
Genomic Insights into the Origin of Parasitism in the Emerging Plant Pathogen
Bursaphelenchus xylophilus is the nematode responsible for a devastating epidemic of pine wilt disease in Asia and Europe, and represents a recent, independent origin of plant parasitism in nematodes, ecologically and taxonomically distinct from other nematodes for which genomic data is available. As well as being an important pathogen, the B. xylophilus genome thus provides a unique opportunity to study the evolution and mechanism of plant parasitism. Here, we present a high-quality draft genome sequence from an inbred line of B. xylophilus, and use this to investigate the biological basis of its complex ecology which combines fungal feeding, plant parasitic and insect-associated stages. We focus particularly on putative parasitism genes as well as those linked to other key biological processes and demonstrate that B. xylophilus is well endowed with RNA interference effectors, peptidergic neurotransmitters (including the first description of ins genes in a parasite) stress response and developmental genes and has a contracted set of chemosensory receptors. B. xylophilus has the largest number of digestive proteases known for any nematode and displays expanded families of lysosome pathway genes, ABC transporters and cytochrome P450 pathway genes. This expansion in digestive and detoxification proteins may reflect the unusual diversity in foods it exploits and environments it encounters during its life cycle. In addition, B. xylophilus possesses a unique complement of plant cell wall modifying proteins acquired by horizontal gene transfer, underscoring the impact of this process on the evolution of plant parasitism by nematodes. Together with the lack of proteins homologous to effectors from other plant parasitic nematodes, this confirms the distinctive molecular basis of plant parasitism in the Bursaphelenchus lineage. The genome sequence of B. xylophilus adds to the diversity of genomic data for nematodes, and will be an important resource in understanding the biology of this unusual parasite.
Published in the journal:
. PLoS Pathog 7(9): e32767. doi:10.1371/journal.ppat.1002219
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002219
Summary
Bursaphelenchus xylophilus is the nematode responsible for a devastating epidemic of pine wilt disease in Asia and Europe, and represents a recent, independent origin of plant parasitism in nematodes, ecologically and taxonomically distinct from other nematodes for which genomic data is available. As well as being an important pathogen, the B. xylophilus genome thus provides a unique opportunity to study the evolution and mechanism of plant parasitism. Here, we present a high-quality draft genome sequence from an inbred line of B. xylophilus, and use this to investigate the biological basis of its complex ecology which combines fungal feeding, plant parasitic and insect-associated stages. We focus particularly on putative parasitism genes as well as those linked to other key biological processes and demonstrate that B. xylophilus is well endowed with RNA interference effectors, peptidergic neurotransmitters (including the first description of ins genes in a parasite) stress response and developmental genes and has a contracted set of chemosensory receptors. B. xylophilus has the largest number of digestive proteases known for any nematode and displays expanded families of lysosome pathway genes, ABC transporters and cytochrome P450 pathway genes. This expansion in digestive and detoxification proteins may reflect the unusual diversity in foods it exploits and environments it encounters during its life cycle. In addition, B. xylophilus possesses a unique complement of plant cell wall modifying proteins acquired by horizontal gene transfer, underscoring the impact of this process on the evolution of plant parasitism by nematodes. Together with the lack of proteins homologous to effectors from other plant parasitic nematodes, this confirms the distinctive molecular basis of plant parasitism in the Bursaphelenchus lineage. The genome sequence of B. xylophilus adds to the diversity of genomic data for nematodes, and will be an important resource in understanding the biology of this unusual parasite.
Introduction
The nematode Caenorhabditis elegans was the first multicellular organism for which a complete genome sequence was available, and subsequent genomics research on a wider range of nematodes has provided information on many important biological processes and is underpinned by the information developed for C. elegans [1]. While C. elegans is a free-living bacterial feeder, nematodes exhibit a wide range of ecological interactions, including important parasites of humans and livestock that have huge agricultural and medical impacts [2].
Plant parasitic nematodes cause damage to crops globally. Within the Nematoda, the ability to parasitise plants has evolved independently on several occasions [3] and nematodes use a wide range of strategies to parasitise plants. Some nematodes are migratory ectoparasites which remain outside the roots and have a very limited interaction with their hosts. Migratory endoparasitic nematodes invade their host and cause extensive damage as they move through the host and feed. Sedentary endoparasitic nematodes, including the cyst nematodes and root knot nematodes, have highly complex biotrophic interactions with their hosts. These are the most damaging plant nematodes and consequently genomes for two species of root knot nematode have been reported [4], [5] with others in progress for cyst nematodes. Currently there is no genome sequence for any migratory endoparasitic nematode.
The pine wood nematode Bursaphelenchus xylophilus is a migratory endoparasite that causes severe damage to forestry and forest ecosystems (reviewed in [6]). B. xylophilus is native to North America such that trees there have evolved tolerance or resistance to the pathogen. However, at the start of the 20th Century it was introduced into Japan and has subsequently spread to other countries in Asia where no natural resistance is present, causing huge damage in an on-going epidemic of pine wilt disease. Despite global quarantine efforts B. xylophilus was recently introduced into Portugal [7] and has now also spread to Spain.
Most species within the Bursaphelenchus genus, including the closest relatives of B. xylophilus [8], are fungal feeders that are transmitted by vector insects only to dead or dying trees during oviposition. B. xylophilus and the few other pathogenic species described to date are unique in their capacity to feed on live trees as well as on the fungi that colonise dead or dying trees, so these species may represent a relatively recent, independent origin of plant parasitism. The nematode is a member of the Aphelenchoididae and belongs to clade 10 [3] while most other major plant parasites including Meloidogyne species belong to clade12 [3] (Figure 1). The life cycle of B. xylophilus is summarised in Figure 2 and the infection and disease process has been reviewed by Mamiya [9] and by Jones et al. [6].


Little was known about the molecular basis of the interactions between B. xylophilus and its host plants. A series of advances have been made in the last few years, underpinned by a relatively small scale Expressed Sequence Tag (EST) project on this nematode [10]. Genes involved in parasitism were identified and characterised, including those encoding a wide range of plant cell wall degrading or modifying proteins [11]–[13]. As in other plant parasitic nematodes, it is now clear that horizontal gene transfer has played an important role in the evolution of plant parasitism in B. xylophilus [14].
In order to shed further light on the mechanisms of parasitism used by B. xylophilus and to investigate the genetic and genomic factors involved in the evolution of parasitism, we have produced a high quality genome sequence from an inbred line of this nematode. We describe the assembly and initial annotation and characterisiation of the genome sequence, then interrogate this dataset to identify genes involved in key biological processes, including those associated with chemosensation, neurotransmission, alimentation, stress-responses and development. Further, we identify genes potentially important in functional genetics such as the RNAi pathway and putative control targets such as G-protein coupled receptors (GPCRs), peptidases and neuropeptide genes. This genome sequence allows a comparison of genes involved in plant parasitism across nematode clades and expands our knowledge of the role played by horizontal gene transfer in the evolution of plant parasitism by nematodes.
Materials and Methods
Biological materials and DNA/RNA extraction
The B. xylophilus Ka4 population, which originated in Ibaraki prefecture Japan and has been maintained for over 15 years in the Nematology Lab in FFPRI was previously used to generate biological material for ESTs. The Ka4C1 inbred line was established by sister-brother mating over 8 generations from the Ka4 population and was used to generate of material for genome sequencing.
Nematodes were cultivated for 10 days on Botrytis cinerea grown on autoclaved barley grains with antibiotics (100 µg/ml streptomycin and 25 µg/ml chloramphenicol). The nematodes were collected using a modified Baermann funnel technique for 3 h at 25°C and cleaned by sucrose flotation [15] followed by three rinses in 1x PBST. The cleaned nematodes were incubated in 1x PBST containing antibiotics (100 µg/ml streptomycin and 25 µg/ml chloramphenicol) at 23°C for 8 hours to allow nematodes to digest fungal residues remaining in their guts before use. Genomic DNA was extracted from nematodes using GenomeTip-100G (Qiagen) following the manufacturer's instructions.
Poly-(A) + RNA was extracted from mixed-stage nematodes or fourth-stage dispersal larva (DL4 or LIV) collected from the vector insect beetle as described previously [10] and used for the construction of EST libraries.
Chromosome number and genome size estimation
To determine the B. xylophilus chromosome number, chromosomes were observed in early embryos by DAPI staining and confocal laser-scanning microscopy [16]. The genome size of B. xylophilus was estimated using real time PCR as described in Welhen et al [17]. Translation elongation factor 1 alpha (genbank no GU130155) was used as a reference. DNA concentration was calculated using Qubit (Invitrogen) and the real time PCR reaction was performed using the StepOnePlus system (Applied BioSystems) with SYBR Green I. These protocols can be extremely sensitive and consequently the experiments were repeated in triplicate.
Sequencing strategy
454/Roche GS FLX (454 FLX) sequencing
A library of single stranded DNA fragments was obtained from B. xylophilus genomic DNA using Roche/454 standard procedures and a total of 993,947 single reads were obtained from this library using a 454 FLX Genome Sequencer. For paired end sequencing, libraries of 8 kb and 20 kb fragments were prepared using the manufacturer's standard protocol after shearing using a Hydroshear device. A total of 1,729,870 reads for the 8 k library and 223,236 reads for the 20 k library were obtained after standard Roche quality trimming. Of these, 89% and 88% were recognized as paired by the Newbler assembler (version 2.3). The 454 FLX sequence data provided 13× coverage of the genome sequence.
Illumina sequencing
Genomic DNA was sequenced on Illumina Genome Analyser I (GAI) using standard procedures. The sequences were 51 bases long. A total of 71,593,088 reads passed the default filter, giving a total coverage of about 48 genome equivalents.
Assembly process
Sequence data from 454 FLX and Illumina GAI were assembled using the Newbler de novo assembler (version 2.3) and Velvet assembler (version 1.0.12) [18] respectively. The result from each assembly was combined using Minimus2 (sourceforge.net/projects/amos/) and contigs supported by both assemblies were used as fake unpaired capillary reads in the subsequent Newbler assembly. The resulting assembly was improved using three different methods: AbacasII [19] to merge small contigs; IMAGE [20] to iteratively map and assemble short reads to close gaps; and iCORN [21] to iteratively correct single base substitutions and small indels.
As an indirect measure of completeness of the assembly, a search for orthologues was performed by CEGMA (ver. 2.0) software using CEGs (core eukaryotic genes); a set of 248 extremely highly conserved genes thought to be present in almost all eukaryotes in a reduced number of paralogues [22].
EST clustering was performed using PartiGene (ver. 3.0), a software pipeline designed to analyze and organize EST data sets [23]. Briefly, EST sequences were clustered into groups (putative genes) on the basis of sequence similarity and then clusters were assembled to yield consensus sequences using Phrap (P. Green, unpublished). Mapping of individual ESTs or clustered ESTs to the genome assembly was performed using PASA software [24].
Protein coding gene prediction
EST data generated in this study and previously obtained by capillary sequencing [10] were used to predict genes in the assembly. The total number of ESTs used was 82,100.
A reference dataset of 565 B. xylophilus protein encoding genes was manually curated from EST clusters and predictions of highly conserved genes using CEGMA [25]. 365 of these were used to train ab-initio gene predictors Augustus [26] and SNAP [27] and 200 were used to evaluate accuracy of the predictions. EVidenceModeler [24] was used to combine all predictions from gene predictors Augustus, SNAP and GeneMark.hmm [28], EST mapping data from PASA [24] and protein homology data against the Pfam database using GeneWise2 [29]. Gene prediction accuracy was computed at the level of nucleotides, exons and complete genes on 200 manually-curated gene models (Figure S1) as described previously [30].
Functional annotation
Initial functional annotation was performed using InterProScan to search against the InterPro protein family database, which included PROSITE, PRINTS, Pfam, ProDom, SMART, TIGRFAMs, PIR SuperFamily and SUPERFAMILY [31]. The latest version Pfam search (ver. 24.0) [32], which uses HMMER3 and is more sensitive than the previous version packed in InterProScan, was also performed independently for B. xylophilus genome. Gene Ontology annotation was derived using Blast2GO software [33] based on the BLAST match against NCBI non-redundant (NR) proteins with an E-value cutoff of 1e-10 and InterProScan results.
Assignments to conserved positions in metabolic and regulatory pathways were performed using KOBAS software [34] based on the KEGG annotation resource [35]. KEGG genes and KO term annotations were assigned based on similarity searches with a 1e-5 E-value cutoff and a rank cutoff 5. Significantly enriched pathway terms between two organisms were identified by frequencies of terms with chi-square and FDR correlation tests.
To study the evolution of gene families across nematodes within the order Rhabditida, we used the predicted protein sets from all 9 genomes available in WormBase release WS221 (www.wormbase.org) – C. elegans, C. brenneri, C. briggsae, C. japonicum, C. remaneri, Meloidogyne hapla, M. incognita, Pristionchus pacificus and Brugia malayi, together with predicted proteins of B. xylophilus. Version 2.0 of the OrthoMCL pipeline [36] was used to cluster proteins into families of orthologous genes, with default settings and the BLAST parameters recommended in the OrthoMCL documentation. We reconstructed the evolution of gene families on a phylogeny for these 10 species, based on aligning amino-acid sequences from single-copy gene families using Muscle v3.6 [37], and constructing coding-sequence nucleotide alignments based on these. Phylogenetic inference was performed using BEAST v.1.6.1 [38] from 10 million Markov Chain Monte Carlo (MCMC) generations under a strict clock model using the SRD [39] model for each gene partition. Three independent MCMC runs converged to the same posterior probability. Birth and death of gene families was inferred under Dollo parsimony using the Dollop program from v3.69 of the Phylip package [40].
To look for potential horizontal gene transfers specifically into the B. xylophilus lineage, we used BLASTP to compare predicted B. xylophilus protein sequences against the NCBI NR database, producing a candidate set of laterally transferred genes that either had no significant BLAST hit (E-value≤10−5) to any nematode sequence or had significant hits only to genes from Aphelenchoidoidea and no other nematode. For each of these candidates, amino acid sequence data was extracted from the NCBI database, aligned using Muscle v3.6 and ML phylogenetic trees estimated using the best-fitting model under the AICc criterion in RaxML v7.2.8. [41]. Clade support was estimated using 100 non-parametric bootstrap replicates in RaxML, and approximately unbiased (AU) statistical tests of tree topology were performed in Consel v1.19 [42].
Non-coding RNA
The detection and annotation of ncRNA molecules was performed using the LeARN pipeline [43]. This pipeline contains four independent methods: tRNAscanSE for transfer RNA (tRNA) gene detection, NCBI-BLASTN versus a ribosomal RNA (rRNA) sequence database, the Rfam database (release 9.0) to detect common ncRNA families and a mirfold-based pipeline using the mirBase library as a source of micro RNA (miRNA) candidates.
Repeat analysis
Transposable elements (TEs) in the assembly were identified using two approaches. The first stage consisted of de novo identification of repeat families in the assembly based on signatures of transposable elements and assuming fragments of TEs are present throughout the genome. Long terminal repeat (LTR) retrotransposons were identified using LTRharvest which searches for two near-identical copies of an LTR flanked by target site duplications that are close to each other. We also used RepeatModeler (http://www.repeatmasker.org/RepeatModeler.html) which aims to construct repeat consensus from two de novo detection programs (RepeatScout and RECON). Repeats present at less than 10 copies in the genome or that were less than 100 bp were excluded from further analysis. The second approach used homology searching of the assembly sequence against curated TEs using TransposonPSI (http://transposonpsi.sourceforge.net/). UCLUST was used to cluster the candidate sequences (with 80% identity) and create a non-redundant library of repeat consensus sequences.
The annotation of repeat candidates involves a search against RepBase and NCBI non-redundant library. Some of these candidates that have some annotations available from program output (for example, from TransposonPSI) were further checked this way. Manual curation of the candidates was carried out to determine coding regions on intact TEs that are potentially active. We found the majority of unannotated elements contained no ORFs longer than 30 bp nor had any significant matches to repetitive elements in the databases.
RepeatMasker (v3.2.8) was used to calculate the distribution of each repeat and its abundance. Custom perl scripts were used to choose the best match from overlapping matches in RepeatMasker output to avoid calculating the same region twice or more when considering repeat content of the genome.
Annotation of carbohydrate active enzymes
The CAZymes Analysis Toolkit (CAT) [44] was used to detect B. xylophilus carbohydrate active enzymes (CAZymes) based on the CAZy database. An annotation method “based on association rules between CAZy families and Pfam domains” was used with an E-value threshold of 0.01, a bitscore threshold of 55 and rule support level 40. The annotation was supplemented and confirmed manually using BLAST search similarities and protein length matches. Expansin-like genes were detected by BLAST search using core modules of known expansin proteins as queries. Putative functions of the proteins were predicted by similarity to known protein modules and presence of catalytic sites using BLASTP search against NCBI's Conserved Domain Database service and InterProScan (www.ebi.ac.uk/Tools/InterProScan).
Peptidase detection
The MEROPS server was used to identify B. xylophilus putative peptidases. The peptidase candidates were derived from MEROPS batch BLAST [45]. The candidates were manually examined in terms of similarity (E-value cutoff 1e-10) to MEROPS proteins and presence of all catalytic sites.
Effector candidates involved in host-parasite interaction
BLASTP was used to search for B. xylophilus homologues of effectors from M. incognita [46], Heterodera glycines [47] and Globodera pallida [48]. An E-value cutoff of 1e-5 was used to identify significant matches. In addition, candidate effectors were sought from the B. xylophilus protein set using a bioinformatic approach. Secreted proteins were identified as those having a potential signal peptide at their N-terminus predicted by SignalP 3.0 [49] and no transmembrane domain within the mature peptide as predicted using TMHMM 2.0 [50]. Novel candidate effectors within this secreted protein dataset were identified as those with no BLASTP matches against the NR protein database (E-value cutoff 0.001).
Embryogenesis
Orthologues of C. elegans genes necessary for unequal cell divisions were identified from the B. xylophilus protein set by BLASTP search using protein sequences retrieved from WormBase as queries. Their structures were confirmed manually using NCBI and WormBase BLAST.
RNAi Pathway
Seventy-eight proteins known to be involved in core aspects of the C. elegans RNAi pathway were identified from the literature. Protein sequences were retrieved from WormBase (release WS206) and used as queries in TBLASTN and BLASTP searches against the predicted protein and contig databases. All primary BLAST hits returning with a bitscore ≥40 and an E-value≤0.01 were manually translated to amino acid sequence in six reading frames (www.expasy.ch/tools/dna.html), and analysed for identity domain structure by BLASTP (through NCBI's Conserved Domain Database service) and InterProScan. The appropriate reading frame in each case (determined empirically on a case by case basis, but usually that with the largest uninterrupted open reading frame) was then subjected to reciprocal TBLASTN and BLASTP against the C. elegans non-redundant nucleotide and protein databases on the NCBI BLAST server (http://www.ncbi.nlm.nih.gov/BLAST), using default settings. The identity of the top-scoring reciprocal BLAST hit was accepted as identity of the relevant primary hit, as long as that identity was also supported by domain structure analysis.
The predicted B. xylophilus AGOs were analysed further through annotation of conserved RNase-H-like catalytic residue sites and the MID sub-domain which was then used for a further BLAST search against the C. elegans nr protein set (NCBI). The MID domain is highly conserved in functionally comparable AGOs [51], [52].
Neuropeptide genes
Neuropeptide genes were identified using the BLAST search tool available through the genome consortium website. FLP and NLP search strings were constructed from orthologous flp and nlp transcript sequences taken from C. elegans in the first instance, or another plant-parasitic nematode where a C. elegans orthologue was unavailable. Search strings were constructed by concatenating the mature peptides (including basic cleavage sites) encoded by each orthologue. Where this concatenation resulted in a search string of less than 20 characters, the string was repeated end-to-end at least once. Search strings were entered into BLASTP searches of the predicted proteins, and tBLASTn searches of the genome scaffolds, with E-value set to 1000. INS search strings were created by concatenating functionally conserved A and B peptide regions from the C. elegans ins gene complement, in addition to a series of mammalian and molluscan orthologues. These were used to search the predicted protein set of B. xylophilus, using a higher E-value threshold of 1,000,000. The BLAST returns were annotated for both A and B peptides, which were isolated, concatenated and used as BLAST search strings against the C. elegans nr protein set (NCBI) with an increased E-value of 1,000,000. All reciprocal BLAST returns with a Bit score ≥25 and an E-value≤10 were annotated for conserved A and B peptides, and the C. elegans orthologue with the highest similarity to these domains was accepted as identity. Putative INS orthologues which did not meet the reciprocal BLAST criteria, but which closely resemble the structure of INS A and B peptide domains are included as putative variant (Var) INS orthologues (≤25, ≥20 bit score; E-value ≥10, ≤20). All hits were analysed by eye for the presence of neuropeptide precursor sequences, dibasic cleavage sites, and analysed for the presence of secretory signal peptides using SignalP 3.0 [49].
Stress response related genes
Orthologues of C. elegans genes were identified from B. xylophilus protein set using BLASTP. The families of GSTs, UGTs, CYPs, and ABC transporters were identified with InterProScan. Their structures were confirmed manually with BLAST on NCBI and WormBase.
Dauer related genes
Protein sequences known to be involved in dauer formation and maintenance were retrieved from WormBase and used as search strings in a series of tBLASTn and BLASTP searches against B. xylophilus genome and protein sequences. An E-value cutoff of 1e-10 was used to identify significant matches.
Chemoreception related genes
Potential chemoreceptors included 7-transmembrane, G-protein coupled receptors (GPCRs), e. g. Str, Sra and Srg gene superfamilies in C. elegans [53], and other receptors with transmembrane structures, e. g., gustatory receptors (GURs, orthologues of insect gustatory receptors) in C. elegans, ionotropic glutamate receptors (IRs) in Drosophila melanogaster [54] and transient receptor potential (TRP) channels [55], [56]. Other putative GPCRs including those for neurotransmitters (e.g., bioorganic amines) and other signal transduction pathways were also searched. BLASTP and InterProScan were used to search for B. xylophilus orthologues of these proteins. All primary BLASTP hits returning with an E-value ≤ 0.0001 and coverage ratio ≥ 0.7 (apart from insect GURs, which included only hits of very low similarity to nematode GURs) were analysed for identity and domain structure by BLASTP (NCBI's Conserved Domain Database service), WormBase (WS221) and TMHMM 2.0 [50].
Molecular phylogenetic trees of serpentine receptor and gustatory receptor proteins were built by maximum likelihood method using MEGA5 with the JTT matrix-based model [57]. A few receptors were removed in some families based on very long branch lengths in a preliminary maximum likelihood tree. A maximum likelihood tree with culled proteins was drawn to a scale, with branch lengths measured in the number of substitutions per site. All positions containing gaps and missing data were eliminated.
Accession Numbers
The contigs resulting from Bursaphelenchus xylophilus assembly were deposited in the EMBL/Genbank/DDBJ databases under accession numbers CADV01000001-CADV01010432.
Results/Discussion
Sequencing and assembly
Our assembly strategy assembled the 6 pairs of nuclear chromosomes (Figure S2) into 1,231 scaffolds, totaling 74.5 Mb, with half of these nucleotides present in scaffolds of at least 1.16 Mb (Table 1). The size of the assembly is in good agreement with our experimental estimate of 69.0±5.5 Mb (see Text S1, Table S1 in Text S2). The genome size of 74.5 Mb is smaller than that of C. elegans and other published nematode genomes except that of Meloidogyne hapla. The GC content of the genome was 40.4%, higher than that of other nematodes except for Pristionchus pacificus. Most of the mitochondrial genome was assembled in a single 13,410 bp scaffold, and shows a similar gene content and organization to that of C. elegans (Figure S3). Analysis of conserved eukaryotic genes (CEGs) showed that 96.77% and 97.98% of CEGs were present as full or partial genes respectively, with an average of 1.08―1.09 genes per CEG (Table 1), suggesting high completeness of the assembly.

The assembly is approximately 22% repetitive, of which only around 1.3% had characteristics of transposable elements (TEs, Table 2). A complete set of tRNA (Table S2 in Text S2) and rRNA genes were found in the genome. In common with other parasitic nematodes, including B. malayi and M. incognita [58], SL2 trans-splicing appears not to exist in Bursaphelenchus, but we find 25 SL1-like sequences, found in the same tandem repeats as the 5S rRNAs, as in C. elegans.

Analysis of chromosomal rearrangements between B. xylophilus and C. elegans identified a similar pattern of macrosynteny to that found between the more distantly related Trichinella spiralis and C. elegans [59]. Large B. xylophilus scaffolds largely contain genes orthologous to those from a single C. elegans chromosome. These genes are, however, interspersed by genes orthologous to those from other chromosomes (see Text S1, Figure 3).

Protein-coding genes and gene families
A total of 18,074 protein coding genes were predicted in the assembly (Table 1). This is fewer than the 20,416 in C. elegans (WormBase WS221) and 19,212 in M. incognita, although it is higher than the number for M. hapla. The average protein length is similar to that of other nematodes, but B. xylophilus displays the largest average exon size (289 bp) and the smallest average number of exons per gene (4.5). The Bursaphelenchus genome shows a number of characteristics of compact parasite genomes, for example having relatively few, short introns like M. hapla, but has a similar repetitive element content to other published nematode genomes, and is overall only slightly smaller.
Our automated annotation of the B. xylophilus proteome assigned some functional information to a total of 12,483 proteins (69%) (see Text S1 Figure S4). The top 20 Pfam hits in B. xylophilus are shown in Figure 4, and compared with hits in C. elegans. As part of our annotation approach, B. xylophilus proteins were mapped to pathways defined by KEGG, and pathways that are under - and over-represented in this genome compared to C. elegans are shown in Table S3 in Text S2 and are discussed in sections focusing on particular biological features below.

The combined predicted proteins from B. xylophilus and 9 other nematode species were grouped into 27,547 families of orthologues and an additional 51,942 singleton proteins. We used a molecular phylogeny based on single-copy gene families to reconstruct the distribution and evolutionary dynamics of these gene families (Figure 5), and find that the B. xylophilus genome has been relatively conserved over the long divergence from other plant parasitic nematodes. The pattern of sharing of gene families between genomes (Figure 6) shows little obvious phylogenetic pattern, but identifies relatively small numbers of genes - 202 genes shared by the two plant parasitic genomes, and 144 genes shared by Pristionchus and Bursaphelenchus that both show a close association with insects during their lifecycle – that could be implicated in these particular specializations.


Cell wall degradation
The plant cell wall is the primary barrier faced by most plant parasites and the production of enzymes able to break down this cell wall is thus of critical importance. A summary of the carbohydrate active enzymes (CAZymes) and expansin-like proteins which may modify plant cell walls detected in B. xylophilus and other nematodes is shown in Table 3. B. xylophilus contains 34 putative plant cell wall modifying enzymes but compared to other plant parasitic nematodes its composition is unique. Most interestingly, glycoside hydrolase family 45 (GH45) cellulases are present only in B. xylophilus. Other plant parasitic nematodes have GH5 proteins that degrade cellulose but no such genes are present in B. xylophilus. In addition, GH30 xylanases, GH43 arabinases and GH28 pectinases were also absent in B. xylophilus.
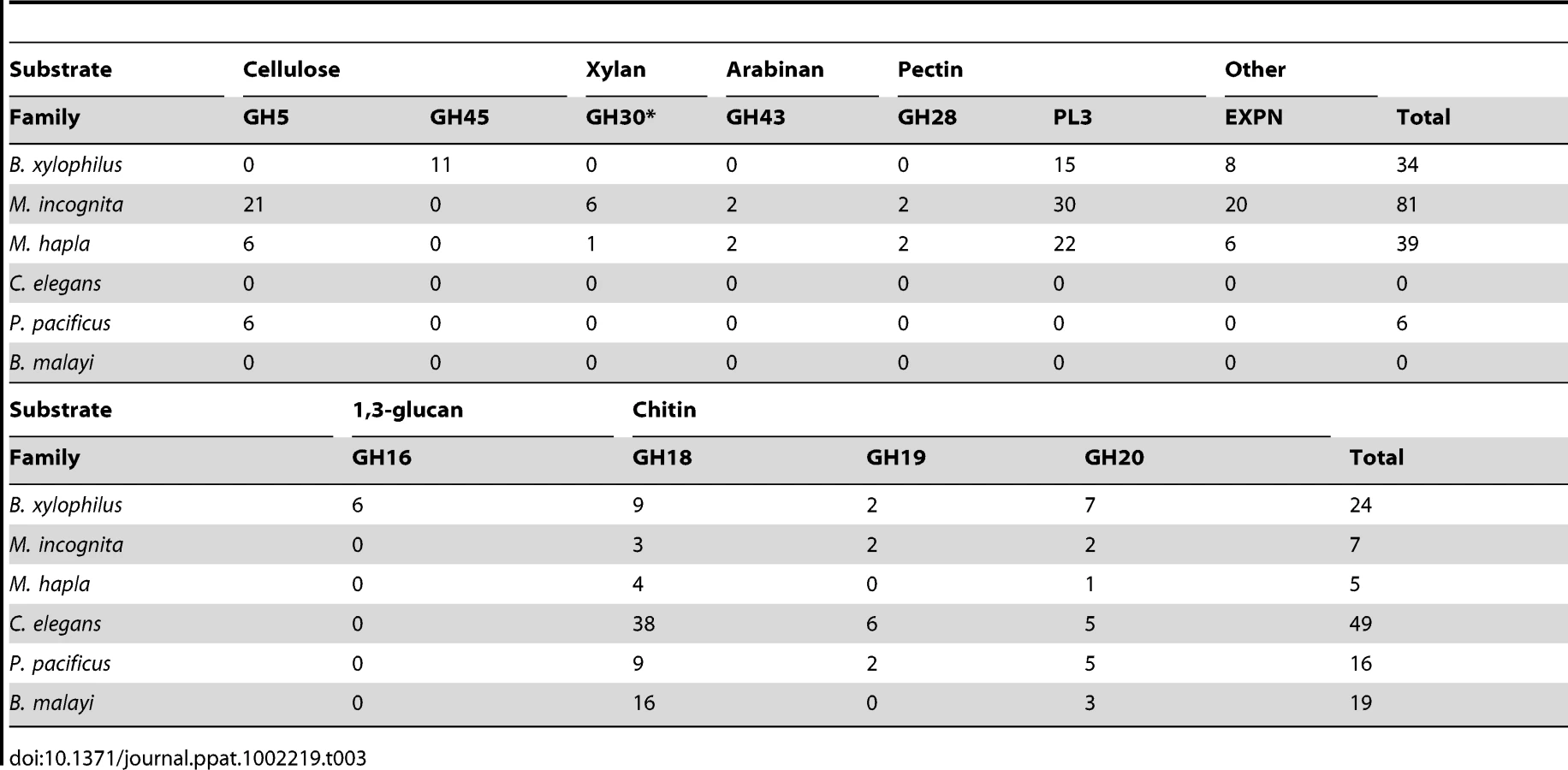
Cell wall degrading CAZymes found in plant parasitic nematodes are thought to have been acquired via horizontal gene transfer (HGT) because similar genes are absent in almost all other nematodes and because they are most similar to genes from bacteria or fungi [60], [61]. GH5 cellulases have been found in many plant parasitic Tylenchoidea including Meloidogyne, Globodera and Heterodera. Recently, the genome sequence of P. pacificus revealed that this nematode also has GH5 cellulases [62]. However phylogenetic analysis suggested that these cellulases were not closely related to those in the Tylenchida and that they are likely to have been acquired independently from different sources [62]. B. xylophilus GH45 cellulases have not been found in any other nematode genus and are most similar to those from fungi. Thus these genes have been hypothesised to be acquired via HGT from fungi [11]. In a phylogenetic analysis all 11 GH45 proteins found in the B. xylophilus genome are grouped in a highly supported monophyletic group and embedded within a clade of fungal homologues (Figure S5). This supports the idea of HGT from fungi with subsequent duplication within the B. xylophilus genome. Recent analysis has revealed that distribution of GH45 proteins is limited to the genus Bursaphelenchus and its sister genus Aphelenchoides (T Kikuchi unpublished results). The absence of GH5 genes in the B. xylophilus genome and the absence of GH45 proteins in Tylenchida nematodes support these hypotheses and suggest that HGT events have repeatedly played important roles in the evolution of plant parasitism in nematodes.
A systematic evolutionary study of plant cell wall modifying genes in Tylenchoidea concluded that those genes were acquired by multiple HGT events from bacteria closely associated with the ancestors of these nematodes followed by gene duplication [61]. B. xylophilus is closely associated with fungi and it is likely that this feeding strategy is ancestral for this group as most Bursaphelenchus species are solely fungal feeders.
In addition to plant cell wall degrading enzymes, CAZymes are present in B. xylophilus which potentially degrade the fungal cell wall. Chitin is one of the main components of fungal cell walls. Proteins related to chitin degradation were identified in the B. xylophilus genome. In comparison to the two Meloidogyne species, P. pacificus and B. malayi the number of chitin-related CAZymes in B. xylophilus is increased, likely reflecting its fungal feeding activity (Table 4). C. elegans has a much larger number of GH18 proteins than B. xylophilus. This may be because those proteins have been used in C. elegans for specific biological features such as bacterial feeding. Interestingly, six GH16 proteins which may degrade beta-1,3-glucan, another core component of the fungal cell wall, have been identified in the genome while no homologues have been found in other nematodes (Table 4). Because the B. xylophilus GH16 β-1,3-glucanase genes are most similar to those from bacteria, it has been suggested that they were acquired from bacteria which were closely associated with its ancestor [63]. This suggests that HGT processes, similar to those associated with plant cell wall modifying enzymes of other parasitic nematodes, have enhanced the ability of Bursaphelenchus spp. to feed on fungi.

Not all of the carbohydrate-active enzymes over-represented in the B. xylophilus genome are involved in cell-wall degradation – several other CAZyme families are substantially expanded in B. xylophilus compared to other nematodes sequenced to date (Table 4). For example, the genome also has more glycosyl transferases family 43 (GT43) genes than other nematodes. These proteins may have beta-glucuronyltransferase activities, but the reason for the increase in these genes in B. xylophilus remains unclear.
Peptidases and lysosome activity
Peptidases (proteases) catalyse the cleavage of peptide bonds within proteins, play important functions in all cellular organisms and are involved in a broad range of biological processes. In nematodes, peptidases play critical roles not only in physiological processes including embryogenesis and cuticle remodeling during larval development but also in parasitic processes such as tissue penetration, digestion of host tissue for nutrition and evasion of the host immune response.
In our analysis 581 peptidase genes were identified in B. xylophilus, which is the largest number in any characterized nematode genome (Table 5), with peptidase families involved in extracellular digestion and lysosomal activities particularly expanded (see Text S1, Table S4 in Text S2). One family of endopeptidases appears to have been acquired by HGT from an ascomycete fungus (Table 6). In addition, B. xylophilus contains an expanded number of GH27 proteins homologous to the gana-1 gene of C. elegans (Table 4), which has α-galactosidase and α-N-acetylgalactosaminidase activities and is localized to lysosomes [64].


The gut granules of intestinal cells in C. elegans are intestine-specific secondary lysosomes, so lysosomal enzymes play important roles in the digestion of food proteins in nematodes. B. xylophilus has an expanded repertoire of peptidases and other digestive enzymes that are either secreted or localised in lysosomes and so may play important roles in food digestion. Genes in the lysosome pathway were the most significantly over-represented in B. xylophilus (Table S3 in Text S2). B. xylophilus uses food sources such as fungi and woody plants that may be difficult to digest and the expansion of digestive peptidases in the nematode is therefore consistent with its unusual life style.
Effector candidates
Plant parasitic nematodes produce a variety of secreted proteins that mediate interactions with their hosts – these encompass a variety of functions and include the cell-wall modifying enzymes discussed above. Such proteins have variously been termed “parasitism genes” or “effectors” and encompass any protein secreted by the nematode into the host that manipulates the host to the benefit of the nematode. For example, cyst nematodes produce effectors that mimic plant peptides and which may help initiate the formation of the biotrophic feeding structures induced by these nematodes [65], as well as proteins that suppress host defence responses [66]. We found that the majority of effectors from other plant parasitic nematodes have no homologues in B. xylophilus. Some significant matches to effectors from all three species were found (Table 7). However, the B. xylophilus sequences that matched these effectors, except cell wall degrading enzymes, either did not have predicted signal peptides or, if a signal peptide was predicted, homologues were also present in a wide range of other species including C. elegans and animal parasitic nematodes. Both these lines of evidence suggest that the B. xylophilus homologues identified in this analysis are not true effectors that play a role in parasitism. These findings are consistent with the differing biology of the various nematode groups; root knot and cyst nematodes are biotrophic species whereas B. xylophilus is a migratory endoparasite that does not rely on biotrophy.

There are two possible exceptions. B. xylophilus contains homologues of venom allergen proteins. These proteins are present in all nematodes investigated to date and are thought to be important for the parasitic process of animal and plant parasites (e.g. [67], [68]). Several venom allergen proteins from B. xylophilus have been characterized and are known to be expressed in the oesophageal gland cells [69]. Our HGT analysis identified a putative cystatin, or cystein protease inhibitor, apparently acquired from a bacterium (Table 6). Cystatins are well known as immunomodulatory pathogenicity factors in the animal parasitic filarial nematodes [70], so this protein could potentially play a role in parasite-host interaction in a plant parasitic nematode. However proteins from this family are involved in regulating a variety of endogenous proteinase activities in many cellular roles and, for example, are described as having anti-fungal properties [71], so the function of this protein will require experimental verification. We also identified a total of 923 predicted secreted proteins in the B. xylophilus genome that show no significant similarity to proteins from other species (Dataset S1), representing a pool of candidates that may play a role in the interaction between B. xylophilus and the other organisms with which it interacts. Very few (5) of these sequences produce matches against other plant parasitic nematode ESTs, consistent with previous studies which have shown that secreted proteins of parasitic nematodes often bear a high proportion of novel genes [72].
Utilisation of host secondary metabolites
Detoxification of potentially damaging compounds is an important process for any organism to cope with its environment and may be particularly crucial for parasitic organisms, which come under attack from host responses to infection. In particular, plant parasites must cope with a wide range of secondary metabolites that plants generate in order to protect their tissues [73].
B. xylophilus principally inhabits the resin canals of its pine hosts. The resin to which it is exposed – a complex mixture of compounds, including terpenoids [74] and cyclic aromatic compounds [75] – is likely to have nematocidal activity and, like the detoxification of xenobiotics by C. elegans [76], would be expected to proceed in three distinct phases: (I) the addition of functional groups to molecules, making them more suitable substrates for downstream; (II) the actual detoxification reactions; and (III) efflux. Cytochrome P450s (CYPs) represent the most important group of phase I proteins, and B. xylophilus encodes a similar number of CYPs to that found in C. elegans (Table 8). Of the two main families of phase II detoxification enzymes – the glutathione S-transferases (GSTs) and UDP-glucuronosyl transferases (UGTs), we identified 41 full-length and 26 partial GSTs, and 60 UGTs, similar numbers to those found in C. elegans (Table 8, Figure S6). The final phase of the detoxification process involves ATP-binding cassette (ABC) transporters actively exporting detoxified xenobiotics. A total of 106 ABC transporters were detected in the B. xylophilus genome; this number was about twice that for C. elegans and about three times that for M. incognita (Table 8), suggesting that B. xylophilus is particularly enriched in genes responsible for the efflux of detoxified molecules.

Finally, we investigated genes involved in regulating the detoxification process. In C. elegans, the transcription factor SKN-1 regulates expression of many detoxification enzymes [77], and SKN-1 activity is in turn controlled by a number of different pathways (see Figure S7). In the presence of oxidative stress or electrophilic compounds, SKN-1 induces the expression of many phase II detoxification enzymes. Orthologues of all these regulatory pathways can be identified in B. xylophilus, suggesting that the regulation of xenobiotic degradation may be conserved in nematodes.
There are other signs that Bursaphelenchus may have an unusual repertoire of genes involved in the defence against or utilisation of complex pine tree metabolites – in our KEGG pathway analysis, xenobiotic and drug metabolism through CYPs were among the pathways showing most significant enrichment in gene copy number over C. elegans, confirming that other genes, likely to be involved downstream of the CYP genes themselves, as well as efflux effectors, are notably enriched (Table S3 in Text S2). Furthermore, our search for carbohydrate active enzymes reported a number of genes classified into the GH109 family of glycosyl hydrolases (Table 5) that on closer inspection proved to be most similar (approx 39% identity and E-values<1E-50) to enzymes displaying trans-1,2-dihydrobenzene-1,2-diol dehydrogenase activity, which is involved in the pathway downstream of cytochrome P450 in the metabolism of naphthalene and other polycyclic aromatic hydrocarbons (PAHs) [78] (Table S3 in Text S2). While PAHs, including naphthalene itself, are known to be produced in small quantities by a few plant species [79], they are not known from pines, and it seems more likely that this enzyme is homologous to naphthalene-degrading enzymes but acts on some of the many other aromatic molecules generated by plants [73].
It seems likely that B. xylophilus has a larger number of detoxification enzymes than other plant parasitic nematodes (M. incognita and M. hapla), with similar or expanded repertoires of such genes to those reported for the free-living C. elegans and the necromenic P. pacificus [62] for the various components of the detoxification process. This expansion in detoxification process components may reflect the variety of stressful environments that it encounters during its life cycle, and perhaps the particular challenges of inhabiting living tissues in a plant host that produces diverse secondary toxic metabolites.
Development
B. xylophilus embryos seem to form the anterior-posterior axis quite differently from those of C. elegans as the point of sperm entry becomes the future anterior end of the animal [80]. Surprisingly, however, other early events in B. xylophilus embryos, such as pronuclear meeting and posterior spindle movement followed by the unequal first cell division are quite similar [80]. Therefore, it is informative to compare and contrast the proteins that control these processes in these two species. Orthologues of the majority of C. elegans proteins involved in these processes were identified in B. xylophilus and appear to be highly conserved. However one putative homologue of the serine/threonine kinase protein PAR-1 was quite distinct in B. xylophilus from that in C. elegans in that the former was considerably smaller (467 AA compared to 1,192 AA in C. elegans); the implications of this difference are unknown.
The formation of dauer (or infective) larvae specialized for surviving adverse conditions or for invading host organisms is an important life stage for many nematodes. In B. xylophilus, we identified orthologues of most genes involved in pathways which regulate dauer larva formation and recovery in C. elegans [81] (Table S5 in Text S2). We also identified orthologues of genes involved in C. elegans dauer pheromone synthesis (see Text S1). As C. elegans, adverse conditions trigger B. xylophilus to enter the third-stage dauer larva (DL3 or dispersal third-stage larva LIII) (Figure 2). Pathways that respond to these environmental cues may be more conserved in B. xylophilus than in other parasitic nematodes, most of which use different cues when forming a dauer (infective) stage. In addition to DL3, B. xylophilus has a specialized stage called the fourth-stage dispersal larva (DL4 or LIV). B. xylophilus DL3 develop into the DL4 when stimulated by the presence of the vector beetle Monochamus alternatus and become ready to board the vector [82], [83]. Previous studies showed that several novel genes are expressed specifically in the DL4 nematodes [10], suggesting that B. xylophilus responds to different environmental stimuli, and likely uses distinct pathways and proteins to control this part of the lifecycle.
Neuropeptide-encoding genes
Nematode neuropeptides are encoded on flp (FMRFamide-like peptide), nlp (neuropeptide-like protein) or ins (insulin-like peptide) genes [84], [85]. Diverse arrays of neuropeptides exist within every nematode species that has been studied, and neuropeptide receptors are promising potential drug targets [86]. The complexity of this peptidergic signalling environment likely aids behavioural diversity and plasticity in spite of the structurally simple nematode nervous system. We find B. xylophilus's flp and nlp gene complements are typical of those seen in other parasitic nematode species [87], [88], although the absence of two flp genes - flp-30 and -31 - is noteworthy (Table S6 in Text S2), as these genes have previously been considered unique to Meloidogyne spp. [4], [87]. Their absence from B. xylophilus suggests they may associate with an obligate parasitic lifestyle. The discovery of seven ins-like orthologues in the B. xylophilus genome is significant as the first description of nematode INS-like peptides outside C. elegans (Table S6 in Text S2, Dataset S2).
Chemoreception
Chemoreception governs essential aspects of the life of many invertebrates, including the search for mates and hosts and the timing of critical steps in their life cycles. Chemoreceptors constitute one interface between the animal and its world, and could be expected to exhibit local adaptations to the specific chemosensory niche of each organism. The main group of putative chemosensory genes in nematodes is represented by serpentine receptors, which are GPCRs, include a large number of families and are also important drug targets. We find representatives of most C. elegans serpentine receptor families in the B. xylophilus genome, but many represent specific expansions, so the two species have related but largely distinct repertoires. The total number of serpentine receptor genes identified from B. xylophilus represents only 10–20% of the number found in C. elegans [53] but 35–45 times of those in M. hapla [5]. It is unclear whether these striking differences represent a reduced and/or expanded chemosensory systems in various nematodes, or whether additional gene families have been expanded to cover some of the chemosensory spectrum in B. xylophilus and other species. Other chemosensory genes identified include gustatory receptors, GPCR receptors for a range of neurotransmitters that could have chemosensory roles, and members of the ionotropic glutamate receptor family [54], among others (see Text S1, Figure S8–S10).
RNAi pathway
The B. xylophilus genome encodes more predicted orthologues of C. elegans RNAi pathway effectors (37 of a potential 78) than found in M. incognita (27) and M. hapla (28) (unpublished data). Whilst B. xylophilus has orthologues of eight of the nine small RNA biosynthetic protein-encoding genes considered, dsRNA uptake and spreading genes are not well represented, e.g. no sid gene orthologues were identified with rsd-3 the only representative gene identified. RNA-dependent RNA polymerases (RdRps) are expanded relative to C. elegans, with four ego-1-, two rrf-1-, and three rrf-3-like orthologues. Sixteen Argonaute (AGO) genes were identified relative to the 27 of C. elegans and, for some of these, there was divergence within the catalytic and RNA-binding MID subdomains; other RNA-induced silencing complex (RISC) cofactors were identified (ain-1, tsn-1 and vig-1). Whilst the short interfering RNA (siRNA) inhibitor eri-1 was not found, microRNA (miRNA) inhibitors (somi-1; xrn-2) were identified. Nuclear effectors were reasonably well represented such that most of the components of a functional RNAi pathway were identified within the B. xylophilus genome. See Text S1 and Table S7-S11 in Text S2 for details.
Concluding remarks
In addition to its status as an economically important plant pathogen, B. xylophilus is remarkable for its unusual biological traits that relate to its complex ecology. During its life cycle it occupies two distinct habitats – an insect and a tree – where it exploits a number of different food sources, including plant tissues and a wide variety of fungi. This adaptability to a number of different niches is reflected in its genome sequence. The presence of a rich repertoire of detoxification enzymes and transporters reflects B. xylophilus' habitat in the resin canals of its host trees, where it is exposed to a cocktail of secondary metabolites, and its elaboration of a narrow subset of carbohydrate metabolizing enzymes reflects it adaptation to break down plant cell walls. The unique complement of genes involved in cellulose degradation, and other catabolic enzymes and the absence of effectors previously known to function at the host-parasite interface, confirms that B. xylophilus has a mode of parasitism that is distinct from other plant parasitic nematodes. This parasitism is mediated by a unique suite of parasitism-related genes, assembled through a combination of gene duplication and horizontal gene transfer. The genome provides strong evidence of multiple independent horizontal gene transfer events and these have shaped the evolution of this group.
Most importantly the genome sequence will act as a foundation for functional studies using a wide range of techniques and will directly inform efforts aimed at controlling this parasite. The identification of genes involved in nematode invasion and feeding from the plant will empower efforts to understand the interaction of B. xylophilus's with its host. One exciting possibility is the potential for genomic information from the hosts of B. xylophilus to facilitate understanding of the host-parasite interaction and associated pathology; host genetics is likely to play a key role in the disease as B. xylophilus is non-pathogenic to American pine species. We have identified genes involved in a range of crucial biological processes, many of which, such as neuropeptides, GPCRs and developmental genes could be viable control targets. The presence of a rich set of RNAi pathway effector genes gives much hope that reverse genetics will underpin future functional genomics efforts in this species. In this way, the genome sequence provides the opportunity to identify and validate putative control targets without the need to rely on C. elegans and make assumptions on conserved functionality/importance between nematodes from different clades.
To our knowledge, only seven nematode genomes have previously been published, and data are available for only a handful more, mostly from the genus Caenorhabditis. Given the breadth of the nematode phylum, genomic information from any new nematode species is an important advance but, B. xylophilus, in particular, is the first species to be sequenced from clade 10, and the first from the order Aphelenchoidea. We hope that with the other imminent nematode genomes being sequenced, B. xylophilus will serve as an important comparator. These data provide a rich resource for those trying to develop novel control strategies directed against B. xylophilus. In addition, the parasite's unusual life cycle makes this genome sequence a unique resource to investigate the association between genome structure and lifestyle, casting new light on the many conserved processes for which the free-living non-parasitic C. elegans remains the pre-eminent model.
Supporting Information
Zdroje
1. The-Caenorhabditis-elegans-Sequencing-Consortium 1998 Genome sequence of the nematode C. elegans: a platform for investigating biology. Science 282 2012 2018
2. BlaxterM 1998 Caenorhabditis elegans is a nematode. Science 282 2041 2046
3. van MegenHvan den ElsenSHoltermanMKarssenGMooymanP 2009 A phylogenetic tree of nematodes based on about 1200 full-length small subunit ribosomal DNA sequences. Nematology 11 927 950
4. AbadPGouzyJAuryJMCastagnone-SerenoPDanchinEG 2008 Genome sequence of the metazoan plant-parasitic nematode Meloidogyne incognita. Nat Biotechnol 26 909 915
5. OppermanCHBirdDMWilliamsonVMRokhsarDSBurkeM 2008 Sequence and genetic map of Meloidogyne hapla: A compact nematode genome for plant parasitism. Proc Natl Acad Sci U S A 105 14802 14807
6. JonesJTMoensMMotaMLiHKikuchiT 2008 Bursaphelenchus xylophilus: opportunities in comparative genomics and molecular host-parasite interactions thol Mol Plant Pa 9 357 368
7. MotaMBraaschHBravoMPenasABurgermeisterW 1999 First report of Bursaphelenchus xylophilus in Portugal and in Europe. Nematology 17 727 734
8. KanzakiN 2008 Taxonomy and Systematics of the Nematode Genus Bursaphelenchus (Nematoda: Parasitaphelenchidae). ZhaoBGFutaiKSutherlandJTakeuchiY Pine Wilt Disease Japan Springer 44 66
9. MamiyaY 1983 Pathology of the pine wilt disease caused by Bursaphelenchus xylophilus. Annu Rev Phytopathol 21 201 220
10. KikuchiTAikawaTKosakaHPritchardLOguraN 2007 Expressed sequence tag (EST) analysis of the pine wood nematode Bursaphelenchus xylophilus and B. mucronatus. Mol Biochem Parasitol 155 9 17
11. KikuchiTJonesJTAikawaTKosakaHOguraN 2004 A family of glycosyl hydrolase family 45 cellulases from the pine wood nematode Bursaphelenchus xylophilus. FEBS Lett 572 201 205
12. KikuchiTShibuyaHAikawaTJonesJT 2006 Cloning and characterization of pectate lyases expressed in the esophageal gland of the pine wood nematode Bursaphelenchus xylophilus. Mol Plant-Microbe Interact 19 280 287
13. KikuchiTLiHKarimNKennedyMWMoensM 2009 Identification of Expansin-like genes from the pine wood nematode Bursaphelenchus xylophilus and evolution of the expansin gene family within the Nematoda. Nematology 11 355 364
14. JonesJTFurlanettoCKikuchiT 2005 Horizontal gene transfer from bacteria and fungi as a driving force in the evolution of plant parasitism in nematodes. Nematology 7 641 646
15. LewisJAFlemingJT 1995 Basic culture methods. Methods Cell Biol 48 3 29
16. HasegawaKMotaMMFutaiKMiwaJ 2006 Chromosome structure and behaviour in Bursaphelenchus xylophilus (Nematoda: Parasitaphelenchidae) germ cells and early embryo. Nematology 8 425 434
17. WilhelmJPingoudAHahnM 2003 Real-time PCR-based method for the estimation of genome sizes. Nucleic Acids Res 31 e56
18. ZerbinoDRBirneyE 2008 Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18 821
19. AssefaSKeaneTMOttoTDNewboldCBerrimanM 2009 ABACAS: algorithm-based automatic contiguation of assembled sequences. Bioinformatics 25 1968 1969
20. TsaiIJOttoTDBerrimanM 2010 Improving draft assemblies by iterative mapping and assembly of short reads to eliminate gaps. Genome Biol 11 R41
21. OttoTDSandersMBerrimanMNewboldC 2010 Iterative Correction of Reference Nucleotides (iCORN) using second generation sequencing technology. Bioinformatics 26 1704 1707
22. ParraGBradnamKNingZKeaneTKorfI 2009 Assessing the gene space in draft genomes. Nucleic Acids Res 37 289 297
23. ParkinsonJAnthonyAWasmuthJSchmidRHedleyA 2004 PartiGene–constructing partial genomes. Bioinformatics 20 1398 1404
24. HaasBJSalzbergSLZhuWPerteaMAllenJE 2008 Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol 9 R7
25. ParraGBradnamKKorfI 2007 CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23 1061 1067
26. StankeMKellerOGunduzIHayesAWaackS 2006 AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res 34 W435 439
27. KorfI 2004 Gene finding in novel genomes. BMC Bioinformatics 5 59
28. LukashinAVBorodovskyM 1998 GeneMark. hmm: new solutions for gene finding. Nucleic Acids Res 26 1107 1115
29. BirneyEClampMDurbinR 2004 GeneWise and genomewise. Genome Res 14 988 995
30. GuigRFlicekPAbrilJFReymondALagardeJ 2006 EGASP: the human ENCODE genome annotation assessment project. Genome Biol 7 Suppl 1 S2. 1 31
31. QuevillonESilventoinenVPillaiSHarteNMulderN 2005 InterProScan: protein domains identifier. Nucleic Acids Res 33 W116 120
32. FinnRDMistryJTateJCoggillPHegerA 2010 The Pfam protein families database. Nucleic Acids Res 38 D211 222
33. GotzSGarcia-GomezJMTerolJWilliamsTDNagarajSH 2008 High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res 36 3420 3435
34. MaoXCaiTOlyarchukJGWeiL 2005 Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 21 3787 3793
35. KanehisaMGotoS 2000 KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28 27 30
36. LiLStoeckertCJRoosDS 2003 OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13 2178 2189
37. EdgarRC 2004 MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32 1792 1797
38. DrummondAJRambautA 2007 BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evolutionary Biology 7 214
39. ShapiroBRambautADrummondAJ 2006 Choosing appropriate substitution models for the phylogenetic analysis of protein-coding sequences. Mol Biol Evol 23 7 9
40. FelsensteinJ 1989 PHYLIP-phylogeny inference package (version 3.2). Cladistics 5 164 166
41. StamatakisA 2006 RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22 2688 2690
42. ShimodairaHHasegawaM 2001 CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics 17 1246 1247
43. NoirotCGaspinCSchiexTGouzyJ 2008 LeARN: a platform for detecting, clustering and annotating non-coding RNAs. BMC Bioinformatics 9 21
44. ParkBHKarpinetsTVSyedMHLeuzeMRUberbacherEC 2010 CAZymes Analysis Toolkit (CAT): Web-service for searching and analyzing carbohydrate-active enzymes in a newly sequenced organism using CAZy database. Glycobiology 20 1574 1584
45. RawlingsNDMortonFR 2008 The MEROPS batch BLAST: a tool to detect peptidases and their non-peptidase homologues in a genome. Biochimie 90 243 259
46. HuangGZGaoBLMaierTAllenRDavisEL 2003 A profile of putative parasitism genes expressed in the esophageal gland cells of the root-knot nematode Meloidogyne incognita. Mol Plant-Microbe Interact 16 376 381
47. GaoBAllenRMaierTDavisELBaumTJ 2003 The parasitome of the phytonematode Heterodera glycines. Mol Plant-Microbe Interact 16 720 726
48. JonesJTKumarAPylypenkoLAThirugnanasambandamACastelliL 2009 Identification and functional characterization of effectors in expressed sequence tags from various life cycle stages of the potato cyst nematode Globodera pallida. Mol Plant Pathol 10 815 828
49. EmanuelssonOBrunakSvon HeijneGNielsenH 2007 Locating proteins in the cell using TargetP, SignalP and related tools. Nature Protocols 2 953 971
50. KroghALarssonBVon HeijneGSonnhammerELL 2001 Predicting transmembrane protein topology with a hidden markov model: application to complete genomes1. J Mol Biol 305 567 580
51. YigitEBatistaPJBeiYPangKMChenCCG 2006 Analysis of the C. elegans Argonaute family reveals that distinct Argonautes act sequentially during RNAi. Cell 127 747 757
52. MalloryACHinzeATuckerMRBouchNGasciolliV 2009 Redundant and specific roles of the ARGONAUTE proteins AGO1 and ZLL in development and small RNA-directed gene silencing. PLoS Genet 5 e1000646
53. ThomasJHRobertsonHM 2008 The Caenorhabditis chemoreceptor gene families. BMC Biol 6 42
54. BentonRVanniceKSGomez-DiazCVosshallLB 2009 Variant ionotropic glutamate receptors as chemosensory receptors in Drosophila. Cell 136 149 162
55. Kahn-KirbyAHBargmannCI 2006 TRP channels in C. elegans. Physiology 68 719 736
56. BargmannCI 2006 Chemosensation in C. elegans. WormBook 25 1 29
57. TamuraKDudleyJNeiMKumarS 2007 MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24 1596 1599
58. PettittJHarrisonNStansfieldIConnollyBM llerB 2010 The evolution of spliced leader trans-splicing in nematodes. Biochem Soc Trans 38 1125 1130
59. MitrevaMJasmerDPZarlengaDSWangZAbubuckerS 2011 The draft genome of the parasitic nematode Trichinella spiralis. Nat Genet 43 228 235
60. JonesJTFurlanettoCKikuchiT 2005 Horizontal gene transfer from bacteria and fungi as a driving force in the evolution of plant parasitism in nematodes. Nematology 7 641 646
61. DanchinEGJRossoMNVieiraPde Almeida-EnglerJCoutinhoPM 2011 Multiple lateral gene transfers and duplications have promoted plant parasitism ability in nematodes. Proc Natl Acad Sci U S A 107 17651 17656
62. DieterichCCliftonSWSchusterLNChinwallaADelehauntyK 2008 The Pristionchus pacificus genome provides a unique perspective on nematode lifestyle and parasitism. Nat Genet 40 1193 1198
63. KikuchiTShibuyaHJonesJT 2005 Molecular and biochemical characterization of an endo-beta-1,3-glucanase from the pinewood nematode Bursaphelenchus xylophilus acquired by horizontal gene transfer from bacteria. Biochem J 389 117 125
64. HujovJSikoraJDobrovolnRPoup tovHLedvinovJ 2005 Characterization of gana-1, a Caenorhabditis elegans gene encoding a single ortholog of vertebrate α-galactosidase and α-N-acetylgalactosaminidase. BMC Cell Biol 6 5
65. WangXMitchumMGGaoBLiCDiabH 2005 A parasitism gene from a plant-parasitic nematode with function similar to CLAVATA3/ESR (CLE) of Arabidopsis thaliana. Mol Plant Pathol 6 187 191
66. RehmanSPostmaWTytgatTPrinsPQinL 2009 A secreted SPRY domain-containing protein (SPRYSEC) from the plant-parasitic nematode Globodera rostochiensis interacts with a CC-NB-LRR protein from a susceptible tomato. Mol Plant-Microbe Interact 22 330 340
67. MaizelsRMGomez-EscobarNGregoryWFMurrayJZangX 2001 Immune evasion genes from filarial nematodes. Int J Parasitol 31 889 898
68. GaoBAllenRMaierTDavisELBaumTJ 2001 Molecular characterisation and expression of two venom allergen-like protein genes in Heterodera glycines* 1. Int J Parasitol 31 1617 1625
69. LinSJianHZhaoHYangDLiuQ 2011 Cloning and characterization of a venom allergen-like protein gene cluster from the pinewood nematode Bursaphelenchus xylophilus. Exp Parasitol 127 440 447
70. SchierackPLuciusRSonnenburgBSchillingKHartmannS 2003 Parasite-specific immunomodulatory functions of filarial cystatin. Infect Immun 71 2422 2429
71. MartinezMDiazI 2008 The origin and evolution of plant cystatins and their target cysteine proteinases indicate a complex functional relationship. BMC Evolutionary Biology 8 198
72. WasmuthJSchmidRHedleyABlaxterM 2008 On the extent and origins of genic novelty in the phylum nematoda. PLoS Negl Trop Dis 2 e258
73. CroteauRKutchanTMLewisNG 2000 Natural products (secondary metabolites). 1250 1318 Biochemistry and Molecular Biology of Plants American Society of Plant Biologists, Rockville, MD
74. LangenheimJH 1994 Higher plant terpenoids: a phytocentric overview of their ecological roles. J Chem Ecol 20 1223 1280
75. TakeuchiYKanzakiNFutaiK 2006 Volatile compounds in pine stands suffering from pine wilt disease: qualitative and quantitative evaluation. Nematology 8 869 879
76. LindblomTHDoddAK 2006 Xenobiotic detoxification in the nematode Caenorhabditis elegans. J Exp Zool A Comp Exp Biol 305 720 730
77. ParkSKTedescoPMJohnsonTE 2009 Oxidative stress and longevity in Caenorhabditis elegans as mediated by SKN-1. Aging Cell 8 258 269
78. AyengarPKHayaishiONakajimaMTomidaI 1959 Enzymic aromatization of 3, 5-cyclohexadiene-1, 2-diol. Biochim Biophys Acta 33 111 119
79. KraussMWilckeWMartiusCBandeiraAGGarciaMVB 2005 Atmospheric versus biological sources of polycyclic aromatic hydrocarbons (PAHs) in a tropical rain forest environment. Environ Pollut 135 143 154
80. HasegawaKFutaiKMiwaSMiwaJ 2004 Early embryogenesis of the pinewood nematode Bursaphelenchus xylophilus. Dev Growth Differ 46 153 161
81. HuPJ 2007 Dauer. WormBook 25 1 29
82. IshibashiNKondoE 1977 Occurrence and survival of the dispersal forms of pine wood nematode, Bursaphelenchus lignicolus Mamiya and Kiyohara. Appl Entomol Zoolog 12 293 302
83. MaeharaNFutaiK 1996 Factors affecting both the numbers of the pinewood nematode, Bursaphelenchus xylophilus (Nematoda: Aphelenchoididae), carried by the Japanese pine sawyer, Monochamus alternatus (Coleoptera: Cerambycidae), and the nematode's life history. Appl Entomol Zoolog 31 443 452
84. MousleyAMcVeighPDalzellJJMauleAG 2011 Progress in the understanding of nematode neuropeptide communication systems. KennedyMWHarnettW Parasitic Nematodes, 2nd Edition: Molecular Biology, Biochemistry and Immunology Wallingford, UK CABI
85. LiCKimK 2010 Neuropeptide Gene Families in Caenorhabditis elegans. Neuropeptide Systems as Targets for Parasite and Pest Control 98 137
86. HielMBLoyTPoelsJVandersmissenHPVerlindenH 2010 Neuropeptide receptors as possible targets for development of insect pest control agents. GearyTGMauleAG Neuropeptide Systems as Targets for Parasite and Pest Control: Springer 211 226
87. McVeighPLeechSMairGRMarksNJGearyTG 2005 Analysis of FMRFamide-like peptide (FLP) diversity in phylum Nematoda. Int J Parasitol 35 1043 1060
88. McVeighPAlexander-BowmanSVealEMousleyAMarksNJ 2008 Neuropeptide-like protein diversity in phylum Nematoda. Int J Parasitol 38 1493 1503
89. BlaxterMLDe LeyPGareyJRLiuLXScheldemanP 1998 A molecular evolutionary framework for the phylum Nematoda. Nature 392 71 75
90. DieterichCSommerRJ 2009 How to become a parasite-Lessons from the genomes of nematodes. Trends Genet 25 203 209
91. ZimniakPSinghSP 2007 Families of Glutathione Transferases. AwasthiYC Toxicology of glutathione transferases: Taylor & Francis 11 26
92. GhedinEWangSSpiroDCalerEZhaoQ 2007 Draft genome of the filarial nematode parasite Brugia malayi. Science 317 1756 1760
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 9
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- HTLV-1 Propels Thymic Human T Cell Development in “Human Immune System” Rag2 gamma c Mice
- Hostile Takeover by : Reorganization of Parasite and Host Cell Membranes during Liver Stage Egress
- Exploiting and Subverting Tor Signaling in the Pathogenesis of Fungi, Parasites, and Viruses
- A Viral Ubiquitin Ligase Has Substrate Preferential SUMO Targeted Ubiquitin Ligase Activity that Counteracts Intrinsic Antiviral Defence
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy