
The Timing of Stimulation and IL-2 Signaling Regulate Secondary CD8 T Cell Responses
Since memory CD8 T cells afford hosts increased protection, extensive research has been devoted to understanding the parameters that affect the generation of these cells. Humans are typically infected with pathogens more than once, thus leading to re-stimulation of existing primary memory CD8 T cell populations. The factors influencing the development of CD8 T cells responding to repetitive antigen stimulations remain unknown. We demonstrate that the time at which primary memory CD8 T cells encounter antigen and are re-stimulated during infection influences the outcome of a secondary pathogen-specific CD8 T cell response. We show that at the time of antigen re-encounter, interleukin-2 cytokine signals received by developing secondary CD8 T cells impact the rate of acquiring secondary memory CD8 T cell characteristics. These data indicate that secondary memory CD8 T cell generation is a process that can be manipulated, which may have implications in the development of consecutive prime-boost immunization strategies.
Published in the journal:
. PLoS Pathog 11(10): e32767. doi:10.1371/journal.ppat.1005199
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1005199
Summary
Since memory CD8 T cells afford hosts increased protection, extensive research has been devoted to understanding the parameters that affect the generation of these cells. Humans are typically infected with pathogens more than once, thus leading to re-stimulation of existing primary memory CD8 T cell populations. The factors influencing the development of CD8 T cells responding to repetitive antigen stimulations remain unknown. We demonstrate that the time at which primary memory CD8 T cells encounter antigen and are re-stimulated during infection influences the outcome of a secondary pathogen-specific CD8 T cell response. We show that at the time of antigen re-encounter, interleukin-2 cytokine signals received by developing secondary CD8 T cells impact the rate of acquiring secondary memory CD8 T cell characteristics. These data indicate that secondary memory CD8 T cell generation is a process that can be manipulated, which may have implications in the development of consecutive prime-boost immunization strategies.
Introduction
Memory CD8 T cells are an important component of the adaptive immune response because of their ability to establish long-lasting protective immunity against recurrent infections [1–6]. Memory CD8 T cells are derived from naïve Ag-specific CD8 T cells that responded to pathogen-derived Ags, underwent robust proliferative expansion, and survived the contraction phase [7,8]. The protection afforded by memory CD8 T cells is due to persistence at higher numbers, unique trafficking abilities and localization in peripheral tissues, and rapid initiation of effector functions after Ag re-encounter [1,9,10]. These characteristics of primary memory (1° M) CD8 T cells distinguish them from the naïve CD8 T cells they are derived from.
Research devoted to understanding the development of memory CD8 T cells suggests that the generation of 1° M CD8 T cells is influenced by a number of factors [2,11–13]. For instance, studies have shown that the number of 1° M CD8 T cells generated correlates with the number of accumulated 1° effector CD8 T cells at the peak of expansion [14,15]. Therefore, parameters influencing 1° effector CD8 T cell expansion and/or survival (e.g. Ag presentation, co-stimulation, and signal 3 cytokines) impact the generation of 1° M CD8 T cells [2,10–12]. Interestingly, these factors have also been shown to influence the rate of 1° M CD8 T cell differentiation [10,11,16]. As an example, naïve CD8 T cells activated in a low-inflammatory environment (e.g. peptide-coated DC vaccination) undergo reduced levels of proliferative expansion but acquire long-term memory characteristics at an accelerated rate [17–19]. Additionally, the modulation of functional Ag presentation (e.g. antibiotic treatment to stop bacterial infection) also impacts the transition of Ag-specific CD8 T cells from effector to memory cells [16,20,21]. Furthermore, naive CD8 T cells activated in the presence of pre-existing memory CD8 T cells of an unrelated Ag specificity acquire memory characteristics at an accelerated rate [22]. Finally, recruitment of naïve Ag-specific CD8 T cells over time into an immune response influences memory CD8 T cell differentiation based on when cognate Ag is encountered [23,24]. This suggests that the process of naïve to 1° M CD8 T cell differentiation is not fixed and that the progression to memory can be manipulated.
Studies have shown that the generation of large numbers of memory CD8 T cells enhances CD8 T cell-mediated protection to re-infection. An effective strategy to increase the absolute numbers of memory CD8 T cells is through prime-boost vaccinations that elicit 2° immune responses [25,26]. Recent studies from our lab have shown that repeated stimulations of Ag-specific CD8 T cells results in differential regulation of a large number of genes in subsequent populations of memory CD8 T cells [27]. Interestingly, similar to 1° M CD8 T cell responses, 2° M CD8 T cell numbers and phenotype are modulated by the type and duration of infection and levels of inflammation [28,29]. However, it has also been previously documented that 2° M CD8 T cells are slower to acquire a long-term memory phenotype compared to 1° M CD8 T cells [30].
Since the timing of stimulation of naïve CD8 T cells has been shown to influence 1° M CD8 T cell differentiation, we devised a model in which 1° M CD8 T cells are recruited at different times into the response relative to the initiation of infection to determine if the timing of recruitment influences the development of 2° CD8 T cell responses. IL-2 signaling impacts the accumulation and differentiation of 1° effector CD8 T cells [31,32], thus we addressed whether the timing of stimulation of 1° M CD8 T cells modulates sensitivity to IL-2 signaling, thereby affecting 2° expansion and 2° CD8 T cell differentiation. By regulating IL-2 signaling in developing 2° CD8 T cells, either by enhancing signaling with stimulatory IL-2 complexes or blocking IL-2 with a neutralizing antibody, we provided direct evidence of the contribution of this signaling mechanism in controlling the generation of 2° CD8 T cell responses. Overall, these data suggest that the process of 1° M to 2° M CD8 T cell generation and differentiation can be manipulated, which may have implications in the development of consecutive prime-boost immunization strategies.
Results
1° M CD8 T cells are not recruited simultaneously after infection
The differentiation of 1° M CD8 T cells has been extensively studied, however much less is known about the factors that influence 2° M CD8 T cell generation. Previous studies have shown that the time at which naïve CD8 T cells recognize Ag during an immune response impacts the rate of acquiring 1° M CD8 T cell characteristics [23,24]. Therefore, we wanted to determine if the timing of stimulation of 1° M CD8 T cells was a factor impacting the generation of 2° M CD8 T cell responses.
First, we asked if recruitment of 1° M CD8 T cells occurs simultaneously or over extended period of time after infection. To test this, LCMV-specific CFSE-labeled TCR-Tg 1° M P14 CD8 T cells (2x106 cells/recipient; Thy1.1) [28,29] were transferred into naïve C57BL/6 (B6; Thy1.2/1.2) recipient mice that were either left uninfected or infected with the Armstrong strain of LCMV (Fig 1A, experimental design). The expression of CD25 and CD69, molecules known to be rapidly unregulated on activated CD8 T cells, was determined on responding 1° M P14 CD8 T cells. By 24 hours p.i., a fraction of 1° M CD8 T cells were found to be activated in the spleen, as determined by the upregulation of CD25 and CD69 (Fig 1B and 1C). By 48 hours p.i., the percentage of 1° M CD8 T cells that had responded to the infection and expressed these markers had increased; however, most of these responding cells were not undergoing division, as they still expressed high levels of CFSE. Interestingly, at this time there were still 1° M CD8 T cells that had not upregulated either activation marker, suggesting that similar to naïve Ag-specific CD8 T cells [23], 1° M CD8 T cells are not recruited simultaneously after infection.

To understand how the time at which 1° M CD8 T cell recognize Ag in an immune response might impact the differentiation of 2° CD8 T cell responses, we modified a model previously used to determine how time of the recruitment of naïve CD8 T cells influences their development into a 1° M CD8 T cell pool [23,24]. In preliminary experiments using our modified approach, we wanted to first confirm that naïve CD8 T cells recognizing Ag late in an immune response rapidly acquire 1° M characteristics. Physiologically low number of naïve P14 CD8 T cells (5x103 cells/recipient; Thy1.1 [33]) were adoptively transferred into naïve B6 (Thy1.2/1.2) recipient mice on the same day (‘early’ group) or 3 days (‘late’ group) after LCMV infection (S1A Fig, experimental design). Staggering infections in this adoptive transfer model enabled us to track the differentiation of 1° Ag-specific CD8 T cell responses in both groups of mice at the same time relative to the day of transfer. In the ‘late’ group, Ag-specific CD8 T cells are prevented from early priming because they are transferred into mice 3 days after infection. As seen previously, naïve CD8 T cells that recognized Ag late in the immune response underwent reduced levels of proliferative expansion (S1B Fig) [23]. However, despite limited expansion and persistence at lower numbers over time (S1C and S1E Fig), these Ag-specific CD8 T cells stimulated late displayed a memory phenotype at an accelerated rate ((increase in CD27 and CD62L and decrease in KLRG1) S1D and S1F Fig). Consistent with the notion that late-stimulation in the immune response does not influence rates of contraction [23], we found that relative to the respective peak of expansion of activated CD8 T cells in ‘early’ and ‘late’ groups, similar proportions of 1° effector CD8 T cells survived the contraction phase (S1G Fig). Similar results were obtained using naive TCR-Tg OT-I CD8 T cells and L. monocytogenes infection (S1H Fig).
The magnitude of proliferative expansion and transcriptional programming of 2° effector CD8 T cells is impacted by the timing of stimulation of 1° M CD8 T cells
Next we explored the extent to which timing of stimulation influenced the development of 2° CD8 T cell responses. To test this, 1° M P14 CD8 T cells (2x104 cells/recipient; Thy1.1) [28,29] were transferred into naïve B6 (Thy1.2/1.2) recipient mice on the same day (‘early’ group) or 3 days after (‘late’ group) LCMV infection (Fig 2A, experimental design). Examination of P14 CD8 T cells in the blood day 7 after transfer revealed that the magnitude of 2° expansion was significantly decreased in mice in the ‘late’ group (Fig 2B). This suggests that the time at which 1° M CD8 T cells encounter Ag in an immune response impacts the accumulation of 2° effector CD8 T cells.
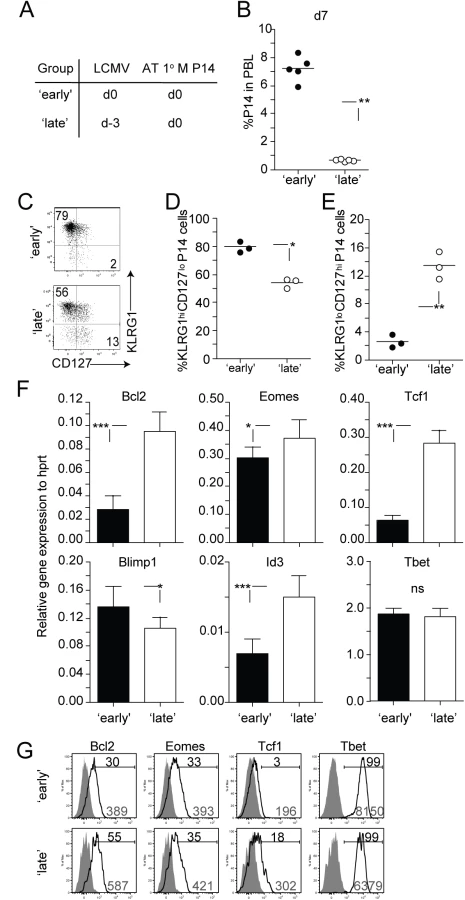
After 1° infection with intracellular pathogens such as LCMV, subsets of differentiating 1° effector CD8 T cells can be distinguished based on the expression of phenotypic markers like KLRG1 and CD127. For example, CD8 T cells exhibiting a KLRG1low CD127hi phenotype at the peak of a 1° anti-LCMV immune response have increased potential to populate the memory CD8 T cell pool [34–36]. Additionally, studies have shown that transcription factors, that play a crucial role in the differentiation of 1° CD8 T cell responses [37,38], are differentially regulated in Ag-specific CD8 T cells based on signals received at early stages of activation. Interestingly, we found that a greater percentage of 2° effector CD8 T cells within the ‘late’ group expressed a KLRG1low CD127hi phenotype (Fig 2C–2E). The mRNA expression of transcription and pro-survival factors within these 2° effector CD8 T cells in mice within the ‘late’ group correlated with this phenotype. Expression of Eomes, Tcf1, and Id3, transcription factors associated with memory formation was significantly increased in 2° effector CD8 T cells derived from the ‘late’ group (Fig 2F) [35,39–42]. Conversely, the expression of Blimp1, known to promote terminal differentiation of CD8 T cells [43], was significantly decreased in these 2° effector CD8 T cells. Finally, expression of the pro-survival factor Bcl2 was increased in CD8 T cells recruited late into the response (Fig 2F). The protein expression of some (Bcl2, Tcf1, and Tbet) but not all (Eomes) transcription and pro-survival factors correlated with levels of mRNA on 2° effector CD8 T cells in ‘early’ and ‘late’ groups, (Fig 2G). Our data suggests that the timing of stimulation of 1° M CD8 T cells influences the phenotypic and transcriptional programs of the developing 2° effector CD8 T cell pool.
1° M CD8 T cells stimulated late in the immune response progress to a long-term 2° M phenotype at an accelerated rate
Although the differentiation state of effector CD8 T cells at the peak of an immune response provides insight on the ability of these cells to populate the memory pool, the generation and maintenance of memory CD8 T cell responses is a time-dependent process. Furthermore, with each additional Ag encounter the rate at which populations of activated CD8 T cells acquire memory characteristics slows substantially [2,44–47]. To extend our observation that the timing of stimulation of 1° M CD8 T cells influences the phenotypic programming of 2° CD8 T cell responses, we determined the fate of 2° M CD8 T cells in ‘early’ and ‘late’ groups at a memory time-point (day 200 after transfer). The number of 2° M CD8 T cells in both groups of mice reflected the initial degree of 2° expansion, as 2° M CD8 T cells persisted at lower frequencies in the PBL of mice in the ‘late’ group (Fig 3A). However, late-entry of 1° M CD8 T cells in an immune response facilitated faster re-expression of the molecules CD27, CD62L and KLRG-1 during 2° M CD8 T cell development (Figs 3B and 3C and S2). To determine the generality of this data, we also employed a model of systemic bacterial infection. 1° M OT-I CD8 T cells (4x104 cells/recipient; Thy1.1) were transferred into naïve B6 (Thy1.2) recipient mice on the same day (‘early’ group) or 3 days after (‘late’ group) infection with attenuated Ova-expressing L. monocytogenes (Att LM-Ova) (S3A Fig, experimental design). Similar to the findings observed with P14 TCR-Tg CD8 T cells and LCMV infection, we found that the timing of stimulation of 1° M OT-I CD8 T cells impacted 2° expansion, numbers of generated 2° M CD8 T cells, and rate of expression of 2° M phenotypic markers (S3B–S3D Fig).

Taken together, these data suggest that the time at which 1° M CD8 T cells recognize Ag during an immune response impacts the rate at which long-term 2° M characteristics are acquired.
The timing of stimulation modulates the function of 2° M CD8 T cells
To address whether the timing of stimulation of 1° M CD8 T cells impacts the function of 2° M CD8 T cells, the tissue distribution of 2° M P14 CD8 T cells in 2° lymphoid organs and tertiary (3°) tissues of mice in ‘early’ and ‘late’ groups 200 days after transfer was first determined. There was a statistically significant decrease in the number of 2° M P14 CD8 T cells recovered from the PBL, spleen, and lung of mice in the ‘late’ group (Fig 4A). In contrast, increased levels of CD62L expression (Fig 3C) allowed 2° M CD8 T cells in the ‘late’ group to preferentially home to the LN (Fig 4A), suggesting that the timing of stimulation impacts the localization of 2° M CD8 T cells. In addition to changes in tissue distribution, the function of 2° M P14 CD8 T cells in ‘early’ and ‘late’ groups was evaluated after ex vivo peptide stimulation. A similar percentage of 2° M CD8 T cells from both groups of mice produced IFNγ (Fig 4B), however 2° M CD8 T cells in the ‘late’ group had a significantly increased ability to co-produce IL-2 (Fig 4C). Given that the timing of stimulation was found to modulate a rapid polyfunctional cytokine response after peptide stimulation, a defining characteristic of memory, we were also interested in determining whether Ag-driven 3° expansion was also modulated. To test this on a per-cell basis, equal numbers (1.5x104 cells/recipient; Thy1.1) of 2° M P14 CD8 T cells from ‘early’ and ‘late’ groups were transferred into new naïve B6 Th1.2 hosts. One day after transfer, both groups of recipient mice were infected with an attenuated strain of L. monocytogenes expressing the LCMV-derived GP33 epitope (Att LM-GP33) (Fig 4D and 4E). Ag-driven 3° accumulation of P14 cells was significantly increased in the PBL of mice that received memory CD8 T cells from the ‘late’ group (Fig 4F and 4G), suggesting that the timing of stimulation also modulates their ability to undergo proliferative expansion in numbers upon additional Ag-encounter.

Taken together, these data show that 1° M CD8 T cells that recognized Ag late during a 2° immune response have an increased capacity to traffic to the LN, produce IL-2, and undergo robust proliferative expansion after Ag re-encounter.
The timing of stimulation of 1° M CD8 T cells regulates IL-2 sensitivity and expression of cell-cycle related proteins
It is well established that early inflammatory signals received by activated CD8 T cells shape subsequent phases of the CD8 T cell response. For example, in response to signal 3 cytokines, either IL-12 or type I IFNs, activated CD8 T cells maintain high-affinity IL-2 signaling in vivo which results in continued expression of cell-cycle associated genes and extended cellular division [48]. In addition to driving optimal accumulation of activated CD8 T cells, IL-2 signaling has also been shown to regulate 1° M CD8 T cell differentiation. Based on the expression of the high-affinity IL-2 receptor α chain (CD25), activated CD8 T cells either favor terminal differentiation in response to strong IL-2 signaling or have an increased potential to become 1° M cells as a result of lower CD25 expression and diminished sensitivity to IL-2 signals [31,32].
We were interested in determining if 1° M CD8 T cells stimulated early or late in the immune response receive signals at the initial expansion phase that initiate unique programs of 2° M CD8 T cell differentiation. Therefore, we determined the extent to which the timing of stimulation of 1° M CD8 T cells affects the duration of expression of CD25 on 2° effector CD8 T cells (Fig 5A). Although an increased frequency of 2° effector P14 CD8 T cells expressed a CD25hi phenotype in the ‘late’ group compared to the ‘early’ group on day 1 after transfer, this marked increase was transient (Fig 5B and 5C). Surface expression of CD25 gradually increased and was sustained on 2° effector P14 CD8 T cells in the ‘early’ group as late as 5 days after transfer (Fig 5A–5D), while expression decreased over time on 2° effector P14 CD8 T cells in the ‘late’ group. Furthermore, in response to stimulation with titrated amounts of recombinant IL-2 in vitro, isolated 2° effector P14 CD8 T cells from ‘early’ mice exhibited more pronounced STAT5 activation at lower concentrations of IL-2 compared to ‘late’ mice (Fig 5E). This suggests that the duration of CD25 expression on 2° effector CD8 T cells correlates with IL-2 sensitivity and is modulated by the timing of stimulation.

Activated 1° CD8 T cells that maintain high-affinity IL-2 signaling show continued expression of cell-cycle associated genes and proteins which drives enhanced proliferative expansion [48]. Since we found that 2° effector P14 CD8 T cells in ‘early’ groups of mice not only undergo increased levels of proliferative expansion (Figs 2B and S3B), but also sustain CD25 expression (Fig 5A–5D), we wanted to determine whether this increased sensitivity to IL-2 signaling enhances the expression of cell-cycle related proteins such as FoxM1, Cyclin A, and Cyclin B1. Expression of these proteins was higher in 2° effector P14 CD8 T cells in the ‘early’ group, which correlated with sustained IL-2 signaling and enhanced ability of these cells to undergo proliferative expansion (Fig 5F). Thus, these data suggest that the timing of stimulation of 1° M CD8 T cells impacts the strength of IL-2 signals received by differentiating 2° effector CD8 T cells as a consequence of differential CD25 expression.
The availability of IL-2 signals impacts the differentiation of 2° CD8 T cells
Since we found that the responsiveness of 2° effector CD8 T cells to IL-2 is dependent on the timing of stimulation, we wanted to determine whether providing sustained IL-2 signaling during early or late recruitment of 1° M CD8 T cells would impact the subsequent differentiation of 2° CD8 T cell responses. It has been previously demonstrated that the biological activity of the IL-2 cytokine is enhanced once bound to a particular anti-IL-2 monoclonal antibody [49]. Notably, the S4B6 clone binds to the region of IL-2 that interacts with the CD25 receptor chain [49]. Stimulatory IL-2/S4B6 antibody complexes therefore selectively target CD122hi cells, independently of CD25, and facilitate IL-2 signaling in memory CD8 T cells and NK cells [49]. During a secondary infection with LCMV, 2° effector P14 CD8 T cells in ‘early’ and ‘late’ groups of mice similarly expressed CD122 over time after transfer (S4 Fig). Therefore, we chose to use these complexes as a tool to provide sustained IL-2 signaling in developing secondary effector CD8 T cells. ‘Early’ and ‘late’ groups of mice were treated with IL-2/S4B6 antibody complexes at days 4–6 post transfer and the magnitude of the secondary expansion was determined in the spleen at day 7 post transfer. Interestingly, enhanced accumulation in the frequency (Fig 6A) and total numbers (Fig 6B) of 2° effector P14 CD8 T cells was only observed in the ‘late’ group of mice. This increase in accumulation also correlated with a modest increase in CD25 expression on 2° effector P14 CD8 T cells in the ‘late’ group (Fig 6C). These data suggest that since 2° effector CD8 T cells in the ‘early’ group are already sensitive to IL-2 signaling, IL-2 stimulation resulted in no additional effect; however, 2° effector CD8 T cells in the ‘late’ group underwent increased accumulation in numbers due to sustained IL-2 signaling provided by the IL-2/S4B6 antibody complexes.
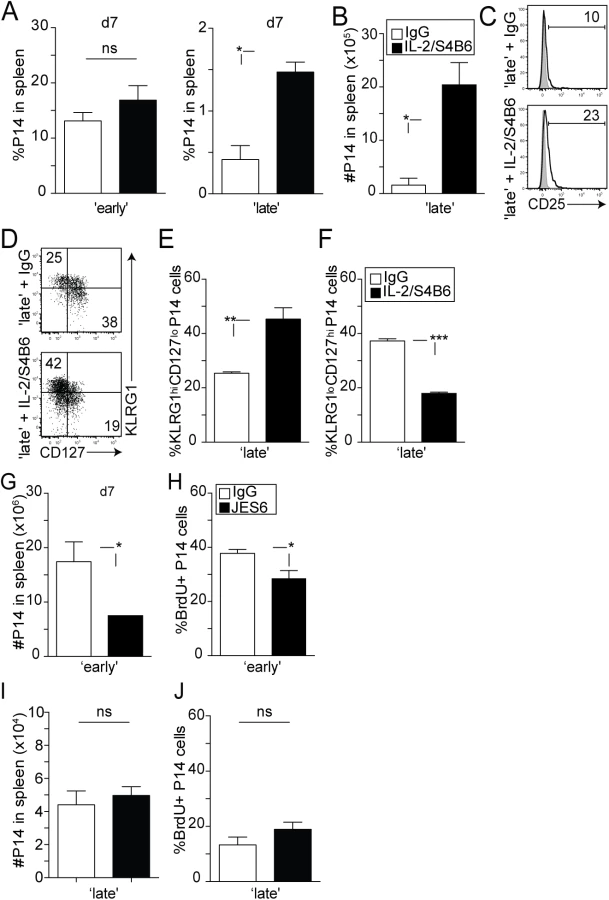
It is important to note that overall increase in 2° CD8 T cell accumulation in the ‘late’ group is facilitated by both antigenic stimulation and increased sensitivity to IL-2 signals following complex treatment, as 1° M CD8 T cells activated with the stimulatory IL-2/S4B6 complexes in the absence of infection undergo a moderate level of homeostatic driven proliferation (S5 Fig). Interestingly, manipulating IL-2 signaling also resulted in an altered differentiation status in 2° effector P14 CD8 T cells of ‘late’ mice. Similar to what has been previously documented by Xue et al. [50], increased IL-2 signaling resulted in downregulated IL-7Rα expression (Fig 6D) in a large fraction of cells in the ‘late’ group, with 40–50% of 2° effector CD8 T cells now displaying a KLRG1hiCD127lo phenotype after complex treatment (Fig 6E and 6F). These data suggest that increased sensitivity to IL-2 signals favors terminal differentiation in responding 2° effector CD8 T cells.
In order to determine if extended IL-2 signaling accounts for increased levels of 2° effector CD8 T cell expansion, we neutralized endogenous IL-2 using a blocking antibody, JES6-1A12 [49]. Importantly, the number of accumulated 2° effector P14 CD8 T cells in ‘early’ mice that had received the blocking antibody was significantly reduced (Fig 6G). These data also correlated with a reduced ability of these proliferating cells to incorporate BrdU (Fig 6H). However, this dampened 2° CD8 T cell response observed in the ‘early’ group following IL-2 blockade was not the result of a triggered regulatory CD4 T cell response. While IL-2 bound to the JES6-1A12 monoclonal antibody in a complex selectively stimulates CD25+ regulatory CD4 T cells ([49] and S6A and S6B Fig) and increases their proliferation [51], the administration of the blocking antibody alone did not impact regulatory CD4 T cells in both ‘early’ and ‘late’ groups of mice (S6C Fig). Furthermore, administration of the IL-2 blocking antibody resulted in no effect on 2° effector CD8 T cell accumulation or Brdu incorporation within the ‘late’ group (Fig 6I and 6J). These data suggest that since sensitivity to IL-2 signals was already diminished in 2° effector CD8 T cells in the ‘late’ group, neutralization of endogenous IL-2 did not result in any further changes. Thus, decreasing the sensitivity of 2° effector CD8 T cells to IL-2 signaling results in reduced accumulation upon secondary antigen encounter.
Collectively these data suggest that IL-2 signaling regulates levels of 2° expansion and 2° CD8 T cell differentiation, a previously unrecognized mechanism controlling the generation of 2° CD8 T cell responses.
Discussion
While the formation of 1° M CD8 T cells has been comprehensively studied, much less is known about the factors influencing the generation of 2° M CD8 T cell responses. Longitudinal analyses of developing 1° M and 2° M CD8 T cells suggests that the rate of acquiring a long-term memory phenotype and function varies substantially between these two populations of cells [2]. Prime-boost protocols are often implemented to increase the overall numbers of memory CD8 T cells, leading to the generation of memory CD8 T cells that have encountered Ag more than once. Therefore, understanding if the factors that impact the differentiation of 1° M CD8 T cells also influence 2° M CD8 T cell responses has implications in the design of future consecutive prime-boost vaccine protocols. Here, we show that during an infection recruitment of pathogen-specific 1° M CD8 T cells is not simultaneous, and timing of entry into an immune response relative to the onset of infection impacts the outcome of the ensuing 2° M CD8 T cell response. Specifically, late-entry of 1° M CD8 T cells into the immune response facilitates accelerated acquisition of 2° M characteristics. We also show that the timing of stimulation of 1° M CD8 T cells differentially regulates IL-2 signaling in differentiating 2° effector CD8 T cells, suggesting that this signaling mechanism contributes to the programming of 2° CD8 T cell responses.
D’Souza and Hedrick previously showed that naïve TCR-Tg CD8 T cells are recruited over time after infection with a live replicating pathogen in vivo [23]. In another study by Fousteri et al., early and late recruitment of naïve TCR-Tg CD8 T cells was modeled using an adoptive transfer system [24]. Kinetic analyses of Ag-specific CD8 T cells in these studies revealed that the timing of stimulation of naïve CD8 T cells impacted the magnitude of proliferative expansion. Moreover, Bousso et al. demonstrated using adoptive transfer experiments that the timing of entry into an immune response dramatically impacts CD8 T cell proliferative expansion [52]. Thus, Ag-specific CD8 T cells entering the immune response late undergo limited expansion. Additionally, it was observed that the timing of stimulation impacts the rate of 1° M CD8 T cell differentiation, with late-entry into the immune response facilitating faster re-expression of CD62L in Ag-specific CD8 T cells. Interestingly, it has also been shown that the time course of Ag presentation impacts the formation of 1° M CD8 T cells. Van Fessen et al. showed that the fate of differentiating CD8 T cells was heavily influenced by the strength and duration of stimuli [53]. Specifically, naïve CD8 T cells activated in an environment where the strength and duration of stimuli was reduced, preferentially upregulated CD62L [53]. Similarly, it was found by Sarkar et al. that the rate of CD62L re-expression was more rapid in P14 CD8 T cells that were exposed to limited stimulation during LCMV infection [54]. These results suggest that environmental signals as well as the strength of stimuli received by naïve Ag-specific CD8 T cells during an immune response influences 1° M CD8 T cell generation and differentiation. Thus, naïve Ag-specific CD8 T cells entering an immune response early or late may be receiving unique environmental cues at the time of activation, thereby influencing the rate at which these cells acquire 1° M CD8 T cell characteristics.
Our lab has recently shown that similar to 1° M CD8 T cells, 2° M CD8 T cell responses are impacted by the type and duration of infection and inflammatory environment [28,29]. Wirth et al. found that infection with either Vaccinia virus, Att L. monocytognes, or virulent L. monocytogenes differentially regulated the kinetics, magnitude, and phenotype of 1° and 2° CD8 T cells in the same host [28]. Additionally in this study it was observed using a peptide-coated DC vaccination strategy that 2° effector CD8 T cells undergo robust proliferation in the presence of systemic inflammation, suggesting that similar to 1° CD8 T cells, 2° CD8 T cell expansion is controlled by the presence of inflammatory cytokines [28]. Given these results, we determined whether the timing of stimulation of 1° M CD8 T cells controls the generation of 2° M CD8 T cell responses. We found that the timing of entry of 1° M CD8 T cells into an immune response impacted the rate at which 2° CD8 T cells transitioned into a long-term 2° M population of cells, however it still remained to be determined what mechanism controlled this unique pattern of differentiation in 2° CD8 T cells.
It has been previously observed that sensitivity to IL-2 signaling at the time of activation impacts 1° CD8 T cell differentiation [49,55,56]. As an example, Williams et al. showed that P14 cells lacking CD25 responded to LCMV infection and developed into populations of long-term memory cells [57]. Interestingly, these CD25-deficient P14 cells readily re-expressed CD62L and CD127, indicating accelerated 1° M CD8 T cells development. However, upon Ag re-encounter these CD25-deficient 1° M P14 cells failed to undergo 2° expansion [57], suggesting that CD25-mediated IL-2 signals during initial activation of CD8 T cells are necessary for 2° responses. In studies by Kalia et al. and Pipkin et al., it has been documented that enhanced IL-2 signaling during activation promotes effector differentiation in 1° CD8 T cell responses [31,32]. Similarly, it was found by Obar et al. that IL-2 signals are important in mediating the formation of effector CD8 T cells with a KLRG1hiCD127lo phenotype [58]. We observed that the timing of stimulation of 1° M CD8 T cells modulated the expression of CD25 on 2° effector CD8 T cells, while CD122 expression remained unchanged. This suggested that susceptibility to IL-2 signaling might be an underlying mechanism controlling the programming of 2° M CD8 T cells. Interestingly, we found that 1° M CD8 T cells activated late in the immune response have decreased sensitivity to IL-2 signaling and undergo reduced 2° proliferative expansion. Yet, the transition of these cells to a long-term 2° M phenotype is accelerated. However, enhanced sensitivity to IL-2 signaling following administration of stimulatory IL-2/S4B6 complexes resulted in increased CD25 expression on 2° effector CD8 T cells and favored terminal differentiation (KLRG1hiCD127lo phenotype). These data suggest that sensitivity to IL-2 signaling contributes to the development of 2° CD8 T cell responses.
In summary, our data suggests that the process of 1° M to 2° M CD8 T cell differentiation can be manipulated, a notion with great relevance for the generation of 2° M CD8 T cells and the design of consecutive prime-boost vaccine strategies intended to elicit secondary immune responses.
Materials and Methods
Ethics statement
All experimental procedures utilizing mice were approved by the University of Iowa Animal Care and Use Committee under the ACURF protocol number 1202050. The experiments performed in this study were done under strict accordance to the Office of Laboratory Animal Welfare guidelines and the PHS Policy on Humane Care and Use of Laboratory Animals.
Mice and pathogens
C57BL/6 (B6; Thy1.2/1.2) and P14 and OT-I (Thy1.1; specific for lymphocytic choriomeningitis virus (LCMV)-derived GP33 and chicken ovalbumin Ova257 epitopes, respectively) T-cell-receptor transgenic (TCR-Tg) mice were bred at the University of Iowa and housed under pathogen-free conditions. All mice were used at 6–10 weeks of age. All animal procedures followed approved Institutional Animal Care and Use Committee (ACURF) protocols. The Armstrong strain of LCMV (LCMV, 2x105 PFU/mouse, i.p.) and attenuated actA-deficient L. monocytogenes strain expressing Ova257 (Att LM-Ova, 5x106 CFU/mouse, i.v.) or GP33 (Att LM-GP33, 1x107 CFU/mouse, i.v.) were grown, injected, and quantified as described [59,60]. Infected mice were housed at the University of Iowa under the appropriate biosafety level.
Adoptive transfer and isolation of lymphocytes from tissues
For 1° CD8 T cell responses, naïve Thy1.1 P14 or OT-I cells were obtained from peripheral blood of young naïve TCR-Tg P14 or OT-I mice. Contaminating memory phenotype (CD44hiCD11ahi) P14 and OT-I cells were always <5%. Naïve P14 or OT-I cells were transferred (5x103 P14 or 1x103 OT-I cells/mouse, i.v.) [33] into recipient Thy1.2/1.2 mice on the day of (‘early’ group) or 3 days after (‘late’ group) LCMV or Att LM-Ova infection. To generate 1° M P14 or OT-I cells for adoptive transfer experiments, 1x103 naïve Thy1.1 P14 or OT-I cells were transferred into Thy1.2 recipients and mice were then immunized with either LCMV (2x105 PFU/mouse, i.p.) or Att LM-Ova (5x106 CFU/mouse, i.v.), respectively. At days 40–75 after infection, 1° M P14 or OT-I cells were isolated from spleens of mice by positive selection for Thy1.1 and transferred (2-4x104 cells/mouse, i.v.) into recipient Thy1.2/1.2 mice on the day of (‘early’ group) or 3 days after (‘late’ group) LCMV or Att LM-Ova infection.
Before removal of secondary lymphoid organs and tertiary tissues, samples of blood were obtained by retro-orbital puncture. Anesthetized mice were then perfused through the left ventricle with cold PBS and tissues were collected. Single-cell suspension from spleen, lung, and lymph nodes (LN) were washed before Ab staining. For experiments determining the expression of CD25 and CD69 on CFSE-labeled P14 CD8 T cells, spleens were cut into small pieces and treated with collagenase D (150U/mL) for 30 min at 37°C before further processing [44].
Antibodies and peptides
Flow cytometry data was acquired on a FACS Canto flow cytometer (Beckton-Dickinson Biosciences) and analyzed with FlowJo software (Tree Star). The following is a list of used mAbs with the indicated specificity and appropriate combinations in flourochromes from eBioscience: CD8 (clone 53–6.7), CD4 (GK1.5) Thy1.1 (HIS51), CD27 (LG.759), CD62L (MEL-14), KLRG1 (2F1), CD25 (PC61.5), CD69 (H1.2F3), CD122 (5H4), Bcl2 (BCL/104C4), Eomes (Dan11mag), Tcf1 (C63D9), Tbet (eBio4B10) BrdU (Bu20a), Foxp3 (FJK-16s) and appropriate isotype controls. Western mAb for Cyclin A (CY-A1; mouse, Sigma), Cyclin B1 (Rabbit, Cell Signaling Technology), FoxM1 (Rabbit, Cell Signaling Technology), and β-actin (Mouse, Santa Cruz Biotechnology) were previously described [48]. Intracellular staining for IFNγ (XMG1.2, Biolegend), TNFα (MP6-XT22, Biolegend), and IL-2 (JES6-5H4, eBioscience) was performed after surface fixation and permeabilization of the cell membrane using cytofix/cytoperm solution. Anti-STAT5 (pY694) was purchased from BD Biosciences. Synthetic GP33-41 peptide was used as previously described [17,61].
Quantification of CD8 T cell responses
P14 and OT-I cell responses in the peripheral blood and tissues were monitored by FACS analysis for Thy1.1-positive CD8 T cells. Cells were incubated with mAb at 4°C for 30 min, washed with FACS buffer (PBS containing 1% FCS and 0.1% NaN3), and then fixed with cytofix/cytoperm solution. The percentage of CD8 T cells producing cytokines after stimulation with GP33 peptide was determined using intracellular cytokine staining for IFNγ and TNFα or IL-2 after 5 h incubation in brefeldin A with or without GP33 peptide.
CFSE labeling and BrdU incorporation
For adoptive transfer experiments of CFSE labeled 1° M CD8 T cells, 106 splenocytes/mL from LCMV immune mice, containing 1° M Thy1.1 P14 cells, were incubated for 15 minutes at 37°C in the presence of 5mM CFSE. CFSE-labeled cells were washed twice with PBS containing 10% fetal calf serum and 1x106 1° M Thy1.1 P14 cells were injected into Thy1.2/1.2 recipient mice. To measure division of 2° effector Thy1.1 CD8 T cells BrdU was injected (2 mg/mouse, i.p.) for ~15 h before spleen harvest. Detection of BrdU incorporation was performed according to manufacturer’s protocol (BrdU Flow kits; BD) [48].
Quantitative RT-PCR
Spleen cells from ‘early’ and ‘late’ groups were harvested at day 7 post-transfer. 2° effector Thy1.1 P14 CD8 T cells were sorted directly into Trizol LS reagent (Invitrogen). Following chloroform extraction, the aqueous phase was mixed with 2 volumes of ethanol and loaded onto a purification column from the RNeasy Mini Kit (Qiagen) for further purification. Total RNA was reverse-transcribed using QuantiTech Reverse Transcription Kit (Qiagen). The resulting cDNA was analyzed for the expression of different genes by quantitative PCR using SYBR Advantage qPCR pre-mix (Clontech) on an ABI 7300 Real Time PCR System (Applied Biosystems) as previously described [62]. The relative gene expression levels in each sample were normalized to the housekeeping gene, hypoxanthine phosphoribosyltransferase (Hprt). The primer sequences include the following:
Bcl2: 5’-GCAGATTGCCCTGGATGT-AT and 5’ - AGAAAAGTCAGCCAGCCAGA;
Eomes: 5’-TCCTAACACTGGCTCCCACT and 5’-GTCACTTCCACGATGTGCAG;
Tcf7: 5’-CAATCTGCTCATGCCCTACC and 5’-CTTGCTTCTGGCTGATGTCC;
Prdm1: 5’-CCAAGGAACCTGCTTTTCAA and 5’-GGCATTCTTGGGAACTGTGT;
Id3: 5’–ATCTCCCGATCCAGACAGC and 5’–GAGAGAGGGTCCCAGAGTCC;
Tbx21: 5’-CAATGTGACCCAGATGATCG and 5’-GCGTTCTGGTAGGCAGTCAC;
Hprt: 5’-GCGTCGTGATTAGCGATGATG and 5’-CTCGAGCAAGTCTTTCAGTCC.
In vitro IL-2 stimulation
2° effector Thy1.1 P14 cells (2x106) from ‘early’ and ‘late’ groups of mice were incubated in vitro in the presence or absence of murine rIL-2 (Peprotech). STAT5-pY694 activation was measured directly following murine rIL-2 stimulation at indicated concentrations using manufacturer’s protocol (BD Phosflow; Cell Signaling) [48].
Immunoblot analysis
2° effector Thy1.1 P14, from ‘early’ and ‘late’ group of mice as well as 1° M Thy1.1 P14 cells were stained with PE-anti-Thy1.1 (Clone OX-7, BD PharMingen), and purified with PE-antibody magnetic beads according to standard AutoMacs protocols. Cells were then immediately lysed in in NP-40 lysis buffer (20mM Hepes, pH 7.9, 100mM NaCl, 5mM EDTA, 0.5 mM CaCl, 1%NP-40, 1mM PMSF, 10μM MG-132) for 15 minutes on ice as previously described [48,63]. Whole-cell lysates were clarified by centrifugation at ~20,000 g for 5 minutes at 4°C. Extracts were then resolved by SDS-PAGE (BIO RAD). Primary antibodies were detected with goat anti-mouse IgG or goat anti-rabbit IgG coupled to horseradish peroxidase (Santa Cruz Biotechnology) and SuperSignal West Pico Chemiluminescence (Thermo Scientific).
In vivo cytokine stimulation/neutralization
IL-2/mAb complexes were generated by incubating murine rIL-2 (Peprotech) with either S4B6 or JES6-1A12 anti-IL-2 monoclonal antibodies at a 2 : 1 molar ratio (1.5μg/mL IL-2 : 50μg/mL S4B6 or JES6-1A12) for 15 minutes at room temperature [48]. IL-2 neutralization was achieved in the ‘early’ group of mice by injecting 500μg JES6-1A12 alone daily from day 2–5 post-transfer. For control, equal amounts of rat IgG (Sigma-Aldrich) were used in an additional group of mice [48].
Statistical analyses
Data were analyzed with Prism4 GraphPad software to determine statistical significance as indicated in figure legends. Statistical significance was assessed using the two-tailed, unpaired student’s T test, with a confidence interval >95% (*p ≤ 0.05, **p ≤ 0.005, ***p ≤ 0.001. and n.s. as no significance). Data generated as scatter dot plots are presented as mean, and data generated as bar graphs are presented as mean + SEM.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Badovinac VP, Harty JT (2006) Programming, demarcating, and manipulating CD8+ T-cell memory. Immunol Rev 211 : 67–80. 16824118
2. Harty JT, Badovinac VP (2008) Shaping and reshaping CD8+ T-cell memory. Nat Rev Immunol 8 : 107–119. doi: 10.1038/nri2251 18219309
3. Kaech SM, Wherry EJ, Ahmed R (2002) Effector and memory T-cell differentiation: implications for vaccine development. Nat Rev Immunol 2 : 251–262. 12001996
4. Sallusto F, Lanzavecchia A, Araki K, Ahmed R (2010) From vaccines to memory and back. Immunity 33 : 451–463. doi: 10.1016/j.immuni.2010.10.008 21029957
5. Seder RA, Darrah PA, Roederer M (2008) T-cell quality in memory and protection: implications for vaccine design. Nat Rev Immunol 8 : 247–258. doi: 10.1038/nri2274 18323851
6. Butler NS, Nolz JC, Harty JT (2011) Immunologic considerations for generating memory CD8 T cells through vaccination. Cell Microbiol 13 : 925–933. doi: 10.1111/j.1462-5822.2011.01594.x 21501363
7. Lefrancois L (2006) Development, trafficking, and function of memory T-cell subsets. Immunol Rev 211 : 93–103. 16824120
8. Badovinac VP, Porter BB, Harty JT (2002) Programmed contraction of CD8(+) T cells after infection. Nat Immunol 3 : 619–626. 12055624
9. Jameson SC, Masopust D (2009) Diversity in T cell memory: an embarrassment of riches. Immunity 31 : 859–871. doi: 10.1016/j.immuni.2009.11.007 20064446
10. Kaech SM, Wherry EJ (2007) Heterogeneity and cell-fate decisions in effector and memory CD8+ T cell differentiation during viral infection. Immunity 27 : 393–405. 17892848
11. Mescher MF, Curtsinger JM, Agarwal P, Casey KA, Gerner M, et al. (2006) Signals required for programming effector and memory development by CD8+ T cells. Immunol Rev 211 : 81–92. 16824119
12. Haring JS, Badovinac VP, Harty JT (2006) Inflaming the CD8+ T cell response. Immunity 25 : 19–29. 16860754
13. Williams MA, Bevan MJ (2007) Effector and memory CTL differentiation. Annu Rev Immunol 25 : 171–192. 17129182
14. Butz EA, Bevan MJ (1998) Massive expansion of antigen-specific CD8+ T cells during an acute virus infection. Immunity 8 : 167–175. 9491998
15. Doherty PC (1998) The numbers game for virus-specific CD8+ T cells. Science 280 : 227. 9565533
16. Badovinac VP, Harty JT (2007) Manipulating the rate of memory CD8+ T cell generation after acute infection. J Immunol 179 : 53–63. 17579021
17. Badovinac VP, Messingham KA, Jabbari A, Haring JS, Harty JT (2005) Accelerated CD8+ T-cell memory and prime-boost response after dendritic-cell vaccination. Nat Med 11 : 748–756. 15951824
18. Pham NL, Badovinac VP, Harty JT (2009) A default pathway of memory CD8 T cell differentiation after dendritic cell immunization is deflected by encounter with inflammatory cytokines during antigen-driven proliferation. J Immunol 183 : 2337–2348. doi: 10.4049/jimmunol.0901203 19635915
19. Pham NL, Pewe LL, Fleenor CJ, Langlois RA, Legge KL, et al. (2010) Exploiting cross-priming to generate protective CD8 T-cell immunity rapidly. Proc Natl Acad Sci U S A 107 : 12198–12203. doi: 10.1073/pnas.1004661107 20616089
20. Badovinac VP, Porter BB, Harty JT (2004) CD8+ T cell contraction is controlled by early inflammation. Nat Immunol 5 : 809–817. 15247915
21. Mercado R, Vijh S, Allen SE, Kerksiek K, Pilip IM, et al. (2000) Early programming of T cell populations responding to bacterial infection. J Immunol 165 : 6833–6839. 11120806
22. Martin MD, Wirth TC, Lauer P, Harty JT, Badovinac VP (2011) The impact of pre-existing memory on differentiation of newly recruited naive CD8 T cells. J Immunol 187 : 2923–2931. doi: 10.4049/jimmunol.1100698 21832161
23. D'Souza WN, Hedrick SM (2006) Cutting edge: latecomer CD8 T cells are imprinted with a unique differentiation program. J Immunol 177 : 777–781. 16818730
24. Fousteri G, Dave A, Juedes A, Juntti T, Morin B, et al. (2011) Increased memory conversion of naive CD8 T cells activated during late phases of acute virus infection due to decreased cumulative antigen exposure. PLoS One 6: e14502. doi: 10.1371/journal.pone.0014502 21253594
25. Nolz JC, Harty JT (2011) Strategies and implications for prime-boost vaccination to generate memory CD8 T cells. Adv Exp Med Biol 780 : 69–83. doi: 10.1007/978-1-4419-5632-3_7 21842366
26. Woodland DL (2004) Jump-starting the immune system: prime-boosting comes of age. Trends Immunol 25 : 98–104. 15102369
27. Wirth TC, Xue HH, Rai D, Sabel JT, Bair T, et al. (2010) Repetitive antigen stimulation induces stepwise transcriptome diversification but preserves a core signature of memory CD8(+) T cell differentiation. Immunity 33 : 128–140. doi: 10.1016/j.immuni.2010.06.014 20619696
28. Wirth TC, Harty JT, Badovinac VP (2010) Modulating numbers and phenotype of CD8+ T cells in secondary immune responses. Eur J Immunol 40 : 1916–1926. doi: 10.1002/eji.201040310 20411564
29. Wirth TC, Martin MD, Starbeck-Miller G, Harty JT, Badovinac VP (2011) Secondary CD8+ T-cell responses are controlled by systemic inflammation. Eur J Immunol 41 : 1321–1333. doi: 10.1002/eji.201040730 21425157
30. Jabbari A, Harty JT (2006) Secondary memory CD8+ T cells are more protective but slower to acquire a central-memory phenotype. J Exp Med 203 : 919–932. 16567385
31. Kalia V, Sarkar S, Subramaniam S, Haining WN, Smith KA, et al. (2010) Prolonged interleukin-2Ralpha expression on virus-specific CD8+ T cells favors terminal-effector differentiation in vivo. Immunity 32 : 91–103. doi: 10.1016/j.immuni.2009.11.010 20096608
32. Pipkin ME, Sacks JA, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, et al. (2010) Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity 32 : 79–90. doi: 10.1016/j.immuni.2009.11.012 20096607
33. Badovinac VP, Haring JS, Harty JT (2007) Initial T cell receptor transgenic cell precursor frequency dictates critical aspects of the CD8(+) T cell response to infection. Immunity 26 : 827–841. 17555991
34. Haring JS, Jing X, Bollenbacher-Reilley J, Xue HH, Leonard WJ, et al. (2008) Constitutive expression of IL-7 receptor alpha does not support increased expansion or prevent contraction of antigen-specific CD4 or CD8 T cells following Listeria monocytogenes infection. J Immunol 180 : 2855–2862. 18292507
35. Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, et al. (2007) Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity 27 : 281–295. 17723218
36. Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, et al. (2003) Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol 4 : 1191–1198. 14625547
37. Kaech SM, Cui W (2012) Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol 12 : 749–761. doi: 10.1038/nri3307 23080391
38. Rutishauser RL, Kaech SM (2010) Generating diversity: transcriptional regulation of effector and memory CD8 T-cell differentiation. Immunol Rev 235 : 219–233. doi: 10.1111/j.0105-2896.2010.00901.x 20536566
39. Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, et al. (2005) Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat Immunol 6 : 1236–1244. 16273099
40. Wherry EJ, Teichgraber V, Becker TC, Masopust D, Kaech SM, et al. (2003) Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol 4 : 225–234. 12563257
41. Zhou X, Xue HH (2012) Cutting edge: generation of memory precursors and functional memory CD8+ T cells depends on T cell factor-1 and lymphoid enhancer-binding factor-1. J Immunol 189 : 2722–2726. doi: 10.4049/jimmunol.1201150 22875805
42. Zhou X, Yu S, Zhao DM, Harty JT, Badovinac VP, et al. (2010) Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity 33 : 229–240. doi: 10.1016/j.immuni.2010.08.002 20727791
43. Rutishauser RL, Martins GA, Kalachikov S, Chandele A, Parish IA, et al. (2009) Transcriptional repressor Blimp-1 promotes CD8(+) T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity 31 : 296–308. doi: 10.1016/j.immuni.2009.05.014 19664941
44. Jabbari A, Legge KL, Harty JT (2006) T cell conditioning explains early disappearance of the memory CD8 T cell response to infection. J Immunol 177 : 3012–3018. 16920937
45. Kaech SM, Hemby S, Kersh E, Ahmed R (2002) Molecular and functional profiling of memory CD8 T cell differentiation. Cell 111 : 837–851. 12526810
46. Martin MD, Condotta SA, Harty JT, Badovinac VP (2012) Population dynamics of naive and memory CD8 T cell responses after antigen stimulations in vivo. J Immunol 188 : 1255–1265. doi: 10.4049/jimmunol.1101579 22205031
47. Masopust D, Ha SJ, Vezys V, Ahmed R (2006) Stimulation history dictates memory CD8 T cell phenotype: implications for prime-boost vaccination. J Immunol 177 : 831–839. 16818737
48. Starbeck-Miller GR, Xue HH, Harty JT (2014) IL-12 and type I interferon prolong the division of activated CD8 T cells by maintaining high-affinity IL-2 signaling in vivo. J Exp Med 211 : 105–120. doi: 10.1084/jem.20130901 24367005
49. Boyman O, Sprent J (2012) The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol 12 : 180–190. doi: 10.1038/nri3156 22343569
50. Xue HH, Kovanen PE, Pise-Masison CA, Berg M, Radovich MF, et al. (2002) IL-2 negatively regulates IL-7 receptor alpha chain expression in activated T lymphocytes. Proc Natl Acad Sci U S A 99 : 13759–13764. 12354940
51. Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J (2006) Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science 311 : 1924–1927. 16484453
52. Bousso P, Levraud JP, Kourilsky P, Abastado JP (1999) The composition of a primary T cell response is largely determined by the timing of recruitment of individual T cell clones. J Exp Med 189 : 1591–1600. 10330438
53. van Faassen H, Saldanha M, Gilbertson D, Dudani R, Krishnan L, et al. (2005) Reducing the stimulation of CD8+ T cells during infection with intracellular bacteria promotes differentiation primarily into a central (CD62LhighCD44high) subset. J Immunol 174 : 5341–5350. 15843531
54. Sarkar S, Teichgraber V, Kalia V, Polley A, Masopust D, et al. (2007) Strength of stimulus and clonal competition impact the rate of memory CD8 T cell differentiation. J Immunol 179 : 6704–6714. 17982060
55. Mitchell DM, Ravkov EV, Williams MA (2010) Distinct roles for IL-2 and IL-15 in the differentiation and survival of CD8+ effector and memory T cells. J Immunol 184 : 6719–6730. doi: 10.4049/jimmunol.0904089 20483725
56. Obar JJ, Lefrancois L (2010) Early signals during CD8 T cell priming regulate the generation of central memory cells. J Immunol 185 : 263–272. doi: 10.4049/jimmunol.1000492 20519649
57. Williams MA, Tyznik AJ, Bevan MJ (2006) Interleukin-2 signals during priming are required for secondary expansion of CD8+ memory T cells. Nature 441 : 890–893. 16778891
58. Obar JJ, Molloy MJ, Jellison ER, Stoklasek TA, Zhang W, et al. (2010) CD4+ T cell regulation of CD25 expression controls development of short-lived effector CD8+ T cells in primary and secondary responses. Proc Natl Acad Sci U S A 107 : 193–198. doi: 10.1073/pnas.0909945107 19966302
59. Brundage RA, Smith GA, Camilli A, Theriot JA, Portnoy DA (1993) Expression and phosphorylation of the Listeria monocytogenes ActA protein in mammalian cells. Proc Natl Acad Sci U S A 90 : 11890–11894. 8265643
60. Pope C, Kim SK, Marzo A, Masopust D, Williams K, et al. (2001) Organ-specific regulation of the CD8 T cell response to Listeria monocytogenes infection. J Immunol 166 : 3402–3409. 11207297
61. Masopust D, Murali-Krishna K, Ahmed R (2007) Quantitating the magnitude of the lymphocytic choriomeningitis virus-specific CD8 T-cell response: it is even bigger than we thought. J Virol 81 : 2002–2011. 17151096
62. Yu S, Zhou X, Steinke FC, Liu C, Chen SC, et al. (2012) The TCF-1 and LEF-1 transcription factors have cooperative and opposing roles in T cell development and malignancy. Immunity 37 : 813–826. doi: 10.1016/j.immuni.2012.08.009 23103132
63. Richer MJ, Nolz JC, Harty JT (2013) Pathogen-specific inflammatory milieux tune the antigen sensitivity of CD8(+) T cells by enhancing T cell receptor signaling. Immunity 38 : 140–152. doi: 10.1016/j.immuni.2012.09.017 23260194
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 10
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Chronobiomics: The Biological Clock as a New Principle in Host–Microbial Interactions
- Interferon-γ: The Jekyll and Hyde of Malaria
- Crosslinking of a Peritrophic Matrix Protein Protects Gut Epithelia from Bacterial Exotoxins
- Antigen-Specific Th17 Cells Are Primed by Distinct and Complementary Dendritic Cell Subsets in Oropharyngeal Candidiasis
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy