-mp1mp2 did not (Fig 1C; Fig 2B; S1A Fig). Thus, the residues 21 to 42 of pUL31 constitute a classical bipartite NLS that can mediate nuclear import. Its relevance for nuclear import of pUL31 was analyzed by transient expression of myc-tagged pUL31 or mutants thereof (Fig 2C and 2D). pUL31 was exclusively located to the nucleus (Fig 2C and 2D) consistent with previous results [25,35,49,63,64]. Mutant pUL31 with either the first (pUL31-mp1) or the second basic patch (pUL31-mp2) mutated were also located to the nucleus (Fig 2C). pUL31-mp1mp2, with three basic residues in each of the two patches being replaced by neutral residues showed a more pancellular distribution, while adding an SV40NLS to its N-terminus restored its nuclear localization (Fig 2C). An additional exchange of a single residue G10R generated a hyperbasic patch (hbp) identical to residues 21 to 25 (Fig 1C). The resulting pUL31-hbpmp1mp2 was located to both cytoplasm and nucleus, similar to pUL31-mp1mp2 (Fig 2C), indicating that such an artificial basic patch did not rescue nuclear import. pUL31ΔN that lacked the N-terminal 44 residues (Fig 1C) remained cytoplasmic; again its nuclear import was rescued by adding an SV40NLS (Fig 2D; [62]). To reveal any potential export activity of pUL31, we used the NEX-TRAP (nuclear export trapped by rapamycin) assay [58]. EYFP-FRB-pUL31 was exclusively located in the nucleus both in the absence or presence of rapamycin (Fig 2E). pUL31 was unable to reach the cytoplasmic gM-FKBP for rapamycin-induced dimerization at the TGN and thus lacked any export activity (Fig 2E), a finding further corroborated by the interspecies heterokaryon assay [58]. In summary, we conclude that HSV-1 pUL31 harbors an import activity within the N-terminal variable domain, but no export activity. The import activity of pUL31 is composed of a classical bipartite NLS and an unrelated import activity that together mediate the very efficient nuclear import of pUL31.

The N-terminal domain of pUL31 regulates the interaction with pUL34
Next we determined the subcellular distribution of the different pUL31 variants in the presence of pUL34. Strep-tagged pUL34 expressed alone was located in cytoplasmic structures and the nuclear envelope (Fig 3A, left; [13,35]). Upon co-expression with pUL31, pUL34 was exclusively targeted to the nuclear envelope while pUL31 was predominantly located in the nucleoplasm (Fig 3A, right) consistent with previous reports [13,35]. Co-expression of pUL31-mp1mp2 and pUL34 resulted in localization of both proteins in the cytoplasm (Fig 3A, right). pSV40NLS-UL31-mp1mp2 co-expressed with pUL34 however was targeted to the nucleoplasm indicating its nuclear import (Fig 3A, right). In contrast, upon co-expression of pUL31ΔN or pSV40NLS-UL31ΔN with pUL34, both proteins were predominantly located in the cytoplasm (Fig 3A, right). Thus, while the addition of an SV40NLS restored nuclear localization of pUL31ΔN in the absence of pUL34 (Fig 2D), this was not the case in the presence of pUL34 (Fig 3A, right).
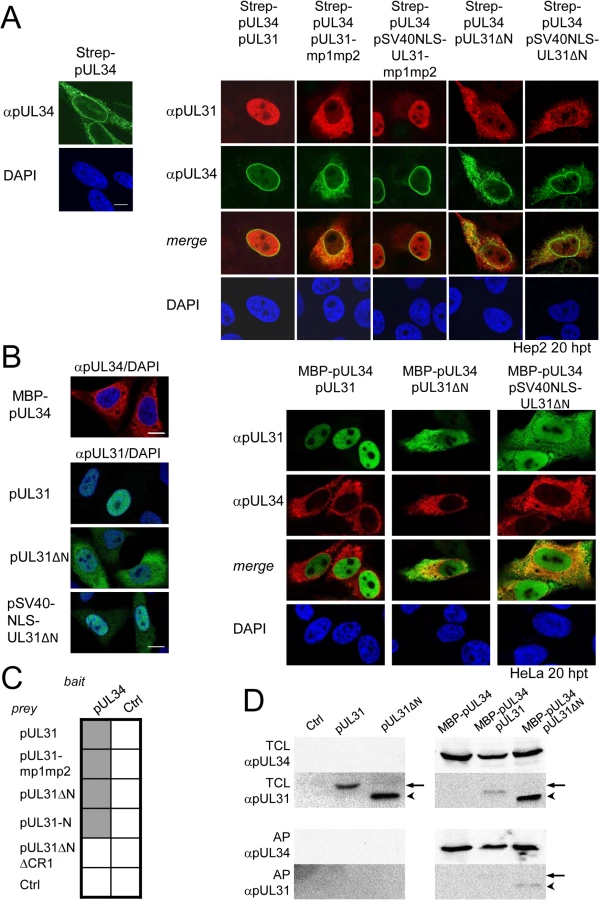
The nucleoplasmic distribution of pUL31 even upon co-expression with pUL34 (Fig 3A, right) suggested that the interaction of pUL31 and pUL34 might be regulated. pUL34 is a tail-anchored membrane protein while pUL31 per se is free to move between cytoplasm and nucleus. The current model for transport of integral membrane proteins to the INM [65] predicts that once anchored in the membrane of the endoplasmic reticulum (ER), pUL34 would be transported laterally along the ER membranes to the outer nuclear membrane (ONM), and the pore membrane (POM), eventually passing the peripheral nuclear pore channels to reach the INM. Transmembrane proteins with cytoplasmic domains above 60 kDa are too large to pass the peripheral nuclear pore channels [65]. To determine the mode of nuclear import of pUL34, pUL34 was fused N-terminally to the maltose-binding protein (MBP) thereby enlarging its cytoplasmic domain to about 60 kDa. Similar to Strep-pUL34 (Fig 3A, left), transiently expressed MBP-pUL34 was targeted to cytoplasmic structures resembling the ER (Fig 3B, left). As shown above, pUL31 expressed alone was exclusively located in the nucleus, pUL31ΔN essentially remained cytoplasmic while pSV40NLS-UL31ΔN was also nuclear (Fig 2D; Fig 3B, left). Upon co-expression of MBP-pUL34 and pUL31, MBP-pUL34 remained cytoplasmic whereas pUL31 was exclusively localized in the nucleus (Fig 3B, right). Thus, MBP-pUL34 was inserted into membranes already in the cytoplasm, but could not enter the nucleus due to its enlarged cytoplasmic domain. In contrast, pUL31 was efficiently imported into the nucleus. Interestingly, a different situation developed upon co-expression of MBP-pUL34 with pUL31ΔN or pSV40NLS-UL31ΔN (Fig 3B, right). With or without an NLS, a considerable amount of either pUL31 protein was retained in the cytoplasm, a finding reminiscent of the results obtained with Strep-pUL34 (Fig 3A, right). This suggested that in the wild type situation, the interaction between pUL34 and pUL31 is prevented in the cytoplasm. In absence of the N-terminal domain however, pUL31ΔN interacted prematurely with pUL34 and/or other components thereby retaining both proteins in the cytoplasm.
To gain further insight into the interaction of pUL31 with pUL34, we used Y2H (Fig 3C) and LUMIER assays (S1B Fig). As expected, pUL31 physically interacted with pUL34 (Fig 3C; S1B Fig). The same was true for pUL31-mp1mp2 and pUL31ΔN (Fig 3C; S1B Fig) consistent with the notion that pUL31-CR1 and potentially other regions of the C-terminal domain contribute to the assembly of the NEC complex [25,26,48,49]. Interestingly, the N-terminal domain of pUL31 also interacted with pUL34, either alone or in co-operation with the neighboring CR1 of pUL31 (Fig 3C). To determine whether pUL31 and pUL31ΔN interacted directly with MBP-pUL34, co-affinity purification was performed. pUL31 or pUL31ΔN were transiently expressed either alone or together with MBP-pUL34. Both myc-pUL31 and myc-pUL31ΔN were co-purified with MBP-pUL34 but not with the Amylose resin alone (Fig 3D). Thus, both proteins had retained the ability to interact with MBP-pUL34 and did so in a specific manner. Interestingly and consistent with previous reports [66], in absence of pUL34 or if spatially separated from it, pUL31 appeared unstable (Fig 3D) while this seemed different with pUL31ΔN (Fig 3D). Taken together, these data show that the NEC proteins pUL34 and pUL31 utilize different transport routes to the nucleus. Most importantly, the presence of a functional N-terminal domain prevents pUL31 from interacting prematurely with pUL34 in the cytoplasm.
The N-terminal domain of pUL31 harboring basic patches is essential for HSV-1 propagation
Previous data suggested a role of the N-terminal domain of pUL31 in viral replication [18]. To analyze the function of the pUL31 N-terminal domain in the context of an HSV-1 infection, we generated pHSV1(17+)Lox-ΔUL31, Lox-UL31ΔN, Lox-SV40NLS-UL31ΔN, Lox-UL31-mp1, Lox-UL31-mp2, Lox-UL31-mp1mp2, Lox-SV40NLS-UL31-mp1mp2, and Lox-UL31-hbpmp1mp2 using BAC mutagenesis (Fig 4A, 4C and 4E). A rescue mutant was generated for HSV1(17+)Lox-ΔUL31/galK-kan (Fig 4B), and revertants were made for Lox-UL31-mp1mp2 as well as Lox-SV40NLS-UL31-mp1mp2 resulting in Lox-UL31-mp1mp2 rev (Fig 4D) and Lox-SV40NLS-UL31-mp1mp2 rev (Fig 4F), respectively. All mutations were verified by restriction digest and sequencing of the mutated regions of the BAC DNAs.

Next, the BAC-DNAs of the respective mutants or the parental pHSV1(17+)Lox were transfected into Vero cells (Fig 5A). pHSV1(17+)Lox readily formed plaques surrounded by cells expressing the HSV-1 immediate early protein ICP0 (Fig 5A). Consistent with an essential function of HSV-1 pUL31 [15,34,67], transfection of pHSV1(17+)Lox-ΔUL31 resulted in single cells expressing ICP0 while no plaques were formed (Fig 5A). Transfection of the pHSV1(17+)Lox-UL31ΔN or Lox-UL31-mp1mp2 gave similar results (Fig 5A), and the N-terminal addition of an SV40NLS did not compensate the growth defect of either mutant (Fig 5A). In contrast, the revertants pHSV1(17+)Lox-UL31-mp1mp2 rev and Lox-SV40NLS-UL31-mp1mp2 rev formed plaques as efficiently as the parental strain thus indicating the integrity of the BAC backbone (Fig 5A). These results furthermore demonstrate that the N-terminal addition of the SV40NLS had not impaired pUL31 function. The 3´coding region of the essential UL32 gene overlaps with the 5´coding region of UL31 (Fig 4A; [68]). When Vero cells had been transfected with pHSV1(17+)Lox, Lox-ΔUL31, or Lox-UL31-mp1mp2, expression of UL32 was comparable and replication compartments appeared normal as indicated by the subcellular localization of ICP8, the HSV-1 single strand DNA binding protein (S2 Fig; [3,5,8]). Thus, pUL31 mutagenesis had not affected pUL32 expression and function. Nevertheless, the UL31 mutant strains were unable to spread and to form plaques. Together these data show that the N-terminal domain of pUL31 and its basic patches are essential for plaque formation. Most importantly, the addition of the SV40NLS to pUL31-mp1mp2 or pUL31ΔN did not restore plaque formation, suggesting that the N-terminal basic patches of pUL31 convey additional functions beyond merely mediating nuclear import.

A context-dependent basic patch restores the function of the N-terminal domain of pUL31
To further analyze the role of the N-terminal basic patches the mutants pHSV1(17+)Lox-UL31-mp1 or Lox-UL31-mp2 were generated (Fig 1C) and transfected into Vero cells; the resulting plaques were comparable to those of the parental BAC (Fig 5A). Thus, either of the authentic single basic patches was sufficient for virus replication. While the addition of the SV40NLS did not compensate the pUL31-mp1mp2 mutation (Lox-SV40NLS-UL31-mp1mp2 in Fig 5A), Lox-UL31-hbpmp1mp2 with the single mutation G10R that generated a sequence identical to basic patch 1 formed plaques (Fig 5A), although they were considerably smaller than those of the parental strain (Fig 5A). Thus, the G10R exchange partially complemented pUL31-mp1mp2 and restored function. Viral reconstitution showed that Lox-UL31-mp1 and Lox-UL31-mp2 replicated to parental titers, while the titers for Lox-UL31-hbpmp1mp2 were at least 2 logs lower (Fig 5B). Taken together, a single basic patch was sufficient to partially restore the crucial functions harbored within the N-terminal region of pUL31. Since the artificial SV40NLS did not compensate, the relative position within the N-terminal domain and the exact amino acid sequence are apparently important for the essential function of pUL31.
pUL31 associates with nucleocapsids and requires its N-terminal domain to direct them to sites of primary envelopment
To further decipher the function(s) of the pUL31 N-terminal domain, Vero cells transfected with the parental pHSV1(17+)Lox or the mutant BACs (Fig 1C; Tables 1 and 4) were analyzed 20 hours post transfection (hpt) using monoclonal antibodies recognizing mature hexon capsid epitopes (mAb 8F5 [69,70] in combination with antibodies directed against pUL31 ([49]; Fig 6A; S3A Fig) or ICP8 (Fig 6B). Confocal fluorescence microscopy analysis showed that all forms of pUL31 were targeted to the nucleoplasm (Fig 6A; S3A Fig). Thus, during HSV-1 infection, both the N-terminal authentic and the SV40NLS were dispensable for nuclear targeting of pUL31. There was no labeling in Vero cells transfected with Lox-ΔUL31 demonstrating the specificity of the anti-pUL31 antibodies (S3A Fig). After transfection with parental pHSV1(17+)Lox, Lox-UL31ΔN or Lox-UL31-mp1mp2 (Fig 6A), the subnuclear localization of pUL31 appeared punctuate and correlated with the capsid protein VP5 detected by antibodies to mature hexon epitopes (Fig 6A; [69]). While wt pUL31 was located to both nucleoplasm and nuclear envelope, pUL31ΔN or pUL31-mp1mp2 co-localized with capsids in the nucleoplasm, but not with the nuclear rim (Fig 6A). In contrast, ICP8, a marker of replication compartments [8], had a different subnuclear localization than the capsids (Fig 6B). Line histograms revealed that pUL31 and capsids largely co-localized while this was not the case for ICP8 and capsids (Fig 6A and 6B, right panels). Thus, pUL31, pUL31ΔN and pUL31-mp1mp2 could associate with capsids. Upon transfection with pHSV1(17+)Lox-UL31ΔN or Lox-SV40NLS-UL31ΔN, pUL31ΔN or SV40NLS-UL31ΔN were partially retained in the cytoplasm (S4A Fig), reminiscent of their localization after co-expression of pUL31ΔN or pSV40NLS-UL31ΔN with pUL34 (Fig 3A and 3B). Thus, the absence of the amino-terminal domain conferred partial cytoplasmic retention of pUL31 while nuclear import and targeting to capsids still occurred. Taken together, pUL31 was targeted to capsids present in the nucleoplasm and the C-terminal domain of pUL31 was sufficient to mediate this association.

To analyze the subcellular distribution of pUL34 upon mutagenesis of the N-terminal domain of pUL31, Vero cells were transfected with pHSV1(17+)Lox or the UL31 BAC mutants (S3B Fig; S4B Fig). As expected, in cells transfected with the parental Lox, pUL34 was located to the nuclear envelope. In cells transfected with Lox-ΔUL31, Lox-UL31-mp1mp2, Lox-UL31ΔN, or Lox-SV40NLS-UL31ΔN, pUL34 was also targeted to the nuclear envelope although its distribution seemed more patchy (S3B Fig; S4B Fig). This suggested that in addition to pUL31, other viral and host factors contribute to targeting of pUL34 to the nuclear envelope, a finding also supported by other recent reports [9,33,34].
Nucleocapsids of HSV1(17+)Lox-UL31-hbpmp1mp2 are impaired in nuclear egress
A single amino-acid exchange within pUL31-mp1mp2 resulting in pUL31-hbpmp1mp2 rescued the functions of the N-terminal domain (Fig 5B). To further define these functions, Vero cells were infected with HSV1(17+)Lox or Lox-UL31-hbpmp1mp2 at an MOI of 1 (Fig 7A–7G). About 80% of the cells infected with either HSV1(17+)Lox or Lox-UL31-hbpmp1mp2 expressed ICP0 at 4 hours post infection (hpi) (Fig 7C). Nevertheless, three phenotypes could be distinguished (Fig 7A and 7B): cells with capsids condensed in the nucleoplasm (category I), cells devoid of cytoplasmic capsids but with a dispersed and speckled appearance of nuclear capsids (category II), and cells with both nuclear and cytoplasmic capsids (category III). To quantify these phenotypes, cells were analyzed at 8 hpi (Fig 7D), 10 hpi (Fig 7E), 12 hpi (Fig 7A, 7B and 7F) or 16 hpi (Fig 7G) with a total of 60 infected cells for each condition (Fig 7D–7G). In the majority of cells infected with the parental strain, the nucleocapsids were dispersed throughout the nucleus with a considerable number of cytoplasmic capsids already at 8 hpi (Fig 7D). Only a few cells fell into category I or II while category III dominated, and this phenotype was further enhanced at later time points. In contrast, upon infection with Lox-UL31-hbpmp1mp2, the majority of cells belonged to category I at 8 hpi (Fig 7D). At 10 and 12 hpi, cytoplasmic capsids were detected in about 50 and 60%, respectively, of the cells (category III; Fig 7E and 7F). At 16 hpi, the percentage of cells in category III remained rather constant, at the same time, nuclei containing dispersed capsids (category II) increased, a phenotype rarely observed with the parental virus (Fig 7G).

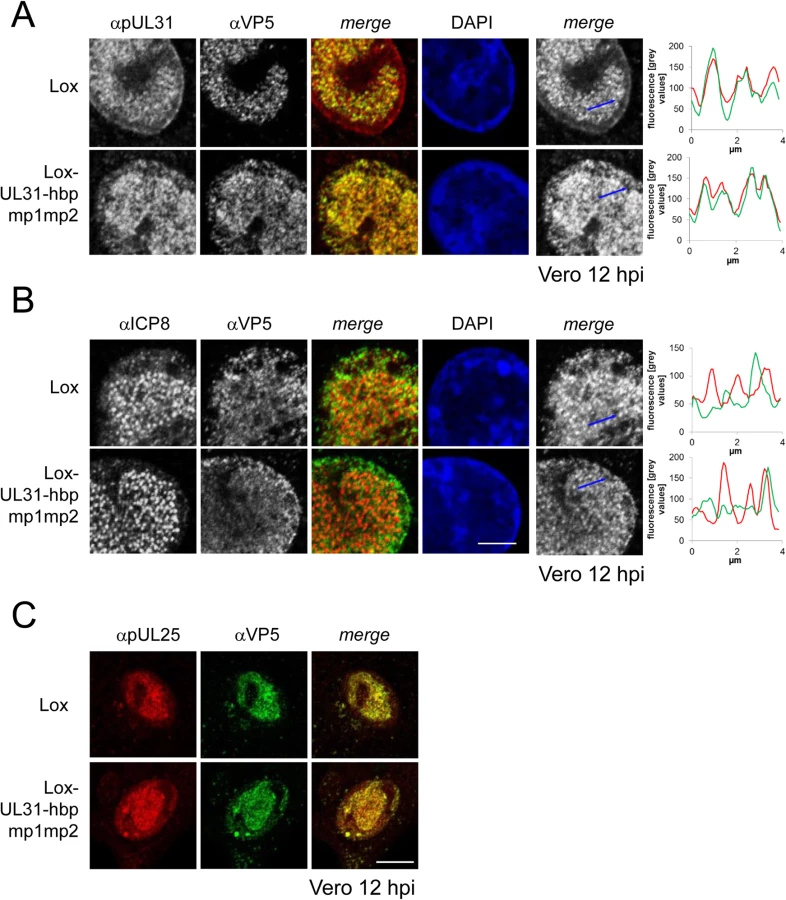
For closer inspection, Vero cells infected with the parental virus were compared to cells infected with Lox-UL31-hbpmp1mp2 (Fig 8A and 8B; S5 Fig). pUL31-hbpmp1mp2 had also been targeted to nucleocapsids (Fig 8A), whereas the nuclear replication compartments containing ICP8 did not co-localize with the nuclear sites of capsid assembly (Fig 8B). Line histograms clearly showed that the capsid protein VP5 co-localized well with pUL31-hbpmp1mp2 but not with ICP8 (Fig 8A and 8B, right panels). Depending on the stage of infection, pUL31-hbpmp1mp2 co-localized with capsids enriched in speckles in close association with the nuclear envelope (Fig 7A and 7B; S5 Fig). However, unlike in cells infected with the parental virus, a clear nuclear rim localization of pUL31 could not be detected [38,39]. The pattern of the subcellular localization of pUL25, a minor-capsid associated protein, was very similar to that of the major capsid protein VP5 and also co-localized with both wt pUL31 and pUL31-hbpmp1mp2 (Fig 8C). Upon infection with Lox-UL31-hbpmp1mp2, pUL34 was as efficiently targeted to the nuclear envelope as with the parental Lox (S6 Fig). To summarize, pUL31 was associated with nucleocapsids that had recruited pUL25 and escorted them to the nuclear periphery, a process that was delayed for Lox-UL31-hbpmp1mp2 concomitant with a defect in viral replication. Thus, basic patches within the N-terminal domain of pUL31 are required for efficient translocation of capsids from the nucleoplasm and to the sites of primary envelopment at the nuclear envelope.
pUL31-hbpmp1mp2 forms mature nucleocapsids delayed in nuclear egress
For high-resolution analysis, Vero cells were infected with Lox-UL31-hbpmp1mp2, fixed at 13 hpi (Fig 9) and analyzed by electron microscopy. Essentially all intracellular stages of virus maturation had been formed. These included mature capsids in the nuclear matrix (Fig 9A, arrowhead), capsids traversing the nuclear envelope (Fig 9B, arrowhead), early stages of secondary envelopment (Fig 9B, white arrow), and fully enveloped virions in vesicles (Fig 9A–9C, black arrows). Fully matured virions (Fig 9D, black arrow) as well as L-particles (Fig 9D, arrowheads) were located at the extracellular surface of the cells consistent with the production of infectious particles. Quantitation of the number of capsids in the nucleus and the cytoplasm revealed that although the cells contained similar amounts of capsids as after infection with the parental HSV1(17+)Lox (Fig 9E), the number of cytoplasmic virus particles was significantly reduced after infection with Lox-UL31-hbpmp1mp2 (Fig 9F). Hence the ratio of nuclear to cytoplasmic capsids was significantly increased in cells infected with the mutant (Fig 9G). We thus conclude that essentially all steps of viral morphogenesis occurred in cells infected with the Lox-UL31-hbpmp1mp2. To summarize, HSV1(17+)Lox-UL31-hbpmp1mp2 formed mature capsids, and pUL31-hbpmp1mp2 co-localized with mature VP5 hexon epitopes and pUL25. However, the escort of the capsids to the nuclear envelope seemed to be delayed for HSV1(17+)Lox-UL31-hbpmp1mp2, consistent with a reduced production of infectious virions. Thus, the N-terminal basic patches of pUL31 were required for efficient translocation of capsids from the nucleoplasm to sites of primary envelopment.

Discussion
The NEC of the herpesviruses is composed of two conserved essential proteins, called pUL34 and pUL31 in HSV-1, that are required for primary envelopment at the INM and for nuclear egress of newly formed capsids [10,11]. In vivo, their co-expression leads to the formation of empty vesicles in the perinuclear space [28,29]. In vitro systems recently revealed that these two proteins represent the minimal virus-encoded membrane-budding machinery that contains intrinsic activity to drive budding and scission of membrane vesicles [30,31]. The observation that a complex formed between pUL34 and pUL31 exhibits membrane budding activity [30], instantly implies that the NEC activity needs to be spatially and temporally controlled and confined to sites of nuclear egress to enable efficient capsid nuclear egress and at the same time prevent perturbations on cytoplasmic membranes.
This study shows that pUL34 and pUL31 utilized separate routes to the nucleus. pUL31 enters the nucleus through the central nuclear pore channels. The N-terminal domain of HSV-1 pUL31 contains a classical bipartite NLS composed of two basic patches that bound importin α and mediated nuclear import congruent with data on the pUL31 orthologs of HSV-2 [51], murine cytomegalovirus (MCMV; [45]), human cytomegalovirus (HCMV; [52]), and pseudorabies virus (PrV; [50]). The bipartite NLS of HSV-1 pUL31, however, was not essential for nuclear import as pUL31-mp1mp2 was also imported into the nucleus, both in absence and presence of other HSV-1 proteins, consistent with results obtained for pUL31 of HSV-2 [51]. Thus, auxiliary modes of nuclear import are likely to exist, in form of additional non-classical import sequences embedded in the amino-terminal domain [51] and likely also by piggy-backing with other viral partners, for example the pUL17/pUL25 complex [38,39,41,71] or ICP22 [34].
In contrast to soluble proteins like pUL31, integral membrane proteins such as pUL34 need to traverse the nuclear pore through its peripheral channels [65] that physically restrict the size of the cyto-/nucleoplasmically exposed domain [65]. pUL31 and pUL34 form a complex of about 60 kDa [30] that, if formed prior to nuclear import, would be too close to the size limitations of the peripheral nuclear pore channels [65]. Thus, a mechanism is required to prevent premature cytoplasmic association of pUL34 and pUL31. Data presented in this study show that pUL34 with its N-terminal domain enlarged by attaching MBP to mimic the size of a pUL34/pUL31 complex, was retained in the cytoplasm, while co-expressed pUL31 was still imported into the nucleus. Recent analysis of the HCMV NEC proteins also shows that pUL53, the ortholog of pUL31, precedes pUL50, the ortholog of pUL34, in nuclear import [52]. Together we conclude that the interaction of pUL31 and pUL34 and their orthologs is prevented in the cytoplasm to allow for their independent import into the nucleus.
Strikingly, we observed that in absence of the 44 N-terminal residues of pUL31, both pUL31 and pUL34 were retained in the cytoplasm, and that the addition of the SV40NLS to pUL31ΔN was unable to confer its nuclear import in the presence of pUL34. Furthermore, we show that pUL31ΔN physically interacted with pUL34, and that both proteins co-localized in the cytoplasm consistent with earlier studies [25,48]. Together these data indicate that pUL31ΔN was retained in the cytoplasm most likely by an interaction with membrane-associated pUL34. A NEC complex preformed and retained in the cytoplasm would be unavailable for its essential nuclear functions. In addition, premature NEC formation in the cytoplasm would unleash the intrinsic activity of the NEC to drive budding and scission of membranes [30] with deleterious effects on membrane integrity and function. Our results here suggest that the N-terminal domain of pUL31 provides a mechanism to prevent premature cytoplasmic NEC activity, and that it is critical in modulating the interaction of pUL31 with pUL34.
While pUL31ΔN and pSV40NLS-UL31ΔN were partially retained in the cytoplasm, significant amounts of them nevertheless had reached the nucleoplasm, most likely because they were expressed prior to pUL34. Despite the nuclear import of these pUL31 variants, there was a drastic reduction in replication for Lox-UL31ΔN and Lox-SV40NLS-UL31ΔN, implying that neither pSV40NLS-UL31ΔN nor pUL31ΔN were able to support capsid nuclear egress. Thus other functions than the NLS are encoded by the N-terminal domain of pUL31 and critical for viral propagation.
We show that once imported into the nucleoplasm, pUL31 was primarily targeted to nucleocapsids consistent with previous reports [35,39,41]. While the nuclear capsids of Lox-UL31ΔN and Lox-UL31-mp1mp2 had recruited the mutated pUL31 proteins, plaque formation was still inhibited. Obviously, the C-terminal domain of pUL31 was sufficient to bind pUL31 to capsids whereas the N-terminal basic patches must contribute essential functions downstream of this event. The mutation G10R generated an artificial basic patch in pUL31-hbpmp1mp2 that partially restored its function: capsids translocated to the nuclear periphery although with lower efficiency than in the parental virus. Thus, a single N-terminal basic patch was critical in promoting capsid translocation and nuclear egress. This mutant thus unveiled a previously unanticipated sequence of events where pUL31 initially interacts with capsids at their assembly sites and then escorts them to the nuclear periphery, a process that is obscured during the fast progression of a natural HSV-1 infection. The basic patches may promote conformational changes of pUL31 and serve as a platform to recruit hitherto unknown viral and/or host proteins that rearrange the host chromatin and mediate capsid transport through the nucleoplasm to the INM [3,5]. Thereby, the capsids may be dispersed and translocated to sites of primary envelopment, as observed in this study and supported by previous findings [3].
The subnuclear localization of HSV-1 pUL31 correlated well with that of capsids harboring mature VP5 hexon epitopes and the minor-capsid associated protein pUL25, a finding further supported by biochemical evidence [38,39,41]. Thus, pUL25 and pUL17 that together form the capsid vertex-specific complex (CVSC) and promote cleavage and packaging of viral genomes into capsids [10,11], could link pUL31 with nucleocapsids to cooperate in maturation [15]. In such a scenario, the C-terminal domain of pUL31 composed of CR1-4 and sufficient for capsid binding may contribute to capsid maturation [38,39,41]. Interestingly, CR2 and CR4 of M53, the MCMV ortholog of pUL31 are also involved in DNA genome cleavage/packaging [37,40]. Furthermore, pUL31 physically interacts with a pUL25/pUL17 complex even in the absence of capsids [38]. Thus, a pre-formed complex of pUL31 and pUL25/pUL17 may bind to capsids during DNA cleavage and packaging. By such a mechanism, the capsid-associated pUL31 may contribute to completion of genome packaging [15,36–41] and selection for primary envelopment at the INM ([10,11], and references therein).
The domains of pUL31 that interact with pUL34 seem to be initially masked but must eventually be exposed to drive NEC formation and activity. Upon infection with Lox-UL31-hbpmp1mp2, capsids with mature VP5 epitopes accumulated in close vicinity to the INM suggesting that primary envelopment was compromised if the authentic basic patches had been neutralized. Thus, the basic patches could serve two functions; to trigger the translocation of capsids from the nucleoplasm to the nuclear envelope, and to promote budding, but only in the presence of capsids. Indeed, in vitro data support an active role of the pUL31 basic patches in regulating the budding process; their deletion abrogated NEC activity but not the formation of the complex and its association with membranes [30]. Interestingly, cytoplasmic membranes harboring pUL34 and pUL31ΔN appeared undisturbed suggesting that a pUL31ΔN/pUL34 complex lacks NEC activity. An attractive scenario suggests that the basic patches in the N-terminal domain of pUL31 promote membrane budding by stabilizing a conformational switch within pUL34. In contrast, the non-essential US3 protein kinase seems not to be critical although it phosphorylates several sites within the N-terminal domain of pUL31 [18]. The severe phenotype of Lox-UL31ΔN and Lox-UL31-mp1mp2 contrasts that of HSV-1 strains lacking US3 [19,32] suggesting that pUL31 phosphorylation has modulatory or cell-type specific effects.
We and others have shown that several regions of HSV-1 pUL31 interact with pUL34. The CR1 of all pUL31 orthologs interacts with pUL34 [45–47] probably involving pUL34 residues 137 to 181 [25,26,49,67]. As an extension of pUL31-CR1, the N-terminal domain could contribute to the interaction with pUL34. Furthermore, the C-terminal domain of pUL31 seems to be functionally linked to the N-terminal domain of pUL34 [25,48,50]. Upon infection with Lox-UL31-hbpmp1mp2, we observed capsid speckles with mature hexon epitopes that accumulated in close vicinity to pUL34 at the INM. These speckles resembled the phenotype of a C-terminal mutant of M53, the pUL31 ortholog of MCMV, that is also impaired in capsid egress [37] suggesting that the N - and C-terminal domains of pUL31 cooperate to coordinate the interaction with pUL34. Upon association with capsids in the nucleoplasm, pUL31 may undergo a conformational switch from a closed conformation with the N - and potentially the C-terminal domain covering pUL34-interacting regions to an open conformation that allows for interaction with pUL34 and thus capsid docking and budding at the INM. The interaction between pUL31 and pUL34 may even involve multiple and sequential conformational changes established during NEC formation and membrane budding, a process that appears to be highly orchestrated [22–27]. Our understanding of the precise molecular mechanism however must await the structural resolution of the NEC.
Our data together with those of others suggest a highly regulated sequence of events during primary envelopment of HSV-1 (Fig 10; [10,11,25–27,35,38,39,41,44]: (A) Newly synthesized, cytoplasmic pUL31 is masked and inhibited by adopting a conformation that prevents premature interaction with pUL34, (B) pUL31 is imported into the nucleus independently of pUL34, but possibly in a complex with other viral and host proteins, (C) pUL31 associates with nucleocapsids and potentially contributes to genome packaging, (D) conformational changes in its N-terminal domain contribute to targeting of mature capsids to the INM, (E) capsid-associated pUL31 interacts with membrane-associated pUL34 to form the NEC, (F) further sequential interactions between several pUL31 and pUL34 molecules induce membrane wrapping until primary capsid envelopment is complete.

In summary, our data not only provide insights into the molecular function of HSV-1 pUL31 during translocation of nucleocapsids from the nucleoplasm to sites of primary envelopment. Given the conservation of the many players involved, the sequence of events described here seems to be applicable to all herpesviruses. Transit of nucleocapsids from the nucleoplasm to the INM is undoubtedly a highly complex process [2–8]. Numerous cellular and viral factors are expected to assist and accompany the nucleocapsids decorated with pUL31 in order to pave their way from the nuclear interior to the INM [33,72]. Thus, the results as well as the tools presented are invaluable to identify proteins and to decipher their function involved in nuclear translocation of capsids, a step decisive for propagation of herpesviruses and potentially other viruses relying on nuclear morphogenesis.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Baines JD (2011) Herpes simplex virus capsid assembly and DNA packaging: a present and future antiviral drug target. Trends Microbiol 19 : 606–613. doi: 10.1016/j.tim.2011.09.001 22000206
2. Monier K, Armas JC, Etteldorf S, Ghazal P, Sullivan KF (2000) Annexation of the interchromosomal space during viral infection. Nature Cell Biology 2 : 661–665. 10980708
3. Simpson-Holley M, Baines J, Roller R, Knipe DM (2004) Herpes simplex virus 1 U(L)31 and U(L)34 gene products promote the late maturation of viral replication compartments to the nuclear periphery. J Virol 78 : 5591–5600. 15140956
4. Reynolds AE, Liang L, Baines JD (2004) Conformational changes in the nuclear lamina induced by herpes simplex virus type 1 require genes U(L)31 and U(L)34. J Virol 78 : 5564–5575. 15140953
5. Simpson-Holley M, Colgrove RC, Nalepa G, Harper JW, Knipe DM (2005) Identification and functional evaluation of cellular and viral factors involved in the alteration of nuclear architecture during herpes simplex virus 1 infection. J Virol 79 : 12840–12851. 16188986
6. Forest T, Barnard S, Baines JD (2005) Active intranuclear movement of herpesvirus capsids. Nat Cell Biol 7 : 429–431. 15803134
7. Feierbach B, Piccinotti S, Bisher M, Denk W, Enquist LW (2006) Alpha-Herpesvirus Infection Induces the Formation of Nuclear Actin Filaments. PLoS Pathog 2: e85. 16933992
8. Chang L, Godinez WJ, Kim IH, Tektonidis M, de Lanerolle P, et al. (2011) Herpesviral replication compartments move and coalesce at nuclear speckles to enhance export of viral late mRNA. Proc Natl Acad Sci U S A 108: E136–144. doi: 10.1073/pnas.1103411108 21555562
9. Bosse JB, Virding S, Thiberge SY, Scherer J, Wodrich H, et al. (2014) Nuclear herpesvirus capsid motility is not dependent on f-actin. MBio 5.
10. Johnson DC, Baines JD (2011) Herpesviruses remodel host membranes for virus egress. Nat Rev Microbiol 9 : 382–394. doi: 10.1038/nrmicro2559 21494278
11. Mettenleiter TC, Muller F, Granzow H, Klupp BG (2013) The way out: what we know and do not know about herpesvirus nuclear egress. Cell Microbiol 15 : 170–178. doi: 10.1111/cmi.12044 23057731
12. Chang YE, Roizman B (1993) The product of the UL31 gene of herpes simplex virus 1 is a nuclear phosphoprotein which partitions with the nuclear matrix. J Virol 67 : 6348–6356. 7692079
13. Ott M, Tascher G, Hassdenteufel S, Zimmermann R, Haas J, et al. (2011) Functional characterization of the essential tail anchor of the herpes simplex virus type 1 nuclear egress protein pUL34. J Gen Virol 92 : 2734–2745. doi: 10.1099/vir.0.032730-0 21832006
14. Schuster F, Klupp BG, Granzow H, Mettenleiter TC (2012) Structural Determinants for Nuclear Envelope Localization and Function of Pseudorabies Virus pUL34. Journal of Virology 86 : 2079–2088. doi: 10.1128/JVI.05484-11 22156520
15. Chang YE, Van Sant C, Krug PW, Sears AE, Roizman B (1997) The null mutant of the U(L)31 gene of herpes simplex virus 1: construction and phenotype in infected cells. J Virol 71 : 8307–8315. 9343183
16. Roller RJ, Zhou Y, Schnetzer R, Ferguson J, DeSalvo D (2000) Herpes Simplex Virus 1 UL34 Gene Product Is Required for Viral Envelopment. J Virol 74 : 117–129. 10590098
17. Marschall M, Feichtinger S, Milbradt J (2011) Regulatory roles of protein kinases in cytomegalovirus replication. Adv Virus Res 80 : 69–101. doi: 10.1016/B978-0-12-385987-7.00004-X 21762822
18. Mou F, Wills E, Baines JD (2009) Phosphorylation of the U(L)31 protein of herpes simplex virus 1 by the U(S)3-encoded kinase regulates localization of the nuclear envelopment complex and egress of nucleocapsids. J Virol 83 : 5181–5191. doi: 10.1128/JVI.00090-09 19279109
19. Ryckman BJ, Roller RJ (2004) Herpes simplex virus type 1 primary envelopment: UL34 protein modification and the US3-UL34 catalytic relationship. J Virol 78 : 399–412. 14671121
20. Bjerke SL, Roller RJ (2006) Roles for herpes simplex virus type 1 UL34 and US3 proteins in disrupting the nuclear lamina during herpes simplex virus type 1 egress. Virology 347 : 261–276. 16427676
21. Mou F, Forest T, Baines JD (2007) US3 of herpes simplex virus type 1 encodes a promiscuous protein kinase that phosphorylates and alters localization of lamin A/C in infected cells. J Virol 81 : 6459–6470. 17428859
22. Granzow H, Klupp BG, Fuchs W, Veits J, Osterrieder N, et al. (2001) Egress of alphaherpesviruses: comparative ultrastructural study. J Virol 75 : 3675–3684. 11264357
23. Mettenleiter TC (2004) Budding events in herpesvirus morphogenesis. Virus Res 106 : 167–180. 15567495
24. Baines JD, Hsieh CE, Wills E, Mannella C, Marko M (2007) Electron tomography of nascent herpes simplex virus virions. J Virol 81 : 2726–2735. 17215293
25. Roller RJ, Bjerke SL, Haugo AC, Hanson S (2010) Analysis of a Charge Cluster Mutation of Herpes Simplex Virus Type 1 UL34 and Its Extragenic Suppressor Suggests a Novel Interaction between pUL34 and pUL31 That Is Necessary for Membrane Curvature around Capsids. Journal of Virology 84 : 3921–3934. doi: 10.1128/JVI.01638-09 20106917
26. Roller RJ, Haugo AC, Kopping NJ (2011) Intragenic and extragenic suppression of a mutation in herpes simplex virus 1 UL34 that affects both nuclear envelope targeting and membrane budding. J Virol 85 : 11615–11625. doi: 10.1128/JVI.05730-11 21900173
27. Kuhn J, Leege T, Klupp BG, Granzow H, Fuchs W, et al. (2008) Partial functional complementation of a pseudorabies virus UL25 deletion mutant by herpes simplex virus type 1 pUL25 indicates overlapping functions of alphaherpesvirus pUL25 proteins. J Virol 82 : 5725–5734. doi: 10.1128/JVI.02441-07 18400859
28. Klupp BG, Granzow H, Fuchs W, Keil GM, Finke S, et al. (2007) Vesicle formation from the nuclear membrane is induced by coexpression of two conserved herpesvirus proteins. Proc Natl Acad Sci U S A 104 : 7241–7246. 17426144
29. Desai PJ, Pryce EN, Henson BW, Luitweiler EM, Cothran J (2012) Reconstitution of the Kaposi's sarcoma-associated herpesvirus nuclear egress complex and formation of nuclear membrane vesicles by coexpression of ORF67 and ORF69 gene products. J Virol 86 : 594–598. doi: 10.1128/JVI.05988-11 22013050
30. Bigalke JM, Heuser T, Nicastro D, Heldwein EE (2014) Membrane deformation and scission by the HSV-1 nuclear egress complex. Nat Commun 5 : 4131. doi: 10.1038/ncomms5131 24916797
31. Lorenz M, Vollmer B, Unsay JD, Klupp BG, Garcia-Saez AJ, et al. (2015) A single herpesvirus protein can mediate vesicle formation in the nuclear envelope. J Biol Chem 290 : 6962–6974. doi: 10.1074/jbc.M114.627521 25605719
32. Purves FC, Longnecker RM, Leader DP, Roizman B (1987) Herpes simplex virus 1 protein kinase is encoded by open reading frame US3 which is not essential for virus growth in cell culture. J Virol 61 : 2896–2901. 3039176
33. Liu Z, Kato A, Shindo K, Noda T, Sagara H, et al. (2014) Herpes Simplex Virus 1 UL47 Interacts with Viral Nuclear Egress factors UL31, UL34 and Us3, and Regulates Viral Nuclear Egress. J Virol 9 : 4657–4667. doi: 10.1128/JVI.00137-14 24522907
34. Maruzuru Y, Shindo K, Liu Z, Oyama M, Kozuka-Hata H, et al. (2014) Role of herpes simplex virus 1 immediate early protein ICP22 in viral nuclear egress. J Virol 88 : 7445–7454. doi: 10.1128/JVI.01057-14 24741100
35. Reynolds AE, Ryckman BJ, Baines JD, Zhou Y, Liang L, et al. (2001) UL31 and UL34 Proteins of Herpes Simplex Virus Type 1 Form a Complex That Accumulates at the Nuclear Rim and Is Required for Envelopment of Nucleocapsids. J Virol 75 : 8803–8817. 11507225
36. Granato M, Feederle R, Farina A, Gonnella R, Santarelli R, et al. (2008) Deletion of Epstein-Barr virus BFLF2 leads to impaired viral DNA packaging and primary egress as well as to the production of defective viral particles. J Virol 82 : 4042–4051. doi: 10.1128/JVI.02436-07 18287246
37. Popa M, Ruzsics Z, Lotzerich M, Dolken L, Buser C, et al. (2010) Dominant negative mutants of the murine cytomegalovirus M53 gene block nuclear egress and inhibit capsid maturation. J Virol 84 : 9035–9046. doi: 10.1128/JVI.00681-10 20610730
38. Yang K, Baines JD (2011) Selection of HSV capsids for envelopment involves interaction between capsid surface components pUL31, pUL17, and pUL25. Proc Natl Acad Sci U S A 108 : 14276–14281. doi: 10.1073/pnas.1108564108 21821792
39. Leelawong M, Guo D, Smith GA (2011) A Physical Link between the Pseudorabies Virus Capsid and the Nuclear Egress Complex. Journal of Virology 85 : 11675–11684. doi: 10.1128/JVI.05614-11 21880751
40. Pogoda M, Bosse JB, Wagner FM, Schauflinger M, Walther P, et al. (2012) Characterization of conserved region 2-deficient mutants of the cytomegalovirus egress protein pM53. J Virol 86 : 12512–12524. doi: 10.1128/JVI.00471-12 22993161
41. Yang K, Wills E, Lim HY, Zhou ZH, Baines JD (2014) Association of Herpes Simplex Virus pUL31 with Capsid Vertices and Components of the Capsid Vertex Specific Complex. J Virol 7 : 3815–3825. doi: 10.1128/JVI.03175-13 24453362
42. Trus BL, Newcomb WW, Cheng N, Cardone G, Marekov L, et al. (2007) Allosteric signaling and a nuclear exit strategy: binding of UL25/UL17 heterodimers to DNA-Filled HSV-1 capsids. Mol Cell 26 : 479–489. 17531807
43. Klupp BG, Granzow H, Keil GM, Mettenleiter TC (2006) The capsid-associated UL25 protein of the alphaherpesvirus pseudorabies virus is nonessential for cleavage and encapsidation of genomic DNA but is required for nuclear egress of capsids. J Virol 80 : 6235–6246. 16775311
44. Kuhn J, Leege T, Granzow H, Fuchs W, Mettenleiter TC, et al. (2010) Analysis of pseudorabies and herpes simplex virus recombinants simultaneously lacking the pUL17 and pUL25 components of the C-capsid specific component. Virus Res 153 : 20–28. doi: 10.1016/j.virusres.2010.06.022 20603164
45. Lotzerich M, Ruzsics Z, Koszinowski UH (2006) Functional domains of murine cytomegalovirus nuclear egress protein M53/p38. J Virol 80 : 73–84. 16352532
46. Milbradt J, Auerochs S, Sevvana M, Muller YA, Sticht H, et al. (2012) Specific residues of a conserved domain in the N terminus of the human cytomegalovirus pUL50 protein determine its intranuclear interaction with pUL53. J Biol Chem 287 : 24004–24016. doi: 10.1074/jbc.M111.331207 22589554
47. Schnee M, Ruzsics Z, Bubeck A, Koszinowski UH (2006) Common and specific properties of herpesvirus UL34/UL31 protein family members revealed by protein complementation assay. J Virol 80 : 11658–11666. 17005637
48. Yamauchi Y, Shiba C, Goshima F, Nawa A, Murata T, et al. (2001) Herpes simplex virus type 2 UL34 protein requires UL31 protein for its relocation to the internal nuclear membrane in transfected cells. Journal of General Virology 82 : 1423–1428. 11369887
49. Liang L, Baines JD (2005) Identification of an essential domain in the herpes simplex virus 1 UL34 protein that is necessary and sufficient to interact with UL31 protein. J Virol 79 : 3797–3806. 15731273
50. Passvogel L, Klupp BG, Granzow H, Fuchs W, Mettenleiter TC (2014) Functional characterization of nuclear trafficking signals in Pseudorabies Virus pUL31. J Virol 4 : 2002–2012.
51. Zhu HY, Yamada H, Jiang YM, Yamada M, Nishiyama Y (1999) Intracellular localization of the UL31 protein of herpes simplex virus type 2. Arch Virol 144 : 1923–1935. 10550666
52. Schmeiser C, Borst E, Sticht H, Marschall M, Milbradt J (2013) The cytomegalovirus egress proteins pUL50 and pUL53 are translocated to the nuclear envelope through two distinct modes of nuclear import. J Gen Virol 94 : 2056–2069. doi: 10.1099/vir.0.052571-0 23740483
53. Schmidt T, Striebinger H, Haas J, Bailer SM (2010) The heterogeneous nuclear ribonucleoprotein K is important for Herpes simplex virus-1 propagation. FEBS Lett 584 : 4361–4365. doi: 10.1016/j.febslet.2010.09.038 20888333
54. Sandbaumhuter M, Dohner K, Schipke J, Binz A, Pohlmann A, et al. (2013) Cytosolic herpes simplex virus capsids not only require binding inner tegument protein pUL36 but also pUL37 for active transport prior to secondary envelopment. Cell Microbiol 15 : 248–269. doi: 10.1111/cmi.12075 23186167
55. Nygardas M, Paavilainen H, Muther N, Nagel CH, Roytta M, et al. (2013) A herpes simplex virus-derived replicative vector expressing LIF limits experimental demyelinating disease and modulates autoimmunity. PLoS One 8: e64200. doi: 10.1371/journal.pone.0064200 23700462
56. Nagel CH, Pohlmann A, Sodeik B (2014) Construction and characterization of bacterial artificial chromosomes (BACs) containing herpes simplex virus full-length genomes. Methods Mol Biol 1144 : 43–62. doi: 10.1007/978-1-4939-0428-0_4 24671676
57. Striebinger H, Koegl M, Bailer SM (2013) A high-throughput yeast two-hybrid protocol to determine virus-host protein interactions. Methods Mol Biol 1064 : 1–15. doi: 10.1007/978-1-62703-601-6_1 23996246
58. Raschbichler V, Lieber D, Bailer SM (2012) NEX-TRAP, a novel method for in vivo analysis of nuclear export of proteins. Traffic 13 : 1326–1334. doi: 10.1111/j.1600-0854.2012.01389.x 22708827
59. Fossum E, Friedel CC, Rajagopala SV, Titz B, Baiker A, et al. (2009) Evolutionarily conserved herpesviral protein interaction networks. PLoS Pathog 5: e1000570. doi: 10.1371/journal.ppat.1000570 19730696
60. Blasche S, Koegl M (2013) Analysis of Protein-Protein Interactions Using LUMIER Assays. Methods Mol Biol 1064 : 17–27. doi: 10.1007/978-1-62703-601-6_2 23996247
61. Schipke J, Pohlmann A, Diestel R, Binz A, Rudolph K, et al. (2012) The C terminus of the large tegument protein pUL36 contains multiple capsid binding sites that function differently during assembly and cell entry of herpes simplex virus. J Virol 86 : 3682–3700. doi: 10.1128/JVI.06432-11 22258258
62. Damelin M, Silver PA, Corbett AH (2002) Nuclear protein transport. Methods Enzymol 351 : 587–607. 12073370
63. Salsman J, Zimmerman N, Chen T, Domagala M, Frappier L (2008) Genome-wide screen of three herpesviruses for protein subcellular localization and alteration of PML nuclear bodies. PLoS Pathog 4: e1000100. doi: 10.1371/journal.ppat.1000100 18617993
64. Xing J, Wang S, Li Y, Guo H, Zhao L, et al. (2011) Characterization of the subcellular localization of herpes simplex virus type 1 proteins in living cells. Med Microbiol Immunol 200 : 61–68. doi: 10.1007/s00430-010-0175-9 20949280
65. Antonin W, Ungricht R, Kutay U (2011) Traversing the NPC along the pore membrane: targeting of membrane proteins to the INM. Nucleus 2 : 87–91. doi: 10.4161/nucl.2.2.14637 21738830
66. Ye G - J, Roizman B (2000) The essential protein encoded by the UL31 gene of herpes simplex virus 1 depends for its stability on the presence of UL34 protein. Proceedings of the National Academy of Sciences 97 : 11002–11007. 11005871
67. Liang L, Tanaka M, Kawaguchi Y, Baines JD (2004) Cell lines that support replication of a novel herpes simplex virus 1 UL31 deletion mutant can properly target UL34 protein to the nuclear rim in the absence of UL31. Virology 329 : 68–76. 15476875
68. Chang YE, Poon AP, Roizman B (1996) Properties of the protein encoded by the UL32 open reading frame of herpes simplex virus 1. J Virol 70 : 3938–3946. 8648731
69. Nagel CH, Dohner K, Fathollahy M, Strive T, Borst EM, et al. (2008) Nuclear egress and envelopment of herpes simplex virus capsids analyzed with dual-color fluorescence HSV1(17+). J Virol 82 : 3109–3124. 18160444
70. Trus BL, Newcomb WW, Booy FP, Brown JC, Steven AC (1992) Distinct monoclonal antibodies separately label the hexons or the pentons of herpes simplex virus capsid. Proc Natl Acad Sci U S A 89 : 11508–11512. 1280828
71. Scholtes L, Baines JD (2009) Effects of major capsid proteins, capsid assembly, and DNA cleavage/packaging on the pUL17/pUL25 complex of herpes simplex virus 1. J Virol 83 : 12725–12737. doi: 10.1128/JVI.01658-09 19812148
72. Henaff D, Remillard-Labrosse G, Loret S, Lippe R (2013) Analysis of the early steps of herpes simplex virus 1 capsid tegumentation. J Virol 87 : 4895–4906. doi: 10.1128/JVI.03292-12 23408623
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 6
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- HIV Latency Is Established Directly and Early in Both Resting and Activated Primary CD4 T Cells
- Battling Phages: How Bacteria Defend against Viral Attack
- A 21st Century Perspective of Poliovirus Replication
- Adenovirus Tales: From the Cell Surface to the Nuclear Pore Complex
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy