
Geminivirus Activates to Accelerate Cytoplasmic DCP2-Mediated mRNA Turnover and Weakens RNA Silencing in
In higher plants, aberrant RNAs generated during virus replication serve as templates to make small interfering RNAs. These small RNAs are used by host as a defense mechanism to cleave viral RNAs thereby blocking virus replication. The anti-virus defense is attenuated by the host cellular mRNA turnover machinery which clears aberrant RNAs. Viruses may use encoded component(s) to activate host cellular mRNA turnover for their own benefits. In this study, we identified ASYMMETRIC LEAVES2 (AS2) as an activator of mRNA decapping and degradation and an endogenous suppressor of virus silencing. We showed that the geminivirus BV1 protein induces AS2 expression, causes nuclear exit of AS2 to activate mRNA decapping activity and renders infected plants more sensitive to viruses. Similar mechanisms may be used by other viral pathogens to weaken antiviral defenses in host plants and also mammals.
Published in the journal:
. PLoS Pathog 11(10): e32767. doi:10.1371/journal.ppat.1005196
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1005196
Summary
In higher plants, aberrant RNAs generated during virus replication serve as templates to make small interfering RNAs. These small RNAs are used by host as a defense mechanism to cleave viral RNAs thereby blocking virus replication. The anti-virus defense is attenuated by the host cellular mRNA turnover machinery which clears aberrant RNAs. Viruses may use encoded component(s) to activate host cellular mRNA turnover for their own benefits. In this study, we identified ASYMMETRIC LEAVES2 (AS2) as an activator of mRNA decapping and degradation and an endogenous suppressor of virus silencing. We showed that the geminivirus BV1 protein induces AS2 expression, causes nuclear exit of AS2 to activate mRNA decapping activity and renders infected plants more sensitive to viruses. Similar mechanisms may be used by other viral pathogens to weaken antiviral defenses in host plants and also mammals.
Introduction
Host-virus interactions entail susceptibility and resistance mechanisms embedded in anti-pathogen strategies of hosts and anti-host resistance strategies mounted by successful viral pathogens. As obligate intracellular parasites viruses have evolved various strategies to hijack host components for their own survival and multiplication in hosts [1].
In higher plants, post-transcriptional gene silencing (PTGS) is an important native antiviral resistance machinery that suppresses viral gene expression through (si)RNA-directed viral RNA cleavage. Aberrant viral RNAs synthesized during replication of RNA viruses or transcribed from DNA viruses serve as a template for RNA-DEPENDENT RNA POLYMERASE6 (RDR6) to synthesize complementary RNAs [1]. The double stranded RNAs (dsRNAs) produced are processed by the plant’s small RNA pathway to generate siRNAs which mediate viral mRNA cleavage and silencing viral gene expression. The importance of the PTGS pathway in plant viral defense has elicited counter defense measures from the viral pathogens to overcome it. Plant viruses have evolved various viral RNA silencing repressors (VSR) to target different PTGS pathway components. For example, the Tomato bushy stunt virus P19 protein binds to siRNAs directly whereas the Cucumber mosaic virus 2b protein blocks the RNA cleavage activity of Argonaute1 (AGO1) in the RNA-induced Silencing Complex [2,3]. In addition to the VSRs, plants also encode endogenous silencing repressor such as the RDR1 from Nicotiana tabacum to target the dsRNA biogenesis step [4].
Aberrant viral RNAs serve as templates for synthesis of dsRNAs by RDR6, and like cellular mRNAs they can be cleared by the host RNA degradation machinery [1]. The clearance of aberrant viral RNAs will deplete substrates for dsRNA synthesis and siRNA production thus attenuating PTGS. Degradation of cytoplasmic RNAs occurs in P-bodies which are conserved and dynamic protein-RNA aggregates. Among protein subunits of the P-body complex investigated so far, DCP2 is the only protein subunit that possesses enzymatic activity for removing 5' cap structure (m7GpppX-) in vitro [5]. The 5' cap structure is an essential feature of eukaryotic mRNA required for mRNA stability and efficient translation [6]. Plant mutants deficient in P-body components display severe postembryonic developmental defects, suggesting that these cytoplasmic bodies play important roles in regulating gene expression in plant developmental process [7]. Furthermore, plant DCP1 and DCP5 have been shown to be translation suppressors [7,8]. Other than DCP1, DCP2 and DCP5, similar phenotypic and functional co-localization analyses suggest that 3 additional factors VARICOSE (VCS), XRN4, and DHH1 are also key components of plant P-bodies [7]. Genetic and biochemical analysis have shown that mutations in XRN4 [9] or DCP2 [10] enhance PTGS presumably by increasing aberrant RNA levels.
In contrast to higher plants, the role of mRNA decapping in animal host defense against viruses is unclear. RNA viruses, including negative stranded RNA viruses and ambiviruses (the Orthomyxoviridae, Bunyaviridae, and Arenaviridae families), and dsRNA virus totivirus L-A, which infects Saccharomyces cerevisiae, provide their mRNAs with a 5′ cap structure via a cap-snatching mechanism. In this mechanism, the viral polymerase cleaves host mRNAs 10–13 nucleotides from the 5′ end and utilizes the capped fragment as a primer to synthesize viral transcripts [11]. Therefore, the decapping machinery of animal cells may act as an important immune system to accelerate turning over of viral mRNAs thereby limiting virus multiplication for this subgroup RNA viral pathogens [11,12]. On the contrary, the decapping machinery in higher plants is used to suppress PTGS thereby promoting virus replication [10]. Little is known about the roles of plant mRNA decapping on virus-plant interaction and virus pathogenesis, possibly because deficiencies in most of the genes encoding mRNA decapping machinery cause postembryonic lethality [7].
Geminiviruses are the largest group of plant DNA viruses whose compact genomes consist of small single-stranded DNA circles of 2–3 kb. Despite their small size, these viruses inflict big damages on many commercially important crops [13]. Owing to their small genome size the encoded genetic information is extremely compact. Among geminiviruses, members of the genus Begomovirus, such as India cassava mosaic virus (ICMV), possess two genomic components, DNA-A and DNA-B [14]. The DNA-A which encodes 5 gene products is involved in virus replication, transcriptional activation of viral genes and encapsulation of the viral genome. The DNA-B component encodes two proteins that are expressed at the late stage of virus infection, the nuclear shuttle protein BV1 (NSP) and the movement protein BC1 (MP), both being required for viral systemic movement. It is known that BV1 facilitates the intracellular trafficking of viral DNA between nuclei and cytoplasm, whereas BC1 potentiates cell-to-cell movement of viral DNA. BV1 is a virulence factor, found to suppress trans-membrane receptor kinase activity in vitro [15]. AC2 which is encoded by the DNA-A component has been reported as a VSR and it trans-activates host and viral genes [16].
We have previously identified a pathogenesis factor βC1 of the monopartite geminivirus TYLCCNV (Tomato yellow leaf curl China virus) that interacts with Arabidopsis ASYMMETRIC LEAVES 1 (AS1) to cause alterations in leaf development resulting in the manifestation of disease symptoms [17]. AS1 is needed for βC1 function as changes in leaf morphology elicited by this viral factor is largely attenuated in as1 mutant. Unexpectedly, βC1 is able to partially complement as2 mutation suggesting that βC1 is a molecular mimic of ASYMMETRIC LEAVES 2 (AS2) [17]. What roles does AS2 play in viral pathogenicity and/or virulence remains to be investigated.
Here, we show that BV1, a geminivirus virulence factor that is expressed late in the virus life cycle, can induce AS2 expression by binding to its promoter region. Over-expression of AS2 both in Arabidopsis thaliana and Nicotiana benthamiana rendered plants more sensitive to geminivirus infection whereas as2 mutant plants were resistant. Moreover, BV1 can induce nuclear export of AS2 to the cytosol where the latter interacted with DCP2 to promote decapping activity, reduce siRNA accumulation and weaken RNA silencing. This viral counter-defense strategy makes plants more sensitive to virus infection and replication. Finally, we provide evidence that cytoplasmic localization of AS2 was necessary for it to function as a negative regulator of host resistance against virus infection.
Materials and Methods
Plant materials, growth conditions, and transformation
Arabidopsis thaliana Wild type (WT) and as1-1, as2-1, and sgs3-1mutants (all in Col-0 background) were used [18–21]. After vernalization for 2 days at 4°C in darkness, seeds were germinated on Murashing and Skoog (MS) medium at 22°C with 16 h light. Plasmids were introduced into Agrobacterium tumefaciens strain AGL1 or EHA105 by electrotransformation. Arabidopsis transformations were performed using the floral-dip method [22].
Nicotiana benthamiana GFP transgene line 16c was kindly provided by Dr. David Baulcombe. Agrobacterium-mediated transient expression in N.benthamiana leaves was performed by pressure infiltration [23]. Arabidopsis thaliana L1 line with silenced 35S-GUS transgene was from Dr HervéVaucheret [24].
Plants were infected with CaLCuV by either micro-particle bombardment or agroinfiltration. Plasmids pCPCbLCVA.007 (GenBank accession no.AY279345) and pCPCbLCVB.002 (GenBank accession no. AY279344) were kindly provided by Dr Dominique Robertson (Turnage et al., 2002). Biolistic PDS-1000/HE System (Biorad) was used for particle delivery and Arabidopsis plants were inoculated at 1,100 psi Capture Disks (Biorad). Inoculated plants were kept in darkness for 12h and then returned to a growth chamber under normal condition. Symptoms of the inoculated plants were recorded 7 days later. For quantitative analysis of viral titer, leaf samples were collected from plants 3–4 weeks after inoculation. Total gDNA and viral DNAs were extracted by CTAB method and quantitative PCR (Q-PCR) was performed by using Actin2 as an internal genomic DNA control [25]. CaLCuV infectious clones (DNA-A and DNA-B) suitable for agroinfiltration [25] were used to infect Arabidopsis plants with 6–8 true leaves.
Constructs
Full-length cDNAs were amplified by PCR using Phusion High-Fidelity DNA Polymerase (FINNZYMES) and subcloned into binary vectors pCAMBIA1300-2X35-3HA, pBA002-3HA or pBA002-6Myc to generate HA-tagged and Myc-tagged constructs under the control of a 35S promoter. Point mutation in specific amino acids of AS2 were generated with Stratagene's QuikChange II Site Directed Mutagenesis Kit (Stratagene, USA) with primers listed in S1 Table.
pCAMBIA1300 vector was used to construct AS2-EGFPfusion gene expressed from a native AS2 promoter (from -2.8kb to +2.0 kb, just before the initiation codon) to generate the plasmid 1300-AS2p(-2.8K):AS2-EGFP for stable transformation. NES from HIV Tat protein (LQLPPLERLTLD) and for NLS from SV40 virus (PKKKRKVKD) were used to make AS2-NES and AS2-NLSfusion genes. AS2 variants (AS2-NES and AS2-NLS) were replaced WT AS2 in the plasmid of 1300-AS2p(-2.8K):AS2-EGFP and used for AS2 native promoter driven AS2 variants expression vector. BV1 of CaLCuV was cloned into the pBA002-CFP to form pBA002-BV1-CFP.
GFP imaging, antibodies and protein gel blot analysis
GFP imaging was as described [23]. To prepare protein samples, a leaf section (50mg) was ground in liquid N2 and extracted with 200 μl of 8M urea. The extract was mixed with 2 x SDS loading buffer and boiled for 10 min. Ten microliters of protein extract was separated by SDS-PAGE. Antibodies against GFP and protein gel blot analysis were as described in [23]. Transient silencing suppressor activity assays in N.benthamiana was performed as described [23]. For co-infiltrations, the OD600 of GFP-carrying agrobacterial strain was 0.8 and that of AS2 was 1.2.
RNA/DNA gel blot analysis
Total RNA was extracted from 12-day-old seedlings using Trizol reagent (Invitrogen) according to the manufacturer’s instructions. Fifteen μg total RNA were fractionated on a 1.2% (w/v) agarose gel and then transferred to a Hybond-XL membrane (GE Biosciences). Hybridization was performed overnight at 65°C in hybridization buffer (0.3 M sodium phosphate at pH 7.0, 10 mM EDTA, 5% SDS, 10% dextran sulfate, 0.15 mg/mL salmon sperm DNA), and signals were detected by autoradiography. For small RNA analysis, 15 μg of total RNA were fractionated on a 15% polyacrylamide gel containing 8 M urea and then transferred to a Hybond-N+ membrane (GE Biosciences). DNA oligonucleotides were end-labeled with [γ-32P] ATP using T4 polynucleotide kinase (New England Biolabs). Hybridization was performed overnight at 42°C using the ULTRAHyb-Oligo hybridization buffer (Ambion) and signals were detected by autoradiography. Southern blot was performed as described previously [26]. Membrane was hybridized with a probe encoding the AC1 (Rep) of CaLCuV DNA-A. The probe was DIG-dUTP-labeled by PCR using a PCR DIG probe synthesis kit (Roche Shanghai, China) and hybridization signals were detected by autoradiography.
RNA extraction and quantitative real-time PCR (qRT-PCR) analysis
RNAs were isolated from leaf and stem samples using Qiagen RNeasy Plant mini kits (Qiagen) with on-column DNase treatment. Plant RNA purification reagent (Invitrogen) was also used for total RNA extraction followed with DNase treatment [27]. RNA concentration was measured by Nanodrop (Thermo, USA). M-MLV reverse transcriptase (Promega, USA) was used for reverse transcription reactions. Real-time PCR was performed with Power SYBR Green PCR Master (Applied Biosystems, USA) and run in ABI7900HT. All samples were run in triplicates and data was analyzed with RQ manager at a pre-set Ct value (Applied Biosystems, USA). PCR primers were listed in S1 Table. Cordycepin treatments and mRNA stability analysis were performed as described before [7]. Twelve-day-old seedlings were incubated in MS medium with cordycepin (3′-deoxyadenosine; Sigma-Aldrich) and DMSO treatment was used as a mock control. Data derived from the mock control was used for normalization for mRNA stability assays. Total RNA was extracted from samples harvested at various time points using Plant RNA extraction reagent (Invitrogen). Actin2 was used as an internal mRNA control.
In vitro pull-down assay
cDNAs encoding full-length CaLCuV BV1, AS1, AS2, DCP1, DCP2, DCP5 and VCS-C terminal were amplified by PCR using Phusion High-Fidelity DNA Polymerase (FINNZYMES) and subcloned to generate GST fusion, MBP fusion and 6His, 6His-SUMO fusion constructs. The yeast SUMO protein tag was used to improve AS2 protein solubility. All constructs were transformed into E. coli BL21(DE3) cells and cultured at 37°C. After the OD600 had reached ∼0.6, isopropyl β-D-thiogalactopyranoside was added to a final concentration of 0.4 mM and the culture incubated overnight at 20°C. Bacterial cells were collected by centrifugation and suspended in a lysis buffer containing proteinase inhibitor cocktail (Roche). After French press treatment, recombinant proteins were purified with specific affinity columns and AKTA FPLC system (GE Biosciences) followed by size-exclusion columns. The eluted proteins were concentrated by Ultracel YM-30 (Millipore). In vitro pull-down assays were performed with 2 μg of GST/MBP/His/His-SUMO fusion proteins. Proteins were incubated in a binding buffer (50 mM Tris-HCl at pH 7.5, 100 mM NaCl, 0.25% Triton X-100, 35 mM β-mercaptoethanol) for 2 h at 4°C, and 30 μl of glutathione sepharose 4B (GE Biosciences) were added and the mix incubated for overnight. After washing with binding buffer for 6 times, pulled-down proteins were separated on 12% SDS–polyacrylamide gel and detected by Western blotting using anti-His, anti-GST or anti-MBP antibody.
Immunoprecipitation
About 3 g of 14-day old Arabidopsis seedlings expressing35S:BV1-ECFP or AS2p(-2.8K):AS2-EGFP was used for ChIP assays. Seedlings were incubated with 50 μM MG132 (Calbiochem) for 12 hr before harvesting. Proteins were extracted in extraction buffer (50 mM Tris-HCl at pH 7.5, 150 mM NaCl, 2 mM MgCl2, 1 mM DTT, 20% glycerol, 0.5% nonident P-40) containing protease inhibitor cocktail (Roche) and protease inhibitor mixture (Sigma). Cell debris was pelleted by centrifugation at 14,000g for 30 min. The supernatant was incubated with GFP-agrose beads (GFPtrack) overnight at 4°C. Beads collected by centrifugation were washed 6 times with washing buffer (50 mM Tris-HCl at pH 7.5, 150 mM NaCl, 2 mM MgCl2, 1 mM DTT, 10% glycerol, 0.5% nonident P-40). Proteins were eluted by 50 μL of 2.5× sample buffer and analyzed by Western blotting using anti-DCP2 rabbit antibody [7].
Chromatin immunoprecipitation (ChIP) assay
About 2 g of 14-day old Arabidopsis seedlings expressing 35S:BV1-ECFP was used. Seedlings treated with 50 μM MG132 overnight were used for chromatin preparation and immunoprecipitation. Immunoprecipitation was performed by adding GFP-agrose beads (GFPtrack). After washing, immune complexes were eluted from protein A beads and reverse cross-linked by incubation for at least 6 h at 65°C. Samples were treated with proteinase K for 1 h at 65°C. DNA was extracted in a final volume of 80 μL using the QIAquick PCR purification kit (Qiagen). ChIP assay was repeated with 3 biological replicates. One microliter of DNA was used for each real-time quantitative PCR with SYBR Premix Ex Taq (Applied Biosystems, USA) in the ABI7900 real-time system (Applied Biosystems, USA). Each sample was assayed in triplicate by PCR. Error bars in each graph indicate standard deviation (SD) of three biological replicates. We used ACTIN2 as an internal control. Primers used for ChIP assays are listed in S1 Table
Bimolecular fluorescence complementation (BiFC)
BiFC was performed using vectors and methods described in [25]. Full length cDNAs encoding BV1 (CaLCuV), AS1, AS2, DCP1, DCP2, DCP5 and VCS-C terminal were cloned into corresponding restriction enzyme sites of BiFC vectors. The resulting cassettes including fusion genes and constitutive promoters were cloned into pGreen binary vector HY105 and transformed into Agrobacterium. For BiFC experiments, leaves of 3-week-old N.benthamiana plants were co-infiltrated with Agrobacterium as previously described. A nuclear protein Aux/IAA from Jatropha curcas was used as a nuclear marker [28]. IAA-ECFP was produced by PCR cloning into the pBA002-ECFP vector. The agrobacterium strain carrying IAA-ECFP was co-inflitrated with strains carrying BiFC vectors into N.benthamiana leaf cells. Images were taken from cells 48 hours post-infiltration using a confocal laser scanning microscope.
In vitro decapping assay
RNAs (121nt-G16) containing a 16-guanine track at the 3′ end were transcribed with the MAXIscript T7 kit (Ambion) from a DNA fragment of At2g38280 corresponding to the 5′ untranslated region (123 nucleotides) of the transcribed mRNA. To generate cap-labeled RNAs, the ScriptCap m7G Capping system (Epicentre) was sequentially used. Decapping assays were performed at 37°C for 30 min with cap-labeled RNA and the indicated amounts of purified proteins. Reaction products were resolved by thin layer chromatography as described by Xu J. et al. [7]. The reactions were performed in in vitro decapping assay buffer (10 mM Tris-Cl, pH 7.5, 100 mMKAc, 2 mM MgAc2, 0.5 mM MnCl2, 2 mM DTT, 0.1 mM spermine, and 25 μg/mL yeast tRNA). TLS PEI cellulose F plates (Merck) were used to resolve products of decapping assays.
Results
Shuttle protein BV1 of CaLCuV activates AS2 expression in Arabidopsis
We infected Arabidopsis WT (Col-0) plants with Cabbage leaf curl Virus (CaLCuV), a model for bipartite geminivirus, and examined the symptoms of infected plants. CaLCuV infects Arabidopsis leaf tissues with high efficiency, inducing symptoms of severe stunting and leaf epinasty, suggesting inference with leaf development (Fig 1A, 1B and 1C, control plant shown in Fig 1D and 1E). To explore the molecular basis of the developmental abnormality, we determined transcript levels of a number of genes implicated in leaf development. Of the 5 genes tested only AS2 transcript levels were obviously induced by more than 4 fold in infected plants (Fig 1F). By contrast, there was no AS1 expression changes in the infected plants, although AS1 shares overlapping function with AS2 in leaf development. These results suggest that AS2 may play a role in geminivirus pathogenesis and/or virulence, in addition to its known functions in leaf stem cell determination. Expression levels of KNOTTED1-like homeobox (KNOX) genes, BREVIPEDICELLUS (BP) and KNAT2, all of which are targets of AS2, were not visibly altered upon virus infection (Fig 1F).

The considerable increase in AS2 transcript levels led us to hypothesize that AS2 may play a role in virus-host interaction. To see which viral component is responsible for the increased AS2 expression, we generated transgenic Arabidopsis plants expressing several individual CaLCuV proteins. Preliminary analysis of transgenic plants identified the virulence factor BV1 as the candidate. Fig 1G and S1 Fig show a positive correlation between BV1 expression and AS2 expression in several independent transgenic lines. This result shows that BV1 alone may induce AS2 expression, independent of any other viral factors.
The effect of BV1 on AS2 expression may be direct or indirect. To test for possible direct binding of BV1 to the AS2 genomic locus we performed Chromatin Immunoprecipitation (ChIP) assays using transgenic 35S:BV1-CFP seedlings. Fig 1H and 1I show the - 2.2 Kb to -1.6Kb (600-bp) of the AS2 promoter was more efficiently precipitated by BV1-CFP demonstrating possible binding of BV1 to this region of the AS2 promoter.
To ascertain the importance of this AS2 genomic region in BV1 interaction we constructed an AS promoter:GUS reporter gene (AS2p:GUS). Owing to the long intron in the 5'UTR of AS2 (Fig 1H) the reporter gene consisted of 4.8Kb AS2 genomic sequences (from -2.8kb to +2.0 kb, just before the initiation codon) fused to a GUS coding sequence downstream (AS2p:GUS). We transformed Arabidopsis plants of different genotypes with the AS2p:GUS fusion gene. GUS activity was high in WT (Col-0) young seedlings (S2A Fig) but weaker in mature leaves and inflorescence stems confirming previous results [20]. Infection experiment verified the AS2p:GUS activity was virus-inducible in a BV1-dependent manner (compare right and left plants in S2C Fig). By contrast, AS2p:GUS activity was low in as2-1 mutant (S2B Fig) suggesting positive auto-regulation by AS2 (Compare uninfected plants shown in S2A and S2B Fig, also infected plants shown in S2D and S2E Fig), in addition to its activation by BV1. BV1-dependent AS2p:GUS activity was much reduced when a 1.6 Kb sequence (-2.8 to -1.2Kb) of AS2 5′ upstream region was deleted from the promoter of the reporter gene (S2F Fig). This result is consistent with the chromatin immunoprecipitation data that BV1 associated with this 1.6 Kb region to activate AS2 promoter activity.
Two opposing hypothesis may be proposed to explain the activation of AS2 by BV1. One possibility is that the host protein may be induced as part of the host defense system to counter and block virus infection. The other possibility is that the AS2 induction may underpin a subversive mechanism used by the virus to compromise host defense. To discriminate between these two possibilities, we infected Arabidopsis WT and mutant plants of different genotypes: as2-1, as1-1, two related leaf developmental mutants (phenotypes shown in S3A Fig) [19] and sgs3-1, an enhanced susceptibility mutant [29]. Fig 2 shows viral infection symptoms were pronounced on WT plants as well as plants of as1-1 and sgs3-1. Moreover, consistent with published results [29] the viral symptomatic onset on sgs3-1 plants was 2–3 days earlier compared with WT control and as1-1. There was no obvious difference in viral symptomatic onset between WT and as1-1 plants. In contrast to sgs3-1, the onset of viral symptoms on as2-1 was 2–3 days later than in WT and as1-1 (Fig 2A, 12 dpi). At the late stage (28 dpi), there was no obvious symptomatic difference between WT and sgs3-1, whereas as1-1 displayed a slightly weaker symptom (Fig 2B). Much milder symptoms including curling of leaves, flowers and siliques and dwarfing of plant stature were found with infected as2-1 plants (Fig 2B and 2C). These results indicate AS2 deficiency attenuates virus replication.

To obtain quantitative data on viral titers we determined viral genomic DNA accumulation levels in CaLCuV-infected plants of different genotypes. Plants of as2-1 were more resistant to the geminivirus as compared to as1-1 and WT plants. The virus titer was much lower in systemic leaf of as2-1 either by Southern blot or quantitative PCR (Q-PCR) analysis, suggesting AS2 interferes with virus replication (S3B Fig and Fig 2D). The results so far suggested that AS2 may function as a negative regulator of host defense mechanisms and its deficiency would lead to virus resistance. If this hypothesis was correct, then AS2 overexpression should produce the opposite results. Fig 2F and S4 Fig show that this was indeed the case both in Arabidopsis and N. benthamiana. 35S:AS2 over-expression Arabidopsis and N. benthamiana plants were much more sensitive to bipartite geminiviruses, CaLCuV and Indian Cassava Mosaic virus (ICMV) respectively, with high viral DNA-A and B levels.
AS2 overexpression can block PTGS in Arabidopsis and N. benthamiana
To directly confirm AS2 suppressor activity, we expressed 35S:AS2 in the L1 line of Arabidopsis thaliana which harbors a silenced GUS gene owing to PTGS. Fig 3A shows that AS2 overepxression reactivated the silenced 35S:GUS transgene in the L1line. In reactivated plants, the increased GUS mRNA levels were accompanied by decreased GUS-related siRNA levels as compared to the parental L1 line (Fig 3B). Similar results were obtained by over-expression of a 35S:AS2-YFP fusion gene (Fig 3A and 3B). We also tested AS2 suppressor activity by transient expression in N. benthamiana using the strong viral suppressor P19 of Tomato bushy stunt virus as a control. S5 Fig confirms the suppressor activity of AS2 and less small interfering RNA (siRNA) accumulation by co-expression of AS2 also shows that its activity was weaker compared to P19. Together, these results indicate that AS2 can function as an endogenous PTGS suppressor in two different assay systems.

Cellular localization of AS2
AS2 was first characterized as a transcription factor which interacts with AS1 to regulate a number of downstream genes involved in leaf development [19]. This observation implies that the AS2 protein should be nuclear localized in order to execute its transcriptional function. On the other hand, our results above indicate that AS2 can function as a suppressor of PTGS, which is known to occur in the cytosol. This discrepancy can be resolved if AS2 can shuttle between the two cellular compartments to execute different functions. In fact, when expressed from its native promoter the AS2 protein was found in both the nuclear and the cytosolic compartment in transgenic Arabidopsis roots (Fig 4A and S6 Fig). Nuclear localization of AS2 has been previously reported [20]. Similar dual subcellular localization of the AS2 protein was seen by transient expression in tobacco leaves (S7 Fig) To identify the cellular compartment in which AS2 carries out its suppressor activity, we fused AS2 with either a Nuclear Localization Signal (NLS, Simian vacuolating virus 40, SV40) or a Nuclear Exporting Signal (NES, human immunodeficiency virus, HIV). The two AS2 localization mutants were tested for their suppressor activity. Fusion of NES did not interfere with AS2 suppressor activity (Compare Fig 5A and 5B, also 5F) but NLS fusion greatly attenuated AS2 suppressor activity (Compare Fig 5A and 5C, also 5H). Furthermore, protein level analysis indicated that AS2 and AS2 variants have comparable stability (Fig 5I). These results provide evidence that cytosolic localization of AS2 is required for its PTGS suppressor activity.

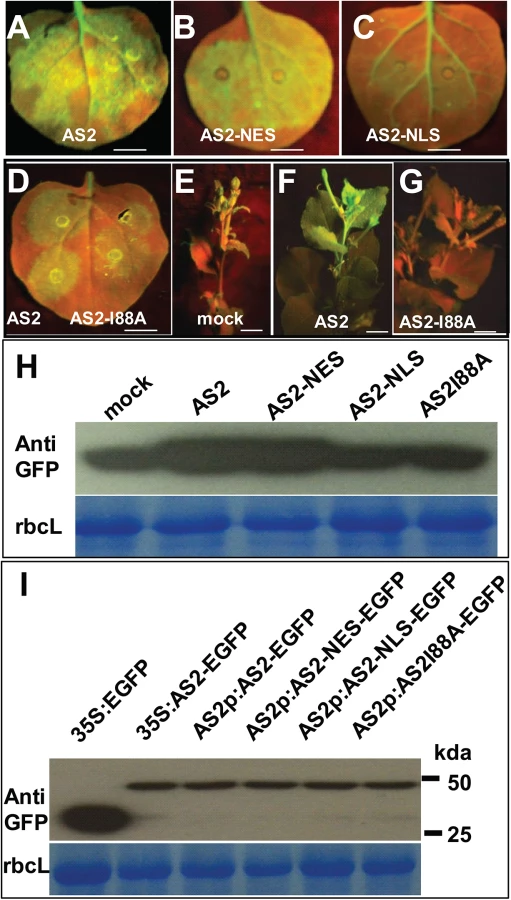
Furthermore, a mutation that converts a conserved amino acid Ile 88 to Ala blocked local and systemic silencing suppressor activity (S8 Fig and Fig 5D to 5I).
AS2 and BV1 interact with decapping proteins
Because BV1 is a nuclear-cytosol shuttle protein we asked whether BV1 was able to bind AS2 in addition to activating its encoding gene. Fig 4B shows BV1 interacted strongly with AS2 but much less with AS1 in pull-down assays in vitro. AS2 was capable of self-interaction forming dimers or even higher-order protein complexes. This self-association property was compromised by the I88A mutation (Fig 4C).
To determine the mechanism of AS2 action in PTGS we screened for possible protein-protein interaction with proteins in the PTGS pathway and also proteins involved in RNA quality control machinery. Arabidopsis DCP2 has been recently reported as a negative regulator for PTGS [10]. This observation prompted us to examine whether BV1 and AS2 may interact with protein subunits of the decapping complex [7]. Using either BV1 or AS2 as a bait we found that AS2 strongly interacted with DCP2 and weakly with DCP1 in vitro (Fig 4D). Moreover BV1 also weakly interacted with the COOH - terminal fragment of VARICOSE (VCS). The strong interaction between either BV1 or AS2 with DCP2 was further confirmed by co-immunoprecipitation experiments (Fig 4E and 4F).
To confirm the observed protein-protein interactions in vivo, we performed Bimolecular Fluorescence Complementation (BiFC) using yellow fluorescent protein (YFP). We generated constructs to express AS2, BV1 or DCP2 fused at their N - or C - termini with the N - or C - terminal portions of YFP (nYFP and cYFP). Expression constructs were introduced into N. benthamiana leaf cells by agroinfiltration, and those with complementary YFP fusions (i.e. nYFP + cYFP) were analyzed by confocal microscopy 48 hours post-infiltration.
Test proteins were examined in all possible combinations. No signal was detected in control experiments in which only one fusion protein was expressed. Co-expression of the paired BV1/AS2 and BV1/DCP2 proteins resulted in YFP fluorescence in nuclei foci indicating complex accumulation in these subcellular locations. AS2/AS2 paired protein signals were found in both nuclear and cytosolic regions (Fig 4G). By contrast, the fluorescence signal of AS2/DCP2 pair was exclusively localized in cytosolic speckles, corresponding most likely to P bodies. No signal was found in the nucleus (Fig 4H).
Next, we examined the effect of the I88A mutation on interaction with DCP2. BiFC analysis showed that amino acid I88 was important for AS2/DCP2 association in N. benthamiana cells (Fig 4H).
Since BV1 is a shuttling protein and interacts with AS2 it is possible that this viral protein may shuttle nuclear AS2 to the cytosol to function as a silencing suppressor. To this end, we co-expressed BV1-CFP in the presence of the AS2/DCP2 BiFC combination. Many cytosol P-bodies like structures with CFP/YFP co-localization were observed (Fig 4I) and the results were confirmed by statistical analysis (Fig 4J). Taken together, our results suggest that the formation of multiple protein bodies (BV1-AS2-DCP2) may play important roles in BV1 and AS2 silencing suppressor activity.
AS2 is able to promote decapping enzyme activity of DCP2
Among the various P body components only DCP2 has been shown to possess decapping activity [13]. Therefore, we examined the effect of AS2 on DCP2 decapping activity in vitro. Fig 6A shows that DCP2 decapping activity was stimulated by the addition of AS2 and this stimulating effect was abolished by the I88A mutation. There was no obvious difference in the stimulating effect on DCP2 decapping activity between WT AS2 and its localization mutant derivatives (AS2-NLS and AS2-NES) (S9 Fig), which still retained the ability to physically interact with DCP2. Taken together, these results indicate that the cytoplasmic localization of AS2 in vivo is indispensable for its silencing suppressor activity. BV1 also showed a moderate stimulating effect on DCP2 decapping activity in vitro (Fig 6A).

We performed qRT-PCR analysis to test whether mRNA turnover was altered in as2-1 and in plants expressing AS2 and derivatives. To this end, we used the native AS2 promoter to express coding sequences for AS2-EGFP, AS2-I88A-EGFP, AS2-NLS-EGFP and AS2-NES-EGFP in the as2-1 mutant background and the resulting transgenic plants were tested for mRNA stability in vivo. Expansin-Like1 (EXPL1) transcript levels were 4-fold in as2-1 mutant than that of in the WT control (S10 Fig). Moreover, Fig 6B shows the estimated half-life of EXPL1 transcripts was about 80 min in as2-1, >120 min in AS2-NLS-EGFP and in AS2-I88A-EGFP plants. These values were at least two times higher than the half-life of 40 min found in WT control samples (Fig 6B). We also observed that the half-life of EXPL1 transcripts was about 40 min for AS2-EGFP and AS2-NES-EGFP plants. These in vivo results support the notion that AS2 increases the decapping capacity of DCP2 in the cytosol to accelerate mRNA decay in vivo.
Virus resistance of AS2 cellular localization mutants
If the ability of AS2 to suppress PTGS is executed mainly in the cytosol then an AS2 mutant that is exclusively localized in the nucleus should be ineffective whereas a mutant that is localized to the cytosol should remain active. To test this hypothesis we used the transgenic lines generated above with appended NLS and NES sequences to direct the AS2 protein to specific subcellular localization. Fig 6C shows that whereas as2-1 mutant was resistant to CaLCuV the complemented line (AS2/as2-1) was as sensitive to the virus as WT. Addition of the NLS sequence to retain AS2 protein in the nucleus resulted in transgenic plants that were as sensitive as as2-1 mutant plants indicating a lack of complementation for PTGS suppression. On the other hand, addition of NES sequence to AS2 produced the same result as WT AS2 indicating that PTGS function was mediated by cytosolic AS2. Quantitative analysis on virus titer in infected plants further confirmed these results (Fig 6C)
Discussion
Roles of RNA decapping in plant virus resistance
Host decapping system plays important roles in host/viral pathogen interaction in several systems but whether increased decapping activity is a host anti-viral defense or a viral strategy to weaken host defense depends on individual cases. For some animal and human viral pathogens, e.g. positive-stranded or negative-stranded RNA viruses, the mRNA decapping machinery is an important host immune system to counter virus infection [11]. On the other hand, mRNA decapping in cytoplasmic P-bodies is also essential for virus to complete their life cycle in animal or human cells, e.g. FHV [30] and therefore, in this case, increased decapping activity would presumably aid the pathogen. So far, the only viral encoded decapping enzyme, the vaccinia D10, is synthesized at a later stage of the virus life cycle, after viral DNA replication. D10 synthesis correlates well with the shutdown of host gene expression, and deletion of this gene has been shown to cause persistence of host and viral mRNAs in infected cells [31]. The vaccina D10 decapping enzyme may help restrict host antiviral responses by accelerating host mRNA degradation during poxvirus infection. The Kaposi's sarcoma-associated herpesvirus encodes a host shutoff factor SOX which commandeers cellular mRNA turnover pathways to destroy host mRNAs following digestion by cellular exonuclease Xrn1. This result suggests that Xrn1 is poised to deplete mRNAs undergoing translation in mammalian cells [32]. Based on these observations, a very likely scenario is that viral component(s) may hijack host cellular mRNA turnover machinery to efficiently destroy or enhance host mRNAs or aberrant viral RNAs transcribed from DNA or RNA viruses. Our data here presents a new mechanism by which plant pathogens mis-regulates host cellular mRNA turnover machinery to attenuate host anti-virus defense. This finding may aid in advancing knowledge on molecular mechanisms of host-virus interaction in animal pathogens. Meanwhile, DCP2 and decapping machinery may also contribute to innate immune response by a negative feedback mechanism to restore normal homeostasis following viral infection [33].
Compromising cytoplasmic or nuclear 5'-3' exoribonuclease function enhances transgene PTGS in Arabidopsis, suggesting that these pathways compete for similar RNA substrates. The Arabidopsis DCP2 is a negative regulator for RNA silencing in Arabidopsis [10] and N. benthamiana (this study, S5 Fig) indicating its inhibitory role on PTGS. Competition between single-stranded RNA substrates between RNA quality control and PTGS ensures appropriate partitioning of RNA substrates among these RNA degradation pathways [34]. Mutants of UPF1, a gene in Nonsense-Mediated Decay pathway, showed hypersensitivity to (+) RNA virus PVX infection, further highlighting the complexity of RNA substrates partitioning for virus immune response [35].
AS2 is a new component of mRNA decapping machinery and functions as an endogenous PTGS suppressor
In higher plants, more recently also in mammals, PTGS has emerged as a vital antiviral resistance machinery that competes with the decapping system for RNA substrates. PTGS suppresses viral gene expression through the production of dsRNAs and siRNA-directed viral RNA cleavage, mainly in the cytoplasm. On the other hand, aberrant RNA substrates are depleted by decapping system inhibiting PTGS. This substrate competition implies that increased decapping activity would block PTGS and is expected to aid virus virulence.
AS2 has been well-characterized as a nuclear factor that transcriptionally regulates several downstream genes involved in leaf development. Here, we identified AS2 as a new regulatory component of Arabidopsis cytosolic P body, which activates DCP2 decapping activity in vitro (Fig 6A) and probably in vivo as reflected by the EXPL1 mRNA stability in planta (Fig 6B). We provide evidence that AS2 also functions as an endogenous PTGS suppressor: 1) AS2 is able to restore expression of a silenced transgene (Fig 3); 2) as2 mutant shows resistance to virus infection (Fig 2 and Fig 6); 3) AS2 overexpression blocks PTGS and promotes virus infection (S5 Fig and S4 Fig); 4) Cytosolic but not nuclear AS2 inhibits PTGS and siRNA accumulation (Fig 5 and S5 Fig); 5) Nuclear-localized AS2 variant is not able to rescue the virus sensitivity phenotype of as2 mutant (Fig 6C); 6) Nuclear-localized AS2 variant is not able to rescue the mRNA turnover defect of as2 (Fig 6B) although this variant retains similar capacity as WT AS2 in promoting decapping activity in vitro (S9 Fig).
The biological activity of the AS2/DCP2 complex is further reflected in similar morphological phenotype of monogenic mutants. The venetion pattern of as2-1 cotyledons was disrupted similar to that found in dcp2-1 suggesting that the two genes, AS2 and DCP2, operate in the same pathway. Furthermore, we found that the AS2 mRNA itself is feed-back regulated by the decapping complex, since AS2 transcript levels are elevated by approximately two-fold in decapping mutants [7]. This feed-back mechanism is also found in dcp1, dcp2 and vcs mutants [7].
In addition to AS2 a few endogenous silencing suppressor for PTGS in plants have been previously identified, including the cytoplasmic exoribonucleases, XRN2, XRN3 and XRN4 and DCP2 [9,10,18,36]. Given the importance of the decapping system in inhibiting PTGS we would expect any potential cellular activators of DCP2 activity to work as endogenous suppressors of PTGS as well. Likely candidates include DCP1 and DCP5 [7,8].
BV1 induces AS2 expression and translocates AS2 into cytosolic decapping body
The begomovirus DNA-B component encodes two proteins, the BV1 (nuclear shuttle protein, NSP) and the BC1 (movement protein, MP), both being required for systemic infection. BV1 which shuttles between the nucleus and the cytoplasm is believed to mediate the intracellular trafficking of viral DNA. BV1 also interacts with jasmonate singalling regulator MYC2 and plant immune receptor NIK1 to counter host resistance to pest and disease [15,25]. Here, we identified the geminivirus BV1 protein as a virulence factor that promotes mRNA decapping efficacy in P bodies so as to indirectly attenuate PTGS. BV1 is a nuclear-cytoplasmic shuttle protein and can bind single or double stranded DNA or RNA with no known sequence preference. The effects of BV1 on decapping activity are both direct and indirect. BV1 can induce expression of the nuclear gene AS2 but the AS2 induction by BV1 or virus does not lead to increased expression levels of downstream genes, e.g. KNAT etc (Fig 1F). This observation suggests that most of the induced and newly-synthesized AS2 are escorted by BV1 into the cytosol to enhance degradation of aberrant RNAs, therefore reducing the RNA substrate for siRNA biogenesis. In addition to this indirect effect, BV1 also weakly activates the activity of RNA decapping, through which some plant transcripts involved in antiviral immunity may be reduced faster. A recent report showed that BV1 may interfere with host antiviral immunity by translation suppression of host proteins [37]. Meanwhile, based on the fact that BV1 mainly interacts with AS2 in the nucleus (Fig 4G), it is also possible that DCP2 drags part of BV1 out of the nucleus
Beside of its involvement in activating decapping we cannot exclude other functions of BV1 on begomovirus pathogenesis, e.g. direct siRNA sequestration, which is dependent or independent of AS2 induction of decapping.
Because of the fundamental roles of dsRNA in initiating and maintenance of PTGS, it is not surprising that SGS3/RDR6 bodies have been identified as a common target for VSRs to suppress PTGS, e.g. the VPg of a (+) RNA virus Potyvirus [38], TRIPLE GENE BLOCK PROTEIN1 (TGBp1) of potexviruses [39], P6 of (-) RNA virus Rhabdovirus [40], p2 of Tenuivirus rice stripe virus (RSV) [41] and V2 protein of tomato yellow leaf curl virus [42]. In addition to the VSRs, plants also encode endogenous silencing repressor such as the RDR1 from Nicotiana tabacum to target the dsRNA biogenesis step [4]. The RNA decapping process could be hijacked and activated by viruses to reduce the template for synthesis of dsRNAs thereby escaping the host PTGS immune surveillance. We expect future work to uncover VSRs from other DNA and RNA viruses that may act directly on other P body components to activate its decapping activity and downregulate PTGS for the benefit of the virus.
Working model
The results in this study can be best summarized by the working model presented in Fig 7. In this model, BV1 of geminivirus translocates into the nucleus to bind and activate AS2 expression (Frame 1). The induced AS2 protein binds to BV1 (Frame 2) which shuttles it into cytoplasmic P bodies where AS2 also promotes decapping efficiency (Frame 3). BV1 can also interact with AS2 and DCP2 to enhance RNA decapping and mRNA turnover. The accelerated aberrant RNA turnover suppresses host PTGS and make plants more sensitive to geminivirus infection and replication (Frame 3).

Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Pumplin N, Voinnet O (2013) RNA silencing suppression by plant pathogens: defence, counter-defence and counter-counter-defence. Nat Rev Microbiol 11 : 745–760. doi: 10.1038/nrmicro3120 24129510
2. Vargason JM, Szittya G, Burgyan J, Hall TM (2003) Size selective recognition of siRNA by an RNA silencing suppressor. Cell 115 : 799–811. 14697199
3. Zhang X, Yuan YR, Pei Y, Lin SS, Tuschl T, et al. (2006) Cucumber mosaic virus-encoded 2b suppressor inhibits Arabidopsis Argonaute1 cleavage activity to counter plant defense. Genes Dev 20 : 3255–3268. 17158744
4. Ying XB, Dong L, Zhu H, Duan CG, Du QS, et al. (2010) RNA-dependent RNA polymerase 1 from Nicotiana tabacum suppresses RNA silencing and enhances viral infection in Nicotiana benthamiana. Plant Cell 22 : 1358–1372. doi: 10.1105/tpc.109.072058 20400679
5. Franks TM, Lykke-Andersen J (2008) The control of mRNA decapping and P-body formation. Mol Cell 32 : 605–615. doi: 10.1016/j.molcel.2008.11.001 19061636
6. Arribas-Layton M, Wu D, Lykke-Andersen J, Song H (2013) Structural and functional control of the eukaryotic mRNA decapping machinery. Biochim Biophys Acta 1829 : 580–589. doi: 10.1016/j.bbagrm.2012.12.006 23287066
7. Xu J, Yang JY, Niu QW, Chua NH (2006) Arabidopsis DCP2, DCP1, and VARICOSE form a decapping complex required for postembryonic development. Plant Cell 18 : 3386–3398. 17158604
8. Xu J, Chua NH (2009) Arabidopsis decapping 5 is required for mRNA decapping, P-body formation, and translational repression during postembryonic development. Plant Cell 21 : 3270–3279. doi: 10.1105/tpc.109.070078 19855049
9. Gazzani S, Lawrenson T, Woodward C, Headon D, Sablowski R (2004) A link between mRNA turnover and RNA interference in Arabidopsis. Science 306 : 1046–1048. 15528448
10. Thran M, Link K, Sonnewald U (2012) The Arabidopsis DCP2 gene is required for proper mRNA turnover and prevents transgene silencing in Arabidopsis. Plant J 72 : 368–377. doi: 10.1111/j.1365-313X.2012.05066.x 22639932
11. Tsai WC, Richard EL (2014) Cytoplasmic RNA Granules and Viral Infection. Annual Review of Virology 1 : 147–170.
12. Hopkins KC, McLane LM, Maqbool T, Panda D, Gordesky-Gold B, et al. (2013) A genome-wide RNAi screen reveals that mRNA decapping restricts bunyaviral replication by limiting the pools of Dcp2-accessible targets for cap-snatching. Genes Dev 27 : 1511–1525. doi: 10.1101/gad.215384.113 23824541
13. Zhou X (2013) Advances in understanding begomovirus satellites. Annu Rev Phytopathol 51 : 357–381. doi: 10.1146/annurev-phyto-082712-102234 23915133
14. Gao S, Qu J, Chua NH, Ye J (2010) A new strain of Indian cassava mosaic virus causes a mosaic disease in the biodiesel crop Jatropha curcas. Arch Virol 155 : 607–612. doi: 10.1007/s00705-010-0625-0 20224893
15. Fontes EP, Santos AA, Luz DF, Waclawovsky AJ, Chory J (2004) The geminivirus nuclear shuttle protein is a virulence factor that suppresses transmembrane receptor kinase activity. Genes Dev 18 : 2545–2556. 15489295
16. Hanley-Bowdoin L, Bejarano ER, Robertson D, Mansoor S (2013) Geminiviruses: masters at redirecting and reprogramming plant processes. Nat Rev Microbiol 11 : 777–788. doi: 10.1038/nrmicro3117 24100361
17. Yang JY, Iwasaki M, Machida C, Machida Y, Zhou X, et al. (2008) betaC1, the pathogenicity factor of TYLCCNV, interacts with AS1 to alter leaf development and suppress selective jasmonic acid responses. Genes Dev 22 : 2564–2577. doi: 10.1101/gad.1682208 18794352
18. Mourrain P, Beclin C, Elmayan T, Feuerbach F, Godon C, et al. (2000) Arabidopsis SGS2 and SGS3 genes are required for posttranscriptional gene silencing and natural virus resistance. Cell 101 : 533–542. 10850495
19. Semiarti E, Ueno Y, Tsukaya H, Iwakawa H, Machida C, et al. (2001) The ASYMMETRIC LEAVES2 gene of Arabidopsis thaliana regulates formation of a symmetric lamina, establishment of venation and repression of meristem-related homeobox genes in leaves. Development 128 : 1771–1783. 11311158
20. Iwakawa H, Ueno Y, Semiarti E, Onouchi H, Kojima S, et al. (2002) The ASYMMETRIC LEAVES2 gene of Arabidopsis thaliana, required for formation of a symmetric flat leaf lamina, encodes a member of a novel family of proteins characterized by cysteine repeats and a leucine zipper. Plant Cell Physiol 43 : 467–478. 12040093
21. Li H, Xu L, Wang H, Yuan Z, Cao X, et al. (2005) The Putative RNA-dependent RNA polymerase RDR6 acts synergistically with ASYMMETRIC LEAVES1 and 2 to repress BREVIPEDICELLUS and MicroRNA165/166 in Arabidopsis leaf development. Plant Cell 17 : 2157–2171. 16006579
22. Clough SJ, Bent AF (1998) Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J 16 : 735–743. 10069079
23. Qu J, Ye J, Fang R (2007) Artificial microRNA-mediated virus resistance in plants. J Virol 81 : 6690–6699. 17344304
24. Elmayan T, Balzergue S, Beon F, Bourdon V, Daubremet J, et al. (1998) Arabidopsis mutants impaired in cosuppression. Plant Cell 10 : 1747–1758. 9761800
25. Li R, Weldegergis BT, Li J, Jung C, Qu J, et al. (2014) Virulence Factors of Geminivirus Interact with MYC2 to Subvert Plant Resistance and Promote Vector Performance. Plant Cell 26 : 4991–5008. doi: 10.1105/tpc.114.133181 25490915
26. Ye J, Qu J, Mao HZ, Ma ZG, Rahman NE, et al. (2014) Engineering geminivirus resistance in Jatropha curcus. Biotechnol Biofuels 7 : 149. doi: 10.1186/s13068-014-0149-z 25352912
27. Ye J, Qu J, Bui HT, Chua NH (2009) Rapid analysis of Jatropha curcas gene functions by virus-induced gene silencing. Plant Biotechnol J 7 : 964–976. doi: 10.1111/j.1467-7652.2009.00457.x 19906247
28. Ye J, Liu P, Zhu C, Qu J, Wang X, et al. (2014) Identification of candidate genes JcARF19 and JcIAA9 associated with seed size traits in Jatropha. Funct Integr Genomics 14 : 757–766. doi: 10.1007/s10142-014-0400-5 25228410
29. Muangsan N, Beclin C, Vaucheret H, Robertson D (2004) Geminivirus VIGS of endogenous genes requires SGS2/SDE1 and SGS3 and defines a new branch in the genetic pathway for silencing in plants. Plant J 38 : 1004–1014. 15165191
30. Gimenez-Barcons M, Alves-Rodrigues I, Jungfleisch J, Van Wynsberghe PM, Ahlquist P, et al. (2013) The cellular decapping activators LSm1, Pat1, and Dhh1 control the ratio of subgenomic to genomic Flock House virus RNAs. J Virol 87 : 6192–6200. doi: 10.1128/JVI.03327-12 23536653
31. Liu SW, Wyatt LS, Orandle MS, Minai M, Moss B (2014) The D10 decapping enzyme of vaccinia virus contributes to decay of cellular and viral mRNAs and to virulence in mice. J Virol 88 : 202–211. doi: 10.1128/JVI.02426-13 24155373
32. Covarrubias S, Gaglia MM, Kumar GR, Wong W, Jackson AO, et al. (2011) Coordinated destruction of cellular messages in translation complexes by the gammaherpesvirus host shutoff factor and the mammalian exonuclease Xrn1. PLoS Pathog 7: e1002339. doi: 10.1371/journal.ppat.1002339 22046136
33. Li Y, Dai J, Song M, Fitzgerald-Bocarsly P, Kiledjian M (2012) Dcp2 decapping protein modulates mRNA stability of the critical interferon regulatory factor (IRF) IRF-7. Mol Cell Biol 32 : 1164–1172. doi: 10.1128/MCB.06328-11 22252322
34. Moreno AB, Martinez de Alba AE, Bardou F, Crespi MD, Vaucheret H, et al. (2013) Cytoplasmic and nuclear quality control and turnover of single-stranded RNA modulate post-transcriptional gene silencing in plants. Nucleic Acids Res 41 : 4699–4708. doi: 10.1093/nar/gkt152 23482394
35. Garcia D, Garcia S, Voinnet O (2014) Nonsense-mediated decay serves as a general viral restriction mechanism in plants. Cell Host Microbe 16 : 391–402. doi: 10.1016/j.chom.2014.08.001 25155460
36. Gy I, Gasciolli V, Lauressergues D, Morel JB, Gombert J, et al. (2007) Arabidopsis FIERY1, XRN2, and XRN3 are endogenous RNA silencing suppressors. Plant Cell 19 : 3451–3461. 17993620
37. Zorzatto C, Machado JP, Lopes KV, Nascimento KJ, Pereira WA, et al. (2015) NIK1-mediated translation suppression functions as a plant antiviral immunity mechanism. Nature: doi: 10.1038/nature14171 25707794
38. Rajamaki ML, Streng J, Valkonen JP (2014) Silencing Suppressor Protein VPg of a Potyvirus Interacts With the Plant Silencing-Related Protein SGS3. Mol Plant Microbe Interact 27 : 1199–1210. doi: 10.1094/MPMI-04-14-0109-R 25099340
39. Okano Y, Senshu H, Hashimoto M, Neriya Y, Netsu O, et al. (2014) In Planta Recognition of a Double-Stranded RNA Synthesis Protein Complex by a Potexviral RNA Silencing Suppressor. Plant Cell 26 : 2168–2183. 24879427
40. Guo H, Song X, Xie C, Huo Y, Zhang F, et al. (2013) Rice yellow stunt rhabdovirus protein 6 suppresses systemic RNA silencing by blocking RDR6-mediated secondary siRNA synthesis. Mol Plant Microbe Interact 26 : 927–936. doi: 10.1094/MPMI-02-13-0040-R 23634838
41. Du Z, Xiao D, Wu J, Jia D, Yuan Z, et al. (2011) p2 of rice stripe virus (RSV) interacts with OsSGS3 and is a silencing suppressor. Mol Plant Pathol 12 : 808–814. doi: 10.1111/j.1364-3703.2011.00716.x 21726383
42. Glick E, Zrachya A, Levy Y, Mett A, Gidoni D, et al. (2008) Interaction with host SGS3 is required for suppression of RNA silencing by tomato yellow leaf curl virus V2 protein. Proc Natl Acad Sci U S A 105 : 157–161. doi: 10.1073/pnas.0709036105 18165314
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 10
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Chronobiomics: The Biological Clock as a New Principle in Host–Microbial Interactions
- Interferon-γ: The Jekyll and Hyde of Malaria
- Crosslinking of a Peritrophic Matrix Protein Protects Gut Epithelia from Bacterial Exotoxins
- Antigen-Specific Th17 Cells Are Primed by Distinct and Complementary Dendritic Cell Subsets in Oropharyngeal Candidiasis
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy