
Exploits a Unique Repertoire of Type IV Secretion System Components for Pilus Assembly at the Bacteria-Host Cell Interface
Colonization of the human stomach by Helicobacter pylori is an important risk factor for development of gastric cancer. The H. pylori cag pathogenicity island (cag PAI) encodes components of a type IV secretion system (T4SS) that translocates the bacterial oncoprotein CagA into gastric epithelial cells, and CagL is a specialized component of the cag T4SS that binds the host receptor α5β1 integrin. Here, we utilized a mass spectrometry-based approach to reveal co-purification of CagL, CagI (another integrin-binding protein), and CagH (a protein with weak sequence similarity to CagL). These three proteins are encoded by contiguous genes in the cag PAI, and are detectable on the bacterial surface. All three proteins are required for CagA translocation into host cells and H. pylori-induced IL-8 secretion by gastric epithelial cells; however, these proteins are not homologous to components of T4SSs in other bacterial species. Scanning electron microscopy analysis reveals that these proteins are involved in the formation of pili at the interface between H. pylori and gastric epithelial cells. ΔcagI and ΔcagL mutant strains fail to form pili, whereas a ΔcagH mutant strain exhibits a hyperpiliated phenotype and produces pili that are elongated and thickened compared to those of the wild-type strain. This suggests that pilus dimensions are regulated by CagH. A conserved C-terminal hexapeptide motif is present in CagH, CagI, and CagL. Deletion of these motifs results in abrogation of CagA translocation and IL-8 induction, and the C-terminal motifs of CagI and CagL are required for formation of pili. In summary, these results indicate that CagH, CagI, and CagL are components of a T4SS subassembly involved in pilus biogenesis, and highlight the important role played by unique constituents of the H. pylori cag T4SS.
Published in the journal:
. PLoS Pathog 7(9): e32767. doi:10.1371/journal.ppat.1002237
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002237
Summary
Colonization of the human stomach by Helicobacter pylori is an important risk factor for development of gastric cancer. The H. pylori cag pathogenicity island (cag PAI) encodes components of a type IV secretion system (T4SS) that translocates the bacterial oncoprotein CagA into gastric epithelial cells, and CagL is a specialized component of the cag T4SS that binds the host receptor α5β1 integrin. Here, we utilized a mass spectrometry-based approach to reveal co-purification of CagL, CagI (another integrin-binding protein), and CagH (a protein with weak sequence similarity to CagL). These three proteins are encoded by contiguous genes in the cag PAI, and are detectable on the bacterial surface. All three proteins are required for CagA translocation into host cells and H. pylori-induced IL-8 secretion by gastric epithelial cells; however, these proteins are not homologous to components of T4SSs in other bacterial species. Scanning electron microscopy analysis reveals that these proteins are involved in the formation of pili at the interface between H. pylori and gastric epithelial cells. ΔcagI and ΔcagL mutant strains fail to form pili, whereas a ΔcagH mutant strain exhibits a hyperpiliated phenotype and produces pili that are elongated and thickened compared to those of the wild-type strain. This suggests that pilus dimensions are regulated by CagH. A conserved C-terminal hexapeptide motif is present in CagH, CagI, and CagL. Deletion of these motifs results in abrogation of CagA translocation and IL-8 induction, and the C-terminal motifs of CagI and CagL are required for formation of pili. In summary, these results indicate that CagH, CagI, and CagL are components of a T4SS subassembly involved in pilus biogenesis, and highlight the important role played by unique constituents of the H. pylori cag T4SS.
Introduction
Helicobacter pylori infection is associated with a significantly increased risk for the development of several gastric diseases, including peptic ulceration, gastric adenocarcinoma, and gastric lymphoma [1]-[4]. H. pylori is a genetically diverse species, and the risk of developing these diseases is dependent in part on characteristics of the H. pylori strain with which an individual is infected. In particular, H. pylori strains harboring the cag pathogenicity island are associated with a higher rate of disease than are strains lacking this determinant [5]–[7].
The cag pathogenicity island is a 40-kilobase chromosomal region that is predicted to encode 27 proteins [8], [9]. One of these proteins, CagA, is an immunodominant antigen that enters gastric epithelial cells and causes numerous cellular alterations [10]–[13]. Additional proteins encoded by the cag PAI comprise a type IV secretion system (T4SS) that translocates CagA into gastric epithelial cells [11], [12], [14]–[17]. Following translocation, CagA is phosphorylated by host cell kinases at tyrosine residues contained within EPIYA motifs in the C-terminal region of the protein [10], [14], [18]. CagA, in both phosphorylated and non-phosphorylated forms, is able to interact with host cell signaling proteins, resulting in an assortment of consequences, including cytoskeletal alterations, disruption of cellular junctions, and altered cellular adhesion and polarity [10]–[12], [19].
When co-cultured with gastric epithelial cells, H. pylori strains containing the cag PAI stimulate gastric epithelial cells to synthesize and secrete proinflammatory cytokines, such as interleukin-8 (IL-8) [20], [21]. One mechanism leading to IL-8 induction involves entry of H. pylori peptidoglycan into the epithelial cell cytoplasm, where it is recognized by the pathogen recognition molecule NOD1; entry of peptidoglycan into the cytoplasm occurs through a cag PAI-dependent process [22]. A second mechanism leading to IL-8 induction involves translocation of CagA. The CagA protein can induce upregulation of IL-8 transcription via activation of the Ras/Raf signaling pathway [23]; upregulation of IL-8 transcription by CagA is detectable in some CagA-positive strains but not others [23]–[26].
T4SSs are utilized by a wide variety of bacterial species for delivery of effector proteins or DNA-protein complexes into an assortment of recipient cells, including mammalian cells, plant cells, fungi, or other bacteria [27]–[31]. Our current understanding of bacterial T4SSs is based in large part on elegant studies of the Agrobacterium tumefaciens T4SS, which translocates bacterial DNA into plant cells [28], [30]. Thus far there has been relatively little study of the H. pylori cag T4SS. Initial identification of bacterial genes required for function of the H. pylori cag T4SS was accomplished by systematic transposon mutagenesis of genes within the cag PAI [21], or by inserting antibiotic cassettes into selected genes within the cag PAI [8], [9], [32]. One study reported that 17 cag genes are required for translocation of CagA into gastric epithelial cells, and 14 cag genes influence the secretion of IL-8 [21]. Several of these genes encode products that exhibit low-level sequence similarity to components found in the T4SSs of A. tumefaciens and other bacterial species [11], [12], [14]. At least 9 of the cag genes that are reported to be required for function of the cag T4SS lack homologs in other bacterial species [12], [21], [33]. The functions of the encoded proteins remain largely uncharacterized. Structural analyses have been undertaken for several components of the cag T4SS [17], and several studies have shown that Cag proteins can interact to form subassemblies [33]–[36]. Overall, there is only a very limited understanding of the structural organization of the cag T4SS apparatus.
Surface-exposed pili are an important feature of T4SSs [28], [37]. The T4SS pili of A. tumefaciens are comprised of VirB2 (the major pilin subunit) and VirB5 (the minor pilin subunit) [28], [37]. When H. pylori is in contact with gastric epithelial cells, the bacteria express pili that are associated with the cag T4SS [38]-[41]. Relatively little is known about the composition and biogenesis of these H. pylori pili. In contrast to T4SS pili from other bacterial species, the H. pylori T4SS pili are reported to be sheathed organelles [38]. H. pylori CagC is reported to be a VirB2 homolog [40], but there is not yet convincing evidence that CagC is a major component of H. pylori pili. Several of the H. pylori proteins (CagY, CagT, and CagX) that have been localized to the pili by immunogold staining [38], [41] are distantly related to core complex components in other T4SS (VirB10, VirB7, and VirB9, respectively). H. pylori CagL has also been localized to pili by immunogold staining and it is suggested that it is a minor pilus component [39]; however, CagL and VirB5 do not exhibit any detectable sequence similarity. Taken together, these findings suggest that the pili associated with the H. pylori cag T4SS are considerably different from the T4SS pili of A. tumefaciens and other known bacterial T4SSs.
CagL is a 26 kDa protein component of the cag T4SS that lacks homologs in other bacterial species. In various studies, CagL has been detected as a constituent of the T4SS pilus [39], on the surface of H. pylori [42], or as a protein localized to the soluble fraction of H. pylori lysates [38], [43]. H. pylori mutants lacking cagL are defective in the ability to translocate CagA into host cells and do not stimulate production of IL-8 by gastric epithelial cells [21], [39]. An important feature of CagL is its ability to bind α5β1 integrin [39]. CagL contains a canonical integrin-binding RGD motif, but there is not uniform agreement about the functional significance of this RGD motif in mediating binding of H. pylori to α5β1 integrin [39], [44]. A recent study reported that recombinantly expressed CagL functionally mimics fibronectin in its ability to induce focal adhesion formation in mouse fibroblasts and stimulate spreading of AGS cells [42]. Another study reported that CagL mediates dissociation of ADAM17 from α5β1 integrin [45]. This leads to NFκB-mediated repression of the gastric H,K-adenosine triphosphate α-subunit (HKα), which may ultimately contribute to transient hypochlorhydria in H. pylori-infected individuals [45]. Recently it was reported that, in addition to CagL, three other H. pylori proteins encoded by the cag PAI (CagA, CagI, and CagY) can interact with β1 integrin [44]. Interactions of these Cag proteins with β1 integrin have been detected using several approaches, including yeast two-hybrid screening, analysis of interactions between these proteins and integrin on the surface of gastric epithelial cells, and surface plasmon resonance analysis [44].
Since CagL is an important component of the cag T4SS that interacts directly with epithelial cells, we reasoned that CagL might physically interact with other T4SS components, and that such interactions might be required for CagL export, localization, stability, or activity. In the current study, we report that two other proteins encoded by the cag PAI (CagH and CagI) co-purify with CagL, and we provide evidence that these three proteins are components of one or more subassemblies associated with the cag T4SS. CagL, CagI, and CagH are not homologous to components of T4SSs in other bacterial species, but we demonstrate that all three proteins are essential for activity of the cag T4SS. We show that all three proteins are detectable on the bacterial surface. Scanning electron microscopy studies reveal that CagI and CagL are required for formation of pili at the interface between H. pylori and gastric epithelial cells, and a ΔcagH mutant strain produces pili with a distorted morphology compared to the pili of wild-type bacteria. This suggests that CagH functions as a regulator of pilus dimensions. Furthermore, we show that CagH, CagI, and CagL proteins contain a conserved C-terminal hexapeptide motif that is critical for T4SS functionality, and in the case of CagI and CagL, this motif is required for T4SS pilus formation. These studies highlight the important functions of these unique H. pylori cag T4SS components and illustrate the marked variation that exists among bacterial T4SSs.
Results
Identification of Cag Proteins that Co-Purify with CagL
Previous studies have shown that CagL is an important component of the H. pylori cag T4SS that can bind α5β1 integrin [21], [39], [44]. We hypothesized that CagL interacts with other components of the cag T4SS to form one or more subassemblies. Therefore, we sought to identify H. pylori proteins that co-purify with CagL. To facilitate these studies, a cagL-deficient H. pylori mutant strain (ΔcagL) was generated as described in Methods. As expected, CagL expression was detected by immunoblot analysis in the wild-type (WT) strain but not in the ΔcagL mutant strain (Figure 1A). Consistent with previous reports [21], [39], [44], the ΔcagL mutant strain was defective in the ability to stimulate IL-8 secretion by gastric epithelial cells (Figure 1B) and defective in the ability to translocate CagA into gastric epithelial cells (Figure 1C). Complementation with an epitope-tagged form of CagL restored the ability of the bacteria to translocate CagA into host cells and induce IL-8 secretion (Figure 1A–D).

We used a polyclonal anti-CagL serum to immunoaffinity-purify CagL from a wild-type H. pylori strain, and a ΔcagL mutant strain was processed in parallel. As expected, immunoblotting studies showed that CagL was immunoaffinity-purified from WT lysate but not ΔcagL lysate (data not shown). The protein content of each sample was analyzed using multidimensional protein identification technology (MudPIT). Numerous proteins were detected in each sample, but we focused in particular on proteins encoded by the cag PAI, since many of these are known or predicted to comprise components of the cag T4SS. CagL was identified by MudPIT in the preparation derived from WT lysate, but was not detected in the preparation derived from ΔcagL lysate (Table 1). In addition, the relative abundance of CagH and CagI peptides detected in affinity-purified preparations derived from the WT strain was significantly higher than the number of CagH and CagI peptides detected in preparations derived from the ΔcagL mutant strain (Table 1). Peptides identified for CagH, CagI, and CagL covered 33% to 51% of the respective proteins (Figure 1E). CagH and CagI co-purified with CagL from the WT strain in six independent experiments (data not shown). CagA and several other Cag proteins were also detected in the affinity purified preparations, but the numbers of spectral counts corresponding to these proteins were not significantly different when comparing preparations derived from WT and ΔcagL mutant strains (Table 1). Many other Cag proteins were detected by mass spectrometric analysis of crude bacterial lysate (Table S1), but were not detected in the affinity purified preparations. These initial experimental results, demonstrating co-purification of CagL, CagH, and CagI, suggested that these three proteins are components of one or more subassemblies associated with the T4SS.

As another approach for identifying proteins that co-purify with CagL, we undertook experiments with a complemented ΔcagL mutant strain expressing CagL with an N-terminal hemagglutinin (HA) epitope tag (Figure 1D). We immunoaffinity-purified CagL-HA from lysate of the strain expressing CagL-HA, using an immobilized monoclonal anti-HA antibody; the WT strain (expressing CagL without an HA tag) was processed in parallel in the same manner. CagL-HA and co-purifying proteins were eluted from the anti-HA affinity column by HA peptide competition, and MudPIT was utilized to analyze the total protein content of the samples. Mass spectrometry identified only three Cag proteins in the sample purified from the strain expressing CagL-HA: the target protein CagL-HA, CagI, and CagH (Table 2). The relative abundance of CagL and CagI peptides detected in the preparation from the CagL-HA-expressing strain was significantly higher than the number of CagL and CagI peptides detected in the preparation from the WT strain. Of note, compared to affinity-purified preparations of CagL obtained using polyclonal antiserum, the preparation of CagL-HA obtained using a monoclonal antibody and peptide elution had a much higher level of purity (Table 2 and Table S2).

To further investigate potential interactions among CagH, CagI, and CagL, we attempted to generate polyclonal antisera against CagH and CagI, but this failed to yield antisera that recognized the corresponding H. pylori proteins. Therefore, we generated unmarked non-polar ΔcagH and ΔcagI mutants (Figure 2A and B), as well as complemented mutant strains expressing epitope-tagged forms of CagH or CagI in a heterologous chromosomal locus (Figure 2C). CagH-HA and CagI-FLAG proteins of the expected masses were detected in the complemented bacteria (but not WT bacteria) by immunoblotting using monoclonal antibodies against the epitope tags (Figure 2D). We immunoaffinity-purified CagH-HA from lysate of the strain expressing this protein, and lysate from the WT strain (expressing CagH without an HA tag) was processed in parallel. The relative abundance of CagH, CagI and CagL detected in the preparation from the CagH-HA-expressing strain was significantly higher than the number of CagH, CagI and CagL peptides detected in the preparation from the WT strain (Table 2 and Table S2).
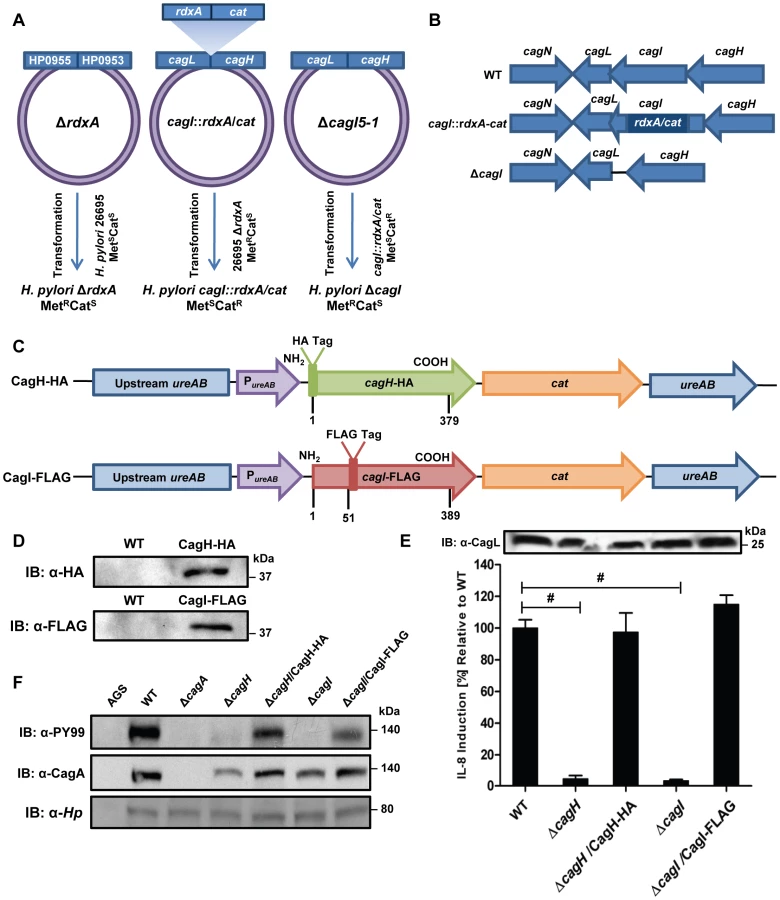
In a similar manner, we purified CagI-FLAG from the strain expressing this epitope-tagged protein. The relative abundance of CagI, CagH, and CagL detected in the preparation from the CagI-FLAG-expressing strain was significantly higher than the number of corresponding peptides detected in the preparation from the WT strain (Table 2). These experiments recapitulated the co-purification of CagH, CagI, and CagL that was observed in initial purifications utilizing polyclonal anti-CagL antisera (Table 1). In individual experiments, we sometimes detected levels of other proteins besides CagH, CagI, and CagL that were significantly increased in affinity-purified preparations compared to control preparations (Table S2), but these results were not consistently reproduced and were not recapitulated when alternate members of a putative CagH-CagI-CagL complex were targeted for purification.
Further analysis revealed that there is weak sequence similarity between the amino acid sequences of CagL and CagH (32% amino acid identity and 47% similarity in the C-terminal region, Figure 1F). CagL, CagI, and CagH are encoded by contiguous, overlapping genes in an operon [46] (Figure 1G). Other genes included in this operon, upstream from cagH, cagI, and cagL, include cagC (encoding the putative T4SS major pilin component [40]), cagE (homologous to virB4, encoding an ATPase required for T4SS function), and cagF (encoding a chaperone protein that binds to CagA) [47]. Since we observed highly reproducible co-purification of CagH, CagI, and CagL, and because genes within an operon often have related functions, we proceeded to undertake further studies of all three proteins.
CagL Stability is Impaired in the Absence of CagH and CagI
Previous studies have reported decreased stability of T4SS components if one or more interacting proteins are absent [36], [37], [43], [48], [49]. Based on the observed co-purification of CagI and CagH with CagL, we hypothesized that CagL stability might be impaired in the absence of CagI or CagH. To assess CagL protein stability, we analyzed bacterial lysates prepared from the WT strain, ΔcagI mutant, and ΔcagH mutant, each cultured for either 24 h or 48 h. CagL could be detected by immunoblotting of lysates from all three strains at 24 h of growth (Figure 3A). However, after 48 h of growth, CagL levels were markedly reduced in lysates from the ΔcagI and ΔcagH mutants compared to levels in WT lysate. CagL stability at the 48 h timepoint was increased by complementation of the mutant strains (Figure 3A). We also evaluated the stability of CagL in the absence of two Cag proteins selected as controls (CagA and CagF). We did not detect significant co-purification of CagA or CagF with CagL in the previous experiments (Table 1), and neither CagA nor CagF is required for functionality of the T4SS, based on analysis of IL-8 induction [21]. Absence of CagA or CagF did not have any detectable effect on CagL stability during early or late phases of growth (Figure 3A).
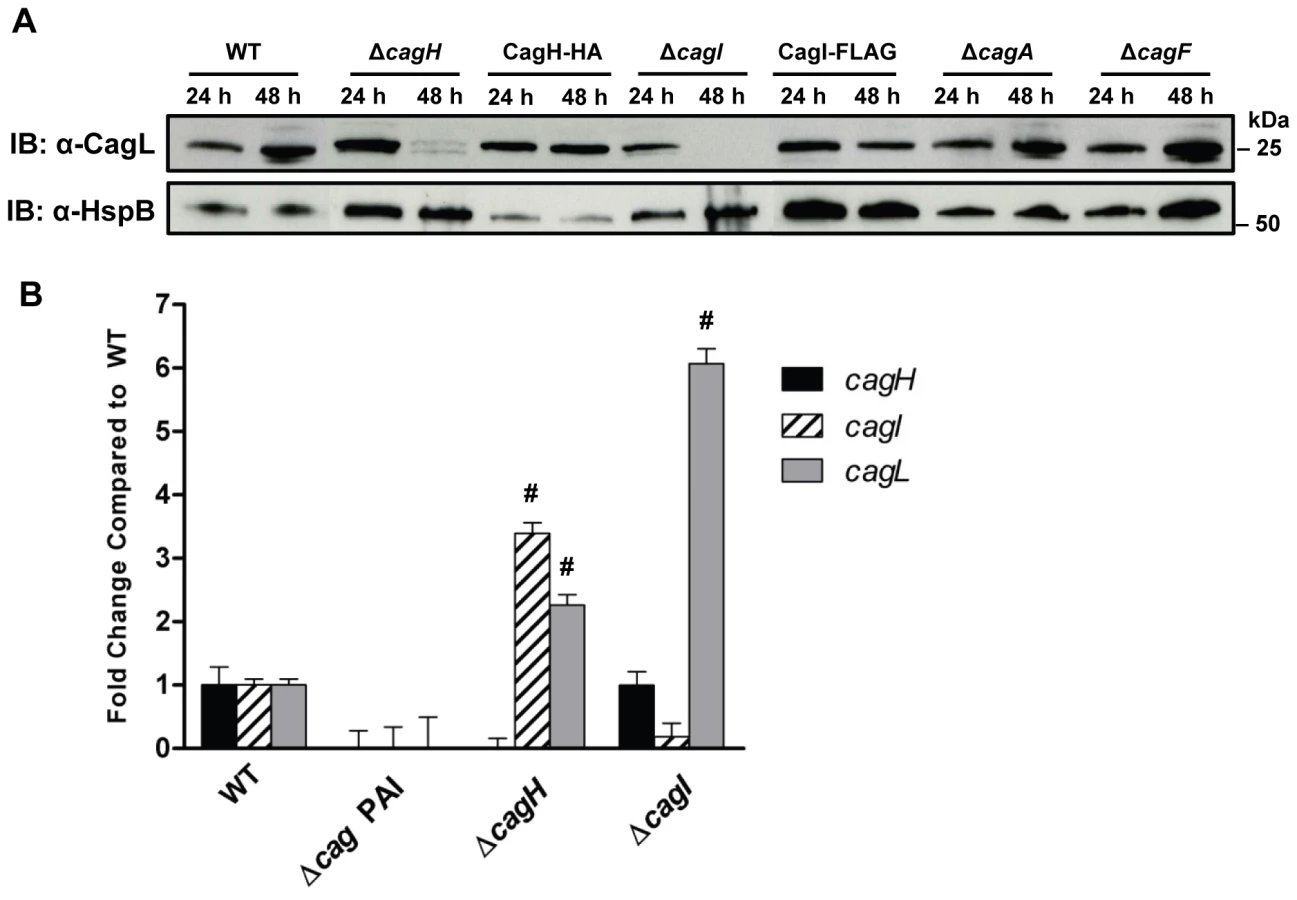
We next investigated the possibility that the observed reductions in CagL protein expression in ΔcagH and ΔcagI mutants might be attributable to a reduction in cagL transcription. Analysis of cagL transcription in each of these strains by real-time PCR indicated that cagL transcription was not diminished, but was in fact increased in the ΔcagH and ΔcagI mutants compared to the WT strain at 48 h of growth (Figure 3B). Thus, the observed reduction in CagL levels in the ΔcagH and ΔcagI mutant strains is attributed to reduced stability of CagL, rather than decreased transcription of cagL.
To further investigate potential relationships among CagL, CagI, and CagH, we analyzed whether co-purification of CagI with CagL was dependent on the presence of CagH. We used polyclonal anti-CagL antiserum to immunoaffinity purify CagL from both the WT strain and a ΔcagH mutant strain, each cultured for 24 h (which permitted stable expression of CagL in both strains). The samples were then analyzed by mass spectrometry. As expected, CagL was immunoaffinity purified from both the WT strain and the ΔcagH mutant (Table S3). CagI was co-purified along with CagL from the WT strain, but not from the ΔcagH mutant (Table S3). In a similar manner, we investigated whether co-purification of CagH with CagL was dependent on the presence of CagI. CagH was co-purified with CagL from both the WT strain and ΔcagI mutant; however, significantly lower levels of CagH were co-purified from the ΔcagI mutant strain (Table S3).
In summary, these experiments indicate that CagL stability is reduced in the absence of CagH or CagI, and co-purification of CagH or CagI with CagL is dependent on the presence of all three proteins. Taken together with the previous results (Table 1 and 2), these data provide evidence that CagL, CagI, and CagH are components of one or more subassemblies associated with the cag T4SS.
Functional Properties of CagH and CagI
A previous study, which analyzed mutant strains containing transposon insertions in individual cag genes, provided an important foundation for identifying genes in the cag PAI that are required for functionality of the H. pylori cag T4SS [21]. This study reported that both CagH and CagI were required for CagA translocation, and that CagH (but not CagI) was required for IL-8 induction [21]. A limitation of the methodologic approach was that unrecognized spontaneous secondary mutations or polar effects associated with transposon insertion into the cag PAI could not be confidently excluded. To determine definitively whether CagH and CagI were required for the function of the cag T4SS, we analyzed the unmarked ΔcagH and ΔcagI deletion mutant strains and complemented mutants described in the previous section.
In comparison to the WT strain, both the ΔcagH and ΔcagI mutants were defective in their ability to induce IL-8 secretion in gastric epithelial cells (Figure 2E). Complementation of these mutants with genes encoding CagH-HA and CagI-FLAG, respectively, rescued the ability of the ΔcagH and ΔcagI mutants to induce IL-8 secretion at WT levels (Figure 2E). Immunoblot analysis indicated that CagL remained intact in the mutant strains throughout the assay (Figure 2E), which indicates that the inability of these mutant strains to induce IL-8 expression is attributable to absence of either CagH or CagI, respectively, rather than instability of CagL. The ΔcagH and ΔcagI mutants were also defective in their ability to translocate CagA into gastric epithelial cells (Figure 2F). The ability of these mutants to translocate CagA at levels comparable to WT was rescued by complementation with epitope-tagged versions of the corresponding proteins (Figure 2F). These results indicate that CagH and CagI, similar to CagL, are required for proper function of the cag T4SS.
Localization of CagH, CagI, and CagL
Since expression of CagH and CagI proteins in H. pylori has not been detected previously, we sought to analyze the subcellular localization of these proteins. As a first approach, we investigated potential surface exposure of CagH, CagI, and CagL by analyzing the susceptibility of these proteins, as well as several controls, to digestion by proteinase K. As expected, incubation of the intact bacteria with proteinase K was sufficient to cleave VacA, a protein known to be localized on the bacterial surface [50], [51] (Figure 4A). In contrast, carbonic anhydrase (CA), a protein known to have a periplasmic domain [52], was not susceptible to cleavage by proteinase K. Similarly, CagV (a VirB8 homolog), which is predicted to have domains in the periplasmic space [16], [17], [43], was also resistant to cleavage by proteinase K (data now shown). CagH-HA, CagI-FLAG, and CagL-HA were each susceptible to cleavage by proteinase K (Figure 4A), but in contrast to VacA, these three Cag proteins were incompletely digested. This suggests that CagH, CagI, and CagL are present on the bacterial surface, and in addition, another pool of these proteins exists in a site that is not susceptible to the protease. Consistent with the susceptibility of CagH-HA, CagI-FLAG, and CagL-HA to cleavage by proteinase K, surface exposure of all three proteins was detected by flow cytometry analysis (Figure 4B). Immunogold labeling studies also revealed localization of CagH-HA, CagI-FLAG, and CagL-HA on the surface of H. pylori (Figure 5). Typically fewer than 10 gold particles per bacterial cell were visualized on the peripheral margins of bacteria, which suggests either that the number of CagH, CagI, and CagL proteins on the bacterial surface under these conditions is relatively small, or that there may be limitations with the use of these monoclonal antibodies for immunogold labeling.


As another approach for analyzing the subcellular localization of CagH, CagI, and CagL, H. pylori strains expressing CagH-HA, CagI-FLAG, or CagL-HA were sonicated and fractionated in the absence of detergent. Soluble fractions (which are expected to contain cytoplasmic and periplasmic proteins) and total membrane fractions were analyzed by immunoblotting using monoclonal antibodies directed against the appropriate epitope tags. In agreement with a previous report [38], [43], the native form of CagL was localized mainly in the soluble fraction, and lesser amounts were present in the membrane fraction; similar results were observed for CagL-HA (Figure 4C, D, E). Both CagH-HA and CagI-FLAG were localized exclusively in the membrane fraction (Figure 4D, E). The insolubility of CagH and CagI in this experiment compared to previous experiments is attributed to the absence of detergent in these fractionated samples and the presence of detergent in earlier samples. We propose that, when complexed together, CagL is loosely tethered to membrane-associated CagH and CagI, and that in the absence of detergent, the shear forces or heat generated by sonication are sufficient to disrupt this interaction and solubilize CagL, but not CagH and CagI. An additional possibility is that the CagH-CagI-CagL complexes include the relatively small portion of membrane-associated CagL.
In summary, these experiments provide evidence that CagL, CagH, and CagI exist in multiple subcellular locations, including the surface of H. pylori. The existence of these proteins in multiple subcellular locations potentially reflects multiple stages of T4SS assembly. Localization of CagL mainly to the soluble fraction as well as on the bacterial surface helps to reconcile apparent contradictions in previous publications, which reported CagL localization to either the soluble fraction or surface-exposed sites [38], [39], [42], [43]. In agreement with a previous study [43], we propose that CagL exists mainly in a periplasmic pool, but it can also be associated with the outer membrane, perhaps loosely tethered to the cell surface. Distribution of at least two other Cag proteins (Cag3 and CagY) into multiple pools with different localizations has been suggested previously [33], [38]. Complexes comprised of CagH, CagI, and CagL could potentially exist in multiple sites, including the bacterial surface and the periplasm (where periplasmic CagL may interact with membrane-associated CagH and CagI proteins).
CagH, CagI, and CagL have Key Roles in T4SS Pilus Formation
To investigate a possible mechanism by which CagH, CagI, and CagL might contribute to activity of the cag T4SS, we used scanning electron microscopy (FESEM) to analyze interactions of H. pylori with AGS gastric epithelial cells. Specifically, we tested the hypothesis that CagH, CagI, or CagL might be required for formation of pili at the bacterial-host cell interface. As expected, we detected pili at the interface between WT H. pylori and AGS cells (Figure 6A), and consistent with previous reports [38], [39], [41], these pili were not visualized when a Δcag PAI mutant (Figure 6B), cagT (virB7 homolog) mutant, or cagE mutant (data not shown) was co-cultured with AGS cells. Adherent WT bacteria exhibited pili on surfaces that were contiguous to the epithelial cells, and we did not observe pili on non-adherent bacteria. The dimensions of the pili were approximately 14 nm in width and 65 nm in length (Table 3), and the pili were distributed along the lateral and polar surfaces of the bacteria. When co-cultured with AGS cells, neither the ΔcagL nor the ΔcagI mutant strains produced detectable pili (Figure 6C, F). This defect was rescued by complementation of the ΔcagI and ΔcagL mutants (Figure 6D, E, G).


In contrast to the ΔcagI and ΔcagL mutant strains, the ΔcagH mutant strain was capable of producing pili. However, there were several striking differences when comparing the ΔcagH mutant strain (Figure 7B, C, D, F) with the WT strain (Figure 7A, E). First, the number of pili produced by the ΔcagH mutant was significantly higher than the number of pili produced by the WT strain (8.0±4.3 vs. 3.8±2.6 visible pili per adherent bacteria, p<0.0001) (Table 3). Second, the length of pili produced by the ΔcagH mutant was significantly greater than that of pili produced by the WT strain (115.1±65.1 nm vs. 65.1±30.7 nm, p<0.0001). Third, the width of pili produced by the ΔcagH mutant was significantly greater than that of pili produced by the WT strain (21.5±4.3 nm vs. 13.7±2.4 nm, p<0.0001). A complemented ΔcagH mutant strain produced pili that were indistinguishable from the pili produced by the WT strain (Figure 7G and Table 3). These results suggest that CagH is a regulator of pilus dimensions. As considered further in the Discussion, an analysis of highly conserved domains within CagH (Figure 7H) provides clues into possible mechanisms by which CagH controls dimensions of the H. pylori T4SS pilus.

In all of the initial experiments (Tables 1, 2, and S2), interactions among CagH, CagI, and CagL were detected in H. pylori that were cultured in the absence of gastric epithelial cells. Our electron microscopy experiments indicate that pili associated with the cag T4SS are not detected when the bacteria are cultured in the absence of gastric epithelial cells (data not shown), and in agreement with previous reports [38], [39], we found that contact of H. pylori with gastric epithelial cells stimulates the formation of pili that are associated with the cag T4SS. This suggests that the cag T4SS is not yet fully assembled when bacteria are cultured in the absence of gastric epithelial cells. To determine whether interactions among CagH, CagI, and CagL are detectable under conditions in which the cag T4SS is fully assembled, we sought to detect the presence of this subassembly in H. pylori that were co-cultured with gastric epithelial cells. We co-cultured bacteria expressing CagH-HA with AGS cells, affinity purified CagH-HA from adherent bacteria, and then analyzed the protein content of the affinity purified preparation. Wild-type bacteria were co-cultured with AGS cells and were processed in parallel as a control. As shown in Table S4, we again observed co-purification of CagH, CagI, and CagL. Therefore, a CagH-CagI-CagL subassembly is detectable not only when bacteria are cultured in the absence of gastric epithelial cells, but also under conditions in which the cag T4SS is fully assembled.
Since CagL was previously detected as a pilus component [39], we hypothesized that CagI and CagH might also localize to pili. To test this hypothesis, we used scanning EM and immunogold labeling studies to analyze H. pylori that were co-cultured with gastric epithelial cells. Using this approach, we were unable to detect localization of CagH or CagI to the pili, and we were also unable to detect CagH or CagI in any other sites. Since we were able to detect surface localization of CagH, CagI, and CagL in transmission EM experiments and flow cytometry experiments but not FESEM experiments, there are probably limitations associated with the use of these monoclonal antibodies for immunogold labeling in the context of FESEM. Specifically, the FESEM methodology requires multiple extra washing steps (a series of seven sequential ethanol dehydration steps and three liquid carbon dioxide washing steps) that are not required for transmission EM, and monoclonal antibodies often are considered suboptimal compared to polyclonal antisera for immunogold EM studies [53].
Analysis of a Conserved C-terminal Motif
Careful inspection of the sequences of CagH, CagI, and CagL revealed that all three proteins contain a conserved C-terminal motif (Figure 8A) consisting of the distal six amino acids of each protein. The C-terminal motifs of CagH, CagI, and CagL are encoded by divergent DNA sequences (Figure 8A). To investigate whether this C-terminal motif is functionally important, we generated H. pylori mutant strains expressing forms of CagH, CagI, or CagL in which this hexapeptide motif was deleted, as described in Methods. Deletion of the C-terminal motif individually in CagH, CagI, and CagL resulted in reduced levels of the mutated proteins compared to levels of the corresponding WT proteins (Figure 8B), along with a marked reduction in ability of the mutant bacteria to induce IL-8 secretion by AGS cells (Figure 8C) and abolishment of CagA translocation (Figure 8D). In contrast to wild-type CagH-HA and CagI-FLAG, which each localize to the membrane fraction (Figures 4 and 8E), CagH-HAΔCT and CagI-FLAGΔCT are partially mislocalized to the soluble fraction (Figure 8E). Furthermore, in the absence of the C-terminal motif of CagH-HA or CagI-FLAG, CagL stability is markedly reduced at the 48 h growth timepoint compared to CagL stability at the 24 h growth timepoint (Figure 8F). This result further supports the conclusion that CagH, CagI, and CagL are all members of a protein subassembly.

Finally, we conducted experiments to determine whether the C-terminal motifs found in CagH, CagI, and CagL were required for formation of pili at the interface between H. pylori and gastric epithelial cells. Mutant strains lacking the C-terminal motif in CagH, CagI, or CagL were co-cultured with AGS cells, and the samples were then analyzed by FESEM. Bacteria expressing CagI-FLAGΔCT or CagLΔCT each failed to produce detectable pili (Figure 9C, D), and thus had a phenotype indistinguishable from ΔcagI and ΔcagL mutants (Figure 6). Bacteria expressing CagH-HAΔCT expressed pili that were indistinguishable from those produced by the WT strain (Figure 9A, B and Table 3). Despite producing normal-appearing pili, the CagH-HAΔCT mutation rendered the cag T4SS non-functional (Figure 8C, D). These experiments indicate that physical contact of these H. pylori pili with the AGS cell surface is not sufficient to induce IL-8 production, and indicate that the C-terminal motif present in CagH, CagI, and CagL is critical for functionality of the T4SS.

Discussion
One of the important mechanisms by which H. pylori infection leads to severe gastric disease is through the actions of the bacterial oncoprotein CagA [4], [10], [11], [54]. Translocation of CagA into gastric epithelial cells occurs through a T4SS-mediated process and requires multiple proteins encoded by the cag PAI [12], [14]–[17], [21]. Several H. pylori proteins required for CagA translocation are distantly related to components of T4SSs in other bacterial species and presumably have conserved functions [12], [21], [28], [43]. In the current study, we provide new insights into three components of the cag T4SS that lack homologs in other T4SSs – CagH, CagI, and CagL.
Prior to the current study, it was known that CagL can bind α5β1 integrin and can cause several alterations in host cells [21], [39], [42], [44], [45]. CagL was localized in various studies to several bacterial subcellular sites, including a soluble bacterial fraction [38], [43], the bacterial surface [42], and pili on the surface of H. pylori [39] We reasoned that CagL might physically interact with other T4SS components, and that such interactions might be required for CagL export, localization, stability, or activity. Therefore, we conducted studies designed to identify H. pylori proteins that interact with CagL. In initial experiments involving affinity purification of CagL, we observed a highly reproducible co-purification of CagH and CagI with CagL, and we subsequently detected the same pattern of co-purification when either CagH or CagI was targeted for purification. We detected evidence of CagH-CagI-CagL interactions not only in experiments involving bacteria grown in pure culture, but also in experiments involving H. pylori that were attached to gastric epithelial cells. We were unable to undertake analysis of direct interactions among CagL, CagI and CagH because recombinant CagI and CagH could not be expressed in a soluble form under non-denaturing conditions. However, several other results provided evidence of a physical relationship among these proteins. Specifically, CagL stability was decreased in the absence of either CagI or CagH, and co-purification of CagI or CagH with CagL was dependent on the presence of all three proteins. All three proteins are required for functional activity of the cag T4SS, and all three proteins have a role in formation of pili at the interface between H. pylori and gastric epithelial cells. Other intriguing relationships among these proteins include their linkage within a single operon in the cag PAI [46], ability of both CagI and CagL to bind β1 integrin [39], [44], and presence of a nearly identical C-terminal motif in all three proteins. Collectively, the data suggest that CagL, CagI, and CagH are components of one or more T4SS subassemblies involved in pilus biogenesis.
Recognition of the relationships among CagH, CagI, and CagL in the current study was facilitated by using immunoaffinity purifications and a robust multidimensional mass spectrometry protein identification technique. In comparison to traditional co-immunoprecipitation experiments, which rely upon the availability of antibodies to detect potential interacting partners of a target protein by immunoblotting, the mass spectrometry-based approach used in the current study allowed a comprehensive analysis that was not limited by the availability of antibodies. In addition, this approach affords advantages compared to two-hybrid screens, because it allows detection of interactions among native proteins expressed in H. pylori, as well as the potential to detect indirect secondary and tertiary interactions. Analysis of the appropriate control strains allowed for assessment of non-specific protein interactions and statistical evaluation of results.
Prior to the current study, CagH and CagI had not been investigated in detail. Neither CagH nor CagI was detected in analyses of H. pylori using 2D gel proteomic methodology [35], [55], and immunologic detection of CagI expression has been hindered by difficulty in raising antibodies against the protein [44]. Similarly, our efforts to raise polyclonal antisera against CagH or CagI proteins failed to yield antisera that recognized the appropriate antigens. However, we were able to detect CagH and CagI expression by mass spectrometry, and we were also able to detect epitope-tagged forms of these H. pylori proteins. One study reported that CagI was capable of binding β1 integrin in a yeast two hybrid assay, and CagI interactions with integrin were confirmed by analyzing binding of a CagI-GST fusion protein to cell lines deficient in β1 integrin expression or cell lines that had been genetically complemented with human β1 integrin [44]. Another study reported that H. pylori mutant strains containing transposon insertions in cagH or cagI were defective in their ability to translocate CagA into host cells, and that CagH (but not CagI) was required for induction of IL-8 synthesis and secretion by cultured gastric epithelial cells [21]. In the current study, we found that cagH and cagI mutant strains were defective in both CagA translocation and IL-8 induction. The discrepancy in results pertaining to CagI and IL-8 induction might be attributable to differences in the mutagenesis methods utilized to generate ΔcagI isogenic mutants. In the current study, we analyzed unmarked strains containing deletions of the relevant cag genes, and complementation of the unmarked mutants confirmed that CagH and CagI are required for CagA translocation and stimulation of IL-8 secretion by cultured gastric epithelial cells.
Localization analyses performed in the current study indicated that CagH, CagI, and CagL can all be exported to the surface of H. pylori, and in addition, provided evidence for the existence of non-surface-exposed pools of these proteins. The existence of these proteins in multiple subcellular locations potentially reflects multiple stages of T4SS assembly. Detection of CagL on the surface of H. pylori in the current study agrees with previous reports, which reported localization of CagL to either the bacterial surface or to pilus structures, based on immunogold EM staining [39], [42]. Other lines of evidence supporting a surface-exposed localization of CagI and CagL include the ability of both proteins to bind β1 integrin [39], [44], signatures of positive selection in the genes encoding these two proteins [56], and the presence of a predicted signal sequence in both proteins (Figure 1E). The detection of a non-surface-exposed pool of CagL in the current study agrees with results of previous studies, which detected CagL in a soluble bacterial fraction consistent with the periplasm [38], [43].
Determining the subcellular location where CagH, CagI, and CagL assemble into complexes is complicated by the presence of these proteins in multiple subcellular sites. All three proteins can localize to the bacterial surface, and this is one site where the formation of CagH-CagI-CagL complexes may be relevant. In addition, since a large proportion of CagL is localized to a soluble fraction, it seems likely that periplasmic CagL may interact with membrane-associated CagH or CagI proteins that have domains in the periplasm. We detected evidence of CagH-CagI-CagL complexes not only in pure bacterial cultures, but also in co-cultures of H. pylori with gastric epithelial cells (Tables 1, 2, S4). Since the CagH-CagI-CagL complexes were detected in pure bacterial cultures (which lack detectable surface pili), the complexes must clearly be localized to sites distinct from pili under these conditions. A previous study used immunogold labeling methods and a polyclonal antiserum to detect localization of CagL to pilus structures [39]. In the current study, we were not able to detect localization of CagH or CagI to pilus structures, probably due to limitations associated with the use of monoclonal antibodies for immunogold labeling in the context of scanning electron microscopy. Specifically, the FESEM methodology requires multiple extra washing steps (a series of seven sequential ethanol dehydration steps and three liquid carbon dioxide washing steps) that are not required for transmission EM, and monoclonal antibodies often are considered suboptimal compared to polyclonal antisera for immunogold EM studies [53]. Therefore, at present it is not known whether CagL localization to pili requires dissociation of CagL from CagH and CagI, or whether all three proteins eventually localize to pili.
An important finding in the current study is the demonstration that both CagL and CagI are required for formation of pili. Complementation of ΔcagL and ΔcagI mutant strains resulted in restoration of pilus formation and T4SS function. In previous studies, CagY, CagT, and VirB11 ATPase were reported to be required for formation of pili [38], [39], [41], but the role of these proteins in H. pylori pilus formation has not yet been verified by testing of complemented mutant strains. CagT, CagY, and the VirB11 ATPase are each homologous to proteins found in T4SSs of other bacteria, but in contrast, CagI and CagL do not have homologues in other T4SSs.
Interestingly, a ΔcagH mutant strain was hyperpiliated and expressed pili that were elongated and thickened in comparison to pili of the WT strain. The increased number of pili visualized in images of the ΔcagH mutant strain might be attributable to their increased thickness and decreased fragility, resulting in a greater likelihood that the structures are preserved and visualized. Complementation of the ΔcagH mutant strain resulted in production of pili with a WT morphology. These results suggest that CagH is a regulator of pilus dimensions. Analysis of the CagH sequence using a conserved domain database search [57] (NCBI) indicates that it contains a flagellar hook-associated protein (FlgK) domain (PRK06945) (Figure 7H). The FlgK domain within CagH is most closely related to the corresponding domains found in FlgK proteins of Burkholderia spp., rather than the corresponding domain found in H. pylori FlgK. The FlgK domain in CagH also exhibits similarity to portions of the FliK flagellar protein of Salmonella and the YscP type III secretion system protein of Yersinia pestis (Figure 7H), two proteins that are known to be determinants of either flagellar hook length [58]-[63] or type III secretion system needle length [64]. We speculate that, analogous to the role of FlgK (a flagellar hook-junction protein) in terminating flagellar hook assembly, CagH may have a role in terminating pilus assembly. Alternatively, analogous to FliK and YscP, CagH may serve as a molecular ruler to control the dimensions of cag T4SS pili in H. pylori. It will be important in future studies to dissect the molecular mechanisms by which CagH regulates pilus dimensions.
Similar to previous studies [38]-[41], we observed that H. pylori contact with gastric epithelial cells stimulated the production of pili, and in agreement with previous studies [39], [41], we observed that Δcag PAI mutant, cagT mutant, and cagE mutant strains failed to produce pili. However, the dimensions of the pili visualized in the current study differed from what was reported previously. Specifically, a previous study [38] reported that the pili were between 45 and 75 nm in width, depending on presence or absence of a sheath structure, whereas in the current study, we found that the pili were much thinner. The same strain of H. pylori was used in both studies, so this difference is probably not attributable to strain-dependent variation. We speculate that the difference in reported dimensions might be attributable to differences in methods for scanning EM, such as the use of a relatively thin layer of gold in the current study and a thicker layer of coating in the previous study.
A striking feature shared by CagH, CagI, and CagL is a conserved C-terminal hexapeptide motif (Figure 8A). This C-terminal motif is not found in any other protein encoded by H. pylori strain 26695. In the current study, we show that this motif is required for functional activity of the T4SS, and that the C-terminal motifs of CagI and CagL are required for pilus formation. Deletion of the C-terminal motif leads to partial mislocalization of CagH and CagI to a soluble fraction instead of a membrane fraction (Figure 8E). Based on the observed mislocalization of C-terminally truncated CagH and CagI proteins, we hypothesize that this C-terminal motif plays a role in protein sorting or localization. In several other bacterial species, C-terminal motifs are required for recognition of proteins by the T4SS machinery. For example, RalF, an effector molecule of the Legionella Dot/Icm system, has a C-terminal motif consisting of hydrophobic residues flanked by lysine/arginine moieties [65]. Mutagenesis studies indicated that deletion of the distal 3 terminal amino acid residues of RalF leads to loss of recognition by the T4SS machinery, and thus loss of translocation [65]. C-terminal translocation signals in T4SS substrates also have been detected in A. tumefaciens and Bartonella henselae [66], [67]. In a similar manner, we propose that the conserved C-terminal motifs in CagH, CagI, and CagL target these proteins to appropriate sites within the H. pylori T4SS. Interestingly, a mutant strain expressing a C-terminally truncated form of CagH produced normal-appearing pili, but this strain was nevertheless defective in both CagA translocation and ability to stimulate IL-8 induction. This result reveals that contact of these H. pylori pili with the AGS cell surface is not sufficient to induce IL-8 production.
T4SSs are a diverse collection of macromolecular machines, found in a broad phylogenetic range of bacteria. It has been proposed that all T4SSs employ a common mechanism for secretion across the cytoplasmic membrane, and that diversity has arisen primarily as a consequence of variation in the types of cell envelopes that must be spanned, along with variation in donor-host cell interactions [29]. Our current understanding of the architecture of T4SSs is based on elegant work with several model systems, including the VirB/VirD4 system of A. tumefaciens and several plasmid conjugation systems. There are limitations, however, when comparing these model systems with the T4SSs found in distantly related bacteria. Several components of the H. pylori cag T4SS are distantly related to components of the VirB/VirD4 system, and as highlighted in the current study, other key constituents of the cag T4SS lack homology to components of T4SSs in other bacterial species. In addition to the three H. pylori proteins analyzed in the current study, cag T4SS functionality probably requires at least six other proteins that lack homology to VirB/VirD4 proteins [21]. In future studies, it will be important to investigate the functional roles of these proteins in further detail, and from a broader perspective, it will be important to investigate the structural correlates of biological diversity among T4SSs.
Methods
Bacterial Strains and Growth Conditions
H. pylori 26695 and its cag isogenic mutant derivative strains were grown on trypticase soy agar plates supplemented with 5% sheep blood or Brucella agar plates supplemented with 5% fetal bovine serum at 37°C in room air containing 5% CO2. H. pylori mutant strains were selected based on resistance to chloramphenicol (5 µg/ml), kanamycin (10 µg/ml), or metronidazole (7.5 to 15 µg/ml). E. coli strain DH5α, used for plasmid propagation, was grown on Luria-Bertani agar plates or in Luria-Bertani liquid medium supplemented with ampicillin (50 µg/ml), chloramphenicol (25 µg/ml), or kanamycin (25 µg/ml), as appropriate.
Generation of Rabbit Polyclonal anti-CagL Serum
CagL derived from H. pylori 26695 (and lacking a putative signal sequence) was expressed as a GST fusion protein from pGEX-6P-1 vector (GE Healthcare, formerly Amersham). CagL-GST was purified using gluthathione beads [35]. Rabbits were then immunized with purified CagL-GST.
Immunoblot Analysis
To detect expression of Cag proteins, individual samples were separated by SDS-PAGE (4-20% gradient), transferred to a nitrocellulose membrane, and subsequently immunoblotted using rabbit polyclonal antiserum raised against recombinant CagL, or mouse monoclonal antibodies reactive with FLAG or HA epitopes (Sigma). To confirm similar loading of samples, immunoblotting using either a monoclonal mouse antibody (Santa Cruz) or a rabbit polyclonal antiserum to H. pylori HspB, a GroEL homolog, was utilized [68]. Horseradish peroxidase-conjugated anti-rabbit IgG or anti-mouse IgG was used as the second antibody. Signals were generated by enhanced chemiluminescence reaction and detection by exposure to X-ray film.
Cell Culture Methods
AGS human gastric epithelial cells were grown in the presence of 5% CO2 in RPMI medium containing 10% FBS, 2 mM L-glutamine, and 10 mM HEPES buffer.
IL-8 Secretion by Gastric Cells in Contact with H. pylori
H. pylori strains were co-cultured with AGS cells at a multiplicity of infection of 100 : 1, and IL-8 secretion was analyzed using an anti-human IL-8 sandwich ELISA (R&D). Levels of IL-8 secreted by AGS cells in contact with isogenic cag mutants were compared to levels secreted by AGS cells in response to infection with WT H. pylori 26695.
CagA Translocation Assay
Translocation of CagA into AGS cells was analyzed by co-culturing H. pylori strains with AGS cells and detecting tyrosine phosphorylation of CagA, as previously described [35], [69]. Briefly, H. pylori and AGS human gastric cells were co-cultured at a MOI of 100 : 1 for 24 h at 37°C. Cells were lysed in NP-40 lysis buffer containing Complete Mini EDTA-free Protease Inhibitor (Roche) and 2 mM sodium orthovanadate, and CagA translocation was assessed by separating the soluble fraction using 7.5% SDS-PAGE and immunoblotting with an anti-phosphotyrosine antibody (α-PY99, Santa Cruz).
Synthesis of a cat-rdxA Cassette
To facilitate introduction of unmarked mutations into H. pylori chromosomal genes of interest, a cat-rdxA cassette was synthesized and cloned into pUC57 vector (Genscript). This cassette confers resistance to chloramphenicol mediated by the chloramphenicol acetyl-transferase (cat) gene from Campylobacter coli, and susceptibility to metronidazole is mediated by an intact rdxA gene (HP0954) from H. pylori 26695. Both genes are in the same orientation, with cat upstream from rdxA. Expression of cat is driven by the C. coli cat promoter, and expression of rdxA is driven by the H. pylori vacA promoter. The two genes are separated by a presumed C. coli cat terminator, and a ФT7 terminator is located at the 3′-distal end of the rdxA gene.
Mutagenesis of H. pylori cag Genes
To construct a ΔcagL mutant strain, we PCR-amplified cagL along with approximately 0.5 kb of flanking DNA from H. pylori 26695 genomic DNA using Amplitaq Gold (ABI), and cloned the PCR product into pGEM-T Easy (Promega). By using this plasmid as a template for inverse PCR and then ligating the PCR product, we generated a modified plasmid lacking cagL and containing a BamHI site at the site of the deletion. A kanamycin resistance cassette was cloned into the BamHI site to yield pCSLK2-4. H. pylori 26695 was transformed with pCSLK2-4 (which is unable to replicate in H. pylori), and single colonies resistant to kanamycin were selected. PCR and sequencing were used to confirm that the kanamycin cassette had inserted into the cagL locus in the same orientation as operon transcription.
To generate unmarked mutant strains, we used a new method that is a variant of counterselection methods used previously in H. pylori (Figure 2) [70], [71]. As a first step, the rdxA gene, which confers resistance to metronidazole, as well as approximately 0.5 kb of flanking DNA on each side, was PCR-amplified from H. pylori 26695 and cloned into pGEM-T Easy to yield pMM670. By using pMM670 as the template for inverse PCR and then ligating the PCR product, we generated a modified plasmid (pMM672) in which the coding region of rdxA was deleted. Transformation of H. pylori 26695 with pMM672 (which is unable to replicate in H. pylori) and selection for metronidazole-resistant colonies resulted in the recovery of a mutant in which the rdxA locus was deleted (H. pylori ΔrdxA) (Figure 2A).
For each cag gene of interest, we PCR-amplified the relevant gene as well as approximately 0.5 kb of flanking DNA on each side from H. pylori 26695 genomic DNA, and cloned these sequences into pGEM-T Easy. By using these plasmids as templates for inverse PCR and then ligating the PCR products, we generated modified plasmids lacking the cag gene of interest and containing a BamHI site at the site of the deletion. A cat-rdxA cassette (described above) was then cloned into the BamHI site. Plasmids containing the cat-rdxA cassette (which are unable to replicate in H. pylori) were transformed into H. pylori ΔrdxA, and single colonies resistant to chloramphenicol and susceptible to metronidazole were selected (Figure 2A, B). In each case, PCR analysis confirmed that the cat-rdxA cassette had inserted into the desired chromosomal site and that the relevant cag gene was deleted. To generate unmarked mutants, strains containing the cat-rdxA cassette were transformed with plasmids harboring the desired mutations, and transformants resistant to metronidazole were selected (Figure 2A, B). A similar approach was used to introduce a gene encoding a C-terminally truncated form of CagL into the endogenous cagL locus.
To complement mutants in cis at a heterologous chromosomal locus, we used a plasmid derived from pAD1 [72], which allows genes of interest to be introduced into the H. pylori chromosomal ureA locus. The modified pAD1 plasmid contains a chloramphenicol resistance cassette, restriction sites to allow cloning of a gene of interest into a site downstream from the ureA promoter and a ribosomal binding site, and flanking sequences derived from the ureA locus. Plasmids were constructed to allow expression of epitope-tagged forms of CagH, CagI, and CagL, and in addition, plasmids were constructed to allow expression of untagged forms of CagI and CagL. CagL was expressed as a protein containing a hemagglutinin (HA) tag introduced at residue 22 immediately following a putative signal sequence (Figure 1D). CagH was expressed as a protein containing an N-terminal HA tag, and CagI was expressed with an internal FLAG epitope introduced at residue 51 in a region downstream of the predicted signal sequence (Figure 2C), which is predicted to be surface exposed based on hydrophilicity plots (ProtScale in Expasy) [73]. H. pylori strains were transformed with these plasmids, and chloramphenicol-resistant colonies were selected. Expression of the epitope-tagged proteins was verified by immunoblotting using monoclonal anti-HA or anti-FLAG antibodies, respectively.
Immunoaffinity Purification of Cag Proteins
WT or ΔcagL isogenic mutant bacteria cultured on solid media were harvested after 48 h of growth, washed once in phosphate-buffered saline (PBS), and lysed overnight at 4°C in RIPA buffer (10 mM Tris, 100 mM NaCL, 1% NP-40, 0.25% deoxycholic acid, Complete Mini EDTA-free Protease Inhibitor (Roche), pH 7.2). Cellular debris was removed by high speed centrifugation. CagL was immunoaffinity purified from bacterial lysate using polyclonal anti-CagL antibodies that had been covalently cross-linked to a Protein A support (Dynal, Invitrogen). Alternatively, CagL-HA and CagH-HA were purified from strains expressing these proteins using monoclonal anti-HA antibodies (Sigma) that had been either covalently cross-linked or non-covalently bound to a Protein G support (Dynal, Invitrogen). Immobilized anti-FLAG M2 antibody (Sigma) was utilized for immunoaffinity purification of CagI-FLAG. Target proteins were immunoaffinity purified at room temperature. Immunoaffinity purified proteins were washed in 100 bed volumes of PBS containing 0.1% Tween-20 (PBST 0.1%) prior to elution by boiling in SDS buffer (0.3 M Tris-HCl, 1% SDS, 10% glycerol, 100 mM DTT, Pierce), or elution by HA or FLAG peptide competition (1 mg/ml in PBS for HA peptide [Sigma], 1 mg/ml in TBS for FLAG peptide [Sigma]). In each case, the presence of the targeted protein in the immunoaffinity purified sample was verified by immunoblotting.
To purify Cag proteins from bacteria attached to gastric epithelial cells, the WT strain and a strain expressing CagH-HA were each co-cultured with AGS cells (80% confluent) at an MOI of 100 for 5 hours prior to the addition of RIPA buffer and mechanical disruption of the cell monolayers. Cells were lysed by 3 pulses of sonication (10 s for each pulse), followed by overnight incubation at 4°C in RIPA buffer. Cleared lysates were incubated with immobilized anti-HA monoclonal antibody (Sigma) at room temperature. Immunoaffinity-purified preparations were washed with 100 bed volumes of PBST 0.1%. Bound proteins were eluted by HA peptide competition.
Mass Spectrometric Analysis of Samples
To provide a comprehensive analysis of the protein content in immunoaffinity purified samples, the samples were analyzed by multidimensional protein identification technology (MudPIT). Purified proteins were eluted from the beads, run about 2 cm into a 10% NuPAGE gel, and then subjected to in-gel digestion with trypsin and peptide extraction. The resulting peptide mixtures were analyzed via MudPIT essentially as described [74]. Briefly, peptides were loaded via pressure cell (New Objective) onto a biphasic pre-column fritted using an Upchurch M-520 filter union (IDEX). This 100-µm fused silica microcapillary column was packed with 3 cm of 5-µm C18 reverse-phase resin (Jupiter, Phenomenex) followed by 5 cm of strong cation-exchange resin (Luna SCX, Phenomenex). Once loaded, it was then placed in-line with a 100 µm×20 cm, C18 packed emitter tip column (Jupiter C18, 3 µm, 300 Å, Phenomonex) coupled to an LTQ ion trap mass spectrometer equipped with an Eksigent NanoLC-AS1 Autosampler 2.08, an Eksigent NanoLC-1D plus HPLC pump, and nanospray source. separations were accomplished using 5 µl autosampled pulses of ammonium acetate in 0.1% formic acid (25, 50*, 75, 100, 150*, 200, 250*, 300, 500*, 750*, 1000* mM pulses - * shorter MudPITs containing these salt pulses were performed on isolated proteins) followed by a 105 min reversed phase gradient from water 0.1% formic acid to 45% acetonitrile 0.1% formic acid. Tandem mass spectra were collected throughout the runs in a data dependent manner using dynamic exclusion to improve data acquisition of lower intensity peptides. These spectra were extracted from the instrument files using ScanSifter and searched using SEQUEST [75] against an H. pylori strain 26695 database that also contained common contaminants and reversed versions of the H. pylori proteins. Identifications were filtered to an estimated desired false discovery rate (FDR) using reverse database hits and collated to proteins using IDPicker [76]. All reported proteins were identified with a minimum of 2 distinct peptides.
Real-time PCR
Total RNA was isolated from H. pylori using Trizol Reagent (Gibco), according to the manufacturer's protocol. RNA samples were refined using the RNeasy Mini Kit (Qiagen), and on-column RNase-free DNase digestion. cDNA synthesis was performed on 100 ng of purified RNA using the iScript cDNA synthesis kit (BioRad). As a control, first strand cDNA reactions were carried out in parallel without reverse transcriptase. Real time PCR was executed in triplicate on an ABI StepOne Real Time PCR machine, using TaqMan MGB chemistry. Abundance of cagH, cagI, and cagL transcripts in cag isogenic mutant strains was calculated using the ΔΔCT method, with each transcript signal normalized to the abundance of the recA internal control and comparison to the normalized transcript levels of WT H. pylori.
Bacterial Subcellular Fractionation
Wild-type H. pylori strain 26695, as well as strains expressing CagL-HA, CagH-HA or CagI-FLAG, were grown on solid media for 24 h prior to harvest, washing, and resuspension in sonication buffer (10 mM Tris-HCL, pH 8.0, Complete Mini EDTA-Free Protease Inhibitor (Roche)). Bacteria were lysed by 5 pulses of sonication. Unbroken bacteria and cellular debris were removed from the lysate by centrifugation at 4500 x g for 10 min. The supernatant was separated by ultracentrifugation (1 h at 250,000 x g, 4°C) into a soluble fraction and a total membrane fraction. Proteins in the soluble fraction were concentrated by methanol-chloroform precipitation [77] prior to solubilization in reducing SDS-PAGE buffer (Pierce). The total membrane fraction was washed three times in sonication buffer, and resuspended in reducing SDS-PAGE buffer (Pierce). Subcellular fractions were immunoblotted with anti-CagL, anti-HA and anti-FLAG antibodies, followed by appropriate secondary antibodies, to detect untagged CagL, CagL-HA, CagH-HA, and CagI-FLAG, respectively.
Cleavage of Surface Proteins by Proteinase K
Susceptibility of surface-exposed H. pylori proteins to digestion with proteinase K was assessed using a modification of previous protocols [68]. H. pylori expressing CagH-HA, CagI-FLAG, or CagL-HA were grown for 24 h on blood agar plates prior to harvesting and washing in PBS. Bacteria were resuspended in RMPI medium, or RPMI medium containing proteinase K (0-20 µg/ml). Bacteria were incubated for 30 min on ice, and proteinase K activity then was abrogated by addition of PMSF (final concentration 2 mM). After washing in RPMI containing 2 mM PMSF, the bacteria were resuspended in SDS sample buffer and analyzed by SDS-PAGE and immunoblotting. CagH-HA, CagI-FLAG, and CagL-HA were detected with monoclonal anti-HA or monoclonal anti-FLAG antibodies. Cleavage of VacA, which is known to be present on the H. pylori surface [50], [51], was assessed using an anti-VacA antiserum [78]. Cleavage of carbonic anhydrase was assessed using an antiserum raised against a periplasmic domain of this protein [52]. Densitometry was accomplished using Image J software [79]; band densities were normalized to HspB and compared to control samples that were not treated with proteinase K.
Flow Cytometry
Bacteria were grown overnight in Brucella broth supplemented with 5% fetal bovine serum. Bacterial cells were fixed in 4% paraformaldehyde before staining with primary antibody (monoclonal anti-HA [Sigma] or monoclonal anti-FLAG [Sigma]) in PBS supplemented with 50 mM EDTA and 0.1% BSA, followed by staining with secondary antibody (goat F(ab')2 anti-mouse IgG labeled with Alexa Fluor 488) in PBS supplemented with 0.1% BSA. Flow cytometry data were collected on a Becton Dickenson LSR2 instrument using Becton Dickenson FACS Diva 6.1.3 software. 20,000 single cell events were collected for each sample and analyzed using Tree Star Flow Jo 8.8.2 software.
Immunoelectron Microscopy
H. pylori cells were grown on blood agar plates for 48 h as described above. Immunogold labeling was performed on whole bacterial cells as previously described with some modifications [41]. Briefly, bacterial cells were lifted onto formvar-coated 200 mesh copper TEM grids (Electron Microscopy Sciences). Cells were washed three times with pre-warmed PBS and incubated in 1% gelatin in PBS for 30 min at 37°C. After blocking, mouse monoclonal primary antibodies (anti-HA or anti-FLAG) were applied to the sample grids and incubated for 30 min at 37°C. Subsequently, grids were washed three times with pre-warmed PBS and a second blocking step was performed using 1% gelatin in PBS for 30 min at 37°C. Then goat anti-mouse secondary antibodies conjugated to 10 nm colloidal gold particles (Ted Pella, Inc.) were applied and incubated for 30 min at 37°C. Afterward, grids were washed three times with prewarmed PBS and cells were negatively stained with 1% ammonium molybdate before being visualized with a Philips CM-12 transmission electron microscope.
FESEM of H. pylori in Contact with Gastric Epithelial Cells
H. pylori and AGS human gastric cells were co-cultured at a MOI of 100∶1 on tissue culture-treated coverslips (BD Biosciences) for 4 h at 37°C in the presence of 5% CO2. Cells were fixed with 2.0% paraformaldehyde, 2.5% glutaraldehyde in 0.05 M sodium cacodylate buffer for 1 h at 37°C. Coverslips were washed with sodium cacodylate buffer and secondary fixation was performed with 1% osmium tetroxide at room temperature for 2 h. Coverslips were washed with sodium cacodylate buffer and dehydrated with sequential washes of increasing concentrations of ethanol. Samples were then dried at the critical point, mounted onto sample stubs, grounded with a thin strip of silver paint at the sample edge, and sputter-coated with gold before viewing with a Zeiss Supra 35V FEG scanning electron microscope. Analysis of pilus dimensions and image analysis was performed using Image J software [79].
Statistical Analysis
The statistical significance of differences in numbers of spectral counts of proteins detected in different immunoaffinity purified preparations was determined by G-test, as previously described [80]. The statistical significance of differences in IL-8 production or cag gene expression when comparing WT H. pylori with mutant strains was determined by one-way ANOVA followed by Dunnett's post-hoc test for multiple comparisons against a single control [81]. Statistical analysis of pilus dimensions was performed using a two-tailed Student's T-Test.
Ethics Statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee of Vanderbilt University School of Medicine (M/07/292).
Accession Numbers
H. pylori 26695 whole genome, GI:15644634; http://genolist.pasteur.fr/PyloriGene/; CagH, NP_207337; CagI, NP_207336; CagL, NP_207335.
Supporting Information
Zdroje
1. SuerbaumSMichettiP 2002 Helicobacter pylori infection. N Engl J Med 347 1175 1186
2. AmievaMREl-OmarEM 2008 Host-bacterial interactions in Helicobacter pylori infection. Gastroenterology 134 306 323
3. BlaserMJAthertonJC 2004 Helicobacter pylori persistence: biology and disease. J Clin Invest 113 321 333
4. CoverTLBlaserMJ 2009 Helicobacter pylori in health and disease. Gastroenterology 136 1863 1873
5. ParsonnetJFriedmanGDOrentreichNVogelmanH 1997 Risk for gastric cancer in people with CagA positive or CagA negative Helicobacter pylori infection. Gut 40 297 301
6. KuipersEJPerez-PerezGIMeuwissenSGBlaserMJ 1995 Helicobacter pylori and atrophic gastritis: importance of the cagA status. J Natl Cancer Inst 87 1777 1780
7. BlaserMJPerez-PerezGIKleanthousHCoverTLPeekRM 1995 Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res 55 2111 2115
8. CensiniSLangeCXiangZCrabtreeJEGhiaraP 1996 cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc Natl Acad Sci U S A 93 14648 14653
9. AkopyantsNSCliftonSWKersulyteDCrabtreeJEYoureeBE 1998 Analyses of the cag pathogenicity island of Helicobacter pylori. Mol Microbiol 28 37 53
10. HatakeyamaM 2004 Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat Rev Cancer 4 688 694
11. BackertSSelbachM 2008 Role of type IV secretion in Helicobacter pylori pathogenesis. Cell Microbiol 10 1573 1581
12. BourzacKMGuilleminK 2005 Helicobacter pylori-host cell interactions mediated by type IV secretion. Cell Microbiol 7 911 919
13. JonesKRWhitmireJMMerrellDS 2010 A tale of two toxins: H. pylori CagA and VacA modulate host pathways that impact disease. Frontiers Microbiol 1 115
14. OdenbreitSPulsJSedlmaierBGerlandEFischerW 2000 Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 287 1497 1500
15. TegtmeyerNWesslerSBackertS 2011 Role of the cag-pathogenicity island encoded type IV secretion system in Helicobacter pylori pathogenesis. FEBS J 278 1190 1202
16. FischerW 2011 Assembly and molecular mode of action of the Helicobacter pylori Cag type IV secretion apparatus. FEBS J 278 1203 1212
17. TerradotLWaksmanG 2011 Architecture of the Helicobacter pylori Cag-type IV secretion system. FEBS J 278 1213 22
18. SegalEDChaJLoJFalkowSTompkinsLS 1999 Altered states: involvement of phosphorylated CagA in the induction of host cellular growth changes by Helicobacter pylori. Proc Natl Acad Sci U S A 96 14559 14564
19. AmievaMRVogelmannRCovacciATompkinsLSNelsonWJ 2003 Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science 300 1430 1434
20. GuilleminKSalamaNRTompkinsLSFalkowS 2002 Cag pathogenicity island-specific responses of gastric epithelial cells to Helicobacter pylori infection. Proc Natl Acad Sci U S A 99 15136 15141
21. FischerWPulsJBuhrdorfRGebertBOdenbreitS 2001 Systematic mutagenesis of the Helicobacter pylori cag pathogenicity island: essential genes for CagA translocation in host cells and induction of interleukin-8. Mol Microbiol 42 1337 1348
22. VialaJChaputCBonecaIGCardonaAGirardinSE 2004 Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol 5 1166 1174
23. BrandtSKwokTHartigRKonigWBackertS 2005 NF-kappaB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc Natl Acad Sci U S A 102 9300 9305
24. KimSYLeeYCKimHKBlaserMJ 2006 Helicobacter pylori CagA transfection of gastric epithelial cells induces interleukin-8. Cell Microbiol 8 97 106
25. ArgentRHHaleJLEl-OmarEMAthertonJC 2008 Differences in Helicobacter pylori CagA tyrosine phosphorylation motif patterns between western and East Asian strains, and influences on interleukin-8 secretion. J Med Microbiol 57 1062 1067
26. SelbachMMoeseSMeyerTFBackertS 2002 Functional analysis of the Helicobacter pylori cag pathogenicity island reveals both VirD4-CagA-dependent and VirD4-CagA-independent mechanisms. Infect Immun 70 665 671
27. FronzesRSchaferEWangLSaibilHROrlovaEV 2009 Structure of a type IV secretion system core complex. Science 323 266 268
28. CascalesEChristiePJ 2003 The versatile bacterial type IV secretion systems. Nat Rev Microbiol 1 137 149
29. Alvarez-MartinezCEChristiePJ 2009 Biological diversity of prokaryotic type IV secretion systems. Microbiol Mol Biol Rev 73 775 808
30. YeoHJWaksmanG 2004 Unveiling molecular scaffolds of the type IV secretion system. J Bacteriol 186 1919 1926
31. FronzesRChristiePJWaksmanG 2009 The structural biology of type IV secretion systems. Nat Rev Micro 7 703 714
32. LiSDKersulyteDLindleyIJDNeelamBBergDE 1999 Multiple genes in the left half of the cag pathogenicity island of Helicobacter pylori are required for tyrosine kinase-dependent transcription of interleukin-8 in gastric epithelial cells. Infect Immun 67 3893 3899
33. Pinto-SantiniDMSalamaNR 2009 Cag3 is a novel essential component of the Helicobacter pylori cag type IV secretion system outer membrane subcomplex. J Bacteriol 191 7343 7352
34. KutterSBuhrdorfRHaasJSchneider-BrachertWHaasR 2008 Protein subassemblies of the Helicobacter pylori Cag type IV secretion system revealed by localization and interaction studies. J Bacteriol 190 2161 2171
35. BuslerVJTorresVJMcClainMSTiradoOFriedmanDB 2006 Protein-protein interactions among Helicobacter pylori Cag proteins. J Bacteriol 188 4787 4800
36. JurikAHausserEKutterSPattisIPrasslS 2010 The coupling protein Cagβ and its interaction partner CagZ are required for type IV secretion of the Helicobacter pylori CagA protein. Infect Immun 78 5244 5251
37. YuanQCarleAGaoCSivanesanDAlyKA 2005 Identification of the VirB4-VirB8-VirB5-VirB2 pilus assembly sequence of type IV secretion systems. J Biol Chem 280 26349 26359
38. RohdeMPulsJBuhrdorfRFischerWHaasR 2003 A novel sheathed surface organelle of the Helicobacter pylori cag type IV secretion system. Mol Microbiol 49 219 234
39. KwokTZablerDUrmanSRohdeMHartigR 2007 Helicobacter exploits integrin for type IV secretion and kinase activation. Nature 449 862 866
40. AndrzejewskaJLeeSKOlbermannPLotzingNKatzowitschE 2006 Characterization of the pilin ortholog of the Helicobacter pylori type IV cag pathogenicity apparatus, a surface-associated protein expressed during infection. J Bacteriol 188 5865 5877
41. TanakaJSuzukiTMimuroHSasakawaC 2003 Structural definition on the surface of Helicobacter pylori type IV secretion apparatus. Cell Microbiol 5 395 404
42. TegtmeyerNHartigRDelahayRMRohdeMBrandtS 2010 A small fibronectin-mimicking protein from bacteria induces cell spreading and focal adhesion formation. J Biol Chem 285 23515 23526
43. KutterSBuhrdorfRHaasJSchneider-BrachertWHaasR 2008 Protein subassemblies of the Helicobacter pylori Cag type IV secretion system revealed by localization and interaction studies. J Bacteriol 190 2161 2171
44. Jiménez-SotoLFKutterSSewaldXErtlCWeissE 2009 Helicobacter pylori type IV secretion apparatus exploits β1 integrin in a novel RGD-independent manner. PLoS Pathog 5 e1000684
45. SahaABackertSHammondCEGoozMSmolkaAJ 2010 Helicobacter pylori CagL activates ADAM17 to induce repression of the gastric H, K-ATPase α subunit. Gastroenterology 139 239 248
46. SharmaCMHoffmannSDarfeuilleFReignierJFindeiszS 2010 The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 464 250 255
47. PattisIWeissELaugksRHaasRFischerW 2007 The Helicobacter pylori CagF protein is a type IV secretion chaperone-like molecule that binds close to the C-terminal secretion signal of the CagA effector protein. Microbiology 153 2896 2909
48. FernandezDSpudichGZhouXChristieP 1996 The Agrobacterium tumefaciens VirB7 lipoprotein is required for stabilization of VirB proteins during assembly of the T-complex transport apparatus. J Bacteriol 178 3168 3176
49. HapfelmeierSDomkeNZambryskiPCBaronC 2000 VirB6 is required for stabilization of VirB5 and VirB3 and formation of VirB7 homodimers in Agrobacterium tumefaciens. J Bacteriol 182 4505 4511
50. IlverDBaroneSMercatiDLupettiPTelfordJL 2004 Helicobacter pylori toxin VacA is transferred to host cells via a novel contact-dependent mechanism. Cell Microbiol 6 167 174
51. FischerWBuhrdorfRGerlandEHaasR 2001 Outer membrane targeting of passenger proteins by the vacuolating cytotoxin autotransporter of Helicobacter pylori. Infect Immun 69 6769 6775
52. MarcusEAMoshfeghAPSachsGScottDR 2005 The periplasmic α-carbonic anhydrase activity of Helicobacter pylori is essential for acid acclimation. J Bacteriol 187 729 738
53. ZerovnikEZavasnik-BergantVKopitar-JeralaNPompe-NovakMSkarabotM 2002 Amyloid fibril formation by human stefin B in vitro: immunogold labelling and comparison to stefin A. Biol Chem 383 859 863
54. HatakeyamaM 2011 Anthropological and clinical implications for the structural diversity of the Helicobacter pylori CagA oncoprotein. Cancer Sci 102 36 43
55. BackertSKwokTSchmidMSelbachMMoeseS 2005 Subproteomes of soluble and structure-bound Helicobacter pylori proteins analyzed by two-dimensional gel electrophoresis and mass spectrometry. Proteomics 5 1331 1345
56. OlbermannPJosenhansCMoodleyYUhrMStamerC 2010 A global overview of the genetic and functional diversity in the Helicobacter pylori cag pathogenicity island. PLoS Genet 6 e1001069
57. Marchler-BauerAAndersonJBChitsazFDerbyshireMKDeWeese-ScottC 2009 CDD: specific functional annotation with the Conserved Domain Database. Nucleic Acids Res 37 D205 D210
58. MuramotoKMakishimaSAizawaS-IMacnabRM 1999 Effect of hook subunit concentration on assembly and control of length of the flagellar hook of Salmonella. J Bacteriol 181 5808 5813
59. ErhardtMHiranoTSuYPaulKWeeDH 2010 The role of the FliK molecular ruler in hook-length control in Salmonella enterica. Mol Microbiol 75 1272 1284
60. KawagishiIHommaMWilliamsAMacnabR 1996 Characterization of the flagellar hook length control protein FliK of Salmonella typhimurium and Escherichia coli. J Bacteriol 178 2954 2959
61. WilliamsAYamaguchiSTogashiFAizawaSKawagishiI 1996 Mutations in fliK and flhB affecting flagellar hook and filament assembly in Salmonella typhimurium. J Bacteriol 178 2960 2970
62. KutsukakeKMinaminoTYokosekiT 1994 Isolation and characterization of FliK-independent flagellation mutants from Salmonella typhimurium. J Bacteriol 176 7625 7629
63. MakishimaSKomoriyaKYamaguchiSAizawaS-I 2001 Length of the flagellar hook and the capacity of the type III export apparatus. Science 291 2411 2413
64. JournetLAgrainClBrozPCornelisGR 2003 The needle length of bacterial injectisomes is determined by a molecular ruler. Science 302 1757 1760
65. NagaiHCambronneEDKaganJCAmorJCKahnRA 2005 A C-terminal translocation signal required for Dot/Icm-dependent delivery of the Legionella RalF protein to host cells. Proc Natl Acad Sci U S A 102 826 831
66. VergunstACvan LierMCMden Dulk-RasAGrosse StüveTAOuwehandA 2005 Positive charge is an important feature of the C-terminal transport signal of the VirB/D4-translocated proteins of Agrobacterium. Proc Natl Acad Sci U S A 102 832 837
67. SchuleinRGuyePRhombergTASchmidMCSchröderG 2005 A bipartite signal mediates the transfer of type IV secretion substrates of Bartonella henselae into human cells. Proc Natl Acad Sci U S A 102 856 861
68. IvieSMcClainMAlgoodHLacyDBCoverT 2010 Analysis of a beta-helical region in the p55 domain of Helicobacter pylori vacuolating toxin. BMC Microbiol 10 60
69. LohJTTorresVJCoverTL 2007 Regulation of Helicobacter pylori cagA expression in response to salt. Cancer Res 67 4709 4715
70. DailidieneDDailideGKersulyteDBergDE 2006 Contraselectable streptomycin susceptibility determinant for genetic manipulation and analysis of Helicobacter pylori. Appl Environ Microbiol 72 5908 5914
71. CopassMGrandiGRappuoliR 1997 Introduction of unmarked mutations in the Helicobacter pylori vacA gene with a sucrose sensitivity marker. Infect Immun 65 1949 1952
72. LohJTForsythMHCoverTL 2004 Growth phase regulation of flaA expression in Helicobacter pylori is luxS dependent. Infect Immun 72 5506 5510
73. HoppTPWoodsKR 1981 Prediction of protein antigenic determinants from amino acid sequences. Proc Natl Acad Sci U S A 78 3824 3828
74. MacCossMJMcDonaldWHSarafASadygovRClarkJM 2002 Shotgun identification of protein modifications from protein complexes and lens tissue. Proc Natl Acad Sci U S A 99 7900 7905
75. YatesJREngJKMcCormackALSchieltzD 1995 Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal Chem 67 1426 1436
76. MaZQDasariSChambersMCLittonMDSobeckiSM 2009 IDPicker 2.0: Improved protein assembly with high discrimination peptide identification filtering. J Proteome Res 8 3872 3881
77. WesselDFlüggeUI 1984 A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem 138 141 143
78. SchrawWLiYMcClainMvan der GootFGCoverTL 2002 Association of Helicobacter pylori vacuolating toxin (VacA) with lipid rafts. J Biol Chem 277 34642 34650
79. AbramoffMDMagelhaesPJRamSJ 2004 Image Processing with ImageJ. Biophotonics Internat 11 36 42
80. ZhangBVerBerkmoesNCLangstonMAUberbacherEHettichRL 2006 Detecting differential and correlated protein expression in label-free shotgun proteomics. J Proteome Res 5 2909 2918
81. ZarJH 1999 Biostatistical Analysis. New Jersey Prentice Hall
82. NakaiKKanehisaM 1991 Expert system for predicting protein localization sites in gram-negative bacteria. Proteins: Structure, Function, and Bioinformatics 11 95 110
83. NielsenHEngelbrechtJBrunakSvon HeijneG 1997 A neural network method for identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Int J Neural Syst 8 581 599
84. CrooksGEHonGChandoniaJ-MBrennerSE 2004 WebLogo: a sequence logo generator. Genome Res 14 1188 1190
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 9
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- HTLV-1 Propels Thymic Human T Cell Development in “Human Immune System” Rag2 gamma c Mice
- Hostile Takeover by : Reorganization of Parasite and Host Cell Membranes during Liver Stage Egress
- Exploiting and Subverting Tor Signaling in the Pathogenesis of Fungi, Parasites, and Viruses
- A Viral Ubiquitin Ligase Has Substrate Preferential SUMO Targeted Ubiquitin Ligase Activity that Counteracts Intrinsic Antiviral Defence
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy