
Structure of Herpes Simplex Virus Glycoprotein D Bound to the Human Receptor Nectin-1
Binding of herpes simplex virus (HSV) glycoprotein D (gD) to a cell surface receptor is required to trigger membrane fusion during entry into host cells. Nectin-1 is a cell adhesion molecule and the main HSV receptor in neurons and epithelial cells. We report the structure of gD bound to nectin-1 determined by x-ray crystallography to 4.0 Å resolution. The structure reveals that the nectin-1 binding site on gD differs from the binding site of the HVEM receptor. A surface on the first Ig-domain of nectin-1, which mediates homophilic interactions of Ig-like cell adhesion molecules, buries an area composed by residues from both the gD N - and C-terminal extensions. Phenylalanine 129, at the tip of the loop connecting β-strands F and G of nectin-1, protrudes into a groove on gD, which is otherwise occupied by C-terminal residues in the unliganded gD and by N-terminal residues in the gD/HVEM complex. Notably, mutation of Phe129 to alanine prevents nectin-1 binding to gD and HSV entry. Together these data are consistent with previous studies showing that gD disrupts the normal nectin-1 homophilic interactions. Furthermore, the structure of the complex supports a model in which gD-receptor binding triggers HSV entry through receptor-mediated displacement of the gD C-terminal region.
Published in the journal:
. PLoS Pathog 7(9): e32767. doi:10.1371/journal.ppat.1002277
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002277
Summary
Binding of herpes simplex virus (HSV) glycoprotein D (gD) to a cell surface receptor is required to trigger membrane fusion during entry into host cells. Nectin-1 is a cell adhesion molecule and the main HSV receptor in neurons and epithelial cells. We report the structure of gD bound to nectin-1 determined by x-ray crystallography to 4.0 Å resolution. The structure reveals that the nectin-1 binding site on gD differs from the binding site of the HVEM receptor. A surface on the first Ig-domain of nectin-1, which mediates homophilic interactions of Ig-like cell adhesion molecules, buries an area composed by residues from both the gD N - and C-terminal extensions. Phenylalanine 129, at the tip of the loop connecting β-strands F and G of nectin-1, protrudes into a groove on gD, which is otherwise occupied by C-terminal residues in the unliganded gD and by N-terminal residues in the gD/HVEM complex. Notably, mutation of Phe129 to alanine prevents nectin-1 binding to gD and HSV entry. Together these data are consistent with previous studies showing that gD disrupts the normal nectin-1 homophilic interactions. Furthermore, the structure of the complex supports a model in which gD-receptor binding triggers HSV entry through receptor-mediated displacement of the gD C-terminal region.
Authors Summary
Herpes simplex virus (HSV) is a widespread human pathogen. Four viral glycoproteins (gD, gB, gH/gL) are required for HSV entry into host cells. gD binding to a cell surface receptor triggers conformational changes in the other viral glycoproteins leading to membrane fusion and viral entry. Two structurally unrelated cellular protein receptors, nectin-1 and HVEM, can mediate HSV entry upon binding to gD. The structure presented here reveals the molecular basis for the stable interaction between HSV-1 gD and the receptor nectin-1. Comparison with the previously determined structures of the gD/HVEM complex and unliganded gD shows that, despite the fact that the two receptors interact with different binding sites, they both cause a similar conformational change in gD. Therefore, our data point to a conserved mechanism for receptor mediated activation of the HSV entry process. In addition, the gD/Nectin-1 structure reveals that the gD-binding site overlaps with a surface involved in nectin-1 homo-dimerization. This observation explains how gD interferes with the cell adhesion function of nectin-1. Finally, the gD/Nectin-1 complex displays similarities with other viral ligands bound to immunoglobulin-like receptors suggesting a convergent mechanism for receptors selection and usage.
Introduction
Herpes simplex virus (HSV) enters cells by fusing its envelope with a membrane of the host cell [1]. Five viral envelope glycoproteins participate in the cell entry process. First, glycoprotein C (gC) and gB promote attachment by interacting with cell surface proteoglycans, then gD binds to a specific receptor [2], [3]. The gD-receptor interaction initiates the process that ultimately leads to gB-mediated membrane fusion [4], [5]. Depending on the cell type, fusion occurs at the cell surface or, after endocytosis of virions, with an endosomal membrane in a low pH-dependent or independent manner [6], [7]. Regardless of the entry pathway gD, gB, gH/gL and a cellular gD receptor are required for entry [8].
Several cell surface molecules can bind gD and mediate HSV entry into human cells [1]. The immune modulator HVEM (herpesvirus entry mediator) is a member of the Tumor Necrosis Factor Receptor (TNFR) family and is used by wild type (wt) HSV type 1 (HSV-1) and HSV-2 [9]. HVEM was the first described herpes simplex virus receptor and is expressed at low levels on fibroblasts and on ocular cell types [10], [11], [12]. However, the cell adhesion molecule nectin-1 (HveC, CD111) is the primary receptor for HSV-1 and HSV-2 on neurons, keratinocytes and epithelial cells [12], [13], [14], [15]. In addition, HSV-1 gD binds to heparan sulfate (HS) specifically modified by 3-O-sulfotransferases (3-OS-HS) [16]. Recently it has been shown that the interaction between gD and its receptor is not only required to trigger the HSV fusion machinery but also to direct the virus to the endocytic pathway in some cell types [6], [17], [18], [19], [20].
We previously reported the structure of HSV-1 gD alone and bound to HVEM [21], [22]. The gD ectodomain consists of 316 residues and is formed by a core with a variable-type immunoglobulin fold (IgV, residues 55 to 185) that is wrapped by a N-terminal extension and a C-terminal proline-rich extension. The first 20 N-terminal residues are flexible and extended in the receptor-free form of gD. However, when gD is bound to HVEM these residues fold back to form a hairpin structure that contains all the amino acids contacting this receptor [21], [23]. The first 32 N-terminal residues of gD are also involved in binding to 3-OS-HS but are dispensable for nectin-1 binding and usage [24].
The C-terminal extension of the gD ectodomain past residue 255 is also flexible and its location and structure were determined after stabilization in a disulfide-bonded gD dimer [22]. In this receptor-free structure, the C-terminal region folds back around the core towards the N-terminus and amino acids 280 to 306 fill the space occupied by the N-terminal first 20 residues of gD in the gD/HVEM complex. In this conformation, the C-terminal gD amino acids were positioned directly over several residues previously implicated in nectin-1 binding [22]. Despite the key insights into the gD/Nectin-1 interaction obtained from mutagenesis studies a structural model for the gD/Nectin-1 complex has been missing.
The nectin-1 ectodomain spans residues 31 to 346 forming three domains with an Ig-like fold (V-C1–C2) and contains 8 potential sites for N-linked oligosaccharides [25], [26]. Chimeric receptor molecules were used to show that the binding site for gD involves mainly the N-terminal V-domain of nectin-1 (residues 31–143) [27], [28] and that residues 64–94 in particular are part of the binding epitope [29]. In addition, linear epitopes of two neutralizing monoclonal antibodies directed at nectin-1, CK6 and CK8, were mapped to residues 80–105 [30].
Nectin-1 form cis-dimers at the cell surface to interact with nectin-1 or other nectin family members in trans (different cells) at cell junctions [31]. The crystal structure of the nectin-1 ectodomain has revealed that the V-domain mediates receptor dimerization [26] and it was proposed that these dimers may be representative of the nectin-1 cis-interaction [26]. Importantly, HSV gD has been shown to interfere with nectin-1 mediated cell-adhesion [28], [32], [33].
Given the importance of the gD/Nectin-1 interaction in HSV entry and the need to gain insights into how two structurally different receptors, nectin-1 and HVEM, trigger HSV entry we sought to obtain a molecular view of the interaction between gD and nectin-1. Here we present the structure of the gD/Nectin-1 complex determined by x-ray crystallography to 4.0 Å resolution and accompanying mutagenesis data. The comparison of the new structure with the previously determined gD/HVEM and unliganded gD structures suggests a common mechanism of receptor-induced conformational changes as a trigger of HSV membrane fusion.
Results
Structure determination and refinement
Truncated forms of the gD ectodomain (gD285t, residues 1–285; gD306t, residues 1 to 306; and gD316t, residues 1 to 316) and the full length nectin-1 ectodomain (residues 31–345; Fig. 1A) were used for complex formation and crystallization. Crystals were obtained for the gD285t/Nectin-1 complex whereas the longer forms of gD (i.e. gD306t or gD316t) failed to produce crystals. Previous experiments have demonstrated a 20–40 fold decreased affinity for nectin-1 by gD306t (KD = 1.8 µM) compared to the shorter gD285t (gD285; KD = 70 nM) [22]. This decreased affinity and the intrinsic flexibility of C-terminus of the gD ectodomain may have hindered crystallization of the longer forms. In addition, crystals from the gD285t/Nectin-1 complex could not be easily reproduced. The nectin-1 ectodomain contains several consensus N-glycosylation sites (Fig. 1A) and heterogeneity in glycosylation may interfere with crystallization [26]. However, attempts to reduce protein heterogeneity by enzymatically deglycosylating nectin-1 produced in insect or mammalian cells did not facilitate crystallization. Ultimately, a 4.0 Å resolution data set was collected and the structure was determined by the molecular replacement method with the program Phaser [34] and refined with Refmac [35] to an Rfree of 28.9% and Rwork of 26.5% (Table S1 and Fig. S1).

Overall structure of the gD/Nectin-1 complex
The crystals belong to the P3221 space group and contain three gD/Nectin-1 complexes per asymmetric unit. Two complexes form a dimer around a two-fold non-crystallographic symmetry (NCS) axis (Fig. S2) and the third complex forms an equivalent dimer with a crystallographically related molecule. Experiments with purified proteins in solution suggest that these dimers might have formed in the crystals as a result of the high protein concentration and crystal packing interactions and are unlikely to be biologically relevant (see below).
The structure shows a direct interaction between one monomer of gD and one of nectin-1 (Fig. 1B). No electron density was observed for residues 1 to 22 and 251 to 285. Both regions are known to be flexible and are presumably disordered in the crystals. In the gD285t/HVEM complex the same C-terminal region of gD was disordered [21]. Moreover, deletions within the first 32 residues of gD do not impair nectin-1 binding and HSV entry in nectin-1 expressing cells [36].
The gD binding site is located exclusively within the N-terminal V-domain of nectin-1, validating previous predictions from in vitro binding and infection assays [27], [28], [37]. The nectin-1 V-domain major axis forms an angle of almost 90° with the long axis of the gD IgV-core while the C1 domain points away from gD. The elbow angle between the V and C1 domains is the same as that observed in the unliganded form of nectin-1 [26] and differs by approximately 15° from what was observed in the structure of the closely related poliovirus receptor (necl-5, CD155) (Fig. S3) [38]. The C2 domain, though present in the crystals of the complex, was not visible in the electron-density maps and is likely to be disordered in the crystals.
The gD/Nectin-1 interface
Nectin-1 interacts with gD mainly through the side chains of exposed residues. A total of 1665 Å2 of surface area, 829 Å2 in nectin-1 and 836 Å2 in gD, are buried in the complex. This value is within the range observed for other protein/protein complexes and viral glycoprotein/receptor complexes [21], [39], [40], [41]. Nectin-1 contacts gD exclusively with one β-sheet of the V-domain (strands C″C′CFG) and the intervening loops (Fig. 1B). This β-sheet forms an extensive interface with residues from the C - and N-terminal extensions of gD. In particular, the upper part of β-strands C″C′ is close to two short gD β-strands (β2, residues 35–38 and β3, residues 219–221) whereas at the bottom part the same strands are in proximity with two short gD helical turns (η1, residues 199–201 and η2, residues 214–217).
Additional contacts are established by the loop connecting strands F and G of the nectin-1 V-domain. In particular, a prominent interaction involves Phe129, at the tip of the FG loop, which protrudes into a pocket formed by residues from the long α3-helix flanking the IgV-like core of gD and the side chain of Phe223 (Fig. 1B and Fig. 2A, B). Direct interaction with the gD IgV-core is limited and involves only nectin-1 strand C″ and the side chains of gD Gln132.

The gD/Nectin-1 structure and previous mutagenesis studies
The structure of the gD/Nectin-1 complex explains the results of previous mutagenesis experiments. On the nectin-1 V-domain, strands C″C′C were predicted to be involved in gD binding based on epitope mapping and chimeric receptor analysis [29], [30], [36], [42] (Fig. 1B). In addition, site-directed mutagenesis identified residues important for nectin-1 binding and usage between positions 64 and 94 [29]. Notably, nectin-1 residues Asn77 and Met85, which mutations to alanine hindered HSV entry [43], are now found at the interface with gD (Fig. 2A). On gD a number of residues have been shown to affect nectin-1 binding (Fig. 2A, B). For instance, gD Tyr38 side chain, a critical residue for nectin-1 binding, is located near the receptor C″ strand and Met85 (Fig. 2A). Furthermore, residues Asp215 and Arg222/Phe223 of gD, contact the receptor β-strands C″C′ and the FG loop respectively. The structural data explain the phenotype of a triple mutant of gD, which combined mutations of these residues prevent nectin-1 usage [36], [44]. In particular, the side chain of Arg222 of gD is in salt bridge distance of nectin-1 Glu125 (Fig. 2A) consistent with the decrease in gD/Nectin-1 binding affinity seen when Arg222 was mutated to alanine [45], [46]. Finally, mutation of nectin-1 Phe129 to leucine, a structurally conservative mutation, was found to impair gD binding, although this form of nectin-1 retained some receptor activity [26], [47]. The gD/Nectin-1 structure demonstrates the critical location of this residue at the interface with gD.
Comparison of the gD/Nectin-1 and Nectin-1/Nectin-1 interface
The structures of the nectin-1 dimer and the gD/Nectin-1 complex reveal similarities between their interfaces. Indeed gD contacts many of the same residues involved in nectin-1 dimerization (Fig. 2A, C) and buries a similar surface area on the receptor (1665 Å2 vs. 1686 Å2) (Fig. 2D). In the nectin-1 homo-dimer several hydrogen bonds, two salt bridges (between Lys75 and Glu135), and a number of van der Waals interactions involving Thr63, Phe129 and Met85 are present [26] (Fig. 2C). In the complex, both Thr63 and Phe129 of nectin-1 establish van der Waals interactions with Phe233 of gD whereas Met85 is in proximity of gD Tyr38. In addition, in the nectin-1 homodimer two inter-molecular salt bridges between Lys75 and Glu135 are present whereas two Glu125 residues make a potentially unfavorable interaction as they are buried at the dimer interface (Fig. 2C). In the complex, the nectin-1 Lys75-Glu135 salt bridge forms intra-molecularly and a new inter-molecular salt bridge is formed between Glu125 of nectin-1 and Arg222 of gD. Therefore, nectin-1 binding to gD leads to the replacement of the potentially unfavorable Glu125-Glu125 interaction at the nectin-1 dimer interface and formation of a new salt bridge (Fig. 2A, C).
Stoichiometry of the gD/Nectin-1 complex and effects of gD binding on nectin-1 homo-dimerization
The β-sheet of the nectin-1 V-domain that contacts gD has been implicated in mediating homophilic and heterophilic trans-interactions in other Ig-superfamily members involved in cell adhesion (e.g. necl-1 [48], CAR [49], JAM [41] and SLAM [50]). The same surface on the nectin-1 V-domain has been shown to mediate its dimerization [26] (Fig. 2D). Together these data are consistent with gD binding preventing nectin-1 from interacting with itself as previously suggested by size exclusion chromatography (SEC) experiments [25], [28].
To analyze the stoichiometry of gD/Nectin-1 binding in solution and to gain further insights into the effects of gD binding on nectin-1 oligomerization and homophilic interaction, we combined SEC with multi-angle light scattering analysis (MALS). Differently from SEC, which is dependent on the hydrodynamic radius of molecules, MALS is not affected by the shape of the molecules and provides with the absolute molar mass of the particles measured. The elution volume of the nectin-1 ectodomain (MW 40 kDa) in SEC experiments suggested an apparent MW of 115 kDa, however MALS revealed a MW of 71 kDa (data not shown). Together these data are consistent with nectin-1 forming an elongated dimer in solution as revealed by the crystal structure [26]. MALS of the gD/Nectin-1 complex yielded a MW of 72 kDa in line with the formation of a 1∶1 complex of monomers (data not shown). These analyses are in agreement with previous experiments demonstrating that gD binding prevents functional nectin-1 homo-dimerization and interferes with nectin-1 mediated cell adhesion [32], [33] and support the notion that the gD/Nectin-1 dimers, seen in the crystals, are not formed in solution (Fig. S2).
Mutations of nectin-1 and effect on gD binding
To gain additional insight into the involvement of specific amino acids in the formation of a stable gD/Nectin-1 complex and to assess the relative contribution of the CC′C″ region versus the FG loop, we targeted four nectin-1 residues located at the interface with gD for mutagenesis. We hypothesized that the CC′C″ region would rely less on single residues for binding and that the protruding phenyl ring of Phe129 was critical for function. Thus we selected Thr66, Asn82, Ser84 in the CC′C″ region and Phe129 at the tip of the FG loop (Fig. 2A). Ser84 was mutated to tyrosine (S84Y) or arginine (S84R), Asn82 was mutated to a threonine (N82T) and Thr66 to glutamine (T66Q). The changes in the size of the side chains at these positions were designed to destabilize the interaction with gD either by steric obstruction or by interfering with the formation of hydrogen bonds. On the other hand, the structure predicted that mutation of Phe129 to serine (F129S) or alanine (F129A) should prevent formation of favorable hydrophobic contacts and potential stacking interactions. In this context F129W is considered a conservative mutation. Importantly, Asn82, Ser84 and Phe129 are located in exposed positions in nectin-1 and we considered it unlikely that their mutation would affect the folding of the receptor.
To analyze the effects of these mutations on binding of nectin-1 to gD in vitro, we produced chimeras comprising the nectin-1 V-domains (aa 31–146) fused at its C-terminus to the maltose binding protein (N1V-MBP). We first analyzed the ability of the N1V-MBP proteins to bind gD by ELISA (Fig. 3A). While S84R and T66Q nectin-1 mutants bound to gD(285t) similarly to wt, S84Y and N82Y mutation markedly decreased binding. Furthermore, the aromatic side chain at Phe129 was critical for gD complex formation as F129S mutation severely compromised gD binding while F129W did not (Fig. 3A).

We next analyzed the ability of mutant N1V-MBP proteins to interact with gD on virions. We tested whether the purified soluble receptors blocked HSV entry into cells by competing with cell surface nectin-1. We used HeLa cells as targets since soluble nectin-1 V-domain produced in baculovirus efficiently blocked entry in these cells [30]. Consistent with the ELISA results, proteins with the mutations F129S or N82Y failed to block entry (Fig. 3B). Mutation S84Y had intermediate blocking activity, which correlated with its reduced binding to gD. The other mutants (F129W, S84R and T66Q), which bound gD like the wt protein by ELISA, retained the ability to block virus entry. These observations indicates that these mutants bind virion gD with sufficient affinity to compete with cell surface nectin-1 for binding to gD on the viral envelope.
Nectin-1 mutations and CK41 MAb binding
We determined the effects of nectin-1 mutations on binding to the conformation-dependent MAb CK41. This antibody blocks binding of nectin-1 to gD and efficiently prevents nectin-1 usage for HSV entry [30]. We therefore anticipated that mutations designed to prevent gD binding would also affect the interaction with CK41. Indeed mutations that affected gD binding (i.e. S84Y, N82Y and F129S) also hindered CK41 binding (Fig. 3C). In addition, since the conserved mutation F129W decreased CK41 recognition by 50%, without affecting gD binding, we infer that Phe129 is likely part of the CK41 epitope. Change of Ser84 to arginine does not affect CK41 detection, while a change to tyrosine decreased binding of this MAb. Overall these data mirror the binding properties of these nectin-1 mutants to gD and are consistent with the prediction that the CK41 epitope involves residues critical for gD binding [30].
Mutations of nectin-1 affect its function as an HSV receptor
To further assess the functional role of individual residues at the gD/Nectin-1 interface, we tested a subset of mutants in a viral entry assay. Mutations were engineered in full-length nectin-1 and transfected into receptor-negative B78H1 mouse melanoma cells [32]. Upon transient expression of wt nectin-1, B78H1 cells become susceptible to HSV entry (Fig. 4A) [32]. While all tested mutants allowed some level entry of HSV-1 KOS tk12 in a dose dependent manner, mutants F129S and F129A exhibited a marked decrease in entry compared to wild type (Fig. 4B). These results are consistent with the reduced binding affinity of the mutated receptor to gD. Nectin-1 mutants N82Y and S84Y were only partially reduced in their ability to support HSV entry despite exhibiting profound decrease in N1V-MBP binding to gD by ELISA. Such a discrepancy between binding and entry function has been previously observed [47], [51]. It is likely that the decrease in affinity observed between the purified proteins is less critical in the context of the cell-virus interaction due to avidity effects (i.e. HSV binding to multiple receptor molecules and to cell surface HS) thus making the entry assays more permissive [47], [51]. Overall, however, the phenotypes of the mutants agreed with their importance for stabilizing the interface indicated by the structure of the complex.

Nectin-1 and HVEM binding sites on gD are mutually exclusive and require the displacement of gD C-terminal region
Comparison of the gD/Nectin-1 and gD/HVEM structures reveals that the two receptors bind to different but overlapping sites. Although each receptor buries a comparable area on gD, for the most part their binding sites do not involve common residues. In fact, the binding site of HVEM is located exclusively within the first 32 N-terminal residues of gD that are folded in a hairpin-like structure [21]. The same hairpin does not form in the nectin-1 complex, rather the first N-terminal 22 residues of gD point towards the solvent (Fig. 5A–D). However, a large portion of the gD surface contacted by nectin-1 would be covered by these N-terminal residues if they were adopting the same hairpin structure observed in the gD/HVEM complex (Fig. 5A–D). Thus the structural comparison reveals that nectin-1 and HVEM bind to different sites on gD but the binding of one receptor should prevent the binding of the other.
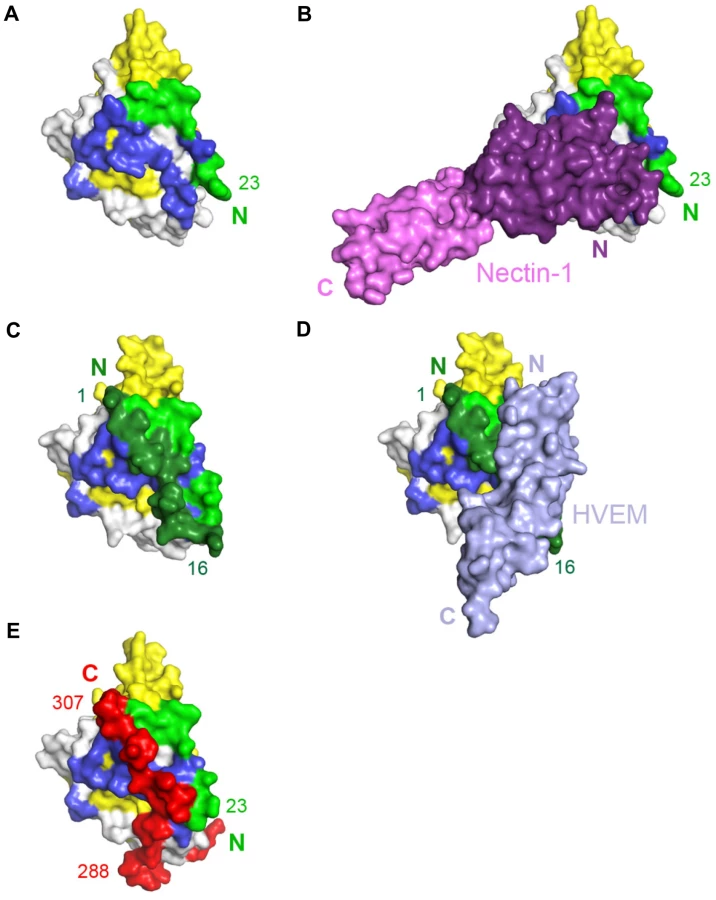
The unliganded gD structure showed the C-terminal residues 268–306 wrapped around the gD core (Fig. 5E) [21], [22]. Notably, in this position, these residues cover a large portion of the nectin-1 binding site (Fig. 5A, B and E). Therefore, formation of the gD/Nectin-1 complex requires disruption of the contacts between the core and the C-terminus of the gD ectodomain [22], [52]. Interestingly, this separation is also required for the formation of the HVEM-binding N-terminal hairpin of gD (Fig. 5C and E) [22].
Discussion
The structure and mutagenesis data reported here provide the molecular basis for the interaction between gD and its cellular receptor human nectin-1. The structure shows the key features of the interface between the nectin-1 V-domain and the core of gD [28]. The binding site for gD overlaps with a nectin-1 dimerization interface explaining how gD interferes with the cell-adhesion function of this receptor. In addition, the gD/Nectin-1 complex reveals structural similarities with other viral ligands bound to receptors with an Ig-like fold, thus pointing to a convergent mechanism for receptor selection and usage by otherwise unrelated viruses. Importantly, comparison of the gD/Nectin-1 structure with the previously determined gD/HVEM structure indicates that despite contacting different amino acids the two receptors compete for binding to gD. Finally, a comparison between the two gD-receptor structures and the unliganded gD structure provides additional insights into the mechanism of receptor-mediated activation of the HSV fusion machinery.
The gD/Nectin-1 interface
The gD/Nectin-1 structure confirms the presence at the interface of a number of gD residues that were previously identified by mutagenesis to be important for nectin-1 usage and HSV entry [36], [44], [46], [53]. gD contacts a large surface on the nectin-1 V-domain composed mainly by residues from the C″C′CFG β-sheet. In other cell adhesion molecules of the Ig-superfamily this same region is involved in homophilic and heterophilic trans-interactions and nectin-1 uses the same surface to homo-dimerize [26]. Therefore, and in agreement with our SEC/MALS data, formation of the gD/Nectin-1 complex requires dissociation of the nectin-1 dimers. Consistent with these findings, gD can prevent nectin-1 mediated cell aggregation [32] and HSV infection is favored by prior disruption of cell junctions [54].
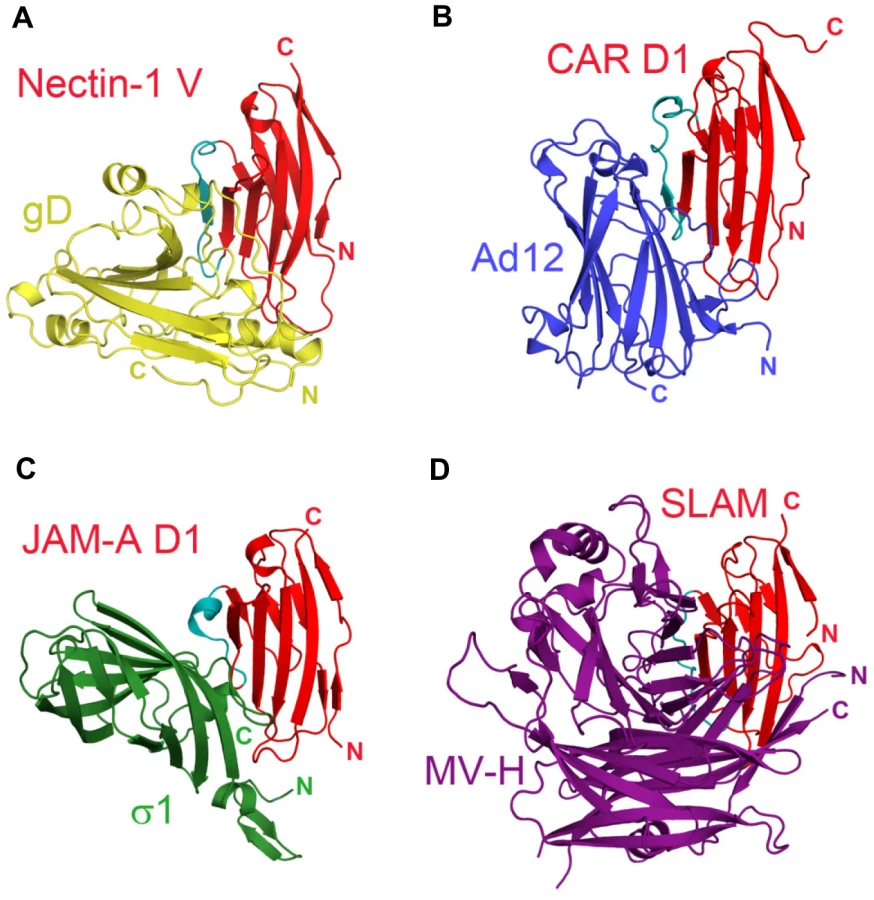
Notably, non-enveloped adenovirus, reovirus and measles virus bind to a similar epitope on their respective receptors, the Ig-like cell-adhesion receptors CAR, JAM-A and SLAM (Fig. 6A–D) [50], [55], [56], [57]). Similarly to HSV, these viruses disrupt the homophilic trans-interactions of their receptors [41], [49]. Binding to cell-adhesion molecules may ultimately favor release of these viruses by opening intercellular junctions [58].
Contribution of nectin-1 phenylalanine 129 to gD binding
On the nectin-1 side, point mutations of residues in the C″C′C region can affect binding to gD (e.g. S84Y, N82Y); however, they have limited effects on nectin-1 as a mediator of HSV entry. This suggests that the interactions established by single amino acids in this region may not be critical for function. Indeed, the same conclusion can be drawn from the gD side where multiple mutations are needed to abolish nectin-1 usage [36], [44]. This is quite different from the role of Phe129 at the tip of the FG loop of the nectin-1 V-domain. Mutations of Phe129 to alanine showed that this residue plays an important role for tight gD binding and for HSV entry. Of note, Phe129 protrudes into a pocket on the gD surface occupied in the unliganded gD by the C-terminal residue Trp294 (Fig. 7). Therefore, nectin-1 Phe129 effectively substitutes for gD Trp294 and provides a key contribution to the stable displacement of the gD C-terminal region.

Importantly, Phe129 is conserved in the poliovirus receptor necl-5 and nectin-2, consistent with the finding that a chimera composed of the necl-5 or nectin-2 V-domain where strands C″C′ were replaced with the corresponding residues of nectin-1 retains binding to gD and allows HSV infection [29], [59]. The equivalent FG loop in necl-5 has been shown to be at the interface with poliovirus and to be important for poliovirus binding [38], pointing to a conserved feature in the interaction between HSV and poliovirus with their respective receptors.
Comparison with the gD/HVEM complex and implication for gD activation
Nectin-1 and HVEM are two gD receptors that belong to different structural families. Consistently, the gD/Nectin-1 complex shares only limited similarity with the gD/HVEM complex (Fig. 5). The binding site of nectin-1 on gD differs dramatically from that of HVEM. The latter contacts only residues within the first N-terminal 32 residues of gD folded in a hairpin-like structure and the interaction involves several hydrogen bonds through main and side chain atoms [21]. The nectin-1 V-domain, instead, contacts a large surface on gD formed mostly by residues from the C-terminal extension and some amino acids from the N-terminal region. However, structural superposition of the two gD-receptor structures reveals that most of the residues involved in nectin-1 binding are buried by the gD-N-terminal residues in the gD/HVEM complex (Fig. 5). Thus, despite their considerably different binding sites, each receptor is likely to interfere with binding of the other. Indeed soluble nectin-1 can block virus entry in HVEM expressing cells [60].
A comparison between the gD-receptor complexes and the structure of unliganded gD provides additional insights in the mechanism of receptor-mediated activation of gD. In the absence of receptors residues from the gD C-terminal region (residues 285–306) are anchored by Trp294 on the core of gD. In this conformation, the C-terminus of gD occludes the nectin-1 binding site and fills the space occupied by gD N-terminal residues in the gD/HVEM complex. This is consistent with the increase receptor affinity of the C-terminally truncated form of gD used in this study compared to the full length molecule [25], [61]. Therefore, for both receptors complex formation requires the displacement of residues from the gD C-terminal region.
Role of the C-terminus of the gD ectodomain
During HSV entry, gD interacts with gH/gL and possibly gB [4], [62], [63] but how gD triggers conformational changes in the other glycoproteins remains unclear. Several observations point to a key role of the gD C-terminal region. A soluble gD molecule encompassing the entire ectodomain allows entry of a gDnull virus (i.e. devoid of envelope gD) in receptor expressing cells, whereas a truncated form of gD lacking residues 260–316 does not [64]. Moreover, mutagenesis data showed that the C-terminal region is essential for virus entry [52], [62], [64], [65], [66]. These data support a role for the gD C-terminal region in activation of the fusion/entry process. They also suggest a critical role for this region for the interaction with other viral glycoproteins. Exposure of the C-terminal region upon receptor binding therefore provides a timely and cell specific trigger for the activation of the HSV entry process. The conformation of the C-terminus in receptor-bound gD has not been determined due to its high flexibility.
Remarkably, gD has been engineered to bind alternate receptors [67], [68], [69], [70]. Heterologous ligands such as uPA, IL13, and a single chain antibody to HER-2 were engineered in at least three different regions in the gD-N-terminus or replaced the entire gD Ig core. All these ligands are able to mediate entry of HSV virions carrying the chimeric forms of gD in cells expressing the respective receptors. In the absence of structural data on such chimeras, one can only speculate on their mechanism of action during entry. Even in the most extreme recombinant [70], the N-terminal and C-terminal extensions to the Ig core are maintained for activity [71]. Thus, these regions likely contain the necessary sites for binding and activation of the other viral glycoproteins. In the engineered gDs the C-terminus may be exposed upon exogenous receptor binding in a way that is very similar to the model supported by our studies for wt HSV. Alternatively, this functional region may already be exposed in these molecules so that exogenous receptor binding would solely allow the close proximity of viral and cell membranes. In such a situation, expressing a pre-activated gD may lead to a decrease in viral fitness by diminishing the selectivity for target cells. It is likely that HSV evolved to unmask the gD C-terminus only upon receptor binding to ensure efficient tropism in the host. Of note, the initial analysis of the structural organization of gD revealed that the Ig-core acts as structural support to the functional C - and N - terminal regions and suggested that it inserted in an ancestor molecule formed by the N - and C - terminal extensions [21]. The results obtained with the above engineered gDs are consistent with such hypothesis.
In summary, our data reveal the molecular basis for the gD/Nectin-1 interaction. This new structure shows how nectin-1 and HVEM, albeit belonging to different structural families and establishing different interactions with gD, similarly cause the disruption of intra-molecular contacts between the gD C-terminal region and the rest of the molecule thus leading to activation of the entry process through a conserved mechanism. The structure of the gD/Nectin-1 complex also provides a frame to design of inhibitors that would block HSV entry and infection.
Materials and Methods
Complex purification
Production of gD-1 KOS (residues 1 to 285, gD(285t)) and human nectin-1 (residues 31–346, gD(346t)) using recombinant baculoviruses and their purification were described previously [21], [25]. The complex used for crystallization experiments was formed by mixing purified gD and nectin-1 in a 1.3∶1 molar ratio. Unbound gD in excess was removed by size exclusion chromatography (SEC) on a analytical Superdex-200 column in 20mM TRIS-HCl pH 8.0 buffer and 300mM NaCl. The complex, which eluted as single peak from the SEC step, was concentrated to 3.5mg/ml and stored at 4°C prior to crystallization.
Crystallization and data processing
The gD/Nectin-1 complex was crystallized by the vapor diffusion method with 1.0M Na2HPO4/KH2PO4 pH 7.2 and 300mM NH4SO4 as precipitating agents. SDS gels and N-terminal sequencing on washed crystals confirmed the presence of the full-sized proteins. The crystals had a very low reproducibility, suffered from strong anisotropy and the great majority diffracted only up to 7–8 Å resolution at different x-ray synchrotron sources. Nevertheless, after screening more than 100 crystals a 4.0 Å data set was collected at the BM14 beam line at the Advanced Photon Source (APS), Argonne National Lab. (Table S1). The HKL suite was used for data integration and scaling [72] and the CCP4 suite was used for further data processing and analysis [73].
Data integration suggested that the crystals belong to the hexagonal P6222 space group, however analysis of the cumulative intensity distribution [73] hinted at the presence of merohedral twinning and as a consequence to a lower symmetry space group. Data analysis with the Merohedral Crystal Twinning server for data integrated in P321 or P312 pointed to the presence of nearly perfect merohedral twinning (α = 0.49) [74]. Due to the difficulty of obtaining similar quality diffraction this data set was used for subsequent structure determination and refinement despite integration in the trigonal space groups resulted in low redundancy (Table S1).
Structure determination and refinement
The structure was determined by the Molecular Replacement (MR) method with the program Phaser [34]. The initial search was carried out in all trigonal and hexagonal space groups with a gD model (PDB entry 2C36: amino acids 27–250) and using all data between 12 and 4.0Å. A clear solution for 3 gD molecules was identified in P3221 whereas no solutions were found in all the other space groups tested. A polyalanine version of the major structural protein of peripheral nerve myelin (1NEU; 27% sequence identity with the V-domain of nectin-1), the C1 domain of the poliovirus receptor necl–5/CD155 (3EOW; 34% identity with the C1 domain of nectin-1) and the perlecan IG3 domain (1GL4 25% identity), after removal of some of the loops, were used as search models for the V, C1 and C2 domains of the receptor, respectively. Keeping the 3 gD molecules fixed, three solutions for the V domain and then for the C1 domains of nectin-1, consistent with 3 gD-nectin-1 complexes in the asymmetric unit, were clearly identified. In addition to the improvements in TFZ, LLG, Rwork and Rfree during all the steps of MR several pieces of evidence supported the correctness of the solution. First, the nectin-1 V-domain was positioned in proximity of gD residues previously implicated in nectin-1 binding; second, the N - and C-termini of the C1 and V-domain, respectively, were at a distance from each other compatible with the number of missing residues connecting the two domains; and third the model agreed with considerations on packing and symmetry relation between the molecules in the crystal (see also Results). However, only a poorly contrasted solution for one of the C2 domains could be identified by MR. Despite this solution positioned the C2 domain in proximity of a C1 domain with reasonable crystal contacts, its addition to the model did not improve the Rfree or the quality of the electron density maps and therefore was excluded from the refinement.
Simulated annealing composite 2mFo-DFc and mFo-DFc omit electron density maps followed by local three fold NCS averaging for each of the nectin-1 domains were calculated with the program Phenix [75] and used during the initial stages of model building. The resulting electron density maps clearly revealed the location of some of the loops in the nectin-1 V-domain and some of the glycans both of which had been excluded from the MR model (Fig. S1). The electron density of the glycans and of residues with large side chains together with the location of disulfide bonds in V and C1 Ig domains were used to confirm the register of the polypeptide chain. The model was refined with Refmac using the ″detwin″ option, overall B-factor refinement and tight non-crystallographic symmetry (NCS) restraints. The twinning fraction estimated by Refmac was of 0.49, in agreement with what reported by the Merohedral Crystal Twinning Server. When the structure was refined without allowing for detwinning of the data the Rfree was ∼10% higher. Following the initial building the crystal structure of nectin-1 became available [26] and was used to modify the V and C1 domains. Additional refinement showed that although most of the structure remained the same between the two models, modest differences were present in between the structures in some of the V1 loops and residues involved in the gD-nectin-1 interface. The final model includes the full V1 domain but does not include some of the loops in the distal part of the nectin-1 C1 domain which did not have interpretable electron density.
Production of nectin-1 V-domain-MBP
A DNA fragment corresponding to the variable immunoglobulin domain (V-type) of nectin-1 was amplified by PCR from a plasmid containing the cDNA form of the full length protein (pCK451) [76], using primers designed to insert a stop codon after residue 146. This fragment was inserted into the bacterial gene fusion vector pMAL-p (New England Biolabs) by a blunt-ended ligation into BamH1 restriction site of the plasmid polylinker. This construct produces a fusion with the maltose-binding protein under the control of the Lac repressor that is exported to the periplasm. Mutants were produced using the Quikchange mutagenesis kit (Stratagene). Correctness of all the constructs was verified by sequencing. For protein purification, E. coli BL21 (DE3*) cells were transformed with the constructs, grown at 37°C in LB to an optical density of 0.7 at 600nm and induced with ITPG for 16 h at 23°C. After an overnight culture cells were resuspended in 40mM TRIS pH 7.5, 300mM NaCl, 10% glycerol, 0.03% β-octyl-glucoside (Buffer A, 100ml/liter of culture) and lysed with a microfluidizer. Debris were removed by centrifugation (13000 rpm) for 45 min at 4°C and the supernatant was incubated with amylose resin (New England Biolabs). After binding, the resin was washed with Buffer A and proteins were eluted with 20mM maltose in Buffer A. Proteins were concentrated and purified by size exclusion chromatography on a Superdex S200 column equilibrated with 40mM Tris pH 8.0 and 150mM NaCl. Fractions containing non-aggregated nectin-1 V-domain MBP fusion protein (N1V-MBP) were pooled, concentrated and dialyzed against PBS. All the N1V-MBP mutants were purified similarly to the wild type protein.
Nectin-1 mutants and gD binding
Purified N1V-MBP fusion proteins were immobilized on a 96 well plate (10 µg/ml in PBS) overnight at 4°C. Plates were blocked with PBS containing 0.05%, Tween20 and 5% nonfat milk (PBS-T milk). Purified gD(306t) (residues 1 to 306) was serially diluted in PBS-T milk and added to the plate [25]. After incubation and washing with PBS-T, bound gD was detected with polyclonal rabbit serum R8 (diluted 1∶500 in PBS-T-milk) and goat anti-rabbit-HRP secondary antibody (KPL Inc). Absorbance at 405 nm was read after addition of ABTS as substrate.
Blocking HSV infection
HSV-1 KOStk12 [9] was diluted in cell culture medium and pre-incubated with purified N1V-MBP proteins at various concentrations for 1 h at 37°C. The mixture was added to confluent HeLa cell cultures in a 96 well plate (5×104 cells/well) and directly incubated at 37°C for 5–6 h. Lysis and reading of β-galactosidase activity was performed as described below for entry assays.
CK41 detection by ELISA
Purified anti-nectin-1 monoclonal antibody CK41 [30] was immobilized on an ELISA plate in PBS (10 µg/ml). After blocking with PBS-T-milk, purified N1V-MBP (20 µg/ml) was added in PBS-T-milk for 2 h at RT. The wells were washed and N1V-MBP was detected with HRP-conjugated anti-MBP antibody MBP-17 (Sigma-Aldrich), followed by goat anti-rabbit-HRP secondary antibody (KPL Inc). Absorbance at 405 nm was read after addition of ABTS as substrate.
Nectin-1 mutants and HSV entry
Mutations were engineered in full-length nectin-1 using the Quikchange mutagenesis kit in plasmid pCK452 expressing full-length human nectin-1α [76]. Plasmids were transfected into B78H1 cells using either Geneporter (2 µg plasmid/ well of subconfluent cells in 6 well plates) or the AMAXA system kit V, 2 µg/106 cells. One day post transfection cells were seeded in 96 well plates (5×104 cells/well). After an overnight culture, cells were infected with dilutions of sucrose-purified HSV-1 KOS tk12. After 6 h incubation at 37°C cells were lysed by adding NP-40 to a final concentration of 0.5%. A 50 µl volume of lysate was mixed with an equal volume of β-galactosidase substrate (chlorophenol red-β-D-galactopyranoside; Roche) and absorbance was read at 595nm for 50 min to record enzymatic activity. Surface expression of nectin-1 mutants was assessed by CELISA using MAb CK6 [30] and was comparable between mutants and wt nectin-1 in transiently transfected B78H1 cells.
Accession number for genes and proteins mentioned in the text
Nectin-1
Swissprot/UniProt: Q15223
NCBI: NP_002846
HVEM
Swissprot/UniProt: Q92956
NCBI: AAQ89238
gD KOS
Swissprot/UniProt: A1Z0Q5
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. HeldweinEEKrummenacherC 2008 Entry of herpesviruses into mammalian cells. Cell Mol Life Sci 65 1653 1668
2. ConnollySAJacksonJOJardetzkyTSLongneckerR 2011 Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat Rev Microbiol 9 369 381
3. EisenbergRJHeldweinEECohenGHKrummenacherC 2011 Recent progress in understanding herpes simplex virus entry: relationship of structure to function. WellerSK Alphaherpesviruses: Molecular and cellular Biology Norwich, UK Horizon Scientific Press 131 152
4. AtanasiuDSawWTCohenGHEisenbergRJ 2010 Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J Virol 84 12292 12299
5. ChowdaryTKCairnsTMAtanasiuDCohenGHEisenbergRJ 2010 Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat Struct Mol Biol 17 882 888
6. MilneRSNicolaAVWhitbeckJCEisenbergRJCohenGH 2005 Glycoprotein D receptor-dependent, low-pH-independent endocytic entry of herpes simplex virus type 1. J Virol 79 6655 6663
7. NicolaAVHouJMajorEOStrausSE 2005 Herpes simplex virus type 1 enters human epidermal keratinocytes, but not neurons, via a pH-dependent endocytic pathway. J Virol 79 7609 7616
8. NicolaAVStrausSE 2004 Cellular and viral requirements for rapid endocytic entry of herpes simplex virus. J Virol 78 7508 7517
9. MontgomeryRIWarnerMSLumBJSpearPG 1996 Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 87 427 436
10. TiwariVClementCScanlanPMKowlessurDYueBY 2005 A role for herpesvirus entry mediator as the receptor for herpes simplex virus 1 entry into primary human trabecular meshwork cells. J Virol 79 13173 13179
11. AkhtarJTiwariVOhMJKovacsMJaniA 2008 HVEM and Nectin-1 are the Major Mediators of Herpes Simplex Virus 1 (HSV-1) Entry into Human Conjunctival Epithelium. Invest Ophthalmol Vis Sci 49 4026 4035
12. KrummenacherCBaribaudFPonce De LeonMBaribaudIWhitbeckJC 2004 Comparative usage of herpesvirus entry mediator A and nectin-1 by laboratory strains and clinical isolates of herpes simplex virus. Virology 322 286 299
13. SimpsonSAManchakMDHagerEJKrummenacherCWhitbeckJC 2005 Nectin-1/HveC Mediates herpes simplex virus type 1 entry into primary human sensory neurons and fibroblasts. J Neurovirol 11 208 218
14. GalenBCheshenkoNTuyamaARamratnamBHeroldBC 2006 Access to nectin favors herpes simplex virus infection at the apical surface of polarized human epithelial cells. J Virol 80 12209 12218
15. HuberMTWisnerTWHegdeNRGoldsmithKARauchDA 2001 Herpes Simplex Virus with Highly Reduced gD Levels Can Efficiently Enter and Spread between Human Keratinocytes. J Virol 75 10309 10318
16. TiwariVClementCXuDValyi-NagyTYueBY 2006 Role for 3-o-sulfated heparan sulfate as the receptor for herpes simplex virus type 1 entry into primary human corneal fibroblasts. J Virol Methods 80 8970 8980
17. DelboyMGPattersonJLHollanderAMNicolaAV 2006 Nectin-2-mediated entry of a syncytial strain of herpes simplex virus via pH-independent fusion with the plasma membrane of Chinese hamster ovary cells. Virol J 3 105
18. StilesKMMilneRSCohenGHEisenbergRJKrummenacherC 2008 The herpes simplex virus receptor nectin-1 is down-regulated after trans-interaction with glycoprotein D. Virology 373 98 111
19. StilesKMKrummenacherC 2010 Glycoprotein D actively induces rapid internalization of two nectin-1 isoforms during herpes simplex virus entry. Virology 399 109 119
20. StilesKMWhitbeckJCLouHCohenGHEisenbergRJ 2010 Herpes simplex virus glycoprotein D interferes with binding of herpesvirus entry mediator to its ligands through downregulation and direct competition. J Virol 84 11646 11660
21. CarfiAWillisSHWhitbeckJCKrummenacherCCohenGH 2001 Herpes simplex virus glycoprotein D bound to the human receptor HveA. Mol Cell 8 169 179
22. KrummenacherCSupekarVMWhitbeckJCLazearEConnollySA 2005 Structure of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. EMBO J 24 4144 4153
23. ConnollySALandsburgDJCarfiAWileyDCCohenGH 2003 Structure-based mutagenesis of herpes simplex virus glycoprotein D defines three critical regions at the gD/HveA interface. J Virol 77 8127 8140
24. YoonMZagoAShuklaDSpearPG 2003 Mutations in the N termini of herpes simplex virus type 1 and 2 gDs alter functional interactions with the entry/fusion receptors HVEM, Nectin-2, and 3-O-sulfated heparan sulfate but not with Nectin-1. J Virol 77 9221 9231
25. KrummenacherCNicolaAVWhitbeckJCLouHHouW 1998 Herpes simplex virus glycoprotein D can bind to poliovirus receptor-related protein 1 or herpesvirus entry mediator, two structurally unrelated mediators of virus entry. J Virol 72 7064 7074
26. NaritaHYamamotoYSuzukiMMiyazakiNYoshidaA 2011 Crystal structure of the cis-dimer of nectin-1: implications for the architecture of cell-cell junctions. J Biol Chem 286 12659 12669
27. CocchiFLopezMMenottiLAoubalaMDubreuilP 1998 The V domain of herpesvirus Ig-like receptor (HIgR) contains a major functional region in herpes simplex virus-1 entry into cells and interacts physically with the viral glycoprotein D. Proc Natl Acad Sci U S A 95 15700 15705
28. KrummenacherCRuxAHWhitbeckJCPonce de LeonMLouH 1999 The first immunoglobulin-like domain of HveC is sufficient to bind herpes simplex virus gD with full affinity while the third domain is involved in oligomerization of HveC. J Virol 73 8127 8137
29. CocchiFLopezMDubreuilPCampadelli FiumeGMenottiL 2001 Chimeric nectin1-poliovirus receptor molecules identify a nectin1 region functional in herpes simplex virus entry. J Virol 75 7987 7994
30. KrummenacherCBaribaudIPonce de LeonMWhitbeckJCLouH 2000 Localization of a binding site for herpes simplex virus glycoprotein D on the herpesvirus entry mediator C by using anti-receptor monoclonal antibodies. J Virol 74 10863 10872
31. TakaiYMiyoshiJIkedaWOgitaH 2008 Nectins and nectin-like molecules: roles in contact inhibition of cell movement and proliferation. Nat Rev Mol Cell Biol 9 603 615
32. KrummenacherCBaribaudISanzoJFCohenGHEisenbergRJ 2002 Effects of herpes simplex virus on structure and function of nectin - 1/HveC. J Virol 76 2424 2433
33. SakisakaTTaniguchiTNakanishiHTakahashiKMiyaharaM 2001 Requirement of interaction of nectin-1alpha/HveC with afadin for efficient cell-cell spread of herpes simplex virus type 1. J Virol 75 4734 4743
34. McCoyAJGrosse-KunstleveRWAdamsPDWinnMDStoroniLC 2007 Phaser crystallographic software. J Appl Crystallogr 40 658 674
35. SkubakPMurshudovGNPannuNS 2004 Direct incorporation of experimental phase information in model refinement. Acta Crystallogr D 60 2196 2201
36. SpearPGManojSYoonMJoggerCRZagoA 2006 Different receptors binding to distinct interfaces on herpes simplex virus gD can trigger events leading to cell fusion and viral entry. Virology 344 17 24
37. GeraghtyRJFridbergAKrummenacherCCohenGHEisenbergRJ 2001 Use of chimeric nectin-1(HveC)-related receptors to demonstrate that ability to bind alphaherpesvirus gD is not necessarily sufficient for viral entry. Virology 285 366 375
38. ZhangPMuellerSMoraisMCBatorCMBowmanVD 2008 Crystal structure of CD155 and electron microscopic studies of its complexes with polioviruses. Proc Natl Acad Sci U S A 105 18284 18289
39. KwongPDWyattRRobinsonJSweetRWSodroskiJ 1998 Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393 648 659
40. BewleyMCSpringerKZhangYBFreimuthPFlanaganJM 1999 Structural analysis of the mechanism of adenovirus binding to its human cellular receptor, CAR. Science 286 1579 1583
41. KirchnerEGuglielmiKMStraussHMDermodyTSStehleT 2008 Structure of reovirus sigma1 in complex with its receptor junctional adhesion molecule-A. PLoS Pathog 4 e1000235
42. MenottiLCerretaniAHengelHCampadelli-FiumeG 2008 Construction of a fully retargeted herpes simplex virus 1 recombinant capable of entering cells solely via human epidermal growth factor receptor 2. J Virol 82 10153 10161
43. MartinezWMSpearPG 2002 Amino acid substitutions in the V domain of nectin-1 (HveC) that impair entry activity for herpes simplex virus types 1 and 2 but not for Pseudorabies virus or bovine herpesvirus 1. J Virol 76 7255 7262
44. UchidaHShahWAOzuerAFramptonARJrGoinsWF 2009 Generation of herpesvirus entry mediator (HVEM)-restricted herpes simplex virus type 1 mutant viruses: resistance of HVEM-expressing cells and identification of mutations that rescue nectin-1 recognition. J Virol 83 2951 2961
45. WhitbeckJCMuggeridgeMIRuxAHouWKrummenacherC 1999 The major neutralizing antigenic site on herpes simplex virus glycoprotein D overlaps a receptor-binding domain. J Virol 73 9879 9890
46. ConnollySALandsburgDJCarfiAWhitbeckJCZuoY 2005 Potential nectin-1 binding site on herpes simplex virus glycoprotein D. J Virol 79 1282 1295
47. StruyfFMartinezWMSpearPG 2002 Mutations in the N-terminal domains of nectin-1 and nectin-2 reveal differences in requirements for entry of various alphaherpesviruses and for nectin-nectin interactions. J Virol 76 12940 12950
48. DongXXuFGongYGaoJLinP 2006 Crystal structure of the V domain of human Nectin-like molecule-1/Syncam3/Tsll1/Igsf4b, a neural tissue-specific immunoglobulin-like cell-cell adhesion molecule. J Biol Chem 281 10610 10617
49. van RaaijMJChouinEvan der ZandtHBergelsonJMCusackS 2000 Dimeric structure of the coxsackievirus and adenovirus receptor D1 domain at 1.7 Å resolution. Structure 8 1147 1155
50. HashiguchiTOseTKubotaMMaitaNKamishikiryoJ 2011 Structure of the measles virus hemagglutinin bound to its cellular receptor SLAM. Nat Struct Mol Biol 18 135 141
51. ConnollySALandsburgDJCarfiAWileyDCEisenbergRJ 2002 Structure-based analysis of the herpes simplex virus glycoprotein D binding site present on herpesvirus entry mediator HveA(HVEM). J Virol 76 10894 10904
52. LazearECarfiAWhitbeckJCCairnsTMKrummenacherC 2008 Engineered disulfide bonds in herpes simplex virus type 1 gD separate receptor binding from fusion initiation and viral entry. J Virol 82 700 709
53. ManojSJoggerCRMyscofskiDYoonMSpearPG 2004 Mutations in herpes simplex virus glycoprotein D that prevent cell entry via nectins and alter cell tropism. Proc Natl Acad Sci U S A 101 12414 12421
54. YoonMSpearPG 2002 Disruption of adherens junctions liberates nectin-1 to serve as receptor for herpes simplex virus and pseudorabies virus entry. J Virol 76 7203 7208
55. BergelsonJMCunninghamJADroguettGKurt-JonesEAKrithivasA 1997 Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 275 1320 1323
56. MendelsohnCLWimmerERacanielloVR 1989 Cellular receptor for poliovirus: molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell 56 855 865
57. BartonESForrestJCConnollyJLChappellJDLiuY 2001 Junction adhesion molecule is a receptor for reovirus. Cell 104 441 451
58. WaltersRWFreimuthPMoningerTOGanskeIZabnerJ 2002 Adenovirus fiber disrupts CAR-mediated intercellular adhesion allowing virus escape. Cell 110 789 799
59. MenottiLCocchiFCampadelli-FiumeG 2002 Critical residues in the CC′ ridge of the human nectin1 receptor V domain enable herpes simplex virus entry into the cell and act synergistically with the downstream region. Virology 301 6 12
60. GeraghtyRJKrummenacherCEisenbergRJCohenGHSpearPG 1998 Entry of alphaherpesviruses mediated by poliovirus receptor related protein 1 and poliovirus receptor. Science 280 1618 1620
61. WillisSHRuxAHPengCWhitbeckJCNicolaAV 1998 Examination of the kinetics of herpes simplex virus glycoprotein D binding to the herpesvirus entry mediator, using surface plasmon resonance. J Virol 72 5937 5947
62. GianniTAmasioMCampadelli-FiumeG 2009 Herpes simplex virus gD forms distinct complexes with fusion executors gB and gH/gL in part through the C-terminal profusion domain. J Biol Chem 284 17370 17382
63. Perez-RomeroPPerezACapulAMontgomeryRFullerAO 2005 Herpes simplex virus entry mediator associates in infected cells in a complex with viral proteins gD and at least gH. J Virol 79 4540 4544
64. CocchiFFuscoDMenottiLGianniTEisenbergRJ 2004 The soluble ectodomain of herpes simplex virus gD contains a membrane-proximal pro-fusion domain and suffices to mediate virus entry. Proc Natl Acad Sci U S A 101 7445 7450
65. ZhouGYeGJDebinskiWRoizmanB 2002 Engineered herpes simplex virus 1 is dependent on IL13Ralpha 2 receptor for cell entry and independent of glycoprotein D receptor interaction. Proc Natl Acad Sci U S A 99 15124 15129
66. ZagoAJoggerCRSpearPG 2004 Use of herpes simplex virus and pseudorabies virus chimeric glycoprotein D molecules to identify regions critical for membrane fusion. Proc Natl Acad Sci U S A 101 17498 17503
67. ZhouGAvitabileECampadelli-FiumeGRoizmanB 2003 The domains of glycoprotein D required to block apoptosis induced by herpes simplex virus 1 are largely distinct from those involved in cell-cell fusion and binding to nectin1. J Virol 77 3759 3767
68. KamiyamaHZhouGRoizmanB 2006 Herpes simplex virus 1 recombinant virions exhibiting the amino terminal fragment of urokinase-type plasminogen activator can enter cells via the cognate receptor. Gene Ther 13 621 629
69. MenottiLCerretaniACampadelli-FiumeG 2006 A herpes simplex virus recombinant that exhibits a single-chain antibody to HER2/neu enters cells through the mammary tumor receptor, independently of the gD receptors. J Virol 80 5531 5539
70. MenottiLNicolettiGGattaVCrociSLanduzziL 2009 Inhibition of human tumor growth in mice by an oncolytic herpes simplex virus designed to target solely HER-2-positive cells. Proc Natl Acad Sci U S A 106 9039 9044
71. ZhouGRoizmanB 2007 Separation of receptor-binding and profusogenic domains of glycoprotein D of herpes simplex virus 1 into distinct interacting proteins. Proc Natl Acad Sci U S A 104 4142 4146
72. OtwinowskiZMinorW 1997 Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276 307 326
73. CCP4 1994 The CCP4 suite: programs for protein crystallography. Acta Crystallogr D 50 760 763
74. YeatesTO 1997 Detecting and overcoming crystal twinning. Methods Enzymol 276 344 358
75. AdamsPDAfoninePVBunkocziGChenVBDavisIW 2010 PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D 66 213 221
76. KrummenacherCBaribaudIEisenbergRJCohenGH 2003 Cellular localization of nectin-1 and glycoprotein D during herpes simplex virus infection. J Virol 77 8985 8999
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 9
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- HTLV-1 Propels Thymic Human T Cell Development in “Human Immune System” Rag2 gamma c Mice
- Hostile Takeover by : Reorganization of Parasite and Host Cell Membranes during Liver Stage Egress
- Exploiting and Subverting Tor Signaling in the Pathogenesis of Fungi, Parasites, and Viruses
- A Viral Ubiquitin Ligase Has Substrate Preferential SUMO Targeted Ubiquitin Ligase Activity that Counteracts Intrinsic Antiviral Defence
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy