
Exposure to the Viral By-Product dsRNA or Coxsackievirus B5 Triggers Pancreatic Beta Cell Apoptosis via a Bim / Mcl-1 Imbalance
The rise in type 1 diabetes (T1D) incidence in recent decades is probably related to modifications in environmental factors. Viruses are among the putative environmental triggers of T1D. The mechanisms regulating beta cell responses to viruses, however, remain to be defined. We have presently clarified the signaling pathways leading to beta cell apoptosis following exposure to the viral mimetic double-stranded RNA (dsRNA) and a diabetogenic enterovirus (Coxsackievirus B5). Internal dsRNA induces cell death via the intrinsic mitochondrial pathway. In this process, activation of the dsRNA-dependent protein kinase (PKR) promotes eIF2α phosphorylation and protein synthesis inhibition, leading to downregulation of the antiapoptotic Bcl-2 protein myeloid cell leukemia sequence 1 (Mcl-1). Mcl-1 decrease results in the release of the BH3-only protein Bim, which activates the mitochondrial pathway of apoptosis. Indeed, Bim knockdown prevented both dsRNA - and Coxsackievirus B5-induced beta cell death, and counteracted the proapoptotic effects of Mcl-1 silencing. These observations indicate that the balance between Mcl-1 and Bim is a key factor regulating beta cell survival during diabetogenic viral infections.
Published in the journal:
. PLoS Pathog 7(9): e32767. doi:10.1371/journal.ppat.1002267
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002267
Summary
The rise in type 1 diabetes (T1D) incidence in recent decades is probably related to modifications in environmental factors. Viruses are among the putative environmental triggers of T1D. The mechanisms regulating beta cell responses to viruses, however, remain to be defined. We have presently clarified the signaling pathways leading to beta cell apoptosis following exposure to the viral mimetic double-stranded RNA (dsRNA) and a diabetogenic enterovirus (Coxsackievirus B5). Internal dsRNA induces cell death via the intrinsic mitochondrial pathway. In this process, activation of the dsRNA-dependent protein kinase (PKR) promotes eIF2α phosphorylation and protein synthesis inhibition, leading to downregulation of the antiapoptotic Bcl-2 protein myeloid cell leukemia sequence 1 (Mcl-1). Mcl-1 decrease results in the release of the BH3-only protein Bim, which activates the mitochondrial pathway of apoptosis. Indeed, Bim knockdown prevented both dsRNA - and Coxsackievirus B5-induced beta cell death, and counteracted the proapoptotic effects of Mcl-1 silencing. These observations indicate that the balance between Mcl-1 and Bim is a key factor regulating beta cell survival during diabetogenic viral infections.
Introduction
Type 1 diabetes (T1D) is a chronic autoimmune disease characterized by the progressive and selective destruction of the insulin-producing pancreatic beta cells [1]. It mainly affects individuals during childhood or adolescence and requires a life-long treatment with insulin, which at the US represents a cost of $14.4 billion per year [2]. Triggering of diabetes results from an interaction between genetical and environmental factors. Most candidate genes for T1D are supposed to act at the immune system level [3], but we have recently shown that nearly 30% of these candidate genes are also expressed in beta cells and may modulate their responses after exposure to potential environmental factors [4]. These findings indicate that beta cells are not only targets, but also actors of T1D pathophysiology [4].
The incidence of T1D is increasing 3.9% per year in Europe, especially among the youngest population (<5 years-old) in which a doubling in the number of new cases is expected between 2005 and 2020 [5]. An explanation for this rapid augment in T1D incidence may be the increased exposure to diabetogenic environmental factors. Viruses are among the potential environmental causes of T1D [6], as suggested by epidemiological [7], experimental [8] and clinical data [9].
Enterovisuses (EV) such as Coxsackievirus B (CVB) [10], are the main candidates. Coxsackievirus B4 was isolated from a child who died of diabetic ketoacidosis and this virus induced diabetes in mice [11]. Among the Coxsackievirus, CVB5 is the most deleterious prototype strain for human pancreatic islets [12]. An increase of T1D incidence has been described after epidemics of CVB5 [13] and these epidemics are frequent in Finland, the country with the highest incidence of T1D [14].
The pathogenic role of viruses in T1D might involve damage to beta cells and the local induction of proinflammatory mediators [1]. CVB-infected pancreatic beta cells can be phagocyted by both macrophages [15] and dendritic cells [16], leading to activation of the immune system, presentation of islet autoantigens and release of cytokines/chemokines. Local injury of beta cells induced by dsRNA, a by-product of viral replication, promotes autoimmune diabetes in animal models [17]. Both the viral mimetic dsRNA [18] and Coxsackievirus [19] induce beta cell apoptosis, but the mechanisms involved remain to be clarified.
Apoptotic cell death can be initiated by two signaling cascades, namely the intrinsic and the extrinsic pathways. The intrinsic pathway is the main pathway for endoplasmic reticulum (ER) stress and cytokine-induced beta cell apoptosis [20], [21], [22], [23]. In this pathway, a disequilibrium between the antiapoptotic and the proapoptotic Bcl-2 family members [24] leads to activation of Bcl-2-associated X protein (BAX) and Bcl-2 antagonist or killer (BAK) and the consequent permeabilization of mitochondrial outer membrane. This allows cytochrome c release to the cytoplasm and the assembly of the apoptosome. The apoptosome then recruits and activates the initiator caspase 9, which cleaves and activates the effector caspase 3, leading to execution of apoptosis [25].
We have presently investigated the molecular pathways involved in dsRNA and viral-induced beta cell apoptosis. Similar to ER stress and cytokine-induced apoptosis [24], the viral by-product dsRNA activates the intrinsic pathway of apoptosis. The nature of the Bcl-2 family members involved is, however, different. Thus, dsRNA-dependent protein kinase (PKR) activation by cytoplasmic dsRNA leads to phosphorylation of the elongation factor eIF2α. The eIF2α phosphorylation promotes inhibition of protein translation, which is followed by an early and progressive decrease in the expression of the antiapoptotic myeloid cell leukemia sequence 1 (Mcl-1) protein. Decreased expression of Mcl-1 allows the proapoptotic protein Bim to activate BAX, leading to cytochrome c release, caspase 9 and 3 activation and beta cell apoptosis. Similar findings were observed during viral infection of pancreatic beta cells by CVB5. These observations clarify the mechanisms involved in viral-induced apoptosis in pancreatic beta cells, and suggest that usage of Bcl-2 proteins is context-dependent during beta cell apoptosis initiated by different stimuli.
Results
Internal dsRNA induces beta cell apoptosis via the intrinsic mitochondrial pathway of apoptosis
Transfection with the synthetic dsRNA polyinosinic-polycytidylic acid (PIC) induced beta cell apoptosis already after 12h, with progressive increase up to 24h (Figure 1A). To characterize the pathways involved in beta cell apoptosis, BAX translocation to the mitochondria was analyzed by immunocytochemistry. Intracellular dsRNA promoted the translocation of BAX (Figure 1B) and the release of cytochrome c from the mitochondria to the cytoplasm (Figures 1C and D). This activated the initiator caspase 9 and effector caspase 3 (Figure 1E), characterizing induction of the intrinsic mitochondrial pathway of apoptosis.

Expression of antiapoptotic Bcl-2 family members regulates beta cell apoptosis and is decreased upon exposure to internal dsRNA
The antiapoptotic Bcl-2 protein Mcl-1 is an important regulator of beta cell death after exposure to proinflammatory cytokines and ER stressors [20]. Mcl-1 protein expression gradually decreased from 6h until 24h after PIC transfection (Figure 2A). This reduction in Mcl-1 protein was a post-transcriptional effect since Mcl-1 mRNA expression was in fact increased at 24h (Figure 2B). Mcl-1 is targeted for degradation at the proteasome [26]. INS-1E cells treated with the proteasome inhibitor MG-132 (same experimental conditions as in [20], [22]) had increased basal Mcl-1 expression, and the inhibitor partially prevented dsRNA-induced decrease in Mcl-1 expression (Figure 2C), suggesting that proteasomal degradation contributes at least in part for the observed Mcl-1 downregulation. Mcl-1 knockdown by two previously validated siRNAs [20] exacerbated cleavage of caspases 9/3 (Figure 2D) and PIC-induced apoptosis in INS-1E cells (Figure 2E) and FACS-purified primary beta cells (Figure 2F). On the other hand, overexpressing rat Mcl-1 by the use of an adenoviral vector (adMcl-1 [20]) prevented the activation of caspases 9/3 and reduced by > 40% apoptosis induced by dsRNA (Figures 2G, H and I). This is a specific effect, since infection with a control adenovirus encoding luciferase (adLuc) did not modify apoptosis (Figures 2H and I) and Mcl-1 silencing or overexpression did not modify expression of Bcl-XL or Bcl-2 (data not shown). These observations indicate a key role for Mcl-1 decrease in dsRNA-induced apoptosis.

The two other key antiapoptotic Bcl-2 proteins in beta cells are Bcl-XL and Bcl-2 [24]. Bcl-XL but not Bcl-2 protein expression was decreased after PIC exposure (Supplemental Figures S1A and B). This downregulation, however, was only detected at later time-points (after 12h and 24h) and to a lesser extent than observed with Mcl-1 (compare Figure 2A and Supplemental Figure S1A). Bcl-XL and Bcl-2 knockdown (Supplemental Figures S1C and D) increased basal beta cell apoptosis from 7% to more than 30% and further increased PIC-induced apoptosis, as compared to siControl (Supplemental Figures S1E and F). As observed with siMcl-1, inhibition of Bcl-XL and Bcl-2 promoted caspases 9 and 3 cleavage in parallel with beta cell apoptosis (Supplemental Figures S1G and H). Similar results were observed with second independent siRNAs against Bcl-XL and Bcl-2 (Supplemental Figure S2). Nevertheless, when the effects of Bcl-XL or Bcl-2 silencing on PIC-induced apoptosis were corrected by their basal levels of apoptosis using the apoptotic index (i.e. siBcl-XL or Bcl-2 + PIC correct by siBcl-XL or Bcl-2 alone [27]) they were not different from the control group (siControl + PIC) (Supplemental Figure S3). This finding suggests that knocking down these proteins has only additive effect to PIC-induced apoptosis. On the other hand, Mcl-1 knockdown using two different siRNAs resulted in increased apoptotic indexes as compared to siControl, indicating that silencing of Mcl-1, different from Bcl-XL and Bcl-2, potentiates apoptosis caused by PIC (Supplemental Figure S3A).
The proapoptotic BH3-only protein Bim regulates pancreatic beta cell apoptosis after dsRNA exposure
The BH3-only protein Bim was previously shown to be an important regulator of glucose-mediated beta cell apoptosis [28] and virus-induced cell death in other cell types [29]. Against this background, we evaluated its role in dsRNA-induced beta cell apoptosis. Time-course experiments with INS-1E cells exposed to intracellular PIC did not show modifications in the expression of the Bim isoforms EL, L and S (Figure 3A). Since Bim may modulate apoptosis independently of changes in its expression [30], we next evaluated whether Bim knockdown affects dsRNA-induced apoptosis. The use of a specific siRNA against Bim inhibited by > 70% its protein expression at both basal and PIC-induced conditions (Figure 3B). Bim knockdown prevented PIC-induced apoptosis in INS-1E cells (Figure 3C) and in primary beta cells (Figure 3H). These results were confirmed with a second siRNA against Bim (Supplemental Figure S4). In agreement with the effects on cell viability, PIC-induced activation of caspases 9 and 3 was also reduced after inhibition of Bim (Figure 3D). We studied two other proapoptotic BH3-only proteins, namely Death Protein-5 (DP5) and p53 Up-regulated Modulator of Apoptosis (PUMA), previously shown to regulate cytokine-induced beta cell apoptosis [21], [31]. We obtained a knockdown of >80% for DP5 and >75% for PUMA mRNA expression (Supplemental Figures S5A and C); however, differently from what was observed following Bim silencing, neither siDP5 nor siPUMA prevented PIC-triggered apoptosis (Supplemental Figures S5B and D).

One of the mechanisms by which the antiapoptotic Bcl-2 proteins act is via binding and inactivating specific proapoptotic BH3-only proteins [32]. We thus performed co-immunoprecipitation studies to evaluate the association between Bim and Mcl-1. Bim was immunoprecipitated using a specific polyclonal antibody and the presence of Mcl-1 in the precipitated material was determined with an anti-Mcl-1 antibody. We observed that at basal condition Bim was bound to Mcl-1, but after 24h of PIC exposure there was a clear decrease in the association between these two proteins, confirming that Bim is liberated from Mcl-1 following dsRNA exposure (Figure 3E).
To evaluate whether the PIC-induced decrease in Mcl-1 protein expression contributes to beta cell apoptosis by hampering Bim inactivation by Mcl-1, we performed double-knockdown (siMcl-1 + siBim) and evaluated apoptosis and cleavage of caspases 9 and 3 in comparison to single knockdown (siMcl-1 or siBim). The combined use of siMcl-1 plus siBim produced a >70% suppression of the target genes, as observed with the use of these siRNAs individually (data not shown). Interestingly, the double-silencing of siMcl-1 + siBim prevented the proapoptotic effect caused by Mcl-1 knockdown in INS-1E cells (Figures 3F and G) and primary beta cells (Figure 3H), suggesting that the release of Bim from the Mcl-1/Bim complex induced by both dsRNA and siMcl-1 is a key effector of beta cell apoptosis.
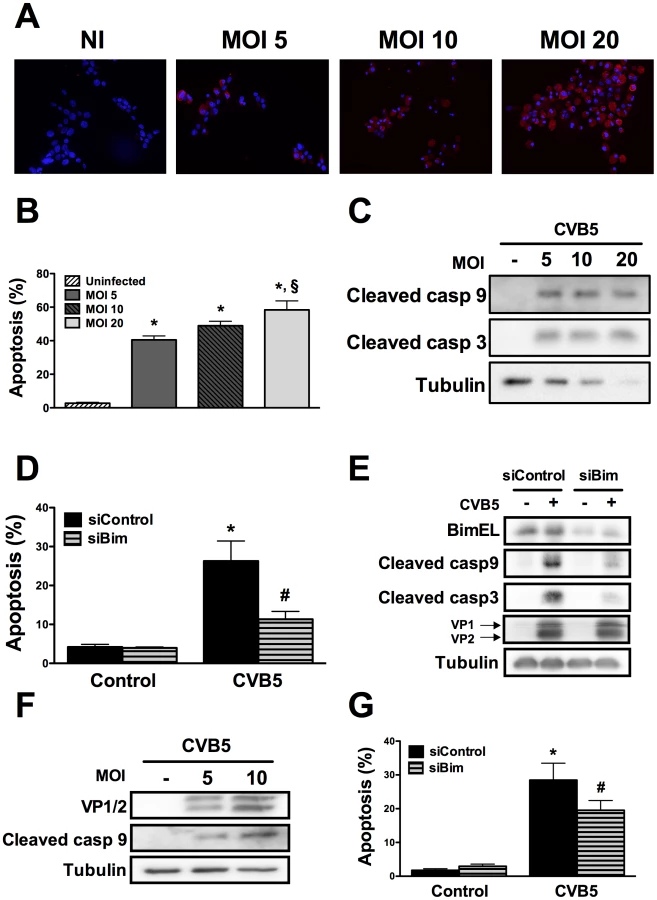
Infection of beta cells by Coxsackievirus B5 induces the mitochondrial apoptosis pathway through the BH3-only protein Bim
To assess whether the above described pathways are also relevant in the context of a viral infection of beta cells, we infected INS-1E cells and FACS-purified rat beta cells with Coxsackievirus B5. First, we confirmed that this enterovirus infects rat beta cells at the different MOIs tested by determining the presence of the viral capsid (VP1 and 2) (Figures 4A, E and F). The viral capsid was already detected in the cells at 8h post-infection and its expression increased further up to 24h (data not shown; Figures 4A and F). Infection of beta cells at these MOIs induced cell death mainly by apoptosis (Figure 4B) via the intrinsic pathway (Figures 4C and F), as observed with internal dsRNA (Figure 1). In line with these observations, Bim silencing significantly decreased virus-induces apoptosis (Figures 4D and G) and caspase 9 and 3 activation (Figure 4E). Importantly, inhibition of virus-induced apoptosis by knocking down Bim did not modify the amount of viral capsids (VP1 and 2; Figure 4E), indicating that, under the present experimental conditions, inhibition of apoptosis did not exacerbate viral replication in beta cells. These observations suggest that both dsRNA and the potentially diabetogenic virus CVB5 induce a similar pathway of apoptosis in beta cells. Interestingly, both dsRNA and CVB5 induced expression of interferon β (Supplemental Figure S6A and B) and interferon α (data not shown), but with different kinetics. Expression of interferon β mRNA was several-fold higher in primary beta cells as compared to INS-1E cells (Supplemental Figure S6C). This increased expression of type I interferons may also contribute to the observed in vitro beta cell apoptosis, as suggested by previous studies [33].
dsRNA-induced decrease in Mcl-1 protein expression depends on eIF2α phosphorylation by dsRNA-dependent protein kinase (PKR)
Two important regulators of Mcl-1 protein expression have been reported in pancreatic beta cells, namely phosphorylation of eIF2α and c-Jun N-terminal Kinase (JNK) activation [20]. Taking this into account we evaluated the time course phosphorylation of eIF2α and JNK in INS-1E cells transfected with PIC. Both eIF2α and JNK were activated by PIC, but with different profiles. Thus, eIF2α phosphorylation started earlier (2h), had a peak at 6h then slowly decreased up to 24h (Figure 5A); JNK phosphorylation started later (6h), reached a plateau at 12h and remained upregulated up to 24h (Figure 5B).

JNK inhibition by a specific chemical inhibitor [34], did not prevent Mcl-1 protein decrease (Supplemental Figure S7). We also evaluated the expression of the BH3-only protein Noxa previously shown to induce proteosomal degradation of Mcl-1 [35], but did not detect its expression in beta cells (data not shown).
PKR-like endoplasmic reticulum kinase (PERK) and PKR were evaluated as two possible mediators of the eIF2α phosphorylation induced by PIC [36], [37]. PERK is a kinase activated by endoplasmic reticulum (ER) stress [36], while PKR is a classic downstream response to dsRNA/viruses [37]. Thapsigargin, a well-know ER stressor [34], was used as positive control for PERK activation. Differently from thapsigargin, PIC did not induce PERK phosphorylation, while it clearly induced eIF2α phosphorylation (Supplemental Figures S8A and B), indicating that another upstream kinase is responsible for this effect. We thus evaluated the role of PKR in this process by using a specific siRNA against PKR which inhibited PKR protein expression by >80% (Figure 6A). Knockdown of PKR almost completely prevented PIC-induced eIF2α phosphorylation and the dsRNA-induced Mcl-1 decrease (Figures 6B and C); there was even an increase in Mcl-1 expression under this condition. In line with the observed increase in Mcl-1 protein expression, silencing of PKR reduced caspases 9 and 3 cleavage (Figure 6D) and PIC-induced apoptosis (Figure 6E). Similar results were obtained with a second siRNA against PKR (data not shown), confirming the key role for PKR in dsRNA-induced Mcl-1 decrease and consequent beta cell apoptosis.

Discussion
We presently show that intracellular dsRNA activates the intrinsic pathway of apoptosis in pancreatic beta cells. Triggering of apoptosis by dsRNA is secondary to an early and sustained downregulation of Mcl-1 protein expression (Figure 7), resulting in the release of the proapoptotic protein Bim. The unbound Bim then activates BAX translocation to the mitochondria, mitochondrial permeabilization, cytochrome c release, caspases 9 and 3 activation and beta cell apoptosis. Key results obtained with dsRNA, an intermediary product of viral replication, where partially confirmed in the context of a beta cell infection by the diabetogenic enterovirus CVB5.
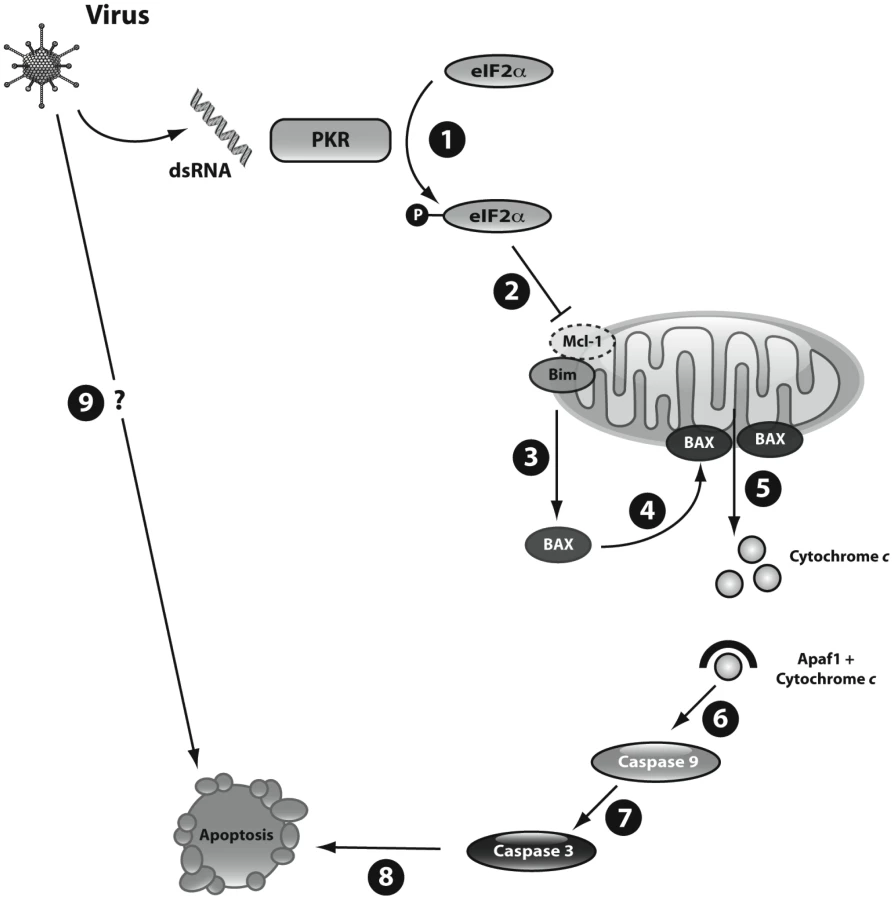
Apoptosis is the main form of cell death in T1D [38]. Several virus-induced human diseases are associated with increased apoptosis in the target cells, including HIV1-associated dementia [39], cytomegalovirus encephalitis [40] and viral myocarditis [41]. Coxsakieviruses can induce beta cell death by different mechanisms, depending on the strain and multiplicity of infection (MOI) used [19]. At higher MOIs (>100) necrosis is the preferential mechanism of cell death, but at lower MOIs a shift towards apoptosis is observed [19]. Viral triggering of apoptosis depends on both the host and the virus [42]. The host regulates viral-induced apoptosis trough the local production of pro-inflammatory cytokines and chemokines [42]. Viral factors leading to apoptosis include dsRNA [43]. This viral nucleic acid is recognized by receptors such as the toll-like receptor 3 (TLR3), the kinase PKR, the helicases melanoma differentiation-associated gene 5 (MDA5), (which is also a candidate gene for T1D [44]) and retinoic-acid-inducible protein 1 (RIG-I); these receptors activate genes involved in both antiviral responses and apoptosis [45]. All these receptors are expressed in pancreatic beta cells [4], [18]. The pathways downstream of viral recognition/signaling leading to beta cell apoptosis, however, remained to be clarified. The present observations provide a coherent hypothesis for the mechanisms triggering beta cell apoptosis following a viral infection (Figure 7).
Mcl-1 is a pro-survival Bcl-2 family member with a short half-life (30–180 min), which makes it specially susceptible to changes in protein translation [35]. Mcl-1 is degraded in the proteasome [26], and use of a proteasome inhibitor prevented, at least in part, dsRNA-induced decrease in Mcl-1 expression (present findings). We also observed that intracellular dsRNA induces an early and sustained phosphorylation of eIF2α which, as previously shown by our group, leads to a decrease in total protein translation [18]. This eIF2α phosphorylation is mediated by PKR, a protein kinase mainly activated by dsRNAs produced during viral replication [37], but not by PERK (present data). Protein translation inhibition contributes to an early and progressive decrease in Mcl-1 protein levels, which is reverted by knocking down PKR. PKR silencing actually increases Mcl-1 protein expression, probably due to the “release” of protein translation combined with an increase in Mcl-1 mRNA induced by dsRNA (Figure 2B). PKR silencing also prevents dsRNA-induced apoptosis, at least partially due to this increase in Mcl-1 protein. Mcl-1 protein stability is partly regulated by multiple sites of phosphorylation, and JNK-mediated phosphorylation of Mcl-1 increases the rate of protein degradation [35]. We recently described that JNK activation contributes to Mcl-1 degradation in beta cells exposed to the cytokines interleukin-1β + interferon-γ [20]. Here, we observed that dsRNA induces JNK phosphorylation in beta cells, but JNK inhibition does not prevent Mcl-1 degradation. These findings indicate that dsRNA mainly regulates Mcl-1 protein expression via inhibition of protein translation.
Mcl-1 functions as a prosurvivor factor by neutralizing specific propapoptotic BH3-only proteins; Bim has been described as a preferential target of Mcl-1 in other cell types [32]. Bim is a BH3-only protein that presents features of both a “sensitizer”, i.e. it binds to antiapoptotic Bcl-2 protein and displaces the effectors BAX and BAK [32], and also as an “activator”; i.e. through directly binding to BAX and BAK it promotes their activation and the induction of the mitochondrial apoptosis pathway [46]. An imbalance between Mcl-1 and Bim has been shown to trigger apoptosis in other cell types in the context of cytokine deprivation [47] and granzyme B [48] activation. We presently observed that Bim is neutralized by Mcl-1 under basal condition. After dsRNA exposure or the use of specific siRNAs against Mcl-1, however, there is an increase in “free” Bim that subsequently activates BAX and triggers apoptosis (Figure 7). This is suggested by the observation that a double knockdown of Bim plus Mcl-1 (Figure 3) reverts the proapoptotic effects of Mcl-1 silencing. Bim can also mediate the pro-apoptotic effects of type I interferons produced during viral infections [49] and it is targeted by viruses to evade apoptosis in the host cells [50]. Type I interferons were induced during both dsRNA and CVB5 exposure (Supplemental Figure S6). Importantly, inhibition of Bim by specific siRNAs prevented caspases 9/3 activation and apoptosis in beta cells infected with the diabetogenic virus CVB5, confirming the key role of Bim during viral infection of beta cells.
Apoptosis is one of the mechanisms by which the host eliminates virus-infected cells and blocks viral spread [51]. In postmitotic and poorly proliferating cells such as neurons and beta cells, apoptosis might also promote functional loss and disease. Indeed, studies in viral-induced neuronal disease demonstrate that inhibition of apoptosis may reduce disease severity without changes in virus titers [52], [53]. In line with these findings, apoptosis prevention in beta cells by knocking down Bim did not modify viral replication in vitro.
In conclusion, we presently show that decreased expression of the anti-apoptotic protein Mcl-1, coupled to activation of the pro-apoptotic protein Bim, contributes to beta cell apoptosis during in vitro exposure to the viral mimetic dsRNA. In case these observations can be confirmed in future in vivo experiments, novel strategies to increase Mcl-1 or decrease Bim expression in pancreatic beta cells [24] may turn to be interesting approaches to protect beta cells during infection by putative “diabetogenic” viruses.
Methods
Ethics statement
This study was carried out in strict accordance with the recommendations in the Belgian Regulations for Animal Care guidelines. The protocol was approved by the Commission d'Ethique du Bien-Être Animal (CEBEA) on the Ethics of Animal Experiments of the Université Libre de Bruxelles (Permit Number: LA 1230351). All procedures was performed under Isoflurane anesthesia, and all efforts were made to minimize suffering.
Culture of INS-1E cells and primary rat beta cells
The rat pancreatic beta cell line INS-1E (passages 57–75; a kind gift from Dr. C. Wollheim, Centre Medical Universitaire, Geneva, Switzerland) was cultured in medium containing RPMI 1640 GlutaMAX-I and 5% heat-inactivated fetal bovine serum (FBS) [54].
Male Wistar rats (Charles River Laboratories, Brussels, Belgium) were housed and used according to the Belgian Regulations for Animal Care guidelines. Rat islets were isolated by collagenase digestion of the pancreases. In order to obtain purified beta cells, the islets were dispersed and submitted to autofluorescence-activated cell sorting (FACS Aria, BD Bioscience, San Jose, USA) as previously described [33], [55]. The preparations used in the present study contained 93±2% of beta cells (n = 10). FACS-purified beta cells were cultured in Ham's F-10 medium containing 10 mM glucose, 5% FBS, 0.5% charcoal-absorbed bovine serum albumin (BSA Fraction V, Boehringer, Indianapolis, USA) [33]. During dsRNA and siRNAs transfection cells were cultured in medium without antibiotics and BSA.
RNA interference
The sequences of the siRNAs used are provide in Supplemental Table S1. As control we used Allstars Negative Control siRNA (Qiagen, Venlo, Netherlands). We have previously shown that this negative control siRNA does not affect beta cell gene expression or insulin release as compared to non-transfected cells [20], [56]. The optimal conditions for siRNAs transfection were determined by using a FITC-coupled siRNA (Thermo Scientific) and functional/viability tests [57]. Cells were transfected overnight with 30 nM of siRNAs using Lipofectamine 2000 or RNAiMAX (Invitrogen, CA, USA) according to the manufacturer's instructions [56]. After a recovery period of 24 to 48 h, cells were treated with a synthetic dsRNA or infected by an enterovirus.
Double-stranded RNA transfection, JNK and proteasome inhibition
The synthetic dsRNA polyinosinic-polycytidylic acid (PIC; Sigma, St. Louis, USA) was used at the concentration of 1 µg/ml [18]. All experiments were made with intracellular PIC, achieved via cell transfection under the same conditions as for siRNAs. Cells exposed to the transfectant alone were used as Control. Since cytoplasmic helicases can recognize dsRNA molecules based on their length [58], a PIC formulation with broad lengths of dsRNAs (10–10,000 bp) was used for the experiments.
The JNK inhibitor SP600125, the ER stressor thapsigargin (Sigma) and the proteasome inhibitor MG-132 were used at concentration of 10 µM, 1 µM and 1 μM respectively [20], [34]. SP600125 was added to the cell medium 4h before PIC transfection and maintained during the whole period of PIC exposure.
Coxsackievirus infection
The prototype strain of enterovirus (CVB5/Faulkner) was obtained from American Type Culture Collection (Manassas, VA). This virus was passaged in Green Monkey Kidney cells. The identity of the enterovirus preparations used was confirmed using a plaque neutralization assay with type-specific antisera [12].
Cells were infected with different multiplicity of infection (MOIs) of the virus preparation diluted in Hanks' Balanced Salt Solution (HBSS, Invitrogen). After adsorption for 1h at 37°C, the inoculum virus was removed, and the cells were washed twice with HBSS. Culture medium was then added to the plates and the virus was allowed to replicate for the indicated periods.
Infection with adenoviral vectors
INS-1E cells and primary FACS-purified rat beta cells were infected for 2 h at different MOIs with an adenovirus encoding Luciferase (Ad-Luc), or with an adenovirus encoding rat Mcl-1 (Vector Biolabs, Philadelphia, PA, USA) [20]. After 48 h of recovery cells were exposed to intracellular PIC for 24h.
Assessment of cell viability
Cells were incubated for 15 minutes with the DNA-binding dyes Propidium Iodide (PI, 5 µg/ml, Sigma) and Hoechst 33342 (HO, 5 µg/ml, Sigma). Subsequently, two independent observers determined the percentage of viable, apoptotic and necrotic cells. One of the observers was unaware of sample identity and the agreement between the results obtained was >90%. At least 500 cells were counted in each experimental condition. Results are presented as percentage of apoptosis, calculated as number of apoptotic cells/total number of cells x 100. This method is quantitative and has been validated for use in primary beta cells and INS-1E cells by systematic comparison with electron microscopy, caspase 3 activation and DNA laddering [33], [34], [57], [59], [60]. The apoptotic index was calculated against the percentage of apoptotic cells in the untreated condition as previously described [27]. In selected experiments, apoptosis was confirmed by analysis of activated (cleaved) caspase 3 and 9, cytoplasmic cytochrome c release and BAX translocation to the mitochondria (see below).
Assessment of cytochrome c release
Cells were harvested in cold PBS, centrifuged (500 g for 2 min), resuspended with 50 µl lysis buffer (75 mM NaCl, 1 mM NaH2PO4, 8 mM Na2PO4, 250 mM sucrose, 21 µg/µl aprotinin, 1 mM PMSF and 0.8 µg/µl digitonin) and vortexed for 30 s. Following centrifugation at 20,000 g for 1 min the supernatant was retrieved as the cytoplasmic fraction. The remaining pellet was resuspended in 50 µl lysis buffer containing a higher digitonin concentration (8 µg/µl), centrifuged at 20,000 g for 1 min and the supernatant retrieved as the mitochondrial fraction. Equal amounts of proteins were then resolved by 14% SDS-PAGE gel [22]. In addition to anti-cytochrome c (BD Biosciences), the immunoblots were probed with antibodies against β-actin and apoptosis-inducing factor (AIF) (Cell Signaling, Danvers, USA) corresponding to a cytoplasmic and a mitochondrial control, respectively.
mRNA extraction and real time PCR
mRNA was obtained using the Dynabeads mRNA DIRECT kit (Invitrogen Dynal, Oslo, Norway), and reverse transcribed to obtain the complementary DNA [33]. This material was analyzed by real time PCR reaction using SYBR Green fluorescence on a LightCycler instrument (Roche, Manheim, Germany) and correlated to a standard curve. Expression of the gene of interest was then corrected for the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The values are normalized by the highest value of each experiment considered as 1. GAPDH was chosen as housekeeping gene because its expression is not modified by PIC treatment in insulin producing cells [33], [61]. Primers sequences are described in Supplemental Table S2.
Western blot analysis
Cells were washed with cold PBS and lysed with either Laemmli buffer or Phospho lysis buffer [4]. Equal amounts of proteins were then resolved by 10–14% SDS-PAGE gel and transferred to a nitrocellulose membrane. Immunoblot analysis was performed with antibodies targeting Mcl-1 (Biovision, CA, USA), PKR, total eIF2α (Santa Cruz Biotechnology, CA, USA), phospho-JNK, cleaved caspases 3 and 9, phospho-eIF2α, phospho-PERK, total PERK, Bcl-XL, Bcl-2, Bim (Cell Signaling), enterovirus-specific polyclonal rabbit antiserum (1/600; KTL-510) [12] or α-tubulin (Sigma), used as housekeeping protein. Horseradish peroxidase-conjugated donkey anti-rabbit or anti-mouse IgG were used as secondary antibodies (Lucron Bioproducts, De Pinte; Belgium). Immunoreactive bands were revealed using a chemiluminescent substrate (Thermo Scientific) and detected by a LAS-3000 CCD camera (Fujifilm, Tokyo, Japan). The densitometry of the bands was evaluated using the Aida Analysis software (Raytest, Straubenhardt, Germany).
Immunoprecipitation
For the immunoprecipitation, INS-1E cells were lysed on ice using immunoprecipitation buffer [31]. Total proteins were quantified and used as the starting material for immunoprecipitations. Equal amounts of proteins were incubated with rabbit anti-BIM antibody overnight at 4°C with gentle rocking. Antibody-protein complexes were retrieved with a 50% protein A-agarose slurry (Santa Cruz Biotechnology), washed with immunoprecipitation buffer, resuspended in SDS sample buffer and then boiled to separate antibody-protein complex from the protein A. Samples were subjected to 14% SDS-PAGE gel and immunoblotted with anti-α-tubulin (Sigma), anti-Mcl-1 (Biovision) or anti-BIM antibody (Cell Signaling).
Immunofluorescence
Beta cells were plated on polylysine-coated glass culture slides (BD Biosciences). Cells were fixed for 15 min in 4% paraformaldehyde, washed with PBS and permeabilized for 5 min in Triton X-100 0.1%. Slides were then blocked using goat serum 5% and incubated overnight at 4°C in the presence of rabbit anti-Bax (1/1000; Santa Cruz Biotechnology) plus mouse anti-ATP Synthase β (mitochondrial marker) (1/2000; BD Biosciences) or an enterovirus-specific rabbit antiserum (1/600; KTL-510). Cells were washed next morning with PBS and incubated for 1 h with the appropriate Alexa fluor 488 or 555-conjugated antibodies (1/1000; Invitrogen). After, cells were stained with Hoechst, mounted and photographed using fluorescence microscopy (Axio Imager, Carl Zeiss, Zaventem, Belgium) [20].
Statistical analysis
Data are presented as mean ± SEM. Comparisons were performed by two-tailed paired Student's t-test or by ANOVA followed by Student's t test with Bonferroni correction, as adequate. A P value < 0.05 was considered as statistically significant.
Gene IDs
Numbers were taken from GenBank at Pubmed: myeloid cell leukemia sequence 1 (Mcl-1) - 60430; BCL2-like 11 (Bim) - 64547; B-cell lymphoma 2 (Bcl-2) - 24224; Bcl-2 extra-large (Bcl-XL) - 24888; dsRNA-dependent protein kinase (PKR) - 54287; p53 Up-regulated Modulator of Apoptosis (PUMA) - 317673; Death Protein-5 (DP5) - 117271; c-jun NH2-terminal kinase (JNK) - 116554; eukaryotic translation initiation factor 2A (eIF2α) - 502531; PRK-like endoplasmic reticulum kinase (PERK) - 29702.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. EizirikDLColliMLOrtisF 2009 The role of inflammation in insulitis and beta cell loss in type 1 diabetes. Nat Rev Endocrinol 5 219 226
2. TaoBPietropaoloMAtkinsonMSchatzDTaylorD 2010 Estimating the cost of type 1 diabetes in the U.S.: a propensity score matching method. PLoS One 5 e11501
3. ConcannonPRichSSNepomGT 2009 Genetics of type 1A diabetes. N Engl J Med 360 1646 1654
4. ColliMLMooreFGurzovENOrtisFEizirikDL 2010 MDA5 and PTPN2, two candidate genes for type 1 diabetes, modify pancreatic beta cell responses to the viral by-product double-stranded RNA. Hum Mol Genet 19 135 146
5. PattersonCCDahlquistGGGyurusEGreenASolteszG 2009 Incidence trends for childhood type 1 diabetes in Europe during 1989-2003 and predicted new cases 2005-20: a multicentre prospective registration study. Lancet 373 2027 2033
6. RoivainenMKlingelK 2010 Virus infections and type 1 diabetes risk. Curr Diab Rep 10 350 356
7. HyotyHHiltunenMKnipMLaakkonenMVahasaloP 1995 A prospective study of the role of coxsackie B and other enterovirus infections in the pathogenesis of IDDM. Childhood Diabetes in Finland (DiMe) Study Group. Diabetes 44 652 657
8. HorwitzMSBradleyLMHarbertsonJKrahlTLeeJ 1998 Diabetes induced by Coxsackie virus: initiation by bystander damage and not molecular mimicry. Nat Med 4 781 785
9. ChehadehWWeillJVantyghemMCAlmGLefebvreJ 2000 Increased level of interferon-α in blood of patients with insulin-dependent diabetes mellitus: relationship with coxsackievirus B infection. J Infect Dis 181 1929 1939
10. DottaFCensiniSvan HalterenAGMarselliLMasiniM 2007 Coxsackie B4 virus infection of beta cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proc Natl Acad Sci U S A 104 5115 5120
11. YoonJWAustinMOnoderaTNotkinsAL 1979 Isolation of a virus from the pancreas of a child with diabetic ketoacidosis. N Engl J Med 300 1173 1179
12. RoivainenMRasilainenSYlipaastoPNissinenRUstinovJ 2000 Mechanisms of coxsackievirus-induced damage to human pancreatic beta cells. J Clin Endocrinol Metab 85 432 440
13. WagenknechtLERosemanJMHermanWH 1991 Increased incidence of insulin-dependent diabetes mellitus following an epidemic of Coxsackievirus B5. Am J Epidemiol 133 1024 1031
14. HoviTStenvikMRosenlewM 1996 Relative abundance of enterovirus serotypes in sewage differs from that in patients: clinical and epidemiological implications. Epidemiol Infect 116 91 97
15. HorwitzMSIlicAFineCBalasaBSarvetnickN 2004 Coxsackieviral-mediated diabetes: induction requires antigen-presenting cells and is accompanied by phagocytosis of beta cells. Clin Immunol 110 134 144
16. SchulteBMKramerMAnsemsMLankeKHvan DoremalenN 2010 Phagocytosis of enterovirus-infected pancreatic beta cells triggers innate immune responses in human dendritic cells. Diabetes 59 1182 1191
17. DevendraDJasinskiJMelanitouENakayamaMLiM 2005 Interferon-α as a mediator of polyinosinic:polycytidylic acid-induced type 1 diabetes. Diabetes 54 2549 2556
18. DogusanZGarciaMFlamezDAlexopoulouLGoldmanM 2008 Double-stranded RNA induces pancreatic beta cell apoptosis by activation of the TLR3 and IRF-3 pathways. Diabetes 57 1236 1245
19. RasilainenSYlipaastoPRoivainenMLapattoRHoviT 2004 Mechanisms of coxsackievirus B5 mediated beta cell death depend on the multiplicity of infection. J Med Virol 72 586 596
20. AllagnatFCunhaDMooreFVanderwindenJMEizirikDL 2011 Mcl-1 downregulation by pro-inflammatory cytokines and palmitate is an early event contributing to beta cell apoptosis. Cell Death Differ 18 328 337
21. GurzovENOrtisFCunhaDAGossetGLiM 2009 Signaling by IL-1β+IFN-γ and ER stress converge on DP5/Hrk activation: a novel mechanism for pancreatic beta-cell apoptosis. Cell Death Differ 11 1539 1550
22. CunhaDALadriereLOrtisFIgoillo-EsteveMGurzovEN 2009 Glucagon-like peptide-1 agonists protect pancreatic beta cells from lipotoxic endoplasmic reticulum stress through upregulation of BiP and JunB. Diabetes 58 2851 2862
23. GrunnetLGAikinRTonnesenMFParaskevasSBlaabjergL 2009 Proinflammatory cytokines activate the intrinsic apoptotic pathway in beta cells. Diabetes 58 1807 1815
24. GurzovENEizirikDL 2011 Bcl-2 proteins in diabetes: mitochondrial pathways of beta cell death and dysfunction. Trends Cell Biol 21 424 431
25. TaitSWGreenDR 2010 Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol 11 621 632
26. Yang-YenHF 2006 Mcl-1: a highly regulated cell death and survival controller. J Biomed Sci 13 201 204
27. CnopMHannaertJCHoorensAEizirikDLPipeleersDG 2001 Inverse relationship between cytotoxicity of free fatty acids in pancreatic islet cells and cellular triglyceride accumulation. Diabetes 50 1771 1777
28. McKenzieMDJamiesonEJansenESScottCLHuangDC 2010 Glucose induces pancreatic islet cell apoptosis that requires the BH3-only proteins Bim and Puma and multi-BH domain protein Bax. Diabetes 59 644 652
29. ChenDWangMZhouSZhouQ 2002 HIV-1 Tat targets microtubules to induce apoptosis, a process promoted by the pro-apoptotic Bcl-2 relative Bim. EMBO J 21 6801 6810
30. KutukOLetaiA 2010 Displacement of Bim by Bmf and Puma rather than increase in Bim level mediates paclitaxel-induced apoptosis in breast cancer cells. Cell Death Differ 17 1624 1635
31. GurzovENGermanoCMCunhaDAOrtisFVanderwindenJM 2010 p53 up-regulated modulator of apoptosis (PUMA) activation contributes to pancreatic beta cell apoptosis induced by proinflammatory cytokines and endoplasmic reticulum stress. J Biol Chem 285 19910 19920
32. ChenLWillisSNWeiASmithBJFletcherJI 2005 Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell 17 393 403
33. RasschaertJLadriereLUrbainMDogusanZKatabuaB 2005 Toll-like receptor 3 and STAT-1 contribute to double-stranded RNA+ interferon-γ-induced apoptosis in primary pancreatic beta cells. J Biol Chem 280 33984 33991
34. CunhaDAHekermanPLadriereLBazarra-CastroAOrtisF 2008 Initiation and execution of lipotoxic ER stress in pancreatic beta cells. J Cell Sci 121 2308 2318
35. ThomasLWLamCEdwardsSW 2010 Mcl-1; the molecular regulation of protein function. FEBS Lett 584 2981 2989
36. EizirikDLCardozoAKCnopM 2008 The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev 29 42 61
37. GarciaMAGilJVentosoIGuerraSDomingoE 2006 Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol Mol Biol Rev 70 1032 1060
38. EizirikDLMandrup-PoulsenT 2001 A choice of death-the signal-transduction of immune-mediated beta cell apoptosis. Diabetologia 44 2115 2133
39. LiWGaleyDMattsonMPNathA 2005 Molecular and cellular mechanisms of neuronal cell death in HIV dementia. Neurotox Res 8 119 134
40. DeBiasiRLKleinschmidt-DeMastersBKRichardson-BurnsSTylerKL 2002 Central nervous system apoptosis in human herpes simplex virus and cytomegalovirus encephalitis. J Infect Dis 186 1547 1557
41. AlterPJobmannMMeyerEPankuweitSMaischB 2001 Apoptosis in myocarditis and dilated cardiomyopathy: does enterovirus genome persistence protect from apoptosis? An endomyocardial biopsy study. Cardiovasc Pathol 10 229 234
42. Baca JonesCvon HerrathM 2011 Pathology and pathogenesis of virus infections. KaufmanSHERouseBTSacksDL The Immune Response to Infection First ed. Washington ASM Press 383 389
43. KiblerKVShorsTPerkinsKBZemanCCBanaszakMP 1997 Double-stranded RNA is a trigger for apoptosis in vaccinia virus-infected cells. J Virol 71 1992 2003
44. SmythDJCooperJDBaileyRFieldSBurrenO 2006 A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat Genet 38 617 619
45. AkiraSUematsuSTakeuchiO 2006 Pathogen recognition and innate immunity. Cell 124 783 801
46. GavathiotisESuzukiMDavisMLPitterKBirdGH 2008 BAX activation is initiated at a novel interaction site. Nature 455 1076 1081
47. Gomez-BougiePBatailleRAmiotM 2004 The imbalance between Bim and Mcl-1 expression controls the survival of human myeloma cells. Eur J Immunol 34 3156 3164
48. HanJGoldsteinLAGastmanBRRabinovitzARabinowichH 2005 Disruption of Mcl-1.Bim complex in granzyme B-mediated mitochondrial apoptosis. J Biol Chem 280 16383 16392
49. BahlKHuebnerADavisRJWelshRM 2010 Analysis of apoptosis of memory T cells and dendritic cells during the early stages of viral infection or exposure to toll-like receptor agonists. J Virol 84 4866 4877
50. TaylorJMQuiltyDBanadygaLBarryM 2006 The vaccinia virus protein F1L interacts with Bim and inhibits activation of the pro-apoptotic protein Bax. J Biol Chem 281 39728 39739
51. ClarkePTylerKL 2009 Apoptosis in animal models of virus-induced disease. Nat Rev Microbiol 7 144 155
52. BeckhamJDGoodyRJClarkePBonnyCTylerKL 2007 Novel strategy for treatment of viral central nervous system infection by using a cell-permeating inhibitor of c-Jun N-terminal kinase. J Virol 81 6984 6992
53. SamuelMAMorreyJDDiamondMS 2007 Caspase 3-dependent cell death of neurons contributes to the pathogenesis of West Nile virus encephalitis. J Virol 81 2614 2623
54. AsfariMJanjicDMedaPLiGHalbanPA 1992 Establishment of 2-mercaptoethanol-dependent differentiated insulin-secreting cell lines. Endocrinology 130 167 178
55. PipeleersDGin't VeldPAVan de WinkelMMaesESchuitFC 1985 A new in vitro model for the study of pancreatic α and β cells. Endocrinology 117 806 816
56. MooreFNaamaneNColliMLBouckenoogheTOrtisF 2011 STAT1 is a master regulator of pancreatic beta cell apoptosis and islet inflammation. J Biol Chem 286 929 941
57. MooreFColliMLCnopMEsteveMICardozoAK 2009 PTPN2, a candidate gene for type 1 diabetes, modulates interferon-γ-induced pancreatic beta cell apoptosis. Diabetes 58 1283 1291
58. KatoHTakeuchiOMikamo-SatohEHiraiRKawaiT 2008 Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med 205 1601 1610
59. KutluBCardozoAKDarvilleMIKruhofferMMagnussonN 2003 Discovery of gene networks regulating cytokine-induced dysfunction and apoptosis in insulin-producing INS-1 cells. Diabetes 52 2701 2719
60. HoorensAVan de CasteeleMKloppelGPipeleersD 1996 Glucose promotes survival of rat pancreatic beta cells by activating synthesis of proteins which suppress a constitutive apoptotic program. J Clin Invest 98 1568 1574
61. LiuDDarvilleMEizirikDL 2001 Double-stranded ribonucleic acid (RNA) induces beta cell Fas messenger RNA expression and increases cytokine-induced beta cell apoptosis. Endocrinology 142 2593 2599
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 9
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- HTLV-1 Propels Thymic Human T Cell Development in “Human Immune System” Rag2 gamma c Mice
- Hostile Takeover by : Reorganization of Parasite and Host Cell Membranes during Liver Stage Egress
- Exploiting and Subverting Tor Signaling in the Pathogenesis of Fungi, Parasites, and Viruses
- A Viral Ubiquitin Ligase Has Substrate Preferential SUMO Targeted Ubiquitin Ligase Activity that Counteracts Intrinsic Antiviral Defence
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy