
Protease-Sensitive Conformers in Broad Spectrum of Distinct PrP Structures in Sporadic Creutzfeldt-Jakob Disease Are Indicator of Progression Rate
The origin, range, and structure of prions causing the most common human prion disease, sporadic Creutzfeldt-Jakob disease (sCJD), are largely unknown. To investigate the molecular mechanism responsible for the broad phenotypic variability of sCJD, we analyzed the conformational characteristics of protease-sensitive and protease-resistant fractions of the pathogenic prion protein (PrPSc) using novel conformational methods derived from a conformation-dependent immunoassay (CDI). In 46 brains of patients homozygous for polymorphisms in the PRNP gene and exhibiting either Type 1 or Type 2 western blot pattern of the PrPSc, we identified an extensive array of PrPSc structures that differ in protease sensitivity, display of critical domains, and conformational stability. Surprisingly, in sCJD cases homozygous for methionine or valine at codon 129 of the PRNP gene, the concentration and stability of protease-sensitive conformers of PrPSc correlated with progression rate of the disease. These data indicate that sCJD brains exhibit a wide spectrum of PrPSc structural states, and accordingly argue for a broad spectrum of prion strains coding for different phenotypes. The link between disease duration, levels, and stability of protease-sensitive conformers of PrPSc suggests that these conformers play an important role in the pathogenesis of sCJD.
Published in the journal:
. PLoS Pathog 7(9): e32767. doi:10.1371/journal.ppat.1002242
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002242
Summary
The origin, range, and structure of prions causing the most common human prion disease, sporadic Creutzfeldt-Jakob disease (sCJD), are largely unknown. To investigate the molecular mechanism responsible for the broad phenotypic variability of sCJD, we analyzed the conformational characteristics of protease-sensitive and protease-resistant fractions of the pathogenic prion protein (PrPSc) using novel conformational methods derived from a conformation-dependent immunoassay (CDI). In 46 brains of patients homozygous for polymorphisms in the PRNP gene and exhibiting either Type 1 or Type 2 western blot pattern of the PrPSc, we identified an extensive array of PrPSc structures that differ in protease sensitivity, display of critical domains, and conformational stability. Surprisingly, in sCJD cases homozygous for methionine or valine at codon 129 of the PRNP gene, the concentration and stability of protease-sensitive conformers of PrPSc correlated with progression rate of the disease. These data indicate that sCJD brains exhibit a wide spectrum of PrPSc structural states, and accordingly argue for a broad spectrum of prion strains coding for different phenotypes. The link between disease duration, levels, and stability of protease-sensitive conformers of PrPSc suggests that these conformers play an important role in the pathogenesis of sCJD.
Introduction
Prions cause a group of fatal and rapidly progressing neurodegenerative diseases, originally described as transmissible spongiform encephalopathies (TSEs) [1], [2]. The most common of these diseases is sporadic Creutzfeldt-Jakob disease (sCJD), which accounts for ∼85% of all CJD cases worldwide [3]. Although 40 years ago sCJD was shown to be transmissible to nonhuman primates [4], its pathogenesis remains enigmatic.
Most researchers today believe that all prion diseases are caused by the accumulation of an aberrantly folded isoform, termed PrPSc, of the prion protein PrP [5]. Having a basic amino acid composition and an unstructured N-terminus, PrP can assume at least two conformations: (1) native, α-helix–rich PrPC and (2) disease-causing, β-sheet–rich PrPSc [6]–[8]. The latter represents a misfolded isoform of the normal cellular prion protein PrPC, which is host-encoded by the chromosomal gene PRNP and expressed at different levels in mammalian cells [9]. Yet despite the impressive progress that has been made in understanding the molecular basis of prion diseases, the molecular mechanism of initial misfolding and the high-fidelity replication of the pathogenic conformation of PrPSc in vivo both remain elusive [2], [10]–[12].
Many lines of evidence from experiments with laboratory prion strains support the view that the phenotype of the disease—its distinctive incubation time, clinical features, and brain pathology—is enciphered in the strain-specific conformation of PrPSc [13]–[17]. Although remarkable progress has been made in understanding the structure of laboratory strains of rodent prions [2], [10], [18]–[20], knowledge of the molecular basis of human prion diseases has lagged behind. Researchers generally agree that the genotype at codon 129 of the chromosomal gene PRNP underlies susceptibility to these diseases and, to some degree, their phenotype [21]. However, in contrast to the experiments with laboratory rodent prion strains, in which the digestion of brain PrPSc with proteolytic enzyme proteinase K (PK) consistently results in a single protease-resistant domain with mass ∼19 kDa, the outcome in sCJD is more complex. Distinctive glycosylation patterns and up to four PK-resistant fragments of the pathogenic prion protein (rPrPSc) found in sCJD brains are easily distinguishable on western blot (WB) [14], [21]–[25]. The WB findings together with PRNP gene polymorphism led Parchi, Gambetti, and colleagues to posit a clinicopathological classification of sCJD into five or six subtypes; notably, the WB characteristics of PrPSc breed true upon transmission to susceptible transgenic mice [14], [21], [22]. An alternative classification of the PrPSc types and their pairing with CJD phenotypes has been proposed by Collinge and collaborators [23], [24], [26], [27]. This classification differs from the previous one in two major aspects: First, it recognizes three (not two) PrPSc electrophoretic mobilities; and second, it identifies also PrPSc isoforms with different ratios of the three PrP glycoforms [26]. Although the disease phenotypes of patients with sCJD are remarkably heterogeneous, 21 kDa fragments of unglycosylated PrPSc (Type 1) frequently differ from the phenotypes associated with the 19 kDa fragments of unglycosylated PrPSc (Type 2) [14], [21], [22], [28].
Cumulatively these findings argue that the PrPSc type represents yet an additional major modifier in human prion diseases; accordingly, WB-based clinicopathologic classifications became an important tool in studies of prion pathogenesis in human brains and in transgenic mice models [14], [26]. Now, inasmuch as two distinct PK cleavage sites in PrPSc Types 1 and 2 most likely stem from distinct conformations, some investigators contend that PrPSc Types 1 and 2 code distinct prion strains [14], [23], [28], [29]. However, the heterogeneity of sCJD, along with a growing number of studies including bioassays, all suggest that the range of prions causing sCJD exceeds the number of categories recognized within the current WB-based clinicopathologic schemes [30]–[32]. Additionally, recent findings revealed the co-occurrence of PrPSc Types 1 and 2 in up to 44% of sCJD cases and thus created a conundrum [33]–[38]. Finally, up to 90% of brain PrPSc in sCJD eludes WB analysis because it is destroyed by proteinase-K treatment, which is necessary to eliminate PrPC. Consequently, the conformation or role of this major protease-sensitive (s) fraction of PrPSc in the pathogenesis of the disease is a subject of speculation [30], [39], [40].
Aiming to advance our understanding of the molecular pathogenesis of human prion diseases, we used the conformation-dependent immunoassay (CDI) [15], [30], [41] to determine the conformational range and strain-dependent molecular features of sCJD PrPSc in patients who were homozygous for codon 129 of the PRNP gene. Even relatively minute variations in a soluble protein structure can be determined by measuring conformational stability in a denaturant such as Gdn HCl [42]. Utilizing this concept, we designed a procedure in which PrPSc is first exposed to denaturant Gdn HCl and then exposed to europium-labeled mAb against the epitopes hidden in the native conformation [15]. As the concentration of Gdn HCl increases, PrPSc dissociates and unfolds from native β-sheet-structured aggregates; and more epitopes become available to antibody binding. These experiments involve insoluble oligomeric forms of PrPSc, and denaturation of this protein is irreversible in vitro; consequently the Gibbs free energy change (ΔG) of PrPSc cannot be calculated [43]. Therefore we chose instead to use the Gdn HCl value found at the half-maximal denaturation ([GdnHCl]1/2) as a measure of the relative conformational stability of PrPSc. The differences in stability reveal evidence of distinct conformations of PrPSc [15], [42], [43]. Because CDI is not dependent on protease treatment, it allowed us to address fundamental questions concerning the concentration and conformation of different isoforms of sCJD PrPSc, including protease-sensitive (s) and protease-resistant (r) PrPSc. We found a broad spectrum of structures that are likely responsible for the phenotypic heterogeneity of sCJD and we identified the structural characteristics of PrPSc that are linked to the duration of the disease.
Results
Diagnostic classification of sCJD patients homozygous for PRNP codon 129 and disease duration
From 340 patients with an unequivocally definite diagnosis of Type 1 or Type 2 sCJD and who were homozygous for codon 129 polymorphism in the PRNP gene, we selected samples from 46 patients. The descriptive statistics and Kaplan-Meier survival curves indicate that these cases are representative of the whole group collected at NPDPSC and are similar to those previously reported by us and others (Compare Figure 1 and Figure S1, Table 1) [21], [38], [44]. As expected, we did not observe statistically significant differences in sex ratio or age at onset of the disease [21], [44]. Kaplan-Meier analyses of survival (Figure 1) demonstrated that patients with PrPSc Type 1 had a significantly shorter disease duration than patients with PrPSc Type 2 (P = 0.002) despite identical codon 129 MM polymorphism, age, and sex distribution (Table 1). Moreover, there is an apparent tendency toward longer survival of patients with Type 2 rPrPSc(129 V) than patients with Type 1 rPrPSc(129 M) (P = 0.017). The difference in survival between patients with Type 2 rPrPSc(129 V) and Type 2 rPrPSc(129 M) was also significant (P = 0.008) with shorter survival of those homozygous for valine (Figure 1).


To ensure that the brain homogenate analyzed by CDI contained only Type 1 or 2 rPrPSc, each brain homogenate underwent a second WB (Figure S2). The results confirmed the original diagnostic classification but we found two atypical patterns: Case #833 (Type 2 PrPSc(129 M) and Case #162 (Type 2 PrPSc(129 V) revealed, in addition to a band of unglycosylated rPrPSc with apparent molecular mass ∼19 kDa, a second band with electrophoretic mobility corresponding to mass ∼17 kDa. The observation of different glycoform patterns of PrPSc in different sCJD cases before protease K treatment and distinct resistance to proteolytic degradation of different glycoforms of PrPSc is interesting and deserves further investigation.
Measurement of PrPSc, sPrPSc, and rPrPSc in sCJD cortex by CDI
To measure the concentration of different forms of PrPSc in the frontal cortex, we used europium-labeled mAb 3F4 [45] for detection and 8H4 mAb (epitope residues 175–185) [46] to capture human PrPSc in a sandwich CDI format (Figure S4) [30], [47]. The analytical sensitivity and specificity of the optimized CDI for detection of both protease-sensitive (s) and protease-resistant (r) conformers of PrPSc was previously reported by us and others in numerous publications [15], [30], [41], [48]–[50] and has been shown to be as low as ∼500 fg (∼20 attomoles) of PrPSc. This sensitivity of CDI is similar to the sensitivity of human prion bioassay in Tg(MHu2M)5378/Prnp0/0 mice [30].
First, we determined the concentration of disease-causing PrPSc in subpopulations of sporadic sCJD patients (Table 1 and Figure 2). We observed wide interindividual variations, and approximately sixfold more accumulated PrPSc in the frontal cortex of patients with Type 2 PrPSc(129 M) than those with Type 1 PrPSc(129 M) or Type 2 PrPSc(129 V). A large portion of PrPSc in all groups is protease-sensitive, constituting a pool of sPrPSc conformers (Table 1 and Figure 3a). The digestion with proteinase K (PK) was performed with 3 IU/ml (100 µg/ml) of 10% brain homogenate containing 1% sarkosyl for one hour at 37°C. The protocol for PrPSc digestion, validated in previously published experiments, was selected according to the following criteria: 1) complete digestion of PrPC determined with CDI in control samples; 2) complete shift of the bands of PrPSc to PrP 27–30 on WBs; 3) unequivocal WB differentiation of Type 1 and Type 2 rPrPSc in all tested samples [15], [30], [38], [40], [47], [51]. Additionally, the complete digestion of the PrPSc N-terminus with PK was monitored on WBs in all samples (Figure S2).

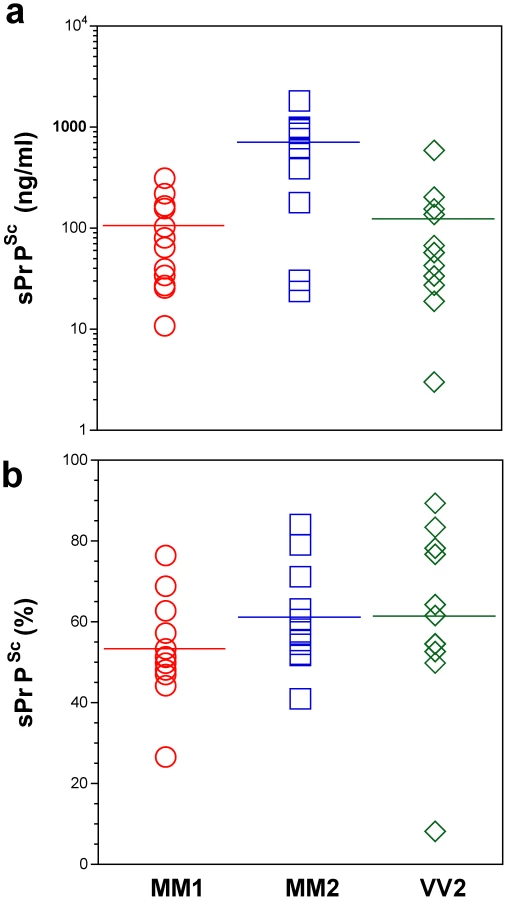
In patients with Type 2 PrPSc(129 M), significantly higher concentrations of total PrPSc and sPrPSc protein (Table 1) are associated with extended duration of disease. However, the concentration of sPrPSc vary greatly between individual patients, with numerous overlapping values between each classification group (Figure 3a). Thus, when the concentration of sPrPSc is expressed as a percentage of total PrPSc, no significant difference between groups appears, and the proportion of sPrPSc varies from 5% to 90% in individual patients (Figure 3b). We concluded from these observations that a major portion of pathogenic sCJD PrPSc is protease-sensitive and that the highest levels of sPrPSc are present in Type 2 PrPSc(129 M). The observed large interindividual differences in PK sensitivity likely indicate a broad range of PrPSc conformers within each PRNP genotype and WB pattern [15], [39]. Since the proteolytic sensitivity of PrPSc is considered a reliable and constant marker of a distinct prion strain, the data support the conclusion that distinct prion structures are present within each classification group.
Monitoring the exposure of epitopes 108–112 and 175–185 in native sCJD PrPSc
The partial exposure of epitopes 108–112 and 175–185 in native pathogenic PrPSc reflects differences in the conformation of native PrPSc [15], [52]. When we adopted this approach previously, we found considerable differences among eight laboratory prion strains passaged in Syrian hamsters [15]. The denatured state is a reference corresponding to the concentration of PrPSc; the ratio between the fluorescence signal of europium-labeled mAb 3F4 reacting with PrPSc in the native (N) or completely denatured (D) state represents a relative measure of the degree of exposure of these epitopes.
The highest D/N PrPSc ratio was found in patients with Type 2 PrPSc(129 M); and despite a large spread of values, the difference is statistically significant (P = 0.002) (Figure 4). PK treatment eliminated most of the exposed 108–112 and 175–185 epitopes in patients with Type 1 PrPSc(129 M) and in patients with Type 2 PrPSc(129 V), resulting in the increased D/N ratios (Figure 4). The opposite trend was observed in patients with Type 2 PrPSc(129 M). After PK treatment the PK-induced differences among the three cohorts proved statistically significant to a remarkable degree (P<0.001). Large variations in D/N values exceed what we expect from our experiments with laboratory prion strains [15] and suggest that a high degree of conformational heterogeneity exists in PrPSc aggregates. Protease treatment change the ratio in all groups and reduced the heterogeneity in MM2 sCJD, and as a result, each group could be reliably differentiated. The increased frequency of exposed epitopes in codon 129 MM samples with Type 2 rPrPSc after PK treatment is unexpected and may indicate one of three possibilities: that the ligand protecting the 3F4 epitope was removed by PK treatment; that epitope 108–112 was protected by the N-terminus of PrPSc; or that conformational transition resulted in more exposed 108–112 epitopes. Whether the epitopes hindrance in undigested PrPSc is the result of lipid, glycosaminoglycan, nucleic acid, or protein binding to the conformers unique to the MM2 sCJDF PrPSc remains to be established.

Dissociation and unfolding of sCJD PrPSc, sPrPSc, and rPrPSc monitored by CDI
First, we asked whether the PTA precipitation has an impact on the stability of PrPSc. This step in the protocol was important for eliminating high concentrations of PrPC and for concentrating PrPSc in brain samples with relatively low levels of PrPSc. (Figure S5). The denaturation curves performed on 5% brain homogenate before PTA precipitation, on PTA pellet and on PTA pellet washed with an excess of H2O, were superimposable, an effect which indicated that PTA quantitatively concentrated all PrPSc conformers and did not influence the stability in CDI. This conclusion accords with numerous previously published data, including bioassays, which indicate that PTA dose not precipitate PrPC and recovers specifically ≥95% of infectious PrPSc in the pellet, regardless of protease sensitivity or prion strain [15], [30], [53]–[55]. The error of the method does not exceed 5% in monitoring [Gdn HCl]1/2 values in the same repeatedly measured brain samples (Figure S5 and Figure S6).
Since the dissociation and unfolding of oligomeric PrPSc may be dependent on protein concentration [42], we first followed the process with CDI at different dilutions of PrPSc (Figure S5). The resulting overlapping dissociation/unfolding curves of PrPSc with variation in Gdn HCl1/2 values <3% indicate that in the 10–250 ng range, the dissociation/unfolding is independent of concentration and is highly reproducible. Furthermore, to ensure the same conditions in all dissociation/unfolding experiments, the PrPSc content in all samples was maintained at a constant 50 ng/ml concentration. As we observed previously with the western blot technique, the Gdn HCl1/2 values obtained with frontal, temporal, parietal, and occipital cortex, thalamus, and cerebellum in three typical sCJD cases were superimposable, indicating that the same conformers of PrPSc are present in different anatomical areas (data not shown) [38].
Next we examined the frontal cortex of individual sCJD patients homozygous for methionine or valine at codon 129 of the PRNP gene. Typical examples of dissociation/unfolding curves are shown in Figures 5a, 5b, and 5c. Comparing all sCJD cases, we found a broad range of Gdn HCl1/2 values ranging from 1.3 to 3.5 M (Figure 6a). Because of the wide spread of values, the difference between the cases with Type 1 and 2 PrPSc(129 M) is only marginally significant (P = 0.040) and there is no statistically significant difference among other groups. The possible cluster of Gdn HCl1/2 values at ∼3.0 M is discernible in cases with Type 1 PrPSc(129 M) (Figure 6a). We concluded from these experiments that PrPSc proteins in different brains of sCJD patients display a vast range of unique conformations within each classification group.
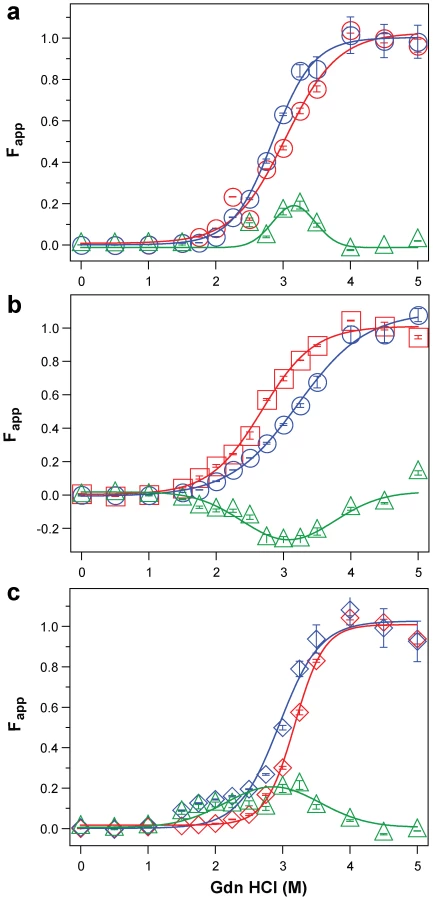

The conformational impact of PK treatment
We next investigated the conformational impact of the proteolytic digestion of sPrPSc conformers and the loss of N-terminal residues in rPrPSc. The proteolysis of PrPSc with PK resulted in increased conformational stability in Type 1 rPrPSc(129 M) and Type 2 rPrPSc(129 V) but did not significantly reduce the range of values (Figure 6a). In contrast, PK treatment uniformly decreased Gdn HCl1/2 values in Type 2 rPrPSc(129 M) (Figure 6a). The marked drop in this group's stability is statistically significant to a high degree (P<0.001). Additionally, there is a discernible cluster of Type 2 PrPSc(129 M) cases at ∼2.6 M (Figure 6a). We interpret the data as providing evidence of a wide range of unique conformations in each subgroup. Proteolytic treatment selects the conformers having a more stable core in Type 1 rPrPSc(129 M) and Type 2PrPSc(129 V). The opposite effect of PK, as well as decreased stability, was observed in samples with Type 2 PrPSc(129 M). These data suggest that PK treatment generates a unique set of conformers in Type 2PrPSc(129 M), characterized by increased exposure of 108–112 and 175–185 epitopes (Figure 5) and, upon PK treatment, decreased stability of the core rPrPSc(129 M).
To investigate the conformational stability of sPrPSc separately from rPrPSc, we subtracted the relative fractional change in stability of rPrPSc after PK treatment from the PrPSc values obtained before PK (Figures 5a, 5b, and 5c). The resulting differential curves exhibit Gaussian distribution with the peak at the median stability of sPrPSc; the height and integrated peak area is proportional to the relative fraction of PK-digested conformers. Overall stability of Type 1 sPrPSc is, as expected, lower than that of rPrPSc and we estimate, from these data alone, that sPrPSc conformers constitute 13–72% of the PrPSc (Figure 6a). A larger spread of positive values obtained with Type 2 sPrPSc(129 V) coincides with a generally larger spread of Gdn HCl1/2 values in this group. In contrast, the negative differential curves for Type 2 sPrPSc(129 M) indicate that sPrPSc is more stable than rPrPSc in this patient group (Figure 6b). Notably, the only positive value in this group came from a sample having an atypical 19 and 17 kDa doublet of unglycosylated rPrPSc on WBs (Figure 6b and Figure S2). Since the stability of sPrPSc and of rPrPSc reflect different initial conformation, the observed spread of values suggests a broad range of unique PrPSc conformers within each PRNP genotype and WB pattern [15], [39], [56].
To determine whether unifying trends exist, we examined which PrPSc characteristics have an impact on duration of the disease in individual patients in all groups using regression analysis. In contrast to analysis of variance (Anova) used to compare MM1, MM2, and VV2 groups (Table 1), the regression analysis is testing the relationship between a dependent variable (duration of the disease) and independent variables (e.g., sPrPSc levels) in individual patients. From concentrations of PrPSc, sPrPSc, and rPrPSc, only the levels of sPrPSc (Figure S7a) correlated significantly with longer duration of the disease. The overall dependency is driven mainly by the higher levels of sPrPSc in Type 2 sPrPSc(129 M) and longer duration of the disease in this subgroup (Table 1). Additionally, the measurement of absolute concentration of sPrPSc is clearly a better indicator of this relationship than the estimate of the relative fraction (percentage) of sPrPSc (Figure 3b). Despite a wide spread of values, this observation corroborates the conclusion, drawn from previous experiments with eight laboratory strains of prion, that incubation time, and by extension duration of the disease, is linked to the higher levels of sPrPSc [15].
We then analyzed the conformational characteristics of PrPSc. The stability of rPrPSc clearly did not correlate with duration of the disease in individual cases (Figure 7a). In contrast, the change in the stability of PrPSc upon PK treatment (Figure S7b) or relative levels of sPrPSc conformers eliminated by PK (Figure 7b) expressed as a fraction of all conformers, both demonstrated better correlation with duration of the disease than did any other parameter in both Type 1 and Type 2 cases. In contrast to simple measurement of sPrPSc concentration, the stability assay performed before and after PK treatment cumulatively determines the shift in the stability of PrPSc, change in the slope of the denaturation curve (dissociation/unfolding rate), and relative levels of the sPrPSc conformers in the total PrPSc pool. This effect leads to the clear separation of Type 1 from Type 2 sPrPSc(129 M) cases (Figure 7b). We interpret these findings as evidence of the differential impact of protease treatment on different conformers, resulting in either increased or decreased stability of the remaining rPrPSc core (PrP 27–30). Taken together, higher levels of more stable sPrPSc conformers are associated with extended duration of the disease. Conversely, lower concentrations of unstable sPrPSc correlate with faster progression of the disease.

Discussion
The discovery of heritable polymorphic PK cleavage sites and glycosylation patterns in PrPSc have been used for the initial diagnostic classifications of sCJD cases. In concert with the codon 129 PRNP haplotype, the different rPrPSc types broadly correlate with distinct disease phenotypes [14], [21], [27]–[29], [57]. The majority of sCJD patients are homozygous for methionine at codon 129 of the PRNP gene; they also accumulate Type 1 rPrPSc and present with so-called classic sCJD, characterized by rapidly progressive dementia, early myoclonus, visual disturbances including cortical blindness, disease duration of approximately 4 months, and fine punctate (synaptic) deposits of PrPSc [21], [30]. In contrast, patients with the second most frequent phenotype are homozygous for valine at codon 129 of the PRNP gene, accumulate Type 2 PrPSc and manifest a different disease course, with early ataxia, predominant extra-pyramidal symptoms, relatively late-onset dementia in the extended course of the disease, and large plaque-like deposits of PrPSc [21].
In the increasing number of subsequent sCJD cases which were examined with more sensitive and specific techniques, investigators began to recognize the extensive variability of the sCJD phenotypes, as well as the extreme complexity of brain immunohistochemistry and western blot patterns of PrPSc [25], [32], [37], [38], [57]–[59]. Although the western blot systems provided early evidence that molecular characteristics of PrPSc are transmissible, evidence regarding the original conformation of PrPSc remains indirect and limited to the most protease-resistant fractions. Because variable fractions of PrPSc are protease-sensitive, we decided to determine the conformational characteristics directly, by using CDI. This method allowed us to compare the conformational features of human PrPSc independently of proteolytic treatment and in addition provided quantitative data on levels of PrPSc, sPrPSc, and rPrPSc [15], [30]. The CDI techniques represent a major improvement over previously used semi-quantitative WB-based methods, the finding that has been independently confirmed by another group [60], [61]. The dissociation and unfolding of PrPSc in a presence of increasing concentration of Gdn HCl can be described as follows: [PrPSc]n→[sPrPSc]n→iPrP→uPrP, where [PrPSc]n are native aggregates of PrPSc, [sPrPSc]n are soluble protease-sensitive oligomers of PrPSc, iPrP is an intermedite, and uPrP is completely unfolded (denatured) PrP [7], [43], [62]. The CDI monitors the global transition from native aggregates to fully denatured monomers of PrPSc. In contrast, the WB based techniques monitor either the partial solubilization of PrPSc [63] or conversion of rPrPSc to protease-sensitive conformers [16] after exposure to denaturant. As a result, the stability data on soluble protease sensitive oligomers and intermediates of PrPs cannot be obtained with WB techniques and lead to the markedly underestimated values [60].
Levels and role of PrPSc isoforms in the pathogenesis of sCJD
The sixfold difference in concentrations of PrPSc between Type 1 and Type 2 PrPSc(129 M) (Figure 2) revealed in the frontal cortex by means of CDI was surprising, even though some variability was to be expected due to differences in the predominantly affected areas in distinct sCJD phenotypes [30]. The average levels of PrPSc are up to 100-fold lower than those in standard laboratory prion models such as Syrian hamsters infected with Sc237 prions [15]; and together with the up to 100-fold variability within each phenotypic group, these lower levels of PrPSc may partially explain why some sCJD cases are difficult to transmit, and why lower endpoint titers are obtained with human prions in transgenic mice expressing human PrPC [14], [26], [30], [64].
As we observed previously, up to 90% of the pathogenic prion protein was protease-sensitive [30]. In this study, we found the highest concentrations in Type 2 PrPSc(129 M). The broad range of absolute and relative levels of rPrPSc and sPrPSc offers evidence of a broad spectrum of PrPSc molecules differing in protease sensitivity in each group with an identical polymorphism at codon 129 of the PRNP gene and an identical WB pattern (Figure 3). Moreover, these findings signal the existence of a variety of sCJD PrPSc conformers; and since protease sensitivity is one of the characteristics of prion strains, they also suggest that distinct sCJD prion strains exist [15], [30], [31], [58], [62], [65].
Structural heterogeneity and origin of sCJD PrPSc
The CJD cases studied in this paper represent 75–90% of all clinical and pathologic diagnostic categories of sCJD [21]. In order to allow unequivocal interpretation of the CDI data, we had to exclude sCJD patients heterozygous for codon 129 polymorphism in the PRNP gene, even though they represent ∼15–20% of sCJD cases. The CDI cannot differentiate PrPSc with codon 129 M from V in a mixture which is present in sCJD heterozygots, and therefore we were unable to differentiate the conformational impact of codon 129 polymorphism. We also excluded the VV1 type of sCJD because of its rarity. This rare form of sCJD constitutes ∼1% of all sCJD cases and we did not collect enough cases to allow statistical comparison with the other groups [21].
The heterogeneity of PrPSc conformations found with CDI within sCJD patients homozygous for codon 129 plymorphism of the PRNP gene is remarkable (Table 1 and Figure 6), with a range corresponding to that of stabilities found in more than ∼30 distinct strains of de-novo and natural laboratory rodent prions studied up to now [15], [16], [66]. The high sensitivity and reproducibility of CDI, together with broad inter-individual variability detected with techniques based on three different principles—PK sensitivity, epitope exposure, and conformational stability—all indicate that the intragroup variations did not originate in the CDI technique but rather reflect differences in the structure of PrPSc in different patients. The intriguing effect of PK treatment on the stability of Type 2 PrPSc(129 M) suggests that the protease-resistant core of Type 2 was profoundly destabilized. Since sCJD cases with Type 2 PrPSc(129 M) have remarkably extended disease durations, the molecular mechanism underlying this effect calls for detailed investigation.
Several theories have been proposed to explain the origin of sCJD. One argues for spontaneous somatic mutations in PRNP; another, for rare stochastic conformational changes in PrPC [26], [67]. Yet a third hypothesis holds that low levels of PrPSc are normally present and cleared, but rise to pathogenic levels when the clearance mechanism fails [40]. Cumulatively, our findings indicate that sCJD PrPSc exhibit extensive conformational heterogeneity. Whether this heterogeneity originates in a stochastic misfolding process that generates many distinct self-replicating conformations [26], [67] or in a complex process of evolutionary selection during development of the disease [17] remains to be established.
Protease-sensitive conformers of PrPSc
We discovered this fraction of PrPSc while developing a conformation-dependent immunoassay (CDI), which does not require proteolytic degradation of ubiquitous PrPC [15]. Although the original definition of sPrPSc was only operational, considerable additional data demonstrate that (1) sPrPSc replicates in vivo and in vitro as an invariant and major fraction of PrPSc; (2) sPrPSc separates from rPrPSc in high speed centrifugation; and (3) the proteolytic sensitivity of PrPSc can reliably differentiate various prion strains [15], [30], [31], [58], [62], [65]. Accumulation of sPrPSc precedes protease-resistant product (rPrPSc) in prion infection [40], [68]; and up to 90% of PrPSc accumulating in CJD brains consists of sPrPSc [30]. Thus, the detection by CDI of sPrPSc as a disease-specific marker is widely regarded as a more reliable basis for diagnosing prion diseases. This improved detection led to the discovery of a new human prion disorder, variably protease-sensitive prionopathy (VPSPr) [15], [30], [39], [69], [70]. It is noteworthy that protease-sensitive synthetic prions generated in vitro during polymerization of recombinant mouse PrP into amyloid fibers produced upon inoculation into wild mice prions composed exclusively of sPrPSc [66].
In laboratory rodent prion models, we found that levels of sPrPSc varied with the incubation time of the disease [15] but the molecular mechanism of this link was unknown [15], [30], [40]. Subsequent experiments with yeast prions indicated that replication rate may be an inverse function of the stability of misfolded protein [71]. The hypothesis based on these experiments posits that the less stable prions replicate faster by exposing more available sites for growth of the aggregates. Additionally, experiments with laboratory and synthetic prions in mouse suggested that the yeast prion principle may apply to mammalian prions as well. However, these experiments were based entirely on the correlation of the shorter incubation time of mouse inoculated with PrPSc that on WBs converted to protease-sensitive isoforms at a lower denaturant concentration, whereas the replication rates of mammalian prions were never determined [72].
In this paper we determined the conformational features and stability of human sPrPSc in sCJD. The data indicate that the levels as well as stability are linked to the progression rate of the disease. Despite the inevitable influence of variable genetic background and the potential difficulties in evaluating initial symptoms, the disease progression rate and incubation time jointly represent an important parameter, which is influenced by replication rate, propagation, and clearance of prions from the brain [2], [40]. The correlations among the levels of sPrPSc, the stability of sPrPSc, and the duration of the disease found in this study all indicate that sPrPSc conformers play an important role in the pathogenesis. When sPrPSc is less stable than rPrPSc, the difference in stability correlates with less accumulated sPrPSc and shorter duration of the disease. Conversely, when sPrP conformers are more stable than rPrPSc, we observe the opposite effect—more accumulated sPrPSc and extended disease duration. It remains to be determined if these effects represent an outcome of different replication rates and clearance, or whether they stem from as yet unknown aspects of the pathogenesis of sCJD.
Materials and Methods
Ethics statement
All procedures were performed under protocols approved by the Institutional Review Board at Case Western Reserve University. In all cases, written informed consent for research was obtained from patient or legal guardian and the material used had appropriate ethical approval for use in this project. All patient's data and samples were coded and handled according to NIH guidelines to protect patients' identities.
Patients and clinical evaluations
We selected 46 representative subjects from a group of 340 patients with definitive diagnosis of sCJD. The criteria for inclusion were (1) availability of clinical diagnosis of CJD according to WHO criteria [73]–[75] and clearly determined and dated initial symptoms upon neurological examination to ascertain the disease duration; (2) methionine or valine homozygous at codon 129 of the human prion protein (PrP) gene (PRNP); (3) unequivocal classification as pure Type 1 or Type 2 sCJD according to WB pattern; (4) unequivocal classification of pathology as definite Type 1 or 2 at the National Prion Disease Pathology Surveillance Center (NPDPSC) in Cleveland, OH; (5) demographic data distribution within 95% confidence interval of the whole group resulting in no difference between selected cases and the whole group in any of the statistically followed parameters.
Retrospective charts review was carried out for all subjects, with particular attention to the documented initial cardinal clinical signs of sCJD such as cognitive impairment, ataxia, and myoclonus [73]–[75]. We also reviewed the findings on electroencephalography, brain magnetic resonance imaging, and CSF markers when available.
Brain samples and PRNP gene sequencing
All Type 1–2 patients or uncertain cases were excluded from this study. DNA was extracted from frozen brain tissues in all cases, and genotypic analysis of the PRNP coding region was performed as described [29], [30], [76]. On the basis of diagnostic pathology, immunohistochemisty, and western blot (WB) examination of 2 or 3 brain regions (including frontal, occipital and cerebellum cortices) with mAb 3F4, the pathogenic PrPSc was classified as (1) Type 1 PrPSc(129 M) (n = 16); (2) Type 2 PrPSc (129 M, n = 16); or (3) Type 2 PrPSc (129 V, n = 14). Patients lacked pathogenic mutations in the PRNP and had no history of familial diseases or known exposure to prion agents. These cases underwent additional detailed WB analyses of the PrPSc so that we could ascertain the accuracy of their original classification and confirm that the same brain homogenate analyzed by CDI contained pure Type 1 PrPSc(129 M), Type 2 PrPSc(129 M), and Type 2 PrPSc(129 V).
Coronal sections of human brain tissues were obtained at autopsy and stored at 80°C. Three 200–350 mg cuts of frontal (superior and more posterior middle gyri) cortex were taken from each brain and used for molecular analyses. The other symmetric cerebral hemisphere was fixed in formalin and used for histologic and immunohistochemical purposes.
Brain homogenates and precipitation of prions with PTA
Slices of tissues weighing 200–350 mg were first homogenized to a final 15% (w/v) concentration in calcium - and magnesium-free PBS, pH 7.4, by 3 75 s cycles with Mini-beadbeater 16 Cell Disrupter (Biospec, Bartlesville, OK). The homogenates were then diluted to a final 5% (w/v) in 1% (v/v) sarkosyl in PBS, pH 7.4 and rehomogenized. After clarification at 500× g for 5 min., one aliquot of the supernatant was treated with protease inhibitors (0.5 mM PMSF and aprotinin and leupeptin at 5 ug/ml, respectively). The second aliquot was treated with 50 µg/ml of proteinase K (Amresco, Solon, OH) for 1 h at 37°C shaking 600 rpm on Eppendorf Thermomixer (Eppendorf, Hauppauge, NY) and PK was blocked with PMSF and aprotinin-leupeptin cocktail. Both aliquots were precipitated with final 0.32% (v/v) NaPTA after 1 h incubation at 37°C as described [15]. The samples were spun 30 min at 14,000× g in Allegra X-22R tabletop centrifuge (Beckman Coulter, Brea, CA) and the pellets were resuspended in 250 ul of deionized water containing protease inhibitors (0.05 mM PMSF, aprotinin and leupeptin at 1 ug/ml each, respectively, and stored for analysis at −80°C.
Western blots
Both PK-treated and untreated samples were diluted 9-fold in 1× Laemmli Buffer (Bio-Rad, Hercules, CA) containing 5% (v/v) beta-mercaptoethanol (ME) and final 115 mM Tris-HCl, pH 6.8. Samples were heated for 5 min at 100°C and ∼2 ng of PrP per lane was loaded onto 1 mm 15% Polyacrylamide Tris-HCl, SDS-PAGE gels (Bio-Rad) mounted in Bio-Rad Western Blot apparatus. After electro-transfer to Immobilon-P Transfer Membranes (Millipore, Bedford, MA), the membranes were blocked with 2% (w/v) BSA in TBS containing 0.1% of Tween 20 (v/v) and 0.05% (v/v) Kathon CG/ICP (Sigma, St. Louis, MO). The PVDF membranes were developed with 0.05 ug/ml of biotinylated mAb 3F4 (Covance, Princeton, NJ) followed by 0.0175 ug/ml Streptavidin-Peroxidase conjugate (Fisher Scientific, Pittsburg, PA) or with ascitic fluid containing mAb 3F4 (kindly supplied by Richard Kascsak) diluted 1∶20,000 followed by Peroxidase-labeled sheep anti-mouse IgG Ab (Amersham, Piscataway, NJ) and diluted 1∶3000. The membranes were developed with the ECL Plus detection system (Amersham) and exposed to Kodak BioMax MR Films (Fisher Scientific) or Kodak BioMax XAR Films (Fisher Scientific).
Conformation-dependent immunoassay (CDI)
The CDI for human PrP was performed as described previously [30], [47], with several modifications. First, we used white Lumitrac 600 High Binding Plates (E&K Scientific, Santa Clara, CA) coated with mAb 8H4 (epitope 175–185) [46] in 200 mM NaH2PO4 containing 0.03% (w/v) NaN3, pH 7.5. Second, aliquots of 20 µl from each fraction containing 0.007% (v/v) of Patent Blue V (Sigma) were directly loaded into wells of white strip plates prefilled with 200 µl of Assay Buffer (Perkin Elmer, Waltham, MA). Finally, the captured PrP was detected by a europium-conjugated [15] anti-PrP mAb 3F4 (epitope 108–112) [45] and the time-resolved fluorescence (TRF) signals were measured by the multi-mode microplate reader PHERAstar Plus (BMG LabTech, Durham, NC). The recHuPrP(90–231,129 M) and PrP(23–231,129 M) used as a calibrant in the CDI was a gift from Witold Surewicz, and preparation and purification have been described previously [77]. The initial concentration of recombinant human PrP(23–231) and PreP(90–231) was calculated from absorbance at 280 nm and molar extinction coefficient 56650 M−1 cm−1 and 21640 M−1 cm−1, respectively. The purified recombinant proteins were dissolved in 4 M GdnHCl and 50% Stabilcoat (SurModics, Eden Prairie, MN), and stored at −80°C. The concentration of PrP was calculated from the CDI signal of denatured samples using calibration cure prepared with either recPrP(23–231) for samples containing full length PrPSc or recPrP(90–231) for samples containing truncated rPrPSc (PrP 27–30) after proteinase-K treatment. This separate calibration was necessary due to the ∼3.5-fold lower affinity of mAb 3F4 with full length hurman PrP(23–231,129 M) compared to PrP(90–231,129 M) (Figure S3).
Monitoring dissociation and unfolding of PrPSc by CDI
The denaturation of human PrPSc was performed as described previously [15], with several modifications. Frozen aliquots of PrPSc were thawed, sonicated 3×5 s at 60% power with Sonicator 4000 (Qsonica, Newtown, CT), and the concentration was adjusted to constant ∼50 ng/ml of PrPSc. The 15 µl aliquots in 15 tubes were treated with increasing concentrations of 8 M GdnHCl containing 0.007% (v/v) Patent Blue V (Sigma, St. Louis, MO) in 0.25 M or 0.5 M increments. After 30 min incubation at room temperature, individual samples were rapidly diluted with Assay Buffer (Perkin Elmer, Waltham, MA) containing diminishing concentrations of 8 M GdnHCl, so that the final concentration in all samples was 0.411 M. Each individual aliquot was immediately loaded in triplicate to dry white Lumitrac 600, High Binding Plates (E&K Scientific, Santa Clara, CA), coated with mAb 8H4, and developed in accordance with CDI protocol using europium-labeled mAb 3F4 for detection [15], [30], [41], [78].
The raw TRF signal was converted into the apparent fractional change of unfolding (Fapp) as follows: F = (TRFOBS−TRFN)/(TRFU−TRFN) where TRFOBS is the observed TRF value, and TRFN and TRFU are the TRF values for native and unfolded forms, respectively, at the given Gdn HCl concentration [7]. To determine the concentration of Gdn HCl where 50% of PrPSc is unfolded ([Gdn HCl]1/2), the data were fitted by least square method with a sigmoideal transition model (Equation 1):
The apparent fractional change (F) in the TRF signal is the function of Gdn HCl concentration(c); c1/2 is the concentration of Gdn HCl at which 50% of PrPSc is dissociated/unfolded and r is the slope constant. To determine the impact of protease treatment on the conformational stability of PrPSc, the values of fractional change after PK were subtracted from Fapp values obtained before PK (ΔFapp = F0−FPK) and then fitted with a Gaussian model to estimate the proportion and average stability of sPrPSc conformers (Equation 2):In this model, the Pk-induced fractional change is ΔF, F0 is fractional change at 0 concentration of Gdn HCl, and c0 is the Gdn HCl concentration at the maximum height A of the peak.
Statistical analysis
We investigated the effect of the following demographic and laboratory variables on survival: sex; age at onset; duration of the disease; electrophoretic Type of PrP 27–30; and the concentration and stability of PrPSc in Gdn HCl before and after PK treatment [15]. Cumulative survival curves were constructed by the Kaplan–Meier method, both overall and by stratifying for each of the above variables. For each type of PrPSc and PRNP gene polymorphism, we report descriptive statistics and the overall survival times stratified for each variable. In the comparison of different patient groups, P values were calculated using Anova. Comparisons of survival curves among groups were carried out by the log rank (Mantel-Cox) and generalized Wilcoxon test. To evaluate the dependency of disease duration upon the concentration and stability of PrPSc in individual CJD cases, the data were analyzed by non-linear regression using the logistic function or the nonlinear models with the best fit. To obtain significance and to compare the relative importance of each characteristic of PrPSc, we used ANOVA and F statistics with regression mean square (MSR) divided by the residual mean square (MSE). All the statistical analyses were performed using SPSS 17 software (SPSS Inc., Chicago, IL).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. GajdusekDCGibbsCJJrAlpersM 1966 Experimental transmission of a kuru-like syndrome to chimpanzees. Nature 209 794 796
2. PrusinerSBScottMRDeArmondSJCarlsonG 2004 Transmission and replication of prions. PrusinerSB Prion Biology and Diseases. 2nd ed Cold Spring Harbor Cold Spring Harbor Laboratory Press 187 242
3. MastersCLGajdusekDCGibbsCJJrBernouilliCAsherDM 1979 Familial Creutzfeldt-Jakob disease and other familial dementias: an inquiry into possible models of virus-induced familial diseases. PrusinerSBHadlowWJ Slow Transmissible Diseases of the Nervous System, Vol 1 New York Academic Press 143 194
4. GibbsCJJrGajdusekDCAsherDMAlpersMPBeckE 1968 Creutzfeldt-Jakob disease (spongiform encephalopathy): transmission to the chimpanzee. Science 161 388 389
5. PrusinerSB 1982 Novel proteinaceous infectious particles cause scrapie. Science 216 136 144
6. PanK-MBaldwinMNguyenJGassetMSerbanA 1993 Conversion of α-helices into β-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci U S A 90 10962 10966
7. SafarJRollerPPGajdusekDCGibbsCJJr 1993 Conformational transitions, dissociation, and unfolding of scrapie amyloid (prion) protein. J Biol Chem 268 20276 20284
8. CaugheyBWDongABhatKSErnstDHayesSF 1991 Secondary structure analysis of the scrapie-associated protein PrP 27–30 in water by infrared spectroscopy. Biochemistry 30 7672 7680
9. OeschBWestawayDWälchliMMcKinleyMPKentSBH 1985 A cellular gene encodes scrapie PrP 27–30 protein. Cell 40 735 746
10. CaugheyBBaronGSChesebroBJeffreyM 2009 Getting a grip on prions: oligomers, amyloids, and pathological membrane interactions. Annu Rev Biochem 78 177 204
11. CobbNJSurewiczWK 2009 Prion diseases and their biochemical mechanisms. Biochemistry 48 2574 2585
12. KimJICaliISurewiczKKongQRaymondGJ 2010 Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J Biol Chem 285 14083 14087
13. BessenRAMarshRF 1994 Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol 68 7859 7868
14. TellingGCParchiPDeArmondSJCortelliPMontagnaP 1996 Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 274 2079 2082
15. SafarJWilleHItriVGrothDSerbanH 1998 Eight prion strains have PrPSc molecules with different conformations. Nat Med 4 1157 1165
16. PeretzDScottMGrothDWilliamsonABurtonD 2001 Strain-specified relative conformational stability of the scrapie prion protein. Protein Sci 10 854 863
17. LiJBrowningSMahalSPOelschlegelAMWeissmannC 2010 Darwinian evolution of prions in cell culture. Science 327 869 872
18. TellingGC 2008 Transgenic mouse models of prion diseases. Methods Mol Biol 459 249 263
19. AguzziAHeikenwalderM 2006 Pathogenesis of prion diseases: current status and future outlook. Nat Rev Microbiol 4 765 775
20. MoralesRAbidKSotoC 2007 The prion strain phenomenon: molecular basis and unprecedented features. Biochim Biophys Acta 1772 681 691
21. GambettiPKongQZouWParchiPChenSG 2003 Sporadic and familial CJD: classification and characterisation. Br Med Bull 66 213 239
22. ParchiPCapellariSChenSGPetersenRBGambettiP 1997 Typing prion isoforms. Nature 386 232 233
23. CollingeJSidleKCLMeadsJIronsideJHillAF 1996 Molecular analysis of prion strain variation and the aetiology of “new variant” CJD. Nature 383 685 690
24. WadsworthJDFHillAFJoinerSJacksonGSClarkeAR 1999 Strain-specific prion-protein conformation determined by metal ions. Nat Cell Biol 1 55 59
25. ZouWQCapellariSParchiPSyMSGambettiP 2003 Identification of novel proteinase K-resistant C-terminal fragments of PrP in Creutzfeldt-Jakob disease. J Biol Chem 278 40429 40436
26. CollingeJClarkeAR 2007 A general model of prion strains and their pathogenicity. Science 318 930 936
27. HillAFDesbruslaisMJoinerSSidleKCLGowlandI 1997 The same prion strain causes vCJD and BSE. Nature 389 448 450
28. MonariLChenSGBrownPParchiPPetersenRB 1994 Fatal familial insomnia and familial Creutzfeldt-Jakob disease: different prion proteins determined by a DNA polymorphism. Proc Natl Acad Sci U S A 91 2839 2842
29. ParchiPCastellaniRCapellariSGhettiBYoungK 1996 Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol 39 767 778
30. SafarJGGeschwindMDDeeringCDidorenkoSSattavatM 2005 Diagnosis of human prion disease. Proc Natl Acad Sci U S A 102 3501 3506
31. Uro-CosteECassardHSimonSLuganSBilheudeJM 2008 Beyond PrP9res) type 1/type 2 dichotomy in Creutzfeldt-Jakob disease. PLoS Pathog 4 e1000029
32. PolymenidouMStoeckKGlatzelMVeyMBellonA 2005 Coexistence of multiple PrPSc types in individuals with Creutzfeldt-Jakob disease. Lancet Neurol 4 805 814
33. PuotiGGiacconeGRossiGCancianiBBugianiO 1999 Sporadic Creutzfeldt-Jakob disease: co-occurrence of different types of PrP(Sc) in the same brain. Neurology 53 2173 2176
34. KovacsGGHeadMWHegyiIBunnTJFlickerH 2002 Immunohistochemistry for the prion protein: comparison of different monoclonal antibodies in human prion disease subtypes. Brain Pathol 12 1 11
35. HeadMWBunnTJBishopMTMcLoughlinVLowrieS 2004 Prion protein heterogeneity in sporadic but not variant Creutzfeldt-Jakob disease: UK cases 1991–2002. Ann Neurol 55 851 859
36. LewisVHillAFKlugGMBoydAMastersCL 2005 Australian sporadic CJD analysis supports endogenous determinants of molecular-clinical profiles. Neurology 65 113 118
37. SchochGSeegerHBogousslavskyJTolnayMJanzerRC 2006 Analysis of prion strains by PrPSc profiling in sporadic Creutzfeldt-Jakob disease. PLoS Med 3 e14
38. CaliICastellaniRAlshekhleeACohenYBlevinsJ 2009 Co-existence of scrapie prion protein types 1 and 2 in sporadic Creutzfeldt-Jakob disease: its effect on the phenotype and prion-type characteristics. Brain 132 2643 2658
39. CronierSGrosNTattumMHJacksonGSClarkeAR 2008 Detection and characterization of proteinase K-sensitive disease-related prion protein with thermolysin. Biochem J 416 297 305
40. SafarJGDeArmondSJKociubaKDeeringCDidorenkoS 2005 Prion clearance in bigenic mice. J Gen Virol 86 2913 2923
41. SafarJGScottMMonaghanJDeeringCDidorenkoS 2002 Measuring prions causing bovine spongiform encephalopathy or chronic wasting disease by immunoassays and transgenic mice. Nat Biotechnol 20 1147 1150
42. ShirleyBA 1995 Protein Stability and Folding: Theory and Practice Totowa, New Jersey Humana Press 377
43. SafarJRollerPPGajdusekDCGibbsCJJr 1994 Scrapie amyloid (prion) protein has the conformational characteristics of an aggregated molten globule folding intermediate. Biochemistry 33 8375 8383
44. PocchiariMPuopoloMCroesEABudkaHGelpiE 2004 Predictors of survival in sporadic Creutzfeldt-Jakob disease and other human transmissible spongiform encephalopathies. Brain 127 2348 2359
45. KascsakRJRubensteinRMerzPATonna-DeMasiMFerskoR 1987 Mouse polyclonal and monoclonal antibody to scrapie-associated fibril proteins. J Virol 61 3688 3693
46. ZanussoGLiuDFerrariSHegyiIYinX 1998 Prion protein expression in different species: Analysis with a panel of new mAbs. Proc Natl Acad Sci U S A 95 8812 8816
47. ChoiEMGeschwindMDDeeringCPomeroyKKuoA 2009 Prion proteins in subpopulations of white blood cells from patients with sporadic Creutzfeldt-Jakob disease. Lab Invest 89 624 635
48. BellonASeyfert-BrandtWLangWBaronHGronerA 2003 Improved conformation-dependent immunoassay: suitability for human prion detection with enhanced sensitivity. J Gen Virol 84 1921 1925
49. ThackrayAMHopkinsLBujdosoR 2007 Proteinase K-sensitive disease-associated ovine prion protein revealed by conformation-dependent immunoassay. Biochem J 401 475 483
50. JonesMPedenAHYullHWightDBishopMT 2009 Human platelets as a substrate source for the in vitro amplification of the abnormal prion protein (PrP) associated with variant Creutzfeldt-Jakob disease. Transfusion 49 376 384
51. TremblayPBallHLKanekoKGrothDHegdeRS 2004 Mutant PrPSc conformers induced by a synthetic peptide and several prion strains. J Virol 78 2088 2099
52. PeretzDWilliamsonRAMatsunagaYSerbanHPinillaC 1997 A conformational transition at the N-terminus of the prion protein features in formation of the scrapie isoform. J Mol Biol 273 614 622
53. GlatzelMAbelaEMaissenMAguzziA 2003 Extraneural pathologic prion protein in sporadic Creutzfeldt-Jakob disease. N Engl J Med 349 1812 1820
54. WadsworthJDJoinerSHillAFCampbellTADesbruslaisM 2001 Tissue distribution of protease resistant prion protein in variant Creutzfeldt-Jakob disease using a highly sensitive immunoblotting assay. Lancet 358 171 180
55. WilleHShanmugamMMurugesuMOlleschJStubbsG 2009 Surface charge of polyoxometalates modulates polymerization of the scrapie prion protein. Proc Natl Acad Sci U S A 106 3740 3745
56. NotariSCapellariSLangeveldJGieseAStrammielloR 2007 A refined method for molecular typing reveals that co-occurrence of PrP(Sc) types in Creutzfeldt-Jakob disease is not the rule. Lab Invest 87 1103 1112
57. WadsworthJDHillAFBeckJACollingeJ 2003 Molecular and clinical classification of human prion disease. Br Med Bull 66 241 254
58. NotariSStrammielloRCapellariSGieseACescattiM 2008 Characterization of truncated forms of abnormal prion protein in Creutzfeldt-Jakob disease. J Biol Chem 283 30557 65
59. ParchiPGieseACapellariSBrownPSchulz-SchaefferW 1999 Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 46 224 233
60. ChoiYPPedenAHGronerAIronsideJWHeadMW 2011 Distinct stability states of disease-associated human prion protein identified by conformation-dependent immunoassay. J Virol 84 12030 12038
61. ChoiYPGronerAIronsideJWHeadMW 2011 Comparison of the level, distribution and form of disease-associated prion protein in variant and sporadic Creutzfeldt-Jakob diseased brain using conformation-dependent immunoassay and Western blot. J Gen Virol 92 727 732
62. TzabanSFriedlanderGSchonbergerOHoronchikLYedidiaY 2002 Protease-sensitive scrapie prion protein in aggregates of heterogeneous sizes. Biochemistry 41 12868 12875
63. PirisinuLDi BariMMarconSVaccariGD'AgostinoC 2011 A new method for the characterization of strain-specific conformational stability of protease-sensitive and protease-resistant PrP. PLoS ONE 5 e12723
64. BishopMTWillRGMansonJC 2010 Defining sporadic Creutzfeldt-Jakob disease strains and their transmission properties. Proc Natl Acad Sci U S A 107 12005 12010
65. PastranaMASajnaniGOniskoBCastillaJMoralesR 2006 Isolation and characterization of a proteinase K-sensitive PrP(Sc) fraction. Biochemistry 45 15710 15717
66. ColbyDWWainRBaskakovIVLegnameGPalmerCG 2010 Protease-sensitive synthetic prions. PLoS Pathog 6 e1000736
67. PrusinerSB 2001 Shattuck Lecture — Neurodegenerative diseases and prions. N Engl J Med 344 1516 1526
68. MallucciGRWhiteMDFarmerMDickinsonAKhatunH 2007 Targeting cellular prion protein reverses early cognitive deficits and neurophysiological dysfunction in prion-infected mice. Neuron 53 325 335
69. GambettiPDongZYuanJXiaoXZhengM 2008 A novel human disease with abnormal prion protein sensitive to protease. Ann Neurol 63 697 708
70. JonesMPedenAProwseCGronerAMansonJ 2007 In vitro amplification and detection of variant Creutzfeldt-Jakob disease PrP(Sc). J Pathol 213 21 26
71. TanakaMCollinsSRToyamaBHWeissmanJS 2006 The physical basis of how prion conformations determine strain phenotypes. Nature 442 585 589
72. LegnameGNguyenH-OBPeretzDCohenFEDeArmondSJ 2006 Continuum of prion protein structures enciphers a multitude of prion isolate-specified phenotypes. Proc Natl Acad Sci U S A 103 19105 19110
73. World Health Organization 1999 WHO infection control guidelines for transmissible spongiform encephalopathies. Geneva 38
74. CollinsSJSanchez-JuanPMastersCLKlugGMvan DuijnC 2006 Determinants of diagnostic investigation sensitivities across the clinical spectrum of sporadic Creutzfeldt-Jakob disease. Brain 129 2278 2287
75. GeschwindMDShuHHamanASejvarJJMillerBL 2008 Rapidly progressive dementia. Ann Neurol 64 97 108
76. ParchiPZouWWangWBrownPCapellariS 2000 Genetic influence on the structural variations of the abnormal prion protein. Proc Natl Acad Sci U S A 97 10168 10172
77. SwietnickiWMorillasMChenSGGambettiPSurewiczWK 2000 Aggregation and fibrillization of the recombinant human prion protein huPrP90–231. Biochemistry 39 424 431
78. SafarJGWilleHGeschwindMDDeeringCLatawiecD 2006 Human prions and plasma lipoproteins. Proc Natl Acad Sci U S A 103 11312 11317
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 9
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- HTLV-1 Propels Thymic Human T Cell Development in “Human Immune System” Rag2 gamma c Mice
- Hostile Takeover by : Reorganization of Parasite and Host Cell Membranes during Liver Stage Egress
- Exploiting and Subverting Tor Signaling in the Pathogenesis of Fungi, Parasites, and Viruses
- A Viral Ubiquitin Ligase Has Substrate Preferential SUMO Targeted Ubiquitin Ligase Activity that Counteracts Intrinsic Antiviral Defence
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy