
Identification of a Novel Lipoprotein Regulator of Spore Germination
Clostridium difficile is a spore-forming bacterium capable of causing severe diarrhea. The dormant spore-form of C. difficile is necessary to cause infection, since vegetative cells of this organism cannot survive in the presence of oxygen. Spores are difficult to eradicate because they can withstand extreme environmental conditions and chemical insults including antibiotics. However, since spores cannot grow, they must transform back into actively replicating cells once the appropriate environmental conditions are sensed through a process called germination. A key step during germination is the break-down of a specialized cell wall layer in the spore known as cortex by the SleC hydrolase. In this paper, we identify GerS as a novel lipid-modified protein that is important for C. difficile germination to occur. GerS is made at high levels during spore formation and gets packaged into mature spores. We show that GerS is required for the cortex hydrolase SleC to degrade the protective cortex layer, since a strain lacking GerS does not lose its cortex layer. Loss of GerS prevents C. difficile from causing infection in a hamster model of infection, suggesting that GerS is a novel target for drug development.
Published in the journal:
. PLoS Pathog 11(10): e32767. doi:10.1371/journal.ppat.1005239
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1005239
Summary
Clostridium difficile is a spore-forming bacterium capable of causing severe diarrhea. The dormant spore-form of C. difficile is necessary to cause infection, since vegetative cells of this organism cannot survive in the presence of oxygen. Spores are difficult to eradicate because they can withstand extreme environmental conditions and chemical insults including antibiotics. However, since spores cannot grow, they must transform back into actively replicating cells once the appropriate environmental conditions are sensed through a process called germination. A key step during germination is the break-down of a specialized cell wall layer in the spore known as cortex by the SleC hydrolase. In this paper, we identify GerS as a novel lipid-modified protein that is important for C. difficile germination to occur. GerS is made at high levels during spore formation and gets packaged into mature spores. We show that GerS is required for the cortex hydrolase SleC to degrade the protective cortex layer, since a strain lacking GerS does not lose its cortex layer. Loss of GerS prevents C. difficile from causing infection in a hamster model of infection, suggesting that GerS is a novel target for drug development.
Introduction
Clostridium difficile is a Gram-positive spore-former capable of causing diarrheal disease that can lead to fatal colitis. Disease symptoms are caused by the production of two toxins, TcdA and TcdB, which are secreted when C. difficile establishes infection in the gastrointestinal tract of mammals [1–3]. C. difficile infections have primarily been associated with individuals undergoing antibiotic therapy, but long hospitalizations, underlying comorbidities, community-acquired infections, and age-related risk factors have also been documented [4–6]. These complications lead to C. difficile disease treatment costs between $1–5 billion per year in the United States [7,8]. Of the 0.5 million C. difficile infections in the United States each year, approximately 30,000 lead to death [9]. These deaths are primarily due to recurrent C. difficile infections, which occur in ~20–30% of people that clear the first infection [9,10].
Since C. difficile is an obligate anaerobe, its endospore, or spore form, is responsible for initiating infection and mediating disease recurrence [11]. Spores are highly resistant, oxygen-tolerant, multi-layered structures composed of a tightly packed, dehydrated inner core surrounded by the inner forespore membrane, a germ cell wall, a thick modified peptidoglycan layer known as cortex, an outer forespore membrane, a series of proteinaceous layers known as the coat, and, in some spore formers, an outermost exosporium layer [12,13]. The specialized packaging of spores confers resistance to many chemical and physical insults and allows them to persist in the environment, and potentially an infected human, for long periods of time [1,14]. The dehydrated core renders spores metabolically dormant and is achieved by the displacement of water by calcium dipicolinic acid (Ca-DPA) in late stages of spore formation [15,16]. The thick cortex layer surrounding the core physically constrains its expansion and prevents hydration [17].
C. difficile infections begin when spores are ingested by a susceptible host and transit to the gastrointestinal (GI) tract [18–20]. In the GI tract, C. difficile spores sense specific bile salts, which induce them to transform into vegetative cells in a process known as germination [18,21]. While germination has been primarily characterized in the model organism Bacillus subtilis and in C. perfringens [13,22], recent studies in C. difficile have revealed that C. difficile uses a unique mechanism to regulate the initiation of spore germination [21,23–26].
While B. subtilis and C. perfringens employ highly conserved inner membrane germinant receptors to sense small molecule nutrients (germinants), which can be amino acids, sugars, and potassium ions [13], C. difficile and related Peptostreptococcaceae family members do not encode inner membrane germinant receptors [22,27]. Instead, C. difficile uses the subtilisin-like serine protease CspC as a germinant receptor [21] to sense bile salt germinants such as taurocholate [18,20,28–30]. Although C. perfringens encodes a CspC homolog and the related Csp family serine proteases, CspA and CspB [25,26], CspC is dispensable for germination in C. perfringens [25] in contrast with C. difficile [21]. Furthermore, C. perfringens CspC is catalytically competent and undergoes autoprocessing similar to other subtilisin-like serine proteases [26], whereas C. difficile CspC carries two mutations in its catalytic triad and lacks autoprocessing activity [21,23]. Unlike the catalytically competent C. perfringens CspA, C. difficile CspA is produced as a pseudoprotease that is fused to a catalytically competent CspB protease [23]. During spore formation, the C. difficile CspBA fusion protein undergoes interdomain processing, and the CspB domain is incorporated into mature spores [23].
Despite these differences, CspB in both C. perfringens and C. difficile functions to process the cortex lytic enzyme (CLE) SleC, which is found in dormant spores as the pro-SleC zymogen [21,23–26,31]. SleC degrades the cortex layer, which is essential for spore germination to proceed [32]. In the Clostridia, SleC targets the cortex-specific modification muramic-δ-lactam (MAL), which allows SleC to avoid degrading the germ cell wall of the outgrowing cell [33,34]. In B. subtilis, the cortex lytic enzymes CwlJ and SleB target MAL [16,35], although these enzymes exhibit little primary sequence homology to clostridial SleC. Cortex hydrolysis in C. difficile was recently shown to be required for Ca-DPA to be released from the core [36,37], whereas in B. subtilis, Ca-DPA is released before the cortex is hydrolyzed and actually activates CwlJ [38,39]. These observations indicate that different regulatory factors and mechanisms control germination in C. difficile relative to B. subtilis and even C. perfringens.
In this report, we describe the identification of a novel regulator of C. difficile spore germination, CD3464 in strain 630, herein referred to as GerS, which is conserved among sequenced Peptostreptococcaceae family members. Using a series of biochemical, genetic, and cell biological assays, we characterize the gerS− phenotype and identify the stage at which spore germination is arrested. We also demonstrate that GerS is essential for virulence in hamsters.
Results
Identification of GerS as a novel regulator of C. difficile spore germination
We previously conducted RNA-Seq analyses of C. difficile sporulation-specific sigma factor mutants to identify gene products that might be required for spore formation and/or germination [40,41]. We hypothesized that highly expressed genes induced during sporulation would likely encode proteins that regulate spore formation and/or germination. gerS (CD3464) and alr2 are the second and sixth most highly expressed, sporulation-induced genes [40,41], respectively, and their gene products have not been previously characterized. Interestingly, alr2 is encoded downstream of gerS (Fig 1A), and these genes are part of a σE-activated operon (S1 Fig, [42]). alr2 encodes a putative alanine racemase that in Bacillus. spp. converts L-alanine to D-alanine and reduces the sensitivity of spores to L-alanine germinant [43–45]. gerS is predicted to encode a lipoprotein that appears to be unique to the Peptostreptococcaceae family (Fig 1B).

To test whether Alr2 or GerS regulate C. difficile sporulation and/or spore germination, we constructed TargeTron gene disruption mutants in alr2 and gerS (S2 Fig). Analysis of the alr2 and gerS mutants by phase contrast microscopy revealed that both strains produced phase-bright spores (Fig 1C). Fluorescence microscopy analyses indicated that alr2− and gerS− forespores appeared to develop similar to wild type (S3 Fig). However, when the alr2− and gerS− strains were tested for functional spore formation, the gerS mutant failed to produce detectable heat-resistant spores, while the alr2 mutant produced wildtype levels of heat-resistant spores (Fig 1C). Western blot analysis confirmed that the gerS mutant was defective in producing GerS, while the alr2 mutant produced wildtype levels of GerS (Fig 1D).
The inability of the gerS mutant to produce heat-resistant spores could be due to heat sensitivity [17,46] or a general defect in spore germination. To distinguish between these possibilities, we isolated spores from wild type and the gerS and alr2 mutants and tested their ability to germinate following heat-treatment using a plate-based assay. No obvious defect in spore morphology was apparent when gerS− and alr2− spores were visualized by phase contrast microscopy (Fig 2A). However, alr2− spores germinated at wildtype levels, whereas gerS− spores exhibited an ~5-log defect in spore germination relative to wild type (Fig 2B). Heating wildtype and alr2− spores to 60°C for 30 min had no impact on spore germination, whereas heat treatment reduced the germination efficiency of gerS− spores by three-fold (p < 0.05, Fig 2B). Although a similar heat treatment potentiates Bacillus sp. spore germination [47–49], this effect has not been observed in C. difficile [37,50]. Western blot analyses verified that GerS is packaged into wildtype and alr2− mutant spores but not gerS− spores (Fig 2C). Taken together, these results strongly suggest that gerS− spores have a significant germination defect that is slightly heat sensitive. Furthermore, the germination defect of gerS mutant spores is unlikely to be caused by polar effects on alr2 expression, since Alr2 itself is dispensable for heat-resistant spore formation.

Complementation of the gerS mutant
To validate that the gerS mutant phenotype was due to absence of GerS, we complemented the mutant in trans by ectopically expressing gerS from its native promoter(s). Since gerS transcription originates from the proximal promoter (P1) directly upstream of gerS [42] and possibly the distal promoter upstream of acpS (P2, S4 Fig), we constructed gerS complementation constructs in which gerS transcription originates from the proximal promoter (P1, single) or from both P1 and P2 promoters (dual, including the two genes upstream of gerS). Heat resistance analyses revealed that the single and dual promoter complementation constructs both restored heat-resistant spore formation to wildtype levels (S4 Fig). Western blot analyses indicated that the dual and single promoter gerS complementation constructs restored GerS production to wildtype levels in the gerS− background (S4 Fig). We chose to use the dual promoter complementation construct, since it produced GerS levels that were most similar to wildtype carrying empty vector.
GerS regulates cortex hydrolysis
We next sought to determine why gerS mutant spores exhibit such a strong germination defect. We first considered that GerS could affect the rate of spore germination, since a lipoprotein, GerD, controls the speed of germination in B. subtilis [51,52]. Loss of B. subtilis GerD results in an ~20-fold germination defect after a 15 hr incubation with germinant; however, after 48 hr, it resembles wild type [52]. Although B. subtilis GerD exhibits no homology to C. difficile GerS, we assessed whether gerS mutants germinated after prolonged incubation. After 48 hrs of germination on BHIS plates containing taurocholate, the change in number of colonies formed following gerS− spore germination was minimal and appeared to arise from spontaneous germination [53].
We next wondered whether GerS regulates germinant accessibility in C. difficile spores, since GerP in B. subtilis and B. anthracis facilitates the interaction of germinants to inner membrane germination receptors, potentially by altering coat permeability [54–56]. Bacillus spp. gerP mutants exhibit slower germination and require higher levels of germinant in order to achieve equivalent levels of germination as wild type. To test whether C. difficile gerS mutant spores are differentially sensitive to germinant, we compared the effect of increasing concentrations of taurocholate germinant on gerS− spores carrying empty vector (gerS−/EV) relative to wildtype carrying empty vector (WT/EV) and gerS− spores carrying the wildtype complementation construct (gerS−/gerS). gerS− spores exhibited a similar dose-dependent germination response to taurocholate as wild type and the gerS complementation spores, although gerS− spores still had an ~5-log defect in spore germination in the presence of 1% taurocholate (Fig 3A), which leads to germination levels equivalent to those obtained by plating on BHIS plates containing 0.1% taurocholate.

This result suggested that gerS− spores can sense germinant similar to wildtype spores. Consistent with this finding, no difference in the levels of CspC germinant receptor and CspB germination protease were observed between the strains by Western blotting (Fig 3B), and no difference in CspB-mediated processing of SleC in response to increasing amounts of germinant was observed. Since CspB-mediated processing of C. perfringens SleC activates its cortex hydrolase function [26], and CspB-mediated processing of C. difficile SleC is required for optimal spore germination [23], these results suggested that GerS acts after SleC-mediated cortex hydrolysis.
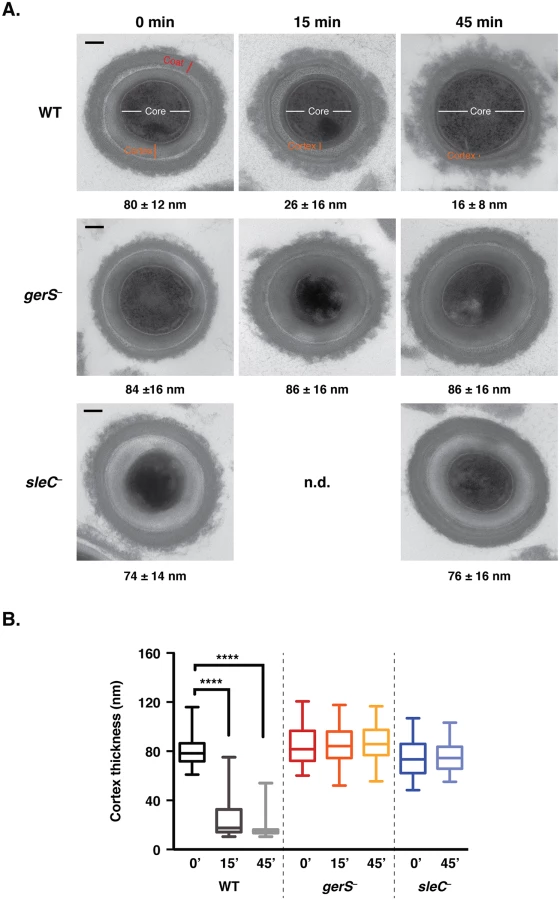
In order to test this hypothesis, we developed a transmission electron microscopy (TEM) assay to visualize and quantify cortex hydrolysis. Although cortex hydrolysis can be measured biochemically [24,36,57], it is difficult to obtain the amount of spores required for these analyses using the JIR8094 strain background. For the TEM assay, wildtype, gerS−, and sleC− spores were exposed to germinant for 45 min, and cortex thickness was measured over time for a minimum of 50 spores per time point (Fig 4). Within 15 min of exposure to germinant, cortex thinning was visible in wildtype spores (Fig 4A), and the average thickness decreased by 3-fold (p < 0.0001, Fig 4B). Cortex thickness decreased even further at 45 min. In contrast with wild type, no change in cortex thickness was observed in either sleC− or gerS− spores even after 45 min of incubation with germinant (Fig 4B). Thus, even though taurocholate induces CspB-mediated pro-peptide removal from the pro-SleC zymogen in gerS− spores (Fig 3), SleC does not appear to be active (Fig 4). These results suggest that GerS may regulate SleC activity through an unknown post-translational mechanism or by altering the availability of the SleC substrate, MAL, in the cortex layer.
If SleC activity is indeed dependent on the presence of GerS, it should be possible to bypass the need for SleC-mediated cortex hydrolysis by artificially germinating gerS− spores. During artificial germination, a reducing agent, thioglycollate, is added to permeabilize the coat layers followed by lysozyme addition to degrade the cortex layer [13,58]. Treatment of wildtype, gerS−, and sleC− spores with thioglycollate and lysozyme restored outgrowth to gerS− and sleC− spores (Fig 5); no statistically significant difference in artificial germination between wildtype, gerS−, sleC− spores was observed. In contrast, wildtype spores germinated much more efficiently than the mutants upon “natural” exposure to taurocholate. The small amount of germination observed in sleC− spores is likely due to spontaneous germination [37,46], which can occur even in the absence of the germinant receptor [21].

Ca-DPA release does not occur in the absence of GerS
Since recent studies have shown that Ca-DPA release immediately follows cortex hydrolysis [36,37], we next tested whether the gerS mutant releases Ca-DPA in response to germinant (S5 Fig). Whereas wildtype spores released ~80% of their Ca-DPA stores in response to germinant, gerS− spores released <5% of their Ca-DPA stores. Since wildtype and gerS− spores contained similar amounts of Ca-DPA (88%, S5 Fig), Ca-DPA storage does not appear to be affected by the gerS mutation, consistent with the recent observation that Ca-DPA release depends on cortex hydrolysis in C. difficile [36,37] in contrast with B. subtilis [13,38].
GerS localizes to a “coat-extractable” (CE) fraction
Having shown that GerS is important for SleC activity, we next wanted to understand how GerS carries out its function. We first tested whether GerS and SleC were present in the “coat-extractable” (CE) fraction. To this end, we subjected wildtype and gerS− mutant spores to a mild boric acid decoating treatment [57] to generate a CE supernatant fraction and a pellet fraction. Western blotting of these fractions revealed that both SleC and GerS co-localized to the CE fraction in wildtype spores but not to the pellet fraction, which consists of decoated spore lysate (Fig 6). Analysis of germination regulators CspC and CspB revealed that they also are concentrated in the CE fraction of wildtype and gerS− spores. These results indicate that the germination regulators GerS, SleC, CspB, and CspC are located in a similar cellular fraction. Since C. perfringens SleC has been shown to localize to the cortex using immunoelectron microscopy [59], and CspB and SleC were recently reported to localize to a CE fraction in C. perfringens [31], these results imply that the CE fraction includes cortex and outer forespore membrane proteins in C. perfringens and likely in C. difficile (although it remains formally possible that SleC does not localize to the cortex region in C. difficile). Importantly, the coat morphogenetic protein SpoIVA [60] localized exclusively to the CE fraction of wildtype and gerS− mutant spores, whereas the forespore-localized germination protease (GPR) [40,61,62] was found exclusively in the pellet fraction of these spores.

Secretion, but not lipidation, of GerS is required for its function
Since GerS is predicted to be a lipoprotein based on the presence of a putative N-terminal signal peptide containing a lipobox [63–65], we tested whether GerS undergoes lipidation and whether its function depends on lipidation and/or secretion. The signal peptide of lipoproteins directs their transport across membranes after which Lgt, a prolipoprotein diacylglyerol transferase, adds a diacylglycerol group to the lipobox cysteine via a thioether bond. Following lipidation, the lipoprotein signal peptidase (Lsp) cleaves off the signal peptide, and the lipoprotein inserts into the plasma membrane in Gram-positive bacteria [63]. Since mutation of the conserved cysteine residue in the lipobox to serine is sufficient to prevent lipidation [63–65], we complemented the gerS mutant with a construct that produces GerS carrying a cysteine 22 to serine (C22S) mutation. We also complemented the gerS mutant with a construct that deletes the GerS signal peptide sequence to prevent secretion (ΔSP, Figs 1B and 7A). The C22S complementation strain produced heat-resistant spores at levels comparable to wild type, whereas the ΔSP complementation strain exhibited a >4-log decrease in functional (heat-resistant) spore formation relative to wildtype (H.R., Fig 7B). These results suggest that secretion but not lipidation is required for GerS to activate cortex hydrolysis. Western blot analyses of the complementation strains revealed that only full-length GerS was detectable in the C22S strain, whereas both full-length and cleaved GerS were observed in wild type and the wildtype gerS complementation strain. These observations strongly suggest that Cysteine 22 is important for cleavage of the signal peptide, similar to other lipoproteins [63–65]. Neither full-length nor cleaved GerS could be detected in ΔSP sporulating cells, implying that loss of the signal peptide leads to destabilization of GerS (Fig 7B). To test this hypothesis, we measured gerS transcript levels in the complementation strains by qRT-PCR relative to wildtype carrying empty vector. The gerS− complementation strains all produced an excess of gerS transcripts relative to wildtype carrying empty vector; this over-expression is likely due to the multi-copy nature of the pMTL83151 plasmid used for complementation (S6 Fig). Thus, GerS lacking its signal peptide appears to be unstable in the mother cell cytosol of sporulating cells.

Consistent with our analyses of sporulating cells, purified spores from the C22S strain germinated at wildtype levels, while ΔSP spores exhibited an ~4-log defect in germination relative to wild type (Fig 7B). Only full-length GerS was detected in C22S spores, whereas only cleaved GerS was detected in wildtype spores carrying empty vector and gerS− spores carrying the wildtype complementation construct. GerS was undetectable in ΔSP spores. Taken together, these analyses suggest that GerS secretion across the mother cell-derived membrane is necessary for GerS function, while lipidation and signal peptide removal are dispensable for GerS to activate cortex hydrolysis.
To test whether alterations to the signal peptide affected the heat sensitivity of gerS− mutant spores, we heated spores for 30 min at 60°C prior to plating on media containing taurocholate germinant. As expected, heat treatment had no impact on the germination of wildtype spores carrying empty vector or wildtype complementation spores (S7 Fig). No difference in spore germination between untreated and heat-treated C22S or ΔSP spores was observed. In contrast, gerS− mutant spores carrying empty vector showed a statistically significant decrease in the number of germinating spores following heat treatment (p < 0.01), similar to results with gerS− spores (Fig 2B).
GerS is necessary to cause disease in hamsters
Since bile acid-mediated germination has previously been shown to be important for C. difficile pathogenesis [21], we tested whether gerS− could cause disease in the hamster model of C. difficile infection (CDI). Hamsters inoculated with gerS− spores carrying empty vector had a 100% survival 7 days post inoculation, whereas wildtype spores carrying empty vector resulted in 50% survival at the same time point (Fig 8). Inoculation with the gerS−/gerS construct resulted in 100% of the hamsters being euthanized by day 5 after inoculation. These results indicate that the gerS mutant’s in vitro germination defect correlates with an inability to cause disease in a hamster model of CDI.

Since a possible explanation for the greater lethality of the gerS−/gerS strain might be a faster rate of spore germination relative to wildtype spores carrying empty vector, we analyzed the rate of germination initiation by measuring the decrease in optical density at 600 nm when spores form the complementation strains were exposed to taurocholate germinant (S8 Fig). This assay revealed that the C22S and gerS−/gerS strains germinated with similar kinetics as wild type, albeit slightly less efficiently, whereas no major change in OD600 was observed for ΔSP and gerS−/EV spores, as expected.
Discussion
Recent studies of C. difficile spore germination have uncovered a unique signaling pathway for sensing bile salt germinants and initiating spore outgrowth relative to previously studied organisms [12]. Although the germination regulators SleC and the Csp family proteases are conserved between C. difficile and C. perfringens [22], they can have different functions and/or activities in these organisms [21,23–26]. In this study, we identified a novel protein specific to C. difficile and related Peptostreptococcaceae family members that functions as a critical regulator of SleC cortex hydrolase activity and is essential for germination in vivo in a hamster model of infection under the conditions tested. While C. difficile strain JIR8094 contains mutations in the flagellar operon that impacts motility and toxin gene expression, a gerS mutant nevertheless causes significantly less disease than wild type JIR8094.
In particular, we showed that GerS regulates SleC activity downstream of CspB-mediated processing of SleC. This processing event had previously been thought to be sufficient to activate SleC’s cortex hydrolase activity, since studies in C. perfringens showed that CspB-mediated cleavage of the pro-SleC zymogen was necessary for SleC to degrade cortex fragments in vitro [26,57], and loss of C. difficile CspB protease activity markedly reduced SleC processing and spore germination [23]. However, unlike C. perfringens SleC, full-length C. difficile SleC can degrade cortex fragments in vitro [33], calling into question why SleC does not automatically degrade cortex in dormant spores. It will be important in future studies to precisely determine the impact of pro-peptide removal in activating SleC function in vitro and in C. difficile.
How then does GerS regulate SleC activity? Our results indicate that gerS is under the control of the mother cell-specific sigma factor σE (Fig 1A) and thus should be produced in the mother cell cytosol [40,41,61]. Deletion of the signal peptide from GerS destabilizes GerS in sporulating cells (Fig 7 and S6 Fig). This observation is consistent with the notion that GerS is transported across the outer forespore membrane into the cortex region during sporulation (Fig 9); more evidence is nevertheless needed to test this hypothesis. Since mutation of the invariant cysteine in the GerS lipobox prevents signal peptide removal but does not affect GerS function (Fig 7), the signal peptide of GerS C22S may insert into the outer forespore membrane where it can apparently function like lipidated wildtype GerS (Fig 7). Although mutation of the invariant lipobox cysteine frequently disrupts lipoprotein function [51,66,67], lipidation of some bacterial lipoproteins can be dispensable for their activity because they remain embedded in the plasma membrane through their signal peptide [63,68]. These observations suggest that GerS may exert its function on the surface of the outer forespore membrane facing the cortex (Fig 9). Notably, SleC activity also appears to be localized to this region, since TEM analyses of germinating wildtype spores revealed that cortex thinning initiates at the outer forespore membrane and radiates inward in C. difficile (Fig 4). While more studies are clearly needed to determine the exact locations of SleC and GerS in mature spores, our results suggest that these germination regulators may be localized to the outer forespore membrane, which likely fractionates with the coat (Fig 9), raising the intriguing possibility that GerS retains SleC at this site. It will be interesting to determine in future work whether GerS acts as a direct or indirect activator of SleC and/or whether GerS is necessary for SleC to recognize its cortex substrate, for example by controlling the predicted modification of NAM residues to muramic acid δ-lactam in the cortex [34,35], particularly since GerS lacks homology to other proteins aside from its lipobox.

Although GerS carries a signal peptide that directs its secretion across mother cell-derived membranes (Fig 9), SleC lacks a canonical N-terminal signal sequence. Thus, it is unclear how SleC is transported across the outer forespore membrane so that it can bind its cortex substrate. Similarly, how CspB is transported across this mother cell-derived membrane to cleave pro-SleC, and how CspC is presumably translocated across this membrane to activate CspB, remains unknown, since both CspB and CspC lack a canonical signal sequence.
Intriguingly, all the known germinant regulators in C. difficile, CspC, CspBA, SleC, and GerS, are produced under the control of mother cell-specific sigma factors [40,42,61]. In contrast, the germination regulators of B. subtilis, the GerAA-AC complex and GerD, are all under the control of the forespore-specific sigma factor σG [69]. These observations suggest that the topology of germination signaling differs significantly between C. difficile and B. subtilis. In B. subtilis, the germinant receptors are located in the inner forespore membrane [70–72], since decoated spores germinate efficiently [73]. Germinant sensing stimulates release of Ca-DPA from the core through the inner forespore membrane-localized channel SpoVAC [13]; Ca-DPA then activates the CwlJ cortex lytic enzyme [46]. In C. difficile, the germinant receptor CspC, the germination protease CspB, the cortex hydrolase SleC, and the lipoprotein GerS, all localize to the CE fraction (Fig 5). Thus, these regulators are unlikely to be associated with the inner forespore membrane in contrast with B. subtilis. Since SleC cortex hydrolase activation precedes Ca-DPA release in C. difficile (S5 Fig, [21,37]), the germinant signal appears to travel from the outside-in, whereas in B. subtilis the signal appears to travel from the inside-out.
While our genetic analyses demonstrated that GerS is a key germination regulator in C. difficile, they also showed that Alr2, a putative alanine racemase, is dispensable for germination (Fig 2). It should be noted that this observation does not exclude the possibility that Alr2 could alter the sensitivity of C. difficile spores to L-alanine, which has been shown to function as a co-germinant for C. difficle in vitro [74]. In B. anthracis and B. cereus, the Alr2 homolog alanine racemase converts L-alanine, a known germinant, to D-alanine to reduce the sensitivity of spores to L-alanine germinant [43,44,75]. Whether Alr2 modulates C. difficile spore germination remains to be determined, in particular whether it functions in suppressing germination. However, Howerton and Abel-Santos have shown that D-alanine is not an inhibitor of C. difficile spore germination [74], suggesting that Alr2 plays little role in C. difficile spore germination or has an as-yet-unknown function.
In summary, in identifying a novel germination regulator conserved in C. difficile and other Peptostreptococcaceae family members, our study reveals yet another difference between the regulation of spore germination in C. difficile relative to B. subtilis and C. perfringens. While many unanswered questions remain, cortex hydrolysis in C. difficile appears to be subject to an additional level of regulation during germination by GerS. Thus, GerS could be a potential target for inhibiting C. difficile disease transmission, especially given its limited conservation in spore-forming organisms.
Materials and Methods
Bacterial strains and growth conditions
C. difficile strains are listed in Table 1 and derive from the parent strain JIR8094, an erythromycin-sensitive derivative of the sequenced clinical isolate 630. C. difficile strains were grown on solid BHIS media, which consists of brain heart infusion media supplemented with yeast extract and 0.1% (w/v) L-cysteine [76]. BHIS media was supplemented with taurocholate (TA; 0.1% w/v), thiamphenicol (5–10 μg/mL), kanamycin (50 μg/mL), cefoxitin (16 μg/mL), FeSO4 (50 μM), and/or erythromycin (10 μg/mL) as indicated. Cultures were grown at 37°C, under anaerobic conditions using a gas mixture containing 85% N2, 5% CO2, and 10% H2.

Sporulation was induced on solid media containing 70% BHIS and 30% SMC (90 g BactoPeptone, 5 g protease peptone, 1 g NH4SO4, 1.5 g Tris base, 15 g agar per liter) [77], as previously described. For strains carrying pMTL83151 derivatives, sporulation was induced on 70 : 30 media containing 5 μg/mL thiamphenicol.
HB101/pRK24 strains were used for conjugations and BL21(DE3) strains were used for protein production. E. coli strains (Table 1) were routinely grown at 37°C, shaking at 225 rpm in Luria-Bertani broth (LB). Media was supplemented with chloramphenicol (20 μg/mL), ampicillin (50 μg/mL), or kanamycin (30 μg/mL) as indicated.
E. coli strain construction
E. coli strains are listed in Table 1; all primers are listed in S1 Table. For disruption of gerS and alr2, a modified plasmid containing the retargeting group II intron, pCE245 (a gift from C. Ellermeier, University of Iowa), was used as the template. Primers for amplifying the targeting sequence from the template carried flanking regions specific for each gene target and are listed as follows: gerS (#1122, 1123, 1124 and 532, the EBS Universal primer (Sigma Aldrich) and alr2 (#1385, 1386, 1385 and 532). The resulting retargeting sequences were digested with BsrGI and HindIII and cloned into pJS107 [21], which is a derivative of pJIR750ai (Sigma Aldrich). The ligations were transformed into DH5α and confirmed by sequencing. The resulting plasmids were used to transform HB101/pRK24.
To construct the dual promoter complementation construct (S4 Fig), primers #1464 and 1466 were used to amplify an ~1.8 kB construct containing acpS, CD3465, gerS, and 360 bp upstream of acpS using 630 genomic DNA as the template. To construct the single promoter complementation construct, primers #1667 and 1466 were used to amplify gerS containing 367 bp upstream of gerS using 630 genomic DNA as the template. The gerS C22S and ΔSP complementation constructs were made using PCR splicing by overlap extension (SOE). For C22S, primer pair #1464 and 1734 was used to amplify the 5’ SOE product (containing the C22S mutation), while primer pair #1733 and 1466 was used to amplify the 3’ SOE product (containing the C22S mutation). The resulting fragments were mixed together, and flanking primers #1464 and 1466 were used to generate the dual promoter complementation construct that encodes the C22S mutation. To construct the ΔSP complementation construct, SOE primers #1464 and 1727 were used to generate a 5’ fragment; primers #1726 and 1466 were used for the 3’ SOE product. The flanking primers #1464 and 1466 were used to amplify the ΔSP complementation construct, which deletes the region encoding residues 2–22. All complementation constructs were digested with NotI and XhoI and ligated into pMTL83151 digested with the same enzymes.
To construct a strain producing GerS for antibody production, primer pairs #1278 and 1173 were used to amplify gerS lacking the signal peptide sequence using genomic DNA as the template. The resulting PCR products were digested with NdeI and XhoI, ligated to pET28a, and transformed into DH5α. The resulting pET28a-gerS plasmid was used to transform BL21(DE3) for protein production. To construct a strain for generating mouse anti-CspC antibodies, primer pairs #1128 and 1166 were used to amplify codon-optimized cspC using pJS148 as the template. The resulting PCR products were digested with NdeI and SacI, ligated to pET22b-CPDSacI [78], and transformed into DH5a. The resulting pET22b-cspC_opt-CPD was transformed into BL21(DE3) for protein production.
Bioinformatic analyses
Homologs of C. difficile 630 GerS (CD3464) were identified using NCBI psi-blast. Homologs identified in Peptostreptococcaceae family members gave an e-value < e-52, whereas the next closest homolog in a Clostridium spp. gave an e-value > e-27. When GerS lacking its N-terminal signal peptide was used in the psi-blast search, the difference in e-value cut-offs was < e-52 for Peptostreptococcaceae family members and the next closest homolog in a Clostridium spp. gave an e-value > e-23.
C. difficile strain construction
C. difficile strains were constructed using TargeTron-based gene disruption as described previously (S2 Fig, [40,79]). TargeTron constructs in pJS107 were conjugated into C. difficile using E. coli HB101/pRK24 as the donor strain. HB101/pRK24 strains containing the appropriate pJS107 construct were grown aerobically to exponential phase in 2.5 mL of LB supplemented with ampicillin (50 μg/mL) and chloramphenicol (10 μg/mL). Cultures were pelleted, transferred into the anaerobic chamber, and resuspended with 1.5 mL of late-exponential phase C. difficile JIR8094 cultures (grown anaerobically in BHIS broth). The resulting cell mixture was plated as seven 100 μL spots onto pre-dried, pre-reduced BHIS agar plates. After overnight incubation, all growth was harvested from the BHIS plates, resuspended in 2.5 mL pre-reduced BHIS, and twenty-one 100 μL spots per strain were plated onto three BHIS agar plates supplemented with thiamphenicol (10 μg/mL), kanamycin (50 μg/mL), and cefoxitin (16 μg/mL) to select for C. difficile containing the pJS107 plasmid. After 24–48 hrs of anaerobic growth, single colonies were patched onto BHIS agar supplemented with thiamphenicol (10 μg/mL), kanamycin (50 μg/mL), and FeSO4 (50 μM) to induce the ferredoxin promoter of the group II intron system. After overnight growth, patches were transferred to BHIS agar plates supplemented with erythromycin (10 μg/mL) for 24–72 hrs to select for cells with activated group II intron systems. Erythromycin-resistant patches were struck out for isolation onto the same media and individual colonies were screened by colony PCR for a 2 kb increase in the size of gerS (primer pair #1212 and 1173) and alr2 (primer pair #1352 and 1359) (S2 Fig).
C. difficile complementation
HB101/pRK24 donor strains carrying the appropriate complementation construct were grown in LB containing ampicillin (50 μg/mL) and chloramphenicol (20 μg/mL) at 37°C, 225 rpm, under aerobic conditions, for 6 hrs. C. difficile recipient strains gerS− and alr2− containing group II intron disruptions, were grown anaerobically in BHIS broth at 37°C with gentle shaking for 6 hrs. HB101/pRK24 cultures were pelleted at 2500 rpm for 5 min and the supernatant was removed. Pellets were transferred to the anaerobic chamber and gently resuspended in 1.5 mL of the appropriate C. difficile culture. The resulting mixture was inoculated onto pre-dried, pre-reduced BHIS agar plates, as seven 100 μL spots for 12 hrs. All spots were collected anaerobically and resuspended in 1 mL PBS. One hundred microliters of the resulting suspension was spread onto pre-dried, pre-reduced BHIS agar plates supplemented with thiamphenicol (10 μg/mL), kanamycin (50 μg/mL), and cefoxitin (10 μg/mL), five plates per conjugation. Plates were monitored for colony growth for 24–72 hrs. Individual colonies were struck out for isolation and analyzed for complementation using the heat resistance assay to test for functional spore formation and Western blot analysis. A minimum of two independent clones from each complementation strain was phenotypically characterized.
Sporulation
C. difficile strains were grown from glycerol stocks on BHIS plates supplemented with TA (0.1% w/v), or with TA and thiamphenicol (5 μg/mL) for strains carrying pMTL83151-derived vectors. Colonies that arose on BHIS agar plates were then used to inoculate 70 : 30 agar plates containing 5 μg/mL thiamphenicol for 17–24 hrs depending on the assay. Sporulating cells were harvested into PBS, pelleted, and resuspended in PBS for visualization by phase contrast microscopy and further processing as needed.
Heat resistance assay
C. difficile strains were induced to sporulate as described above and functional (heat-resistant) spore formation was measured as previously described [41] with the following exceptions. After 24 hrs of growth, cells were harvested into 600 μL of pre-reduced PBS. The sample was divided into two tubes. One tube was exposed to 60°C for 25–30 minutes. Heat-treated and untreated cells were serially diluted, and dilutions were plated on pre-reduced BHIS-TA plates. After ~20 hrs colonies were counted, and cell counts were determined. The percent of heat-resistant spores was determined based on the ratio of heat-resistant cells to total cells, and heat-resistance efficiencies were determined based on the ratio of heat-resistant cells for a strain compared to wildtype. Results are based on a minimum of three biological replicates. The raw data for the heat resistance assay is provided in S2 Table.
Spore purification
Sporulation was induced by growing C. difficile strains on 70 : 30 plates (with 5 μg/mL thiamphenicol when appropriate for 2–3 days, and spores were harvested in ice-cold water as previously described [23,76] with the following modifications. Spores were incubated on ice overnight following multiple water washes. The following day, they were pelleted and treated with DNase (New England Biolabs) at 37°C for 30 minutes. Following DNAse treatment, the spores were purified on a HistoDenz (Sigma Aldrich) gradient, evaluated for purity by phase contrast microscopy, and the optical density of the suspension was measured at OD600. Spores were stored in water at 4°C.
Germination assay
Approximately 1 x 107 spores (equivalent of 0.35 OD600 units) were re-suspended in 100 μL of water. Ten microliters of the suspension was serially diluted in PBS, and dilutions were plated onto pre-reduced BHIS-TA. After ~22 hrs, colonies arising from germinated spores were counted. Germination efficiency represents the number of CFUs produced by germinating spores of a given strain relative to wild type. Results are based on a minimum of three biological replicates. The remaining 90 μL of the spores were pelleted and resuspended in EBB buffer for Western blot analyses.
To assess the effect of heat treatment on spore viability, the procedure above was followed, with the exception that 2 x 107 spores were re-suspended in 200 μL of water (equivalent of 0.7 OD600 units) and the sample was divided into two. One half was incubated at 60°C for 30 min, while the other half was left untreated.
The effect of taurocholate concentration on spore germination efficiency was determined by re-suspending ~4 x 107 spores (~1.4 OD600 units) in 160 μL of water in triplicate. Two hundred microliters of BHIS was added to each spore suspension. Ninety microliters of this suspension was added to either 10 μL of water, 0.1% TA, 1% TA, or 10% TA (to give a final concentration of 0.01% TA, 0.1% TA, or 1% TA). The samples were incubated for 20 min at 37°C, and a 10 μL aliquot was removed for 10-fold serial dilutions into PBS. Ten microliters of the serial dilutions were plated on BHIS to determine the number of spores that had initiated germination. The serial dilutions for untreated and 1% TA-treated spores were also plated on BHIS-TA plates to determine the maximum level of spore germination. Spore germination was maximal following exposure to 1% TA. The remaining samples were pelleted for Western blot analysis.
Antibody production
The anti-GerS used in this study was raised against GerS-His6 lacking its signal peptide in rabbits by Cocalico Biologicals (Reamstown, PA). The anti-CspC mouse antibodies were raised against recombinant untagged CspC in mice by Cocalico Biologicals (Reamstown, PA). GerS-His6 was purified from E. coli strains #1112 using Ni2+-affinity resin as previously described [23]. Recombinant, untagged CspC was purified using the autoprocessing CPD tag as previously described [78] followed by gel filtration [23].
Western blot analyses
C. difficile cell pellets were processed as previously described [40,60]. Briefly, pellets were freeze-thawed three times, diluted in EBB buffer (8 M urea, 2 M thiourea, 4% (w/v) SDS, 2% (v/v) β-mercaptoethanol), and incubated at 95°C for 20 min with vortexing every 5 min. C. difficile spores (~1 x 106) were resuspended in EBB buffer, which can extract proteins in all layers of the spore including the core. Samples were centrifuged for 5 min at 15,000 rpm and 4X sample buffer (40% (v/v) glycerol, 1 M Tris pH 6.8, 20% (v/v) β-mercaptoethanol, 8% (w/v) SDS, and 0.04% (w/v) bromophenol blue), was added. Samples were incubated again at 95°C for 5–15 minutes with vortexing followed by centrifugation for 5 min at 15,000 rpm. The samples were resolved by SDS-PAGE and transferred to Millipore Immobilon-FL membrane. The membranes were blocked in Odyssey Blocking Buffer. Rabbit polyclonal rabbit anti-GerS or anti-GPR [40] antibodies were used at a 1 : 1,000 dilution; anti-CspB [23] antibodies were used at a 1 : 2,500 dilution, and the anti-SleC [23] antibody was used at a 1 : 5,000 dilution. Polyclonal mouse anti-SpoIVA [80] and anti-CspC antibodies were used at 1 : 2,500 dilutions. IRDye 680CW and 800CW infrared dye-conjugated secondary antibodies were used at 1 : 20,000 dilutions. The Odyssey LiCor CLx was used to detect secondary antibody infrared fluorescence emissions.
RNA processing
RNA from WT/EV, gerS−/EV, gerS−/gerS, gerS−/C22S, and gerS−/ΔSP strains grown for 24 hrs on 70 : 30 sporulation media containing thiamphenicol (5 μg/mL) was extracted for qRT-PCR analyses of the gerS transcript. RNA was extracted using a FastRNA Pro Blue Kit (MP Biomedical) and a FastPrep-24 automated homogenizer (MP Biomedical). Contaminating genomic DNA was depleted using three successive DNase treatments, and mRNA enrichment was done using an Ambion MICROBExpress Bacterial mRNA Enrichment Kit (Invitrogen). Samples were tested for genomic DNA contamination using quantitative PCR for rpoB. Enriched RNA was reverse transcribed using Super Script First Strand cDNA Synthesis Kit (Invitrogen) with random hexamer primers.
Quantitative RT-PCR
Transcript levels of gerS and rpoB (housekeeping gene) were determined from cDNA templates prepared from 3 biological replicates of WT/EV, spo0A−/EV, gerS−/EV, gerS−/gerS, gerS−/C22S, and gerS−/ΔSP strains. Gene-specific primer pairs for gerS (#1278 and #1173), alr2 (#1668 and #1356), and rpoB [40] were used. Quantitative real-time PCR was performed (as described by [41]. Briefly, using MaximaTM SYBRTM Green qPCR Master Mix (Thermo Scientific), 50 nM of gene specific primers, and an ABI PRISM 7900HT Sequence Detection System (Applied Biosystems). Transcript levels were normalized to the housekeeping gene rpoB using the standard curve method.
Fluorescence microscopy
For live cell fluorescence microscopy studies, C. difficile strains were harvested in PBS, pelleted, and resuspended in PBS. For characterization of mutant phenotypes, cells were resuspended in PBS containing 1 μg/mL FM4-64 (Molecular Probes) and 15 μg/mL Hoechst 33342 (Molecular Probes). All live bacterial suspensions (4 μL) were added to a freshly prepared 1% agarose pad on a microscope slide, covered with a 22 x 22 mm #1 coverslip and sealed with VALAB (1 : 1:1 of vaseline, lanolin, and beeswax) as previously described [41,81].
DIC and fluorescence microscopy was performed using a Nikon PlanApo Vc 100x oil immersion objective (1.4 NA) or a Nikon PlanApo Vc 60x oil immersion objective (1.4 NA) on a Nikon Eclipse Ti2000 epifluorescence microscope. Multiple fields for each sample were acquired with an EXi Blue Mono camera (QImaging) with a hardware gain setting of 1.0 and driven by NIS-Elements software (Nikon). Images were subsequently imported into Adobe Photoshop CS6 for minimal adjustments in brightness/contrast levels and pseudocoloring.
Artificial germination assay
Artificial germination was determined using thioglycollate and lysozyme treatment. About 1 x 106 spores were pelleted at 8,000 RPM for 3 min, resuspended in 250mM thioglycollate, and incubated at 50°C for 30 min based on previously methods developed [21,58]. Spores were washed with 150 μL of PBS, pelleted, resuspended in 150 μL (2 mg/mL) lysozyme and incubated at 37°C for 15 min. Equivalent numbers of spores for each strain were incubated at the indicated temperatures without thioglycollate or lysozyme treatment for the untreated sample. The spore samples were plated on either BHIS or BHIS-TA. Natural germination represents the number of spores in the untreated sample that outgrew to form colonies on BHIS-TA media. Artificial Germination represents the number of thioglycollate/lysozyme-treated spores that germinated and outgrew to form colonies on BHIS media.
Decoating assay
About 1 x 107 spores (~0.35 OD600) were pelleted and resuspended in 30 μL of decoat buffer (0.1 M H3BO3 pH 10.0, 1% SDS, 2% β-ME) [57]. The sample was incubated for 30 min at 37°C and then pelleted. The supernatant, representing the “coat-extractable” (CE) fraction, was removed, and the pellet was washed in 20 μL decoat buffer and incubated for 10 min. The sample was re-pelleted, and the supernatant was added to the CE; 40 μL of EBB was added to the pooled fractions. The decoated spores were re-suspended in 90 μL EBB to produce the cell lysate (pellet) fraction. For the input fraction, representing whole spore lysate, equal numbers of spores were pelleted and resuspended in 90 μL EBB. All samples were boiled for a minimum of 15 min, followed by centrifugation and sample resuspension. Fractions were pelleted one more time before the samples were resolved by SDS-PAGE and analyzed by Western blotting as described above.
Cortex hydrolysis assay
Cortex hydrolysis was analyzed by transmission electron microscopy for untreated spores (0’) and 15’ and 45’ after germinant addition. About 4 x 107 spores (1.4 OD600 units) were resuspended in 160 μL of water in triplicate. Two hundred microliters of BHIS was added to each spore suspension. Forty microliters of water was added to one sample, while 40 μL of 10% taurocholate (w/v) was added to the remaining samples. The spores were incubated under anaerobic conditions for the indicated time point after which a small sample was removed for visualizing by phase-contrast microscopy and plating on BHIS and BHIS-TA. The remainder of the sample was pelleted and re-suspended in osmium tetroxide fixative for TEM analysis as previously described [60]. TEM grids for each sample analyzed were prepared as previously described [41]. A minimum of 50 spore pictures chosen at random were analyzed for each strain observed. To account for asymmetrical spore shapes, two orthologous cortex lengths were measured such that a minimum and maximum cortex thickness was obtained for every spore. Cortex length was defined as the distance between the outer most germ cell wall and the cortex outer edge. The minimum and maximum measurements were averaged for each spore and the upper and lower values were discarded. The cortex length reported represents the average of these measurements.
Ca-DPA release assay
To evaluate the amount of Ca-DPA released in response to germinant relative to the total amount of Ca-DPA observed in the spore core, a modified Ca-DPA release assay was adopted from [21]. About 2x107 spores from each strain were re-suspended in (i) 1 mL of germination buffer (0.3 m M (NH4)2SO4, 6.6 mM KH2PO4, 15 mM NaCl, 59.5 mM NaHCO3, and 35.2 mM Na2HPO4) and incubated at 37°C for 30 min (background); (ii) 1 mL of germination buffer containing 10% freshly prepared taurocholate and 10 mM glycine and incubated at 37°C for 30 min (DPA release); (iii) 1 mL of germination buffer and incubated at 100°C for 1 hr (total DPA). After incubation, samples were spun down at 15,000 RPM for 2 min. 700 μL was transferred to UV clear cuvettes, and the A270 was determined. The % Ca-DPA release was determined by subtracting the background DPA release value from the germinant containing DPA release value and dividing by total DPA. Total DPA measured in wild type was set as 100% total DPA.
O.D. kinetics assay
Approximately 1 x 107 spores (0.48 OD600 units) were resuspended in BHIS to a total volume of 1100. The sample was divided into two: 540 μL was added to a cuvette containing 60 μL of water, while the other sample was added to a cuvette containing 60 μL of 10% taurocholate. The samples were mixed, and the OD600 was measured every 3–6 mins for 45 min.
Virulence studies
All animal studies were performed with prior approval from the Texas A&M University Institutional Animal Care and Use Committee. Female Syrian golden hamsters (80–120 g) were housed and tested for C. difficile susceptibility as previously described [21,82]. To induce C. difficile infection, hamsters were gavaged with 30 mg/kg clindamycin prior to C. difficile spore inoculum. After 5 days, 10 hamsters per C. difficile strain tested were gavaged with 1,000 spores and monitored for signs of disease. Hamsters showing disease symptoms (wet tail, poor fur coat and lethargy) were euthanized by CO2 asphyxia followed by thoracotomy as a secondary means of death.
Ethics statement
All animal procedures were performed with prior approval from the Texas A&M Institutional Animal Care and Use Committee under the approved Animal Use Protocol number 2014–0085. Animals showing signs of disease were euthanized by CO2 asphyxia followed by thoracotomy as a secondary means of death, in accordance with Panel on Euthanasia of the American Veterinary Medical Association. Texas A&M University’s approval of Animal Use Protocols is based upon the United States Government’s Principles for the Utilization and Care of Vertebrate Animals Used in Testing, Research and Training and complies with all applicable portions of the Animal Welfare Act, the Public Health Service Policy for the Humane Care and Use of Laboratory Animals, and all other federal, state, and local laws which impact the care and use of animals.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Carroll K, Bartlett J (2011) Biology of Clostridium difficile: implications for epidemiology and diagnosis. Annual review of microbiology 65 : 501–521. doi: 10.1146/annurev-micro-090110-102824 21682645
2. Rupnik M, Wilcox M, Gerding D (2009) Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nature reviews Microbiology 7 : 526–536. doi: 10.1038/nrmicro2164 19528959
3. Warny M, Pepin J, Fang A, Killgore G, Thompson A, et al. (2005) Toxin production by an emerging strain of Clostridium difficile associated with outbreaks of severe disease in North America and Europe. Lancet 366 : 1079–1084. 16182895
4. Badger VO, Ledeboer NA, Graham MB, Edmiston CE Jr. (2012) Clostridium difficile: epidemiology, pathogenesis, management, and prevention of a recalcitrant healthcare-associated pathogen. JPEN J Parenter Enteral Nutr 36 : 645–662. doi: 10.1177/0148607112446703 22577120
5. Freeman J, Bauer M, Baines S, Corver J, Fawley W, et al. (2010) The changing epidemiology of Clostridium difficile infections. Clinical microbiology reviews 23 : 529–549. doi: 10.1128/CMR.00082-09 20610822
6. Gupta A, Khanna S (2014) Community-acquired Clostridium difficile infection: an increasing public health threat. Infect Drug Resist 7 : 63–72. doi: 10.2147/IDR.S46780 24669194
7. (CDC) CfDC (2013) Antibiotic Resistance Threats in the United States. http://wwwcdcgov/drugresistance/threat-report-2013:.
8. Dubberke E (2012) Clostridium difficile infection: the scope of the problem. J Hosp Med 7 Suppl 3: S1–4.
9. Lessa FC, Mu Y, Bamberg WM, Beldavs ZG, Dumyati GK, et al. (2015) Burden of Clostridium difficile infection in the United States. N Engl J Med 372 : 825–834. doi: 10.1056/NEJMoa1408913 25714160
10. Louie TJ, Miller MA, Mullane KM, Weiss K, Lentnek A, et al. (2011) Fidaxomicin versus vancomycin for Clostridium difficile infection. N Engl J Med 364 : 422–431. doi: 10.1056/NEJMoa0910812 21288078
11. Deakin LJ, Clare S, Fagan RP, Dawson LF, Pickard DJ, et al. (2012) The Clostridium difficile spo0A gene is a persistence and transmission factor. Infect Immun 80 : 2704–2711. doi: 10.1128/IAI.00147-12 22615253
12. Paredes-Sabja D, Shen A, Sorg JA (2014) Clostridium difficile spore biology: sporulation, germination, and spore structural proteins. Trends Microbiol 22 : 406–416. doi: 10.1016/j.tim.2014.04.003 24814671
13. Setlow P (2014) Germination of spores of Bacillus species: what we know and do not know. J Bacteriol 196 : 1297–1305. doi: 10.1128/JB.01455-13 24488313
14. Kelly C, LaMont J (2008) Clostridium difficile—more difficult than ever. The New England journal of medicine 359 : 1932–1940. doi: 10.1056/NEJMra0707500 18971494
15. Errington J (2003) Regulation of endospore formation in Bacillus subtilis. Nature reviews Microbiology 1 : 117–126. 15035041
16. Peter S (2003) Spore germination. Current Opinion in Microbiology 6.
17. Setlow P (2006) Spores of Bacillus subtilis: their resistance to and killing by radiation, heat and chemicals. Journal of applied microbiology 101 : 514–525. 16907802
18. Koenigsknecht MJ, Theriot CM, Bergin IL, Schumacher CA, Schloss PD, et al. (2015) Dynamics and establishment of Clostridium difficile infection in the murine gastrointestinal tract. Infect Immun 83 : 934–941. doi: 10.1128/IAI.02768-14 25534943
19. Lawley T, Clare S, Walker A, Goulding D, Stabler R, et al. (2009) Antibiotic treatment of Clostridium difficile carrier mice triggers a supershedder state, spore-mediated transmission, and severe disease in immunocompromised hosts. Infection and immunity 77 : 3661–3669. doi: 10.1128/IAI.00558-09 19564382
20. Theriot CM, Koenigsknecht MJ, Carlson PE Jr., Hatton GE, Nelson AM, et al. (2014) Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat Commun 5 : 3114. doi: 10.1038/ncomms4114 24445449
21. Francis MB, Allen CA, Shrestha R, Sorg JA (2013) Bile acid recognition by the Clostridium difficile germinant receptor, CspC, is important for establishing infection. PLoS Pathog 9: e1003356. doi: 10.1371/journal.ppat.1003356 23675301
22. Paredes-Sabja D, Setlow P, Sarker M (2011) Germination of spores of Bacillales and Clostridiales species: mechanisms and proteins involved. Trends Microbiol 19 : 85–94. doi: 10.1016/j.tim.2010.10.004 21112786
23. Adams CM, Eckenroth BE, Putnam EE, Doublie S, Shen A (2013) Structural and functional analysis of the CspB protease required for Clostridium spore germination. PLoS Pathog 9: e1003165. doi: 10.1371/journal.ppat.1003165 23408892
24. Paredes-Sabja D, Setlow P, Sarker MR (2009) SleC is essential for cortex peptidoglycan hydrolysis during germination of spores of the pathogenic bacterium Clostridium perfringens. J Bacteriol 191 : 2711–2720. doi: 10.1128/JB.01832-08 19218389
25. Paredes-Sabja D, Setlow P, Sarker MR (2009) The protease CspB is essential for initiation of cortex hydrolysis and dipicolinic acid (DPA) release during germination of spores of Clostridium perfringens type A food poisoning isolates. Microbiology 155 : 3464–3472. doi: 10.1099/mic.0.030965-0 19628563
26. Shimamoto S, Moriyama R, Sugimoto K, Miyata S, Makino S (2001) Partial characterization of an enzyme fraction with protease activity which converts the spore peptidoglycan hydrolase (SleC) precursor to an active enzyme during germination of Clostridium perfringens S40 spores and analysis of a gene cluster involved in the activity. Journal of bacteriology 183 : 3742–3751. 11371539
27. Yutin N, Galperin MY (2013) A genomic update on clostridial phylogeny: Gram-negative spore formers and other misplaced clostridia. Environ Microbiol 15 : 2631–2641. doi: 10.1111/1462-2920.12173 23834245
28. Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, et al. (2015) Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517 : 205–208. doi: 10.1038/nature13828 25337874
29. Giel J, Sorg J, Sonenshein A, Zhu J (2010) Metabolism of bile salts in mice influences spore germination in Clostridium difficile. PLoS One 5.
30. Sorg JA, Sonenshein AL (2008) Bile salts and glycine as cogerminants for Clostridium difficile spores. J Bacteriol 190 : 2505–2512. doi: 10.1128/JB.01765-07 18245298
31. Banawas S, Korza G, Paredes-Sabja D, Li Y, Hao B, et al. (2015) Location and stoichiometry of the protease CspB and the cortex-lytic enzyme SleC in Clostridium perfringens spores. Food Microbiol 50 : 83–87. doi: 10.1016/j.fm.2015.04.001 25998819
32. Burns DA, Heap JT, Minton NP (2010) SleC is essential for germination of Clostridium difficile spores in nutrient-rich medium supplemented with the bile salt taurocholate. J Bacteriol 192 : 657–664. doi: 10.1128/JB.01209-09 19933358
33. Gutelius D, Hokeness K, Logan SM, Reid CW (2013) Functional Analysis of SleC from Clostridium difficile: an essential lytic transglycosylase involved in spore germination. Microbiology.
34. Makino S, Moriyama R (2002) Hydrolysis of cortex peptidoglycan during bacterial spore germination. Med Sci Monit 8: RA119–127. 12070452
35. Popham DL, Helin J, Costello CE, Setlow P (1996) Muramic lactam in peptidoglycan of Bacillus subtilis spores is required for spore outgrowth but not for spore dehydration or heat resistance. Proc Natl Acad Sci U S A 93 : 15405–15410. 8986824
36. Francis MB, Allen CA, Sorg JA (2015) Spore cortex hydrolysis precedes DPA release during Clostridium difficile spore germination. J Bacteriol.
37. Wang S, Shen A, Setlow P, Li YQ (2015) Characterization of the dynamic germination of individual Clostridium difficile spores using Raman spectroscopy and differential interference contrast microscopy. J Bacteriol.
38. Paidhungat M, Ragkousi K, Setlow P (2001) Genetic requirements for induction of germination of spores of Bacillus subtilis by Ca(2+)-dipicolinate. J Bacteriol 183 : 4886–4893. 11466292
39. Setlow P (2003) Spore germination. Curr Opin Microbiol 6 : 550–556. 14662349
40. Fimlaid KA, Bond JP, Schutz KC, Putnam EE, Leung JM, et al. (2013) Global Analysis of the Sporulation Pathway of Clostridium difficile. PLoS Genet 9: e1003660. doi: 10.1371/journal.pgen.1003660 23950727
41. Pishdadian K, Fimlaid KA, Shen A (2015) SpoIIID-mediated regulation of sigma(K) function during Clostridium difficile sporulation. Mol Microbiol 95 : 189–208. doi: 10.1111/mmi.12856 25393584
42. Saujet L, Pereira FC, Serrano M, Soutourina O, Monot M, et al. (2013) Genome-wide analysis of cell type-specific gene transcription during spore formation in Clostridium difficile. PLoS Genet 9: e1003756. doi: 10.1371/journal.pgen.1003756 24098137
43. Chesnokova ON, McPherson SA, Steichen CT, Turnbough CL Jr. (2009) The spore-specific alanine racemase of Bacillus anthracis and its role in suppressing germination during spore development. J Bacteriol 191 : 1303–1310. doi: 10.1128/JB.01098-08 19074397
44. Venir E, Del Torre M, Cunsolo V, Saletti R, Musetti R, et al. (2014) Involvement of alanine racemase in germination of Bacillus cereus spores lacking an intact exosporium. Arch Microbiol 196 : 79–85. doi: 10.1007/s00203-013-0946-y 24346000
45. Yan X, Gai Y, Liang L, Liu G, Tan H (2007) A gene encoding alanine racemase is involved in spore germination in Bacillus thuringiensis. Arch Microbiol 187 : 371–378. 17165028
46. Paidhungat M, Setlow B, Driks A, Setlow P (2000) Characterization of spores of Bacillus subtilis which lack dipicolinic acid. J Bacteriol 182 : 5505–5512. 10986255
47. Leuschner RG, Lillford PJ (1999) Effects of temperature and heat activation on germination of individual spores of Bacillus subtilis. Lett Appl Microbiol 29 : 228–232. 10583749
48. Levinson HS, Hyatt MT (1970) Effects of temperature on activation, germination, and outgrowth of Bacillus megaterium spores. J Bacteriol 101 : 58–64. 4983656
49. Yi X, Setlow P (2010) Studies of the commitment step in the germination of spores of Bacillus species. J Bacteriol 192 : 3424–3433. doi: 10.1128/JB.00326-10 20435722
50. Dembek M, Stabler RA, Witney AA, Wren BW, Fairweather NF (2013) Transcriptional analysis of temporal gene expression in germinating Clostridium difficile 630 endospores. PLoS One 8: e64011. doi: 10.1371/journal.pone.0064011 23691138
51. Mongkolthanaruk W, Robinson C, Moir A (2009) Localization of the GerD spore germination protein in the Bacillus subtilis spore. Microbiology 155 : 1146–1151. doi: 10.1099/mic.0.023853-0 19332816
52. Pelczar PL, Igarashi T, Setlow B, Setlow P (2007) Role of GerD in germination of Bacillus subtilis spores. J Bacteriol 189 : 1090–1098. 17122337
53. Heeg D, Burns DA, Cartman ST, Minton NP (2012) Spores of Clostridium difficile clinical isolates display a diverse germination response to bile salts. PLoS One 7: e32381. doi: 10.1371/journal.pone.0032381 22384234
54. Behravan J, Chirakkal H, Masson A, Moir A (2000) Mutations in the gerP locus of Bacillus subtilis and Bacillus cereus affect access of germinants to their targets in spores. J Bacteriol 182 : 1987–1994. 10715007
55. Butzin XY, Troiano AJ, Coleman WH, Griffiths KK, Doona CJ, et al. (2012) Analysis of the effects of a gerP mutation on the germination of spores of Bacillus subtilis. J Bacteriol 194 : 5749–5758. doi: 10.1128/JB.01276-12 22904285
56. Carr KA, Janes BK, Hanna PC (2010) Role of the gerP operon in germination and outgrowth of Bacillus anthracis spores. PLoS One 5: e9128. doi: 10.1371/journal.pone.0009128 20161744
57. Kumazawa T, Masayama A, Fukuoka S, Makino S, Yoshimura T, et al. (2007) Mode of action of a germination-specific cortex-lytic enzyme, SleC, of Clostridium perfringens S40. Biosci Biotechnol Biochem 71 : 884–892. 17420590
58. Kamiya S, Yamakawa K, Ogura H, Nakamura S (1989) Recovery of spores of Clostridium difficile altered by heat or alkali. J Med Microbiol 28 : 217–221. 2926793
59. Miyata S, Kozuka S, Yasuda Y, Chen Y, Moriyama R, et al. (1997) Localization of germination-specific spore-lytic enzymes in Clostridium perfringens S40 spores detected by immunoelectron microscopy. FEMS microbiology letters 152 : 243–247. 9231416
60. Putnam EE, Nock AM, Lawley TD, Shen A (2013) SpoIVA and SipL are Clostridium difficile spore morphogenetic proteins. J Bacteriol 195 : 1214–1225. doi: 10.1128/JB.02181-12 23292781
61. Pereira FC, Saujet L, Tome AR, Serrano M, Monot M, et al. (2013) The Spore Differentiation Pathway in the Enteric Pathogen Clostridium difficile. PLoS Genet 9: e1003782. doi: 10.1371/journal.pgen.1003782 24098139
62. Sussman MD, Setlow P (1991) Cloning, nucleotide sequence, and regulation of the Bacillus subtilis gpr gene, which codes for the protease that initiates degradation of small, acid-soluble proteins during spore germination. J Bacteriol 173 : 291–300. 1840582
63. Hutchings MI, Palmer T, Harrington DJ, Sutcliffe IC (2009) Lipoprotein biogenesis in Gram-positive bacteria: knowing when to hold 'em, knowing when to fold 'em. Trends Microbiol 17 : 13–21. doi: 10.1016/j.tim.2008.10.001 19059780
64. Zuckert WR (2014) Secretion of bacterial lipoproteins: through the cytoplasmic membrane, the periplasm and beyond. Biochim Biophys Acta 1843 : 1509–1516. doi: 10.1016/j.bbamcr.2014.04.022 24780125
65. Buddelmeijer N (2015) The molecular mechanism of bacterial lipoprotein modification—how, when and why? FEMS Microbiol Rev 39 : 246–261. doi: 10.1093/femsre/fuu006 25670733
66. Cooper GR, Moir A (2011) Amino acid residues in the GerAB protein important in the function and assembly of the alanine spore germination receptor of Bacillus subtilis 168. J Bacteriol 193 : 2261–2267. doi: 10.1128/JB.01397-10 21378181
67. Pelczar PL, Setlow P (2008) Localization of the germination protein GerD to the inner membrane in Bacillus subtilis spores. J Bacteriol 190 : 5635–5641. doi: 10.1128/JB.00670-08 18556788
68. Leskela S, Wahlstrom E, Kontinen VP, Sarvas M (1999) Lipid modification of prelipoproteins is dispensable for growth but essential for efficient protein secretion in Bacillus subtilis: characterization of the Lgt gene. Mol Microbiol 31 : 1075–1085. 10096076
69. Wang S, Setlow B, Conlon E, Lyon J, Imamura D, et al. (2006) The forespore line of gene expression in Bacillus subtilis. Journal of molecular biology 358 : 16–37. 16497325
70. Hudson KD, Corfe BM, Kemp EH, Feavers IM, Coote PJ, et al. (2001) Localization of GerAA and GerAC germination proteins in the Bacillus subtilis spore. J Bacteriol 183 : 4317–4322. 11418573
71. Korza G, Setlow P (2013) Topology and accessibility of germination proteins in the Bacillus subtilis spore inner membrane. J Bacteriol 195 : 1484–1491. doi: 10.1128/JB.02262-12 23335419
72. Paidhungat M, Setlow P (2001) Localization of a germinant receptor protein (GerBA) to the inner membrane of Bacillus subtilis spores. J Bacteriol 183 : 3982–3990. 11395462
73. Paidhungat M, Setlow P (2000) Role of ger proteins in nutrient and nonnutrient triggering of spore germination in Bacillus subtilis. J Bacteriol 182 : 2513–2519. 10762253
74. Howerton A, Ramirez N, Abel-Santos E (2011) Mapping interactions between germinants and Clostridium difficile spores. J Bacteriol 193 : 274–282. doi: 10.1128/JB.00980-10 20971909
75. Yasuda Y, Kanda K, Nishioka S, Tanimoto Y, Kato C, et al. (1993) Regulation of L-alanine-initiated germination of Bacillus subtilis spores by alanine racemase. Amino Acids 4 : 89–99. doi: 10.1007/BF00805804 24190560
76. Sorg JA, Dineen SS (2009) Laboratory maintenance of Clostridium difficile. Curr Protoc Microbiol Chapter 9: Unit 9A 1.
77. Permpoonpattana P, Tolls E, Nadem R, Tan S, Brisson A, et al. (2011) Surface layers of Clostridium difficile endospores. Journal of bacteriology 193 : 6461–6470. doi: 10.1128/JB.05182-11 21949071
78. Shen A, Lupardus PJ, Morell M, Ponder EL, Sadaghiani AM, et al. (2009) Simplified, enhanced protein purification using an inducible, autoprocessing enzyme tag. PLoS One 4: e8119. doi: 10.1371/journal.pone.0008119 19956581
79. Heap JT, Pennington OJ, Cartman ST, Carter GP, Minton NP (2007) The ClosTron: a universal gene knock-out system for the genus Clostridium. J Microbiol Methods 70 : 452–464. 17658189
80. Kevorkian Y, Shirley DJ, Shen A (2015) Regulation of Clostridium difficile spore germination by the CspA pseudoprotease domain Biochimie. Epub ahead of print (S0300-9084(15)00231-X)
81. Fimlaid KA, Jensen O, Donnelly ML, Siegrist MS, Shen A (2015) A conserved channel regulates multiple stages of Clostridium difficile spore morphogenesis. PLoS Genet Accepted.
82. Sambol SP, Tang JK, Merrigan MM, Johnson S, Gerding DN (2001) Infection of hamsters with epidemiologically important strains of Clostridium difficile. J Infect Dis 183 : 1760–1766. 11372028
83. Dineen SS, Villapakkam AC, Nordman JT, Sonenshein AL (2007) Repression of Clostridium difficile toxin gene expression by CodY. Mol Microbiol 66 : 206–219. 17725558
84. Heap J, Pennington O, Cartman S, Minton N (2009) A modular system for Clostridium shuttle plasmids. Journal of microbiological methods 78 : 79–85. doi: 10.1016/j.mimet.2009.05.004 19445976
85. Underwood S, Guan S, Vijayasubhash V, Baines S, Graham L, et al. (2009) Characterization of the sporulation initiation pathway of Clostridium difficile and its role in toxin production. Journal of bacteriology 191 : 7296–7305. doi: 10.1128/JB.00882-09 19783633
86. Griffiths KK, Zhang J, Cowan AE, Yu J, Setlow P (2011) Germination proteins in the inner membrane of dormant Bacillus subtilis spores colocalize in a discrete cluster. Mol Microbiol 81 : 1061–1077. doi: 10.1111/j.1365-2958.2011.07753.x 21696470
87. Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, et al. (2011) Integrative genomics viewer. Nat Biotechnol 29 : 24–26. doi: 10.1038/nbt.1754 21221095
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 10
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Chronobiomics: The Biological Clock as a New Principle in Host–Microbial Interactions
- Interferon-γ: The Jekyll and Hyde of Malaria
- Crosslinking of a Peritrophic Matrix Protein Protects Gut Epithelia from Bacterial Exotoxins
- Antigen-Specific Th17 Cells Are Primed by Distinct and Complementary Dendritic Cell Subsets in Oropharyngeal Candidiasis
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy