
Analysis of Bottlenecks in Experimental Models of Infection
article has not abstract
Published in the journal:
. PLoS Pathog 11(6): e32767. doi:10.1371/journal.ppat.1004823
Category:
Pearls
doi:
https://doi.org/10.1371/journal.ppat.1004823
Summary
article has not abstract
What Are Bottlenecks?
The metaphor of a bottleneck has been used in a variety of fields to describe the critical constraints that limit a system’s performance or capacity. In biology, particularly in studies of population dynamics and evolution, the bottleneck concept is often used in reference to events that sharply limit population size [1]. Such events frequently produce stochastic changes in the genetic composition of a population (Fig 1), referred to as genetic drift. In extreme cases, population-reducing events can eliminate genotypes from a gene pool (Fig 1), even genotypes not associated with reduced fitness.
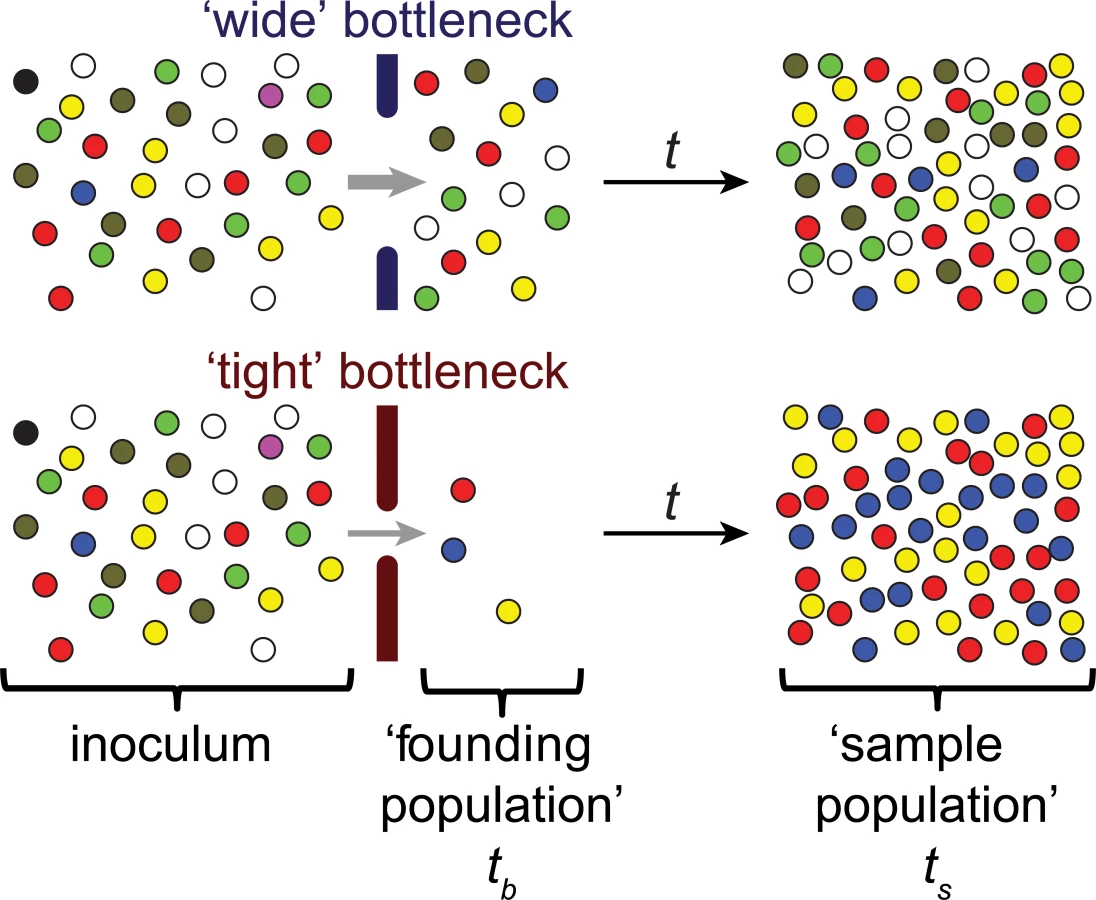
If the limited number of these surviving organisms found a population in a new environment, such as the colonization or infection of a host by microorganisms, those few organisms determine the genetic composition of subsequent generations, creating a “founder effect” within the post-bottleneck population [1]. These changes in genotype frequency are an important driver of evolutionary change and speciation. In infection biology, bottlenecks shape genetic diversity of epidemics and have been shown to have an important influence on the effect of recombination and horizontal gene transfer, as well as the evolution of drug resistance [2–6]. Furthermore, bottlenecks may reduce pathogen virulence and adaptability to new hosts, as they increase the rate at which attenuating mutations become fixed in a population [7,8].
Traditionally, population biologists have taken advantage of natural genetic variation to investigate transmission bottlenecks, e.g., during HIV transmission (reviewed in [9]). In addition, with pathogens that contain or accumulate high amounts of genetic diversity over a small timescale, such as HIV, it is possible to investigate bottlenecks within a single host [5]. However, many pathogens do not possess sufficient natural genetic variation for quantification of bottlenecks in this setting. Measuring bottleneck sizes in animal models allows experimental access to valuable information regarding the anatomical sites, sizes, and causes of population restrictions, which can provide key insights into the nature of host–pathogen interactions. Here, we focus on recent new methods that rely on introduction of artificial genetic variation to quantify bottleneck events during experimental infection, enabling more precise understanding of pathogen population dynamics.
How Are Bottlenecks in Experimental Infections Measured?
In principle, the size of an infection bottleneck can be measured by simply counting the number of organisms (e.g., viral particles, bacterial colony forming units [CFU]) that survive a population-restriction process. However, enumeration of these organisms (sometimes referred to as the founding population) must occur prior to changes in their number, e.g., by replication or migration (Fig 1), and great experimental efforts and a large number of experimental animals are often required to pinpoint the sites and times of population constrictions [10]. Furthermore, when there is complex migration of the pathogen within the host, rather than a linear infection “pathway,” direct counting of individual infectious agents may not be feasible.
An alternative way to measure bottleneck sizes makes use of the stochastic changes in the genetic composition (diversity) of the infecting population that usually accompany population size reductions (Fig 1). This approach is employed in studies of natural populations, including certain viral pathogens, in which there is substantial genetic diversity [11–13]. However, for experimental studies of infection, often there is insufficient natural diversity in the inoculum for meaningful analyses. To circumvent this limitation, genetic variation has been introduced artificially into pathogen populations. Using inheritable, distinguishable markers that ideally do not alter pathogen fitness, changes in marker prevalence between the inoculum (i.e., the population prior to reduction by bottlenecks) and experimental samples can be used to estimate bottleneck sizes. Different genetic markers that have been used for this purpose include antibiotic resistances, lacZ+/lacZ-, fluorescent proteins, transposon insertions, restriction sites, and sequence tags [14–27]. The number of distinct markers is a major factor that limits the resolution of these assays, with a greater number of markers enabling greater resolution. When bottlenecks are very small, the assay limits of resolution are less of an issue; however, when there is a wide bottleneck, the size of the founding population cannot be accurately determined using a inoculum with low marker diversity [27]. Insertion of short DNA sequences into neutral loci in the genome, thereby generating wild-type isogenic tagged strains (WITS) [17], allows for easy creation of a large number of distinguishable strains. Sequence tags have been detected by a variety of methods, including hybridization, PCR, and most recently, DNA sequencing [25–27]. The availability of relatively inexpensive high-throughput sequencing that enables accurate quantification of a large number of sequence tags makes sequencing the current best approach to measure bottleneck sizes.
Several analytic approaches have been developed to identify bottleneck sizes based on differences in marker representation at two time points. These approaches include (i) probabilistic methods that analyze the stochastic loss of tagged strains [15,17,19,23,25], (ii) mathematical modeling [21,22], and (iii) population genetic approaches [18,27]. Analyses based on the presence or absence of individual marked strains are the most commonly performed because many experimental techniques can provide qualitative data about marker presence or absence. The major drawback of these analyses is their limited resolving power because the maximum bottleneck size that can be measured strongly depends on the number of distinguishable markers in the infecting population. Furthermore, since this approach is based on a stochastic model, relatively large numbers of repetitions are required for accurate measurements, usually necessitating the use of many experimental animals [25]. When more detailed knowledge about a pathogen’s behavior during infection is available, e.g., migration pattern in the host, mathematical models that explicitly describe this behavior can be used to estimate pathogen population size as well as the speed of migration between compartments [21]. These complex models can provide high-quality estimations but are dependent on knowledge of parameters that may not be available for less well-characterized systems. Finally, approaches that are based on population genetic theory [28,29] can be applied to estimate bottleneck sizes without knowledge of pathogen spatiotemporal dynamics within the host. These methods infer the bottleneck size by comparing allele frequencies (rather than just absence or presence of an allele) before and after bottleneck events (Fig 1). Two approaches can be distinguished: (i) those requiring data from several experiments, which yield the average bottleneck size across a series of hosts and are unable to distinguish between technical and biological variation between experiments [18,22], and (ii) those allowing bottleneck size determination in a single experiment with high accuracy [27]. The latter, recently developed method requires the use of a large number of tagged strains, but it allows the comparison of bottlenecks between individual hosts and, thereby, makes it possible to analyze the biological variance between hosts.
What Are the Factors and/or Mechanisms That Restrict Pathogen Populations during Infections?
The nature of host bottlenecks, as well as pathogens’ strategies for circumventing them, continues to be a central topic in studies of host–pathogen interactions. All host defenses that counteract infection can be thought of as bottlenecks, as can some intrinsic features of the host environment. Impediments to infection can include physical barriers, innate and adaptive immune defenses, nutritional limitations, competing microorganisms, and niche availability (environments permissive for colonization and replication). The presence and mode of action of some barriers, such as the size of the pore in the squid light organ, low stomach pH, antimicrobial peptides, and low iron availability, have long been known. However, much recent and current research is devoted to uncovering new mechanisms of innate defense. Processes whereby the commensal microbiota restrict pathogen populations (e.g., competition for nutrients or colonization sites, or modulation of host immune development) are also only beginning to come into focus. Investigations of the specific mechanisms that mediate interbacterial competition and/or killing, such as Type VI secretion, will shed light on novel types of bottlenecks.
On a more abstract level, host restrictions can either be toxic to pathogens or merely reflect limited resources that can only sustain a finite number of organisms. These different types of restriction will lead to different effects on the pathogen population. When there are finite resources available, there is an “absolute” limit to the size of the bottleneck; i.e., the bottleneck size is independent of the inoculum size, resulting in an upper limit to the size of the founding population (Fig 2A). In contrast, toxic host defenses can result in a “fractional” bottleneck, such that a proportion, rather than number, of the organisms survive (Fig 2B). For example, if a fraction of the inoculum is phenotypically resistant to the restricting conditions, then a definable percentage of the inoculum may survive. Alternatively, it is possible that host defenses can control a limited number of pathogens. In this “limited” bottleneck scenario, a high number of infecting organisms can exhaust host defenses, so that excess pathogens (above the saturating level) survive (Fig 2C). For fractional and limited bottlenecks, there is no upper limit to the size of the founding population; however, there is a lower limit to the inoculum size for establishment of infection. In addition, more complex biological mechanisms, e.g., quorum-sensing-regulated virulence [30,31], can result in complex inoculum size to founding population size relationships (Fig 2D). In reality, bottlenecks likely reflect a combination of such mechanisms (Fig 2E).

Bottlenecks Can Confound Genome-Wide Analyses of Virulence and Transmission Studies
Bottlenecks can be a major technical challenge for the analysis of high-throughput (e.g., transposon-insertion sequencing) studies of genes required for infection. For example, genome-wide transposon insertion studies can be confounded by the existence of tight bottlenecks, which can limit the complexity of libraries that can effectively be analyzed. Moreover, when stochastic loss of transposon mutants due to bottlenecks overshadows the effects of fitness defects, many false positive classifications result [32]. Genetic drift that results from bottleneck events can also lead to pathogen populations that are very different between primary and secondary host. Such random differences may severely impact the accuracy and reliability of transmission chains constructed based on the genetic similarity between pathogen populations in different hosts [33]. Therefore, it is critical for high-throughput studies to account for bottleneck effects.
Intriguing Questions That Can Be Addressed Using New Approaches for Measuring Bottlenecks
Are bottleneck sizes constant during infection? Our recent work revealed that the size of the Vibrio cholerae founding population in the intestines of infected rabbits changes as infection progresses [27]. It will be fascinating to investigate the dynamics of the processes by which pathogens counteract host restrictions during infection.
How and by what mechanisms does the composition of the microbiota (or co-infecting pathogens) modulate bottleneck sizes?
Do virulence factors enable pathogens to overcome bottlenecks?
Can we take advantage of bottlenecks to design improved regimens for antimicrobial administration or vaccination that limit emergence of resistance and have enhanced therapeutic efficacy?
Overall, the development of new tools to analyze infection bottlenecks creates a new way to understand host–pathogen interactions during infection.
Zdroje
1. Reece JB (2014) Campbell biology. Tenth edition. Boston: Pearson.
2. Levin BR (1981) Periodic selection, infectious gene exchange and the genetic structure of E. coli populations. Genetics 99 : 1–23. 7042453
3. Levin BR, Perrot V, Walker N (2000) Compensatory mutations, antibiotic resistance and the population genetics of adaptive evolution in bacteria. Genetics 154 : 985–997. 10757748
4. Grenfell BT, Pybus OG, Gog JR, Wood JLN, Daly JM, et al. (2004) Unifying the epidemiological and evolutionary dynamics of pathogens. Science 303 : 327–332. 14726583
5. Kouyos RD, Althaus CL, Bonhoeffer S (2006) Stochastic or deterministic: what is the effective population size of HIV-1? Trends Microbiol 14 : 507–511. 17049239
6. Kouyos RD, Silander OK, Bonhoeffer S (2007) Epistasis between deleterious mutations and the evolution of recombination. Trends Ecol Evol 22 : 308–315. 17337087
7. Ebert D (1998) Experimental evolution of parasites. Science 282 : 1432–1435. 9822369
8. Yuste E, Sánchez-Palomino S, Casado C, Domingo E, López-Galíndez C (1999) Drastic fitness loss in human immunodeficiency virus type 1 upon serial bottleneck events. J Virol 73 : 2745–2751. 10074121
9. Shaw GM, Hunter E (2012) HIV transmission. Cold Spring Harb Perspect Med 2.
10. Angelichio MJ, Spector J, Waldor MK, Camilli A (1999) Vibrio cholerae intestinal population dynamics in the suckling mouse model of infection. Infect Immun 67 : 3733–3739. 10417131
11. Edwards CTT, Holmes EC, Wilson DJ, Viscidi RP, Abrams EJ, et al. (2006) Population genetic estimation of the loss of genetic diversity during horizontal transmission of HIV-1. BMC Evol Biol 6 : 28. 16556318
12. Derdeyn CA, Decker JM, Bibollet-Ruche F, Mokili JL, Muldoon M, et al. (2004) Envelope-constrained neutralization-sensitive HIV-1 after heterosexual transmission. Science 303 : 2019–2022. doi: 10.1126/science.1093137 15044802
13. Wang GP, Sherrill-Mix SA, Chang K - M, Quince C, Bushman FD (2010) Hepatitis C virus transmission bottlenecks analyzed by deep sequencing. J Virol 84 : 6218–6228. doi: 10.1128/JVI.02271-09 20375170
14. Moxon ER, Murphy PA (1978) Haemophilus influenzae bacteremia and meningitis resulting from survival of a single organism. Proc Natl Acad Sci U S A 75 : 1534–1536. 306628
15. Barnes PD, Bergman MA, Mecsas J, Isberg RR (2006) Yersinia pseudotuberculosis disseminates directly from a replicating bacterial pool in the intestine. J Exp Med 203 : 1591–1601. 16754724
16. Pfeiffer JK, Kirkegaard K (2006) Bottleneck-mediated quasispecies restriction during spread of an RNA virus from inoculation site to brain. Proc Natl Acad Sci U S A 103 : 5520–5525. 16567621
17. Grant AJ, Restif O, McKinley TJ, Sheppard M, Maskell DJ, et al. (2008) Modelling within-host spatiotemporal dynamics of invasive bacterial disease. PLoS Biol 6: e74. doi: 10.1371/journal.pbio.0060074 18399718
18. Monsion B, Froissart R, Michalakis Y, Blanc S (2008) Large bottleneck size in Cauliflower Mosaic Virus populations during host plant colonization. PLoS Pathog 4: e1000174. doi: 10.1371/journal.ppat.1000174 18846207
19. Schwartz DJ, Chen SL, Hultgren SJ, Seed PC (2011) Population dynamics and niche distribution of uropathogenic Escherichia coli during acute and chronic urinary tract infection. Infect Immun 79 : 4250–4259. doi: 10.1128/IAI.05339-11 21807904
20. Fu Y, Waldor MK, Mekalanos JJ (2013) Tn-Seq analysis of Vibrio cholerae intestinal colonization reveals a role for T6SS-mediated antibacterial activity in the host. Cell Host Microbe 14 : 652–663. doi: 10.1016/j.chom.2013.11.001 24331463
21. Kaiser P, Slack E, Grant AJ, Hardt W - D, Regoes RR (2013) Lymph Node Colonization Dynamics after Oral Salmonella Typhimurium Infection in Mice. PLoS Pathog 9: e1003532.
22. Li Y, Thompson CM, Trzciński K, Lipsitch M (2013) Within-host selection is limited by an effective population of Streptococcus pneumoniae during nasopharyngeal colonization. Infect Immun 81 : 4534–4543. doi: 10.1128/IAI.00527-13 24082074
23. Troy EB, Lin T, Gao L, Lazinski DW, Camilli A, et al. (2013) Understanding barriers to Borrelia burgdorferi dissemination during infection using massively parallel sequencing. Infect Immun 81 : 2347–2357. doi: 10.1128/IAI.00266-13 23608706
24. Lam LH, Monack DM (2014) Intraspecies Competition for Niches in the Distal Gut Dictate Transmission during Persistent Salmonella Infection. PLoS Pathog 10: e1004527. doi: 10.1371/journal.ppat.1004527 25474319
25. Lim CH, Voedisch S, Wahl B, Rouf SF, Geffers R, et al. (2014) Independent bottlenecks characterize colonization of systemic compartments and gut lymphoid tissue by salmonella. PLoS Pathog 10: e1004270. doi: 10.1371/journal.ppat.1004270 25079958
26. Varble A, Albrecht RA, Backes S, Crumiller M, Bouvier NM, et al. (2014) Influenza a virus transmission bottlenecks are defined by infection route and recipient host. Cell Host Microbe 16 : 691–700. doi: 10.1016/j.chom.2014.09.020 25456074
27. Abel S, Abel Zur Wiesch P, Chang H-H, Davis BM, Lipsitch M, et al. (2015) Sequence tag-based analysis of microbial population dynamics. Nat Methods 12 : 223–226. doi: 10.1038/nmeth.3253 25599549
28. Wright S (1938) Size of population and breeding structure in relation to evolution. Science 87 : 430–431.
29. Krimbas CB, Tsakas S (1971) The Genetics of Dacus oleae. V. Changes of Esterase Polymorphism in a Natural Population Following Insecticide Control-Selection or Drift? Evolution 25 : 454–460.
30. De Kievit TR, Iglewski BH (2000) Bacterial Quorum Sensing in Pathogenic Relationships. Infect Immun 68 : 4839–4849. 10948095
31. Parsek MR, Greenberg EP (2000) Acyl-homoserine lactone quorum sensing in Gram-negative bacteria: A signaling mechanism involved in associations with higher organisms. Proc Natl Acad Sci 97 : 8789–8793. 10922036
32. Pritchard JR, Chao MC, Abel S, Davis BM, Baranowski C, et al. (2014) ARTIST: High-Resolution Genome-Wide Assessment of Fitness Using Transposon-Insertion Sequencing. PLoS Genet 10: e1004782. doi: 10.1371/journal.pgen.1004782 25375795
33. Worby CJ, Lipsitch M, Hanage WP (2014) Within-host bacterial diversity hinders accurate reconstruction of transmission networks from genomic distance data. PLoS Comput Biol 10: e1003549. doi: 10.1371/journal.pcbi.1003549 24675511
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 6
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- HIV Latency Is Established Directly and Early in Both Resting and Activated Primary CD4 T Cells
- Battling Phages: How Bacteria Defend against Viral Attack
- A 21st Century Perspective of Poliovirus Replication
- Adenovirus Tales: From the Cell Surface to the Nuclear Pore Complex
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy