
BCG Skin Infection Triggers IL-1R-MyD88-Dependent Migration of EpCAM CD11b Skin Dendritic cells to Draining Lymph Node During CD4+ T-Cell Priming
The arrival of bacilli in the lymph node is a bottleneck for initiating T cell responses to mycobacteria but remains poorly studied. To address this we used a mouse model to track the entry of cells and bacteria into the lymph node during skin infection with Mycobacterium bovis BCG, the live tuberculosis vaccine. We identified a population of migratory Dendritic cells that transport bacilli from the skin into the lymph node and which engage CD4+ T cells therein. The mobilization of Dendritic cells from the skin, and with these cells the transport of mycobacteria into the lymph node, was regulated by cytokines, in particular Interleukin–1. This was also dependent on MyD88, an adaptor molecule downstream of the Interleukin–1 receptor. We also found a requirement for MyD88 in driving Dendritic cells to the lymph node that was both inherent and extrinsic to the migrating cell. These findings bare consequences for our understanding of how T-cell responses are initiated during microbial challenge in the skin and hold promises for improving vaccines of low-to-modest efficacy such as BCG, which rely on such responses.
Published in the journal:
. PLoS Pathog 11(10): e32767. doi:10.1371/journal.ppat.1005206
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1005206
Summary
The arrival of bacilli in the lymph node is a bottleneck for initiating T cell responses to mycobacteria but remains poorly studied. To address this we used a mouse model to track the entry of cells and bacteria into the lymph node during skin infection with Mycobacterium bovis BCG, the live tuberculosis vaccine. We identified a population of migratory Dendritic cells that transport bacilli from the skin into the lymph node and which engage CD4+ T cells therein. The mobilization of Dendritic cells from the skin, and with these cells the transport of mycobacteria into the lymph node, was regulated by cytokines, in particular Interleukin–1. This was also dependent on MyD88, an adaptor molecule downstream of the Interleukin–1 receptor. We also found a requirement for MyD88 in driving Dendritic cells to the lymph node that was both inherent and extrinsic to the migrating cell. These findings bare consequences for our understanding of how T-cell responses are initiated during microbial challenge in the skin and hold promises for improving vaccines of low-to-modest efficacy such as BCG, which rely on such responses.
Introduction
Lymph nodes (LNs) make use of lymphatic drainage and a specialized microanatomy to facilitate productive encounters between antigen-laden Dendritic cells (DCs) and naïve T cells [1]. As sentinel phagocytes that reconnoiter for infection, DCs employ an array of pattern-recognition receptors (PRRs) to sense microbes or their metabolites [2]. Microbial triggering of PRRs unleashes an intracellular signaling cascade in DCs that culminates in enhanced antigen presentation, up-regulation of co-stimulatory molecules and cytokine production. This activation process enables DCs upon engaging a naïve T cell clone to direct the expansion and differentiation of that clone into an armed, effector T-cell population [3]. These dynamic cellular interactions that unfold in the LN mark the initiation of cell-mediated immunity that are critical for host resistance to infection.
Mycobacterium tuberculosis, the etiological agent of tuberculosis, is second only to HIV/AIDS as the largest cause of death in the World due to a single microorganism [4]. Host resistance to mycobacteria relies heavily on cell-mediated immunity and IFN-γ [5,6]. Mounting a robust immune response mediated by Th1 effector cells is thus an important outcome for successful tuberculosis vaccination. In this context, cutaneous infection with the attenuated strain of M. bovis called Bacille Calmette-Guérin (BCG) is the only vaccination available against M. tuberculosis, but is lacking in clinical efficacy. Improving BCG is a valid strategy to promote global control of tuberculosis [7] and understanding how CD4+ T cells are primed by BCG in situ sets a rational basis towards the latter. However, there is a paucity of information regarding the initial steps that ensue upon BCG infection and which culminate in the generation of a Th1 response. In particular, the channeling of antigen from the BCG-inoculation site in the skin to the draining LN (DLN) remains poorly studied.
A large body of data implicate DCs in the active transport of antigen from the periphery to the DLN [1] but the identification of several DC sub-populations in the skin [8] has added to the complexity of this event. Although molecular mechanisms of motility have been investigated in several studies [9,10], the in vivo contribution of PRRs, their signaling pathways and cytokines await full elucidation during DC migration triggered by infection. Here we developed a method to track the movement of cells and mycobacteria from the footpad to the popliteal DLN to study this during BCG infection. We found that migratory EpCAMlow CD11bhigh skin DCs were the main DC sub-population mobilized during the transport of BCG to the DLN. This process, associated with the priming of antigen-specific CD4+ T cells, was dependent on Interleukin–1 receptor (IL-1R) and the Toll-like receptor (TLR)/IL-1R adaptor molecule MyD88. In addition, MyD88 played both a DC-intrinsic and -extrinsic role in BCG-triggered migration.
Results
Priming of BCG-specific CD4+ T cells is concentrated to the DLN
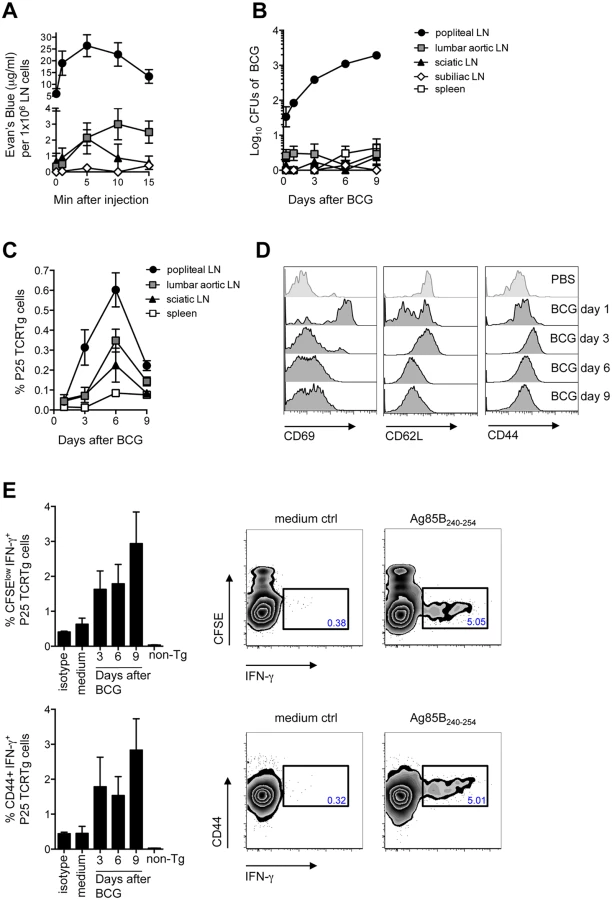
To begin dissecting the early events following BCG infection, we established a model where C57BL/6 mice are inoculated with BCG in the footpad skin and immune responses assessed in the DLN. The popliteal LN (pLN) was established as the DLN after injection of Evan’s Blue in the footpad, with secondary drainage to lumbar aortic and sciatic LNs (Fig 1A). In accordance with the above, inoculation of BCG in the footpad lead to a major detection of bacilli in the draining, pLN (Fig 1B). To examine the outcome of mycobacterial antigen-specific CD4+ T-cell responses in this setting, the fate of naïve P25 TCRTg cells was followed in vivo. These CD4+ T cells have a transgenic T-cell receptor specific for peptide 25 of mycobacterial antigen 85B (Ag85B240–254) [11]. The expansion of P25 TCRTg cells was first evident in the pLN, which was also the dominant site for P25 TCRTg cell expansion (Fig 1C). Up-regulation of CD69 and down-regulation of CD62L were clearly evident on P25 TCRTg cells 1 day after BCG infection, prior to T-cell expansion (Fig 1D). The expansion of P25 TCRTg cells correlated with sequential dilution of CFSE labeling in this population and was corroborated by an increase in the absolute number of activated P25 TCRTg cells in the DLN (S1A and S1C Fig). As reported by others, [12], surface expression of CD69 on transgenic cells was progressively down-regulated with cell division while cells that had divided expressed more CD44 compared to undivided cells (S1B Fig). Moreover, P25 TCRTg cells were found to produce IFN-γ upon recall with Ag85B240–254. IFN-γ+ cells were CFSElow and CD44+ (Fig 1E). Overall, our observations suggest that the priming of P25 TCRTg cells is concentrated to the DLN where also the majority of culturable bacilli are found.
EpCAMlow CD11bhigh DCs are the main DC population relocating from the skin to the DLN after BCG infection
The above did not establish whether active cell migration from the site of BCG inoculation in the skin coincided with T-cell priming in the DLN. A migration assay was thus developed to address this. CFSE was directly injected into the same footpad previously inoculated with BCG and the draining, pLN analyzed 24hrs later for CFSE-labeled cells using flow cytometry. Given their central role in T-cell priming, we focused our investigations on DCs. Indeed, a large portion of CFSE labeling in the pLN was found on MHC-IIhigh and CD11c+/low cells (Fig 2A), consistent with skin DCs that have migrated to DLN [8]. Both the frequency and total numbers of CFSE-labeled, MHC-IIhigh CD11c+/low cells were increased in BCG-infected animals as compared to PBS-injected controls (Fig 2B and 2C), indicating that BCG prompts mobilization of these cells to the DLN. CFSE-labeled migratory skin DCs had increased expression of CD80 and CD86 regardless if isolated from BCG - or PBS-injected animals (S2 Fig). This suggests that skin DCs arriving into the DLN during the 24hr period of our migration assay constitute an activated cell population, consistent with previous findings from skin explant cultures and FITC skin painting [13]. Importantly, LN-resident DCs (MHC-II+ CD11chigh cells) where negative for CFSE (Fig 2D), suggesting that CFSE labeling occurred primarily in the footpad, as expected. In line, plasmacytoid DCs (MHC-IIlow CD11clow PDCA–1+ cells), known to enter inflamed LNs through high endothelial venules (HEVs) [14], were also negative for CFSE, as were monocytes (CD11clow CD11bhigh Ly6Glow cells) (Fig 2D). Contrary to a previous report [15], BCG did not trigger the migration of neutrophils (CD11clow CD11bhigh Ly6Ghigh cells) from the skin to the DLN (Fig 2D), suggesting that in our particular model, skin DCs predominate over neutrophils in this regard.

Since MHC-IIhigh CD11c+/low cells in skin DLNs encompass multiple DC sub-populations [8], this subset was further characterized using CD103, EpCAM (CD326), and CD11b, markers previously employed in combination to define subsets of migratory skin DCs [16–19]. In this manner, a sub-population of EpCAMlow CD11bhigh cells was identified as the main migratory DC subset in response to BCG infection in our model (Fig 3).

CFSE+ cells contain BCG and localize to the T-cell area of the LN
Next, the distribution of these migratory skin DCs was assessed in the DLN. The CFSE-based migration assay was performed and sections of pLN from PBS - and BCG-infected animals were subjected to confocal microscopy. CFSE-labeled cells localized exclusively to the T-cell area of the LN (Fig 4A) and in increased numbers following BCG but not PBS injection (Fig 4B), the latter corroborating our flow cytometry data. The presence of BCG within this migratory cell-population was confirmed at all time-points analyzed by using DsRed-expressing bacilli (BCG-RFP) [15] (Fig 4C and 4D). To test whether migrating skin DCs were capable of engaging BCG-specific CD4+ T cells, naïve P25 TCRTg cells expressing ECFP were adoptively transferred into recipient mice. The latter were then infected with BCG-RFP in the footpad and the CFSE-based migration assay performed. Several CFSE+ cells were found in apposition to P25 TCRTg cells (Fig 4C and 4D), suggesting a possible role for this migratory DC sub-population in priming CD4+ T cells to BCG. Interestingly, such positioning was independent of whether or not CFSE+ cells were directly infected with BCG.

Inhibition of skin DC migration reduces BCG load in DLN and mutes CD4+ T-cell priming
To further study the contribution of migratory skin DCs in T-cell priming, Pertussis toxin (PTx) was injected in the footpad of mice prior to BCG infection as a means of blocking DC migration [20]. Such treatment with PTx lead to a total ablation of BCG-triggered skin DC mobilization to DLN (Fig 5A) and a dramatic reduction in the number of BCG Colony-forming units (CFUs) in the DLN (Fig 5B). Importantly, PTx treatment muted the early expansion of naïve P25 TCRTg cells in the DLN (Fig 5C). PTx did not seem to have a major, direct impact on P25 TCRTg cells as the activation profile of this population in PTx-treated, BCG-infected animals was similar to that of infected controls (S4A and S4B Fig). Similarly, injection of PTx 3 days after BCG infection, when skin DCs and mycobacteria had already reached the DLN, did not alter the activation of naïve P25 TCRTg cells transferred at the time of PTx treatment (S4C Fig). A direct effect of PTx on BCG growth was also ruled out since addition of PTx to BCG cultures did not affect mycobacterial growth in 7H11 agar (S4D Fig). Thus, upon localized injection in the footpad, PTx seems mainly to inhibit skin DC migration to DLN and consequently, the entry of BCG to the DLN. These observations support a role for migratory skin DCs in channeling BCG to the DLN and in doing so, the priming of CD4+ T cells is unleashed therein.

Skin DC migration and BCG entry into DLN is regulated by IL-1R, MyD88, IL-12p40 and TNFR-I
Next, the mechanism behind BCG-triggered skin DC migration to DLN was studied. Given the prominent role of the IL-1R/TLR adaptor molecule MyD88 in the activation of antigen-presenting cells and the initiation of Th1 responses to mycobacteria [21,22], we decided to investigate this molecule in our model by using MyD88-/- mice. Indeed, MyD88-/- mice revealed a major reduction in BCG-triggered skin DC migration to pLN as compared to WT controls (Fig 6A). MyD88-/- mice also displayed an important reduction in BCG CFUs in the pLN compared to WT controls (Fig 6B), providing additional support for skin DCs in the transport of BCG to the DLN. Since mycobacteria-triggered DC production of IL-12p40 and TNF-α are MyD88-dependent in murine DC cultures [22–24], we also decided to investigate the role of these cytokines in migration. Interestingly, a deficit in DC mobilization and BCG entry into the DLN was observed in IL-12p40-/- and TNFR-I-/- mice (Fig 6A and 6B). These findings corroborate the well-established role of TNF-α as a trigger of skin DC migration [25] and support intriguing but isolated reports that IL-12p40 homodimer regulates DC migration during bacterial infection [26–28]. However, TLR2 and TLR9, two of the main TLRs engaged in mycobacterial sensing by DCs, were not required for skin DC mobilization or BCG transport to DLN (Fig 6C and 6D). On the other hand, IL-1R-I-/- mice displayed a reduction in BCG-triggered skin DC migration and BCG CFUs in the DLN (Fig 6E and 6F) similar to that of MyD88-/- mice, suggesting that the DC and BCG relocation defects in the DLN of MyD88-/- mice reflect a defect in IL-1R rather than TLR signaling. In line with MyD88 and IL-1R-I being important for skin DC migration and BCG entry into DLN, we found that the early activation of naïve P25 TCRTg cells transferred into MyD88-/- and IL-1R-I-/- recipients was reduced compared to P25 TCRTg cells that were transferred into BCG-infected WT controls (S5 Fig). Furthermore, it was also observed that administration of IL-12p40 homodimer but not TNF-α to footpads restores BCG-triggered skin DC migration in IL-1R-I-/- mice (S6 Fig), suggesting that the ability of TNF-α to regulate migration in our model may be dependent on IL-1R.

DC migration requires DC-intrinsic and -extrinsic expression of MyD88
The above experiments establish a critical contribution for MyD88 in regulating DC traffic and BCG entry into DLN. We therefore wanted to examine if MyD88 signaling was an intrinsic requirement of the migrating DC. To do so, an adoptive transfer approach with bone marrow-derived DCs (BMDCs) was used where CFSE in vitro-labeled BMDCs where injected in the footpad of naïve, recipient mice which where then inoculated 2hrs later with BCG or PBS in the same footpad; the number of transferred DCs arriving in the draining, pLN were then assessed 3 days later by flow cytometry. Although BMDCs tend to migrate poorly in this assay [29], we observed an increased number of transferred DCs in the DLN of BCG-infected animals compared to PBS-injected controls (S7A Fig). To normalize for differences between BMDC culture preparations, we expressed the number of DCs that migrated to the DLN after BCG infection relative to the number of DCs from the same cultures that migrated to the DLN after an injection of PBS (S7A Fig). Using this approach we found that MyD88-/- DCs were muted in their ability to reach the DLN of WT recipients (Fig 7A), suggesting an intrinsic need for MyD88 signaling in the migrating DC. A similar observation was made with IL-12p40-/- and TNFR-I-/- DCs transferred into WT recipients (Fig 7A). Importantly, these findings could not be explained by inherent differences in expression of CCR7, the main chemokine receptor regulating DC entry into LNs [1], as baseline levels of CCR7 mRNA were similar between the various DC cultures investigated (S7B Fig). Indeed, both WT and MyD88-/- BMDCs could migrate across transwells in response to the CCR7 ligand CCL19 (S7C Fig). Interestingly, WT DCs transferred into MyD88-/-, IL-12p40-/- or TNFR-I-/- recipients were also unable to migrate efficiently to DLN after BCG footpad infection (Fig 7B). The latter reveals an additional contribution of MyD88 in fueling DC migration where the need for MyD88 is not inherent to the migrating DC itself.

To investigate if this DC-extrinsic requirement for MyD88 was associated to expression of MyD88 in hematopoietic cells, we adoptively transferred WT BMDCs into MyD88-/- bone-marrow radiation chimeric mice. Restoring hematopoietic expression of MyD88 in MyD88-/- mice certainly enhanced BCG-triggered DC migration, implicating the bone marrow in MyD88-dependent DC migration (Fig 7C). On the other hand, rendering the hematopoietic compartment of WT mice deficient in MyD88 did not attenuate BCG-triggered DC migration (Fig 7C). Thus, although MyD88 expression in hematopoietic cells plays an important role in driving migration of adoptively transferred DCs, stromal or radio-resistant cells can at least sustain DC migration in a WT host reconstituted with MyD88-/- bone marrow. These findings reveal an unexpected, interplay between myeloid and stromal cells in MyD88-dependent mobilization of transferred DCs to DLN in response to BCG.
Discussion
Although DCs play a central role in the initiation of protective Th1 responses to mycobacteria [30], many questions remain unanswered with regards to the priming of CD4+ T cells. A central caveat is the transport of mycobacteria from the effector site to the DLN where T-cell responses are initiated. During immunization with model antigens, the magnitude and quality of CD4+ T-cell priming is proportional to the number of antigen-bearing DCs reaching the DLN [29]. Indeed, the arrival of increased DC numbers to the DLN can enhance the magnitude of the CD4+ T-cell response by favoring the recruitment of multiple T-cell clones and allowing T cells to successively engage multiple DCs [31]. We show in a BCG footpad infection model that migratory EpCAMlow CD11bhigh skin DCs are important for this channeling of antigen by transporting live bacilli to the DLN in an IL-1R-MyD88-dependent manner. The arrival of BCG in the DLN was associated with the priming of mycobacteria-specific P25 TCRTg cells at that site and supports an accumulating body of evidence that live bacilli in the DLN are needed for the initiation of T-cell responses to mycobacteria [32–34].
In spite of similarities, there exist also differences in T-cell priming between BCG and M. tuberculosis. The priming of CD4+ T cells in our BCG model is certainly faster than in M. tuberculosis [33,35]. The lower bacterial inoculum may delay T-cell activation during M. tuberculosis [33,35], but is not thought to be the singular determinant of this delay [32]. M. tuberculosis may initially reside in a static, cellular compartment, which could consequently postpone the transport of bacilli to DLN. Moreover, M. tuberculosis-infected lung DCs seem to be limited in their capacity to relocate to DLN [35]. On the contrary, our data suggests that transmission of BCG to DLN in skin DCs is rapid and continuous. This provides a possible explanation as to why T-cell priming to BCG is quicker than to M. tuberculosis.
Our study reveals a role for migratory skin DCs in the priming of CD4+ T cells to BCG inoculation, a complex, “antigenic preparation” of microbial debris, secreted microbial products and live bacilli capable of infecting phagocytes. Even after cutaneous injection of a soluble antigen accessible to DCs in the LN via lymphatic drainage, migratory skin DCs that acquire antigen at the injection site are still needed for full-scale priming of CD4+ T cells in the DLN [36]. This is likely due to the higher antigenic load found in migratory skin DCs [36]. On the other hand, during infection with Herpes simplex virus (HSV) in the skin, transfer of antigen from migratory to LN-resident DCs is needed for priming CD8+ T cells in the DLN [37]. By carrying live bacilli to the DLN, EpCAMlow CD11bhigh migratory skin DCs may promote priming both directly and indirectly. Apposition between DCs and naive P25 TCRTg cells in the T-cell area of the LN provides support for the former scenario. However, it is also possible that DCs that mobilize from the site of antigen deposition to the T-cell area promote priming indirectly by facilitating the transfer of BCG or its antigens to other DC populations in the LN. In this regard, LN-resident, CD8α+ DCs can produce copious amounts of IL–12 needed for Th1 differentiation, and excel as an early source of IL-12p40 in BCG-infected spleen [38]. Interestingly, antigen transfer from migratory to LN-resident DCs has been shown to optimize CD4+ T-cell priming to M. tuberculosis in lung DLN [39]. Other, recent studies show that CD11b+ LN-resident DCs lining the lymphatic sinus can capture lymph-borne, particulate antigen and prime CD4+ T cells, independent of migratory DCs [40]. It is unclear to what degree BCG gains direct access to lymphatics to merit the initiation of T-cell responses via this novel route. Similarly, it remains to be determined whether antigen exchange between DCs in skin DLN also favors the priming of Th1 responses to BCG, or for that matter, if there exists a DC sub-population in the LN that champions the priming of CD4+ T cells to mycobacteria. Antigen-presentation assays comparing subsets of migratory skin DCs and LN-resident DCs in priming CD4+ T cells will help unravel this. Likewise, assessment of BCG-triggered Th1 responses in transgenic mice where defined DC subsets are absent or can be depleted, as recently investigated during Candida albicans infection [41], may unfold their relative contribution to initiating T-cell responses to mycobacteria.
Nevertheless, there is an emerging role for migratory EpCAMlow CD11bhigh skin DCs in immune responses to infection. Corroborating our data, a previous study showed that this subset expands in skin DLN after intradermal infection with BCG and E. coli [19]. Importantly, this same subset of migratory skin DCs can trigger CD8+ T-cell priming to a live, adenoviral vaccine delivered via microneedle arrays [42]. CD11bhigh (Langerinneg CD103neg) skin DCs have also been demonstrated to engulf Leishmania major in the dermis [43]. Earlier studies show that submucosal DCs migrate to DLN after intravaginal inoculation of HSV and trigger Th1 responses [20,44]. It is possible that EpCAMlow CD11bhigh skin DCs are at least functionally related to the aforementioned population, or the CD11bhigh subset of migratory lung DCs that can transport M. tuberculosis to DLN [35]. Thus, our study together with the above, support a role for EpCAMlow CD11bhigh skin DCs as a migratory cell population capable of relaying antigen to DLN to promote T-cell priming.
The importance of IL-1R-MyD88 signaling in regulating DC migration in our model is in line with the central role for MyD88 in mycobacterial-induced DC activation and the dominant role of IL-1R over TLR signaling in immunity to M. tuberculosis [45,46]. Our data extend on these findings by showing a role for this pathway in promoting DC and BCG entry into DLN. Interestingly, this contribution was partial, as DC traffic and BCG transport to DLN were not entirely ablated in IL-1R-I-/- and MyD88-/- mice. Additional signaling pathways may thus be involved or come into play in the absence of MyD88. Caspase recruitment domain-containing protein (CARD) 9 could be one such candidate. This adaptor molecule is coupled to several C-type lectin receptors that sense mycobacteria [47] and it has recently been shown to regulate production of IL–1β, TNF-α and IL-12p40 in M. tuberculosis-infected macrophages [48].
DCs also release IL–1β, TNF-α, and IL-12p40 upon co-culture with mycobacteria, and are in fact the primary source of IL-12p40 in response to M. tuberculosis and BCG in vivo [38]. IL-12p40 and TNFR-I were both necessary in our model for BCG-triggered DC mobilization to DLN. IL-12p40 homodimer has been shown to promote DC migration to DLN in the lungs of M. tuberculosis - and Francisella tularensis LVS-infected mice [26,28] respectively, and to promote the transmigration of Yersinia pestis-infected DCs towards CCL19 in transwell assays [27]. Our findings in IL-12p40-/- mice, which are deficient in IL–12, IL–23 and IL-12p40, do not prove that IL-12p40 homodimers regulate DC migration in our model. This is very likely though, given the above studies and the fact that a direct role for IL–12 or IL–23 in DC migration is not well established. Our studies thus build on the idea that IL-12p40 (probably as a p40 homodimer) has an important role in initiating a Th1 response to bacteria that precedes that of bona fide IL–12.
It is unclear if an injection of IL-12p40 suffices to mobilize DCs from the skin as previously shown for IL–1β and TNF-α [49]. TNF-α-mediated DC mobilization in particular, is associated to down-regulation of E-cadherin on DCs and increased expression of adhesion molecules and CCL21 on lymphatics [9]. We show that administration of IL-12p40 homodimer can restore BCG-triggered skin DC migration in IL-1R-I-/- mice while treatment with TNF-α at concentrations that suffice to mobilize skin DCs in other models [49] [29] does not. It could thus be that TNF-α acts in our model by inducing IL–1 production rather than as a consequence of IL-1R signaling. Additional studies are needed to clarify this.
Our adoptive transfer experiments show that functional MyD88 signaling is needed not only in the migrating DC itself, but also in the recipient, i.e., extrinsic to the migrating DC. Understanding the impact of the above for T-cell priming is a complex undertaking that merits attention in future investigations aimed at unfolding these mechanisms. In addition, specific depletion of MyD88 on DCs may help clarify DC-intrinsic requirements. In regards to DC migration, it is possible that transferred DCs are already “primed” to migrate as both WT and MyD88-/- BMDCs expressed sufficient levels of CCR7 to sustain migration in response to CCL19. This suggests that BCG infection can play a dual role; on the one hand directly activating the migrating DC and on the other, activating the local effector site to make migration more efficient by other means, e.g. by reactivating the lymphatic endothelium or increasing production of pro-inflammatory cytokines. Indeed, TNF-α can condition a vaccination site and enhance T-cell priming [29]. Another possibility is that the DLN itself may influence the recruitment of DCs from the skin to its microenvironment once the DLN becomes exposed to BCG or its products.
DC transfers in bone-marrow radiation chimeras also reveal a requirement for MyD88 signaling in myeloid cells for relocating the transferred DCs to DLN. This suggests that a migrating DC can receive MyD88-dependent signals for migration, e.g. IL–1, TNF-α and/or IL-12p40, from neighboring myeloid cells. These signals could be derived from other DCs in the skin, neutrophils which home to the skin after BCG infection [15], and/or mast cells, recently shown to regulate skin DC migration [50]. Furthermore, results from our bone marrow chimeras also suggest that stromal cells may play a role in sustaining DC migration. In line, factors derived from lymphatic endothelial cells have recently been shown to at least moderately enhance DC migration in response to inflammatory stimuli [51].
In summary, we identified a migratory skin DC sub-population that relocates to DLN together with BCG in an IL-1R-MyD88-dependent manner and which can engage naïve CD4+ T cells during priming. Our findings also implicate myeloid and stromal cells in driving DCs from a BCG effector site to the DLN. These results bare consequences for the quality and magnitude of T-cell priming during infection or cell-based therapy with DCs and as such hold promises for modifying CD4+ T-cell responses to BCG, other vaccines of low-to-modest efficacy, and to the implementation of adjunct therapies to infection and tumors.
Materials and Methods
Mice
MyD88-/- [52], IL-12p40-/- [53], TNFR-I-/- [54], TLR2-/-/TLR9-/- [55], all on a C57BL/6 background, were obtained from the MTC breeding unit, Karolinska Institutet, Stockholm, Sweden. Congenic CD45.1+ mice on a C57BL/6 background (B6.SJL/Ptprca) originally from Charles River Laboratories, were also obtained from the MTC breeding unit. IL-1R-I-/- mice [56] on a C57BL/6 background were purchased from the Jackson Laboratory. P25 TCRTg RAG–1-/- mice expressing ECFP were generated by crossing P25 TCRTg RAG–1-/- mice [11] with ECFP mice on a RAG–1-/- background (kindly provided by Dr. Ronald Germain, NIAID, NIH). C57BL/6 mice were used as wild-type (WT) controls. All animals were maintained under specific pathogen-free conditions at the MTC breeding unit. Both male and female mice between 8 and 12 wks old were used.
Ethics statement
Animal were housed and handled at MTC, Karolinska Institutet, according to the directives and guidelines of the Swedish Board of Agriculture, the Swedish Animal Protection Agency, and Karolinska Institutet (djurskyddslagen 1988 : 534; djurskyddsförordningen 1988 : 539; djurskyddsmyndingeheten DFS 2004 : 4). Experiments were approved by the Stockholm North Animal Ethics Council under the Animal Study Proposals N381/11, N431/12, N397/13 and N171/14.
Mycobacteria
Mycobacterium bovis Bacille Calmette-Guérin (BCG) strain Pasteur (1173P2) and BCG Pasteur expressing the red fluorescent protein DsRed (BCG-RFP, kindly provided by Dr. Nathalie Winter, INRA, Tours, France) [15] were expanded in 7H9 broth as previously described [38]. Quantification of mycobacterial Colony-forming units (CFUs) for bacterial stocks and determination of bacterial load in organs was performed by culture on 7H11 agar supplemented with OADC (Difco).
Inoculation of mice
Animals were inoculated in the hind footpad with 1x106 CFUs of BCG in 30 μl of PBS. Control animals received an equal volume of PBS. For assessment of cell migration from the footpad skin, animals previously injected with BCG or PBS where injected 24hrs before sacrifice in the same footpad with 20 μl of 0.5 mM 5 - and 6-carboxyfluorescein diacetate succinimidyl ester (CFSE) (Invitrogen). In select experiments animals were injected with 50 ng or 300 ng rTNF-α or 2 μg rIL-12p40 homodimer (R&D Systems) in the same footpad that 2hrs earlier was inoculated with BCG or PBS. For assessment of lymphatic drainage to LNs, 20 μl 5% Evan’s blue (Sigma) was injected in the footpad and LNs isolated at different time points after injection. For assessment of CD4+ antigen-specific T-cell responses, 1x105 LN cells from P25 TCRTg RAG–1-/- mice were first labeled with 1 μM CFSE and then injected i.v. in the tail vein of congenic CD45.1+ recipients in a final volume of 200 μl. For DC adoptive transfer experiments, 2x106 naïve BMDCs generated from WT or transgenic mice were labeled with 3 μM CFSE and then injected into the footpad in a final volume of 40 μl. Two hrs after DC transfer, the same footpads were inoculated with 30 μl PBS or 1x106 CFUs of BCG.
Generation of single-cell suspensions from tissue
LN, spleen or bone marrow were aseptically removed and processed as previously described [38]. Tissues were either gently homogenized through a 100 micron cell strainer or using a tissue grinder. Homogenates were resuspended in PBS and pelleted at 1200 rpm for 5 min. For spleens, erythrocytes were lysed using ACK lysis buffer unless used for determination of CFUs on 7H11 agar. Resulting single-cell suspensions were washed and counted by Trypan blue exclusion prior to further processing. For quantification of Evan’s blue in LNs, LNs were aseptically removed and incubated for 24hrs in 100 μl formamide solution (Sigma) at 56°C and OD650nm measured on a spectrophotometer (Molecular Devices).
Flow cytometric staining
Single-cell suspensions from tissues were incubated with various combinations of flourochrome-conjugated rat anti-mouse monoclonal antibodies specific for CD4 (L3T4), CD11b (M1/70), CD11c (HL3), MHC-II I-A/I-E (M5/114.15.2), Ly6G (1A8), CD44 (IM7), CD45.2 (104), CD62L (MEL14), CD69 (H1.2F3), Vβ11 (RR3-15) (BD Biosciences), CD326/EpCAM (G8.8), CD103 (2E7) (Biolegend), CD4 (RM4-5), CD45.1 (A20), CD317/PDCA–1 (927) (eBiosciences), for 45 min in FACS buffer (2% FCS in 5 mM EDTA, 0.1% azide) containing 0.5 mg/ml anti-mouse FcγIII/II receptor (2.4G2) (BD Biosciences). For analysis of intracellular cytokine production, cells were stimulated ex vivo for 6hrs with 1 μM Ag85B240–254 peptide in the presence of 10 μg/ml Brefeldin A (Sigma). Cells were washed and surface stained with CD4, CD44, CD45.2, CD62L and CD69. Cells were fixed in 2% paraformaldehyde (Electron Microscopy Sciences), permeabilized in 1% saponin (Sigma) and stained with anti-IFN-γ (XMG1.2) (BD Biosciences). Flow cytometry was performed on a LSR-II (BD Biosciences) and analyzed with FlowJo software (Tree Star).
Confocal microscopy
LNs were excised from naïve and infected animals, fixed overnight with 4% paraformaldehyde/PBS followed by dehydration in 30% sucrose/PBS prior to embedding in Tissue-Tek OCT freezing media (Sakura Finetek). 16 to 20 micron-thick sections were cut on a HM 550 cryostat (Thermo Scientific) and adhered to Superfrost Plus slides (VWR). Sections were permeabilized and blocked in PBS containing 0.3% Triton X–100 (Sigma) and 10% goat serum (Jackson Immunoresearch). This was followed by incubation with AlexaFluor 647-conjugated rat anti-CD3 (17A2) (BD Biosciences). Incubation with unconjugated rat anti-PNAd (MECA–79) (BD Biosciences) was followed by staining with AlexaFlour 647-conjugated secondary antibodies (Invitrogen). Slides were mounted with Prolong Gold (Invitrogen). 3D image stacks of LN sections were acquired on a LSM 780 confocal microscope (Carl Zeiss MicroImaging). Images are displayed as 2D maximum intensity projections using Imaris software (Bitplane).
Radiation bone marrow chimeras
MyD88-/- or congenic CD45.1+ C57BL/6 recipients were sub-lethally irradiated with 2 x 500 Rads and each animal inoculated i.v. with 1 x106 bone marrow cells from MyD88-/- or congenic CD45.1+ C57BL/6 mice. Mice were provided with antibiotics in the drinking water for 3 wks and used for experiments 18 to 22 wks after engraftment. Reconstitution was assessed by flow cytometric analysis of blood where no significant differences in engraftment were observed between groups. The frequency of engraftment was approximately 90%.
Generation of bone marrow-derived Dendritic cells (BMDCs)
BMDCs were generated using recombinant GM-CSF (Biosource) from 6 day-old cultures as previously described [38]. These cells were further enriched for CD11c expression by magnetic selection (Miltenyi) that yielded a population of approximately 98–99% CD11c+ cells. Naïve BMDCs were labeled with 3 μM CFSE for adoptive transfer experiments in mice or stimulated overnight at 37°C, 5% CO2, with BCG at a multiplicity of infection of 1, for gene expression analysis or transwell migration assays.
CCR7 gene expression measurement by Realtime PCR
Total RNA was extracted from BMDCs using TRIZOL reagent (Sigma) and reverse transcribed into cDNA using M-MLV reverse transcriptase (Promega). Real-time PCR was performed on an ABI PRISM 7500 sequence detection system (Applied Biosystems) using SYBR Green (Sigma). The relative expression of CCR7 was determined by the 2(-ΔCT) method in which samples were normalized to hypoxanthine phosphoribosyl transferase (HPRT). The following primers pairs were used:
- HPRT forward:
CCC AGC GTC GTG ATT AGC
- HPRT reverse:
GGA ATA AAC ACT TTT TCC AAA TCC
- CCR7 forward:
ACC ATG GAC CCA GGG AAA C
- CCR7 reverse:
GGT ATT CTC GCC GAT GTA G
Transwell migration assay
Transwell inserts with an 8 micron pore-size (VWR) were placed on 24-well plates containing 1 μg/ml recombinant CCL19 (Peprotech). Naïve or BCG-stimulated BMDCs (3 x 105) were added to the transwell inserts and incubated at 37°C, 5% CO2. Transwells were removed 2hrs later and the number of DCs that reached the bottom of the plate were enumerated by Trypan blue exclusion.
Statistical analyses
The significance of differences in data group means was analyzed by Student’s t test or Anova where appropriate, with a cut-off of p < 0.05. In some experiments, outliers were excluded from analysis following Grubbs’s test for outliers (GraphPad, quickcals).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Platt AM, Randolph GJ (2013) Dendritic cell migration through the lymphatic vasculature to lymph nodes. Adv Immunol 120 : 51–68. doi: 10.1016/B978-0-12-417028-5.00002-8 24070380
2. Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140 : 805–820. doi: 10.1016/j.cell.2010.01.022 20303872
3. Walsh KP, Mills KH (2013) Dendritic cells and other innate determinants of T helper cell polarisation. Trends Immunol 34 : 521–530. doi: 10.1016/j.it.2013.07.006 23973621
4. WHO (2015) Tuberculosis. fact sheet 104.
5. Flynn JL, Chan J (2001) Immunology of tuberculosis. Annu Rev Immunol 19 : 93–129. 11244032
6. Cooper AM (2009) Cell-mediated immune responses in tuberculosis. Annu Rev Immunol 27 : 393–422. doi: 10.1146/annurev.immunol.021908.132703 19302046
7. Doherty TM, Andersen P (2005) Vaccines for tuberculosis: novel concepts and recent progress. Clin Microbiol Rev 18 : 687–702. 16223953
8. Henri S, Guilliams M, Poulin LF, Tamoutounour S, Ardouin L, et al. (2010) Disentangling the complexity of the skin dendritic cell network. Immunol Cell Biol 88 : 366–375. doi: 10.1038/icb.2010.34 20231850
9. Alvarez D, Vollmann EH, von Andrian UH (2008) Mechanisms and consequences of dendritic cell migration. Immunity 29 : 325–342. doi: 10.1016/j.immuni.2008.08.006 18799141
10. Heuze ML, Vargas P, Chabaud M, Le Berre M, Liu YJ, et al. (2013) Migration of dendritic cells: physical principles, molecular mechanisms, and functional implications. Immunol Rev 256 : 240–254. doi: 10.1111/imr.12108 24117825
11. Tamura T, Ariga H, Kinashi T, Uehara S, Kikuchi T, et al. (2004) The role of antigenic peptide in CD4+ T helper phenotype development in a T cell receptor transgenic model. Int Immunol 16 : 1691–1699. 15477229
12. Lambrecht BN, Pauwels RA, Fazekas De St Groth B (2000) Induction of rapid T cell activation, division, and recirculation by intratracheal injection of dendritic cells in a TCR transgenic model. J Immunol 164 : 2937–2946. 10706680
13. Henri S, Vremec D, Kamath A, Waithman J, Williams S, et al. (2001) The dendritic cell populations of mouse lymph nodes. J Immunol 167 : 741–748. 11441078
14. Diacovo TG, Blasius AL, Mak TW, Cella M, Colonna M (2005) Adhesive mechanisms governing interferon-producing cell recruitment into lymph nodes. J Exp Med 202 : 687–696. 16147979
15. Abadie V, Badell E, Douillard P, Ensergueix D, Leenen PJ, et al. (2005) Neutrophils rapidly migrate via lymphatics after Mycobacterium bovis BCG intradermal vaccination and shuttle live bacilli to the draining lymph nodes. Blood 106 : 1843–1850. 15886329
16. Bedoui S, Whitney PG, Waithman J, Eidsmo L, Wakim L, et al. (2009) Cross-presentation of viral and self antigens by skin-derived CD103+ dendritic cells. Nat Immunol 10 : 488–495. doi: 10.1038/ni.1724 19349986
17. Kumamoto Y, Linehan M, Weinstein JS, Laidlaw BJ, Craft JE, et al. (2013) CD301b+ dermal dendritic cells drive T helper 2 cell-mediated immunity. Immunity 39 : 733–743. doi: 10.1016/j.immuni.2013.08.029 24076051
18. Mollah SA, Dobrin JS, Feder RE, Tse SW, Matos IG, et al. (2014) Flt3L Dependence Helps Define an Uncharacterized Subset of Murine Cutaneous Dendritic Cells. J Invest Dermatol 134 : 2850–2851.
19. Ochiai S, Roediger B, Abtin A, Shklovskaya E, Fazekas de St Groth B, et al. (2014) CD326lo CD103lo CD11blo dermal dendritic cells are activated by thymic stromal lymphopoietin during contact sensitization in mice. J Immunol 193 : 2504–2511. doi: 10.4049/jimmunol.1400536 25057004
20. Lee HK, Zamora M, Linehan MM, Iijima N, Gonzalez D, et al. (2009) Differential roles of migratory and resident DCs in T cell priming after mucosal or skin HSV–1 infection. J Exp Med 206 : 359–370. doi: 10.1084/jem.20080601 19153243
21. Feng CG, Scanga CA, Collazo-Custodio CM, Cheever AW, Hieny S, et al. (2003) Mice lacking myeloid differentiation factor 88 display profound defects in host resistance and immune responses to Mycobacterium avium infection not exhibited by Toll-like receptor 2 (TLR2) - and TLR4-deficient animals. J Immunol 171 : 4758–4764. 14568952
22. Bafica A, Scanga CA, Feng CG, Leifer C, Cheever A, et al. (2005) TLR9 regulates Th1 responses and cooperates with TLR2 in mediating optimal resistance to Mycobacterium tuberculosis. J Exp Med 202 : 1715–1724. 16365150
23. von Meyenn F, Schaefer M, Weighardt H, Bauer S, Kirschning CJ, et al. (2006) Toll-like receptor 9 contributes to recognition of Mycobacterium bovis Bacillus Calmette-Guerin by Flt3-ligand generated dendritic cells. Immunobiology 211 : 557–565. 16920494
24. Rothfuchs AG, Bafica A, Feng CG, Egen JG, Williams DL, et al. (2007) Dectin–1 interaction with Mycobacterium tuberculosis leads to enhanced IL-12p40 production by splenic dendritic cells. J Immunol 179 : 3463–3471. 17785780
25. Cumberbatch M, Kimber I (1992) Dermal tumour necrosis factor-alpha induces dendritic cell migration to draining lymph nodes, and possibly provides one stimulus for Langerhans' cell migration. Immunology 75 : 257–263. 1551688
26. Khader SA, Partida-Sanchez S, Bell G, Jelley-Gibbs DM, Swain S, et al. (2006) Interleukin 12p40 is required for dendritic cell migration and T cell priming after Mycobacterium tuberculosis infection. J Exp Med 203 : 1805–1815. 16818672
27. Robinson RT, Khader SA, Locksley RM, Lien E, Smiley ST, et al. (2008) Yersinia pestis evades TLR4-dependent induction of IL–12(p40)2 by dendritic cells and subsequent cell migration. J Immunol 181 : 5560–5567. 18832714
28. Slight SR, Lin Y, Messmer M, Khader SA (2011) Francisella tularensis LVS-induced Interleukin–12 p40 cytokine production mediates dendritic cell migration through IL–12 Receptor beta1. Cytokine 55 : 372–379. doi: 10.1016/j.cyto.2011.05.017 21669537
29. Martin-Fontecha A, Sebastiani S, Hopken UE, Uguccioni M, Lipp M, et al. (2003) Regulation of dendritic cell migration to the draining lymph node: impact on T lymphocyte traffic and priming. J Exp Med 198 : 615–621. 12925677
30. Tian T, Woodworth J, Skold M, Behar SM (2005) In vivo depletion of CD11c+ cells delays the CD4+ T cell response to Mycobacterium tuberculosis and exacerbates the outcome of infection. J Immunol 175 : 3268–3272. 16116218
31. Celli S, Day M, Muller AJ, Molina-Paris C, Lythe G, et al. (2012) How many dendritic cells are required to initiate a T-cell response? Blood 120 : 3945–3948. doi: 10.1182/blood-2012-01-408260 22995897
32. Wolf AJ, Desvignes L, Linas B, Banaiee N, Tamura T, et al. (2008) Initiation of the adaptive immune response to Mycobacterium tuberculosis depends on antigen production in the local lymph node, not the lungs. J Exp Med 205 : 105–115. 18158321
33. Reiley WW, Calayag MD, Wittmer ST, Huntington JL, Pearl JE, et al. (2008) ESAT-6-specific CD4 T cell responses to aerosol Mycobacterium tuberculosis infection are initiated in the mediastinal lymph nodes. Proc Natl Acad Sci U S A 105 : 10961–10966. doi: 10.1073/pnas.0801496105 18667699
34. Biot C, Rentsch CA, Gsponer JR, Birkhauser FD, Jusforgues-Saklani H, et al. (2012) Preexisting BCG-specific T cells improve intravesical immunotherapy for bladder cancer. Science translational medicine 4 : 137ra172.
35. Wolf AJ, Linas B, Trevejo-Nunez GJ, Kincaid E, Tamura T, et al. (2007) Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. J Immunol 179 : 2509–2519. 17675513
36. Itano AA, McSorley SJ, Reinhardt RL, Ehst BD, Ingulli E, et al. (2003) Distinct dendritic cell populations sequentially present antigen to CD4 T cells and stimulate different aspects of cell-mediated immunity. Immunity 19 : 47–57. 12871638
37. Allan RS, Waithman J, Bedoui S, Jones CM, Villadangos JA, et al. (2006) Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity 25 : 153–162. 16860764
38. Rothfuchs AG, Egen JG, Feng CG, Antonelli LR, Bafica A, et al. (2009) In situ IL–12/23p40 production during mycobacterial infection is sustained by CD11bhigh dendritic cells localized in tissue sites distinct from those harboring bacilli. J Immunol 182 : 6915–6925. doi: 10.4049/jimmunol.0900074 19454688
39. Srivastava S, Ernst JD (2014) Cell-to-cell transfer of M. tuberculosis antigens optimizes CD4 T cell priming. Cell Host Microbe 15 : 741–752. doi: 10.1016/j.chom.2014.05.007 24922576
40. Gerner MY, Torabi-Parizi P, Germain RN (2015) Strategically localized dendritic cells promote rapid T cell responses to lymph-borne particulate antigens. Immunity 42 : 172–185. doi: 10.1016/j.immuni.2014.12.024 25607462
41. Igyarto BZ, Haley K, Ortner D, Bobr A, Gerami-Nejad M, et al. (2011) Skin-resident murine dendritic cell subsets promote distinct and opposing antigen-specific T helper cell responses. Immunity 35 : 260–272. doi: 10.1016/j.immuni.2011.06.005 21782478
42. Bachy V, Hervouet C, Becker PD, Chorro L, Carlin LM, et al. (2013) Langerin negative dendritic cells promote potent CD8+ T-cell priming by skin delivery of live adenovirus vaccine microneedle arrays. Proc Natl Acad Sci U S A 110 : 3041–3046. doi: 10.1073/pnas.1214449110 23386724
43. Ribeiro-Gomes FL, Peters NC, Debrabant A, Sacks DL (2012) Efficient capture of infected neutrophils by dendritic cells in the skin inhibits the early anti-leishmania response. PLoS Pathog 8: e1002536. doi: 10.1371/journal.ppat.1002536 22359507
44. Zhao X, Deak E, Soderberg K, Linehan M, Spezzano D, et al. (2003) Vaginal submucosal dendritic cells, but not Langerhans cells, induce protective Th1 responses to herpes simplex virus–2. J Exp Med 197 : 153–162. 12538655
45. Fremond CM, Togbe D, Doz E, Rose S, Vasseur V, et al. (2007) IL–1 receptor-mediated signal is an essential component of MyD88-dependent innate response to Mycobacterium tuberculosis infection. J Immunol 179 : 1178–1189. 17617611
46. Mayer-Barber KD, Barber DL, Shenderov K, White SD, Wilson MS, et al. (2010) Caspase–1 independent IL-1beta production is critical for host resistance to Mycobacterium tuberculosis and does not require TLR signaling in vivo. J Immunol 184 : 3326–3330. doi: 10.4049/jimmunol.0904189 20200276
47. Marakalala MJ, Graham LM, Brown GD (2010) The role of Syk/CARD9-coupled C-type lectin receptors in immunity to Mycobacterium tuberculosis infections. Clin Dev Immunol 2010 : 567571. doi: 10.1155/2010/567571 21274433
48. Dorhoi A, Desel C, Yeremeev V, Pradl L, Brinkmann V, et al. (2010) The adaptor molecule CARD9 is essential for tuberculosis control. J Exp Med 207 : 777–792. doi: 10.1084/jem.20090067 20351059
49. Cumberbatch M, Dearman RJ, Kimber I (1997) Langerhans cells require signals from both tumour necrosis factor-alpha and interleukin–1 beta for migration. Immunology 92 : 388–395. 9486113
50. Otsuka A, Kubo M, Honda T, Egawa G, Nakajima S, et al. (2011) Requirement of interaction between mast cells and skin dendritic cells to establish contact hypersensitivity. PLoS One 6: e25538. doi: 10.1371/journal.pone.0025538 21980488
51. Vigl B, Aebischer D, Nitschke M, Iolyeva M, Rothlin T, et al. (2011) Tissue inflammation modulates gene expression of lymphatic endothelial cells and dendritic cell migration in a stimulus-dependent manner. Blood 118 : 205–215. doi: 10.1182/blood-2010-12-326447 21596851
52. Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, et al. (1998) Targeted disruption of the MyD88 gene results in loss of IL-1 - and IL-18-mediated function. Immunity 9 : 143–150. 9697844
53. Magram J, Connaughton SE, Warrier RR, Carvajal DM, Wu CY, et al. (1996) IL-12-deficient mice are defective in IFN gamma production and type 1 cytokine responses. Immunity 4 : 471–481. 8630732
54. Vieira LQ, Goldschmidt M, Nashleanas M, Pfeffer K, Mak T, et al. (1996) Mice lacking the TNF receptor p55 fail to resolve lesions caused by infection with Leishmania major, but control parasite replication. J Immunol 157 : 827–835. 8752935
55. Sorensen LN, Reinert LS, Malmgaard L, Bartholdy C, Thomsen AR, et al. (2008) TLR2 and TLR9 synergistically control herpes simplex virus infection in the brain. J Immunol 181 : 8604–8612. 19050280
56. Glaccum MB, Stocking KL, Charrier K, Smith JL, Willis CR, et al. (1997) Phenotypic and functional characterization of mice that lack the type I receptor for IL–1. J Immunol 159 : 3364–3371. 9317135
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 10
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Chronobiomics: The Biological Clock as a New Principle in Host–Microbial Interactions
- Interferon-γ: The Jekyll and Hyde of Malaria
- Crosslinking of a Peritrophic Matrix Protein Protects Gut Epithelia from Bacterial Exotoxins
- Antigen-Specific Th17 Cells Are Primed by Distinct and Complementary Dendritic Cell Subsets in Oropharyngeal Candidiasis
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy