
Variations in TcdB Activity and the Hypervirulence of Emerging Strains of
Hypervirulent strains of Clostridium difficile have emerged over the past decade, increasing the morbidity and mortality of patients infected by this opportunistic pathogen. Recent work suggested the major C. difficile virulence factor, TcdB, from hypervirulent strains (TcdBHV) was more cytotoxic in vitro than TcdB from historical strains (TcdBHIST). The current study investigated the in vivo impact of altered TcdB tropism, and the underlying mechanism responsible for the differences in activity between the two forms of this toxin. A combination of protein sequence analyses, in vivo studies using a Danio rerio model system, and cell entry combined with fluorescence assays were used to define the critical differences between TcdBHV and TcdBHIST. Sequence analysis found that TcdB was the most variable protein expressed from the pathogenicity locus of C. difficile. In line with these sequence differences, the in vivo effects of TcdBHV were found to be substantially broader and more pronounced than those caused by TcdBHIST. The increased toxicity of TcdBHV was related to the toxin's ability to enter cells more rapidly and at an earlier stage in endocytosis than TcdBHIST. The underlying biochemical mechanism for more rapid cell entry was identified in experiments demonstrating that TcdBHV undergoes acid-induced conformational changes at a pH much higher than that of TcdBHIST. Such pH-related conformational changes are known to be the inciting step in membrane insertion and translocation for TcdB. These data provide insight into a critical change in TcdB activity that contributes to the emerging hypervirulence of C. difficile.
Published in the journal:
. PLoS Pathog 6(8): e32767. doi:10.1371/journal.ppat.1001061
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1001061
Summary
Hypervirulent strains of Clostridium difficile have emerged over the past decade, increasing the morbidity and mortality of patients infected by this opportunistic pathogen. Recent work suggested the major C. difficile virulence factor, TcdB, from hypervirulent strains (TcdBHV) was more cytotoxic in vitro than TcdB from historical strains (TcdBHIST). The current study investigated the in vivo impact of altered TcdB tropism, and the underlying mechanism responsible for the differences in activity between the two forms of this toxin. A combination of protein sequence analyses, in vivo studies using a Danio rerio model system, and cell entry combined with fluorescence assays were used to define the critical differences between TcdBHV and TcdBHIST. Sequence analysis found that TcdB was the most variable protein expressed from the pathogenicity locus of C. difficile. In line with these sequence differences, the in vivo effects of TcdBHV were found to be substantially broader and more pronounced than those caused by TcdBHIST. The increased toxicity of TcdBHV was related to the toxin's ability to enter cells more rapidly and at an earlier stage in endocytosis than TcdBHIST. The underlying biochemical mechanism for more rapid cell entry was identified in experiments demonstrating that TcdBHV undergoes acid-induced conformational changes at a pH much higher than that of TcdBHIST. Such pH-related conformational changes are known to be the inciting step in membrane insertion and translocation for TcdB. These data provide insight into a critical change in TcdB activity that contributes to the emerging hypervirulence of C. difficile.
Introduction
Clostridium difficile is a gram-positive, spore-forming anaerobe, first described by Hall and O'Toole over 75 years ago [1]; however, the organism was not associated with human disease until 1978 [2], [3]. Over the past three decades C. difficile has become a major nosocomial pathogen and is the leading cause of diarrhea in hospitalized patients [4]. C. difficile associated disease (CDAD) is routinely treated by supportive therapy and regimens of vancomycin and metronidazole, but treatment of CDAD has become more difficult due to the emergence of hypervirulent (NAP1/BI/027) strains of C. difficile [5], [6], [7]. Elucidating the major differences between historical strains of C. difficile and the NAP1/BI/027-related strains of C. difficile is critical to understanding how this serious human pathogen continues to emerge.
The phenotypes of hypervirulent and historical strains of C. difficile are different [7], [8], [9]. C. difficile NAP1/BI/027 produces more toxin and sporulates with higher efficiency than historical strains [6], [7], [8], [9], [10]. NAP1/BI/027 strains also produce a binary toxin, CDT, which is thought to enhance colonization of C. difficile by triggering the formation of microtubule protrusions on cells of the gastrointestinal epithelium [11], [12], [13]. Finally, C. difficile NAP1/BI/027 strains are resistant to fluoroquinolones due to mutations in DNA gyrase genes [7], [14], [15], [16]. The extent to which one or more of these differences between the two strains contributes to hypervirulence has not been determined.
Recent work from Stabler and colleagues identified several genetic variations between epidemic and historical strains of C. difficile [17]. For example, the historical C. difficile strain, 630, was found to contain 505 unique coding sequences compared to hypervirulent strains. This analysis also identified differences in flagellar genes, metabolic genes, phage islands, and transcriptional regulators. Of interest to our work was the finding that TcdB from C. difficile hypervirulent strains had a greater cytopathic effect on a variety of cell types than TcdB isolated from a C. difficile historical strain. The steps in cellular intoxication that account for these differences in TcdB activity, and whether in vivo tropism varies between the historical and hypervirulent TcdB have not been reported.
TcdB (∼269 kDa) is a 2366 residue single polypeptide toxin encoded on a C. difficile pathogenicity locus (PaLoc) that also includes genes for two regulators (TcdC and TcdR) of toxin expression, a putative holin (TcdE), and TcdA [18], [19]. TcdB has at least four functional domains that contribute to cell entry and glucosylation of small-GTPases within the cytosol of the cell [20]. TcdB's glucosyltransferase domain is included in the first 516 residues of the toxin, which also includes a conserved DXD motif (Asp286/Asp288) and Trp102, which form a complex with Mn2+ and UDP-Glucose [21], [22], [23], [24], [25]. A substrate recognition domain is located between residues 365–516 [26]. The cysteine protease domain at residues 544–955 is necessary for autoproteolytic activity and delivery of the enzymatic domain into the cytosol [27], [28], [29]. A putative membrane-spanning domain resides between residues 956–1128, yet whether this domain is required for intoxication is not known. Finally, the fourth functional domain of TcdB is located within the carboxy-terminal region of the toxin, and is predicted to interact with receptors on target cells [30], [31], [32], [33].
Sequence variations in one or more of the functional domains of TcdB could account for the differences in cytotoxicity between historical and hypervirulent isolates. In the current work we test this hypothesis and demonstrate that TcdB from hypervirulent strains exhibits broader tropism in vivo. We also demonstrate TcdB from hypervirulent C. difficile undergoes hydrophobic conformational changes at a higher pH than toxin from the historical strain, and this correlates with more rapid cell entry. These findings provide insight into a possible mechanism through which hypervirulent C. difficile causes more severe illness than historical strains of this organism.
Results
Sequence comparison of the functional domains of TcdB from a historical strain (TcdBHIST) and TcdB from a hypervirulent strain (TcdBHV)
The carboxy-terminal sequence of TcdB varies between isolates of C. difficile, including hypervirulent and historical strains [17], [34]. Yet, whether sequence variations are more extensive in TcdB compared to other genes in the PaLoc or if the sequences outside of the carboxy-terminal domain of TcdB also varied among different strains of C. difficile has not been reported.
We compared the sequences of proteins encoded within the PaLoc of C. difficile 630 (a non-NAP1/BI/027 strain) and C. difficile R20291 (a 027 strain). The sequence of TcdR, a positive regulator of toxin expression was found to be 100% identical between the two strains of C. difficile. TcdE, the putative holin encoded in the middle of the PaLoc exhibited 99% identity and 100% similarity between the two strains of C. difficile. The enterotoxin, TcdA, exhibited 98% identity and 99% similarity between the two strains. The gene encoding TcdC from the hypervirulent strain encodes a stop codon and contains a deletion, which made it difficult to precisely compare this protein in the two strains. However, at the DNA level the gene was 95% homologous in the intact coding regions of tcdC. In contrast to these almost exact identities of TcdR, TcdE, and TcdA from the two strains, the amino-acid sequence of TcdB from the two strains was found to have the most variation with 92% identity and 96% similarity.
We next compared the functional regions of TcdBHIST and TcdBHV (Fig. 1). The enzymatic region of TcdB (encompassing residues 1–543) was found to be 96% identical and 98% similar between the two strains of C. difficile. Residues critical for catalytic activity, W102 and the DXD motif, did not vary between the two forms of TcdB (Fig. 1A). The substrate specificity domain of TcdB (residues 365 to 516) [26] exhibited 99% identity and 100% similarity (Fig. 1A). The autoproteolytic region (residues 544 to 955) was found to contain 96% identity and 98% similarity. Moreover, the reported catalytic triad (D587, H653, and C698) was conserved between the two forms of TcdB. Interestingly however, the analysis found a rearrangement of a second cysteine residue in this region of TcdB. TcdBHIST contains a cysteine at residue 870, but this residue is a tyrosine in TcdBHV (Fig. 1B). Conversely, TcdBHV has a cysteine residue at 1477, but this was found to be a glycine residue in TcdBHIST. The third putative functional domain of TcdB is between residues 956 and 1644, and encodes a hydrophobic region thought to mediate membrane insertion. Comparison of this region found 91% identity and 96% similarity (Fig. 1C).

In line with earlier reports [17], [34] the carboxy-terminal region, encompassing residues 1645 to 2366, exhibited the highest degree of sequence variation in the toxin. The carboxy-terminal region showed 88% identity and 95% similarity between the two forms of TcdB. The number of CROP regions is identical, with TcdBHIST and TcdBHV containing 24 regions based on the YF consensus motif [30], [32], [35], [36]. However, eight of these regions in TcdBHV were found to exhibit less than 80% sequence identity to TcdBHIST (Fig. 1D).
Fig. 1E shows an SDS-PAGE analysis of TcdBHIST and TcdBHV purified from wild-type strains of C. difficile as described in the materials and methods. Both forms of the toxin were obtained at greater than 95% purity based on minimal detection of contaminating proteins.
In vivo assessment of TcdBHIST and TcdBHV
We next used a zebrafish model to compare the in vivo effects of the two forms of this toxin. Our group has previously utilized the zebrafish embryo as a model to examine the effects of TcdBHIST in real time, and found that this toxin had potent cardiotoxic effects [37]. The zebrafish provides a distinct advantage for the purpose of examining tissue damage and tropism because it is possible to visualize these events directly with this model.
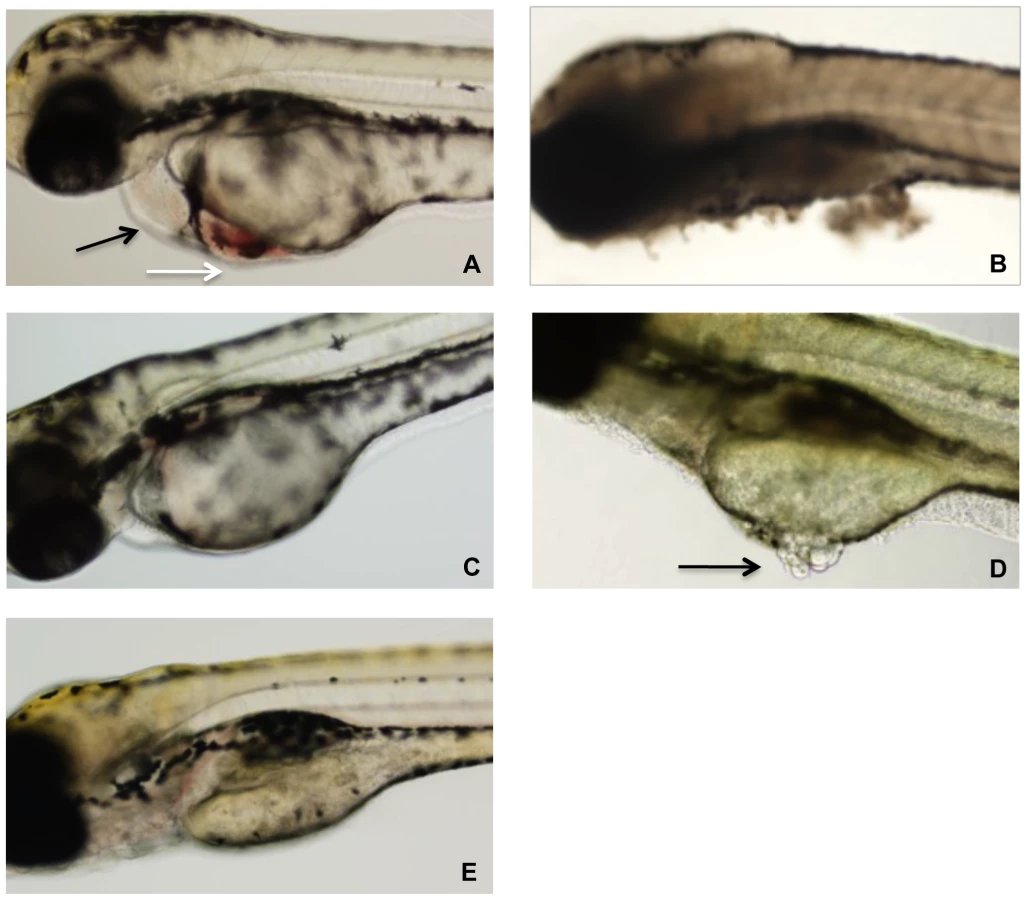
Zebrafish embryos were arrayed in a 48-well plate in embryo water and TcdBHIST or TcdBHV across a range of concentrations was applied to the individual wells. At 24 h following treatment, a minimum of 20 zebrafish larvae per condition were examined by light microscopy for physiological changes, tissue damage, and viability (Fig. 2). Extensive necrosis was evident in all embryos exposed to TcdBHV, with broad tissue damage caused to the yolk sac, body, and head at concentrations as low as 1 nM (Fig. 2B and 2D). Furthermore, all zebrafish treated with TcdBHV succumbed to the effects of the toxin within 48 h. In contrast, treatment with TcdBHIST resulted in more specific damage at the cardiac region in approximately 75% of embryos, and was not immediately lethal (Fig. 2A). Incubation with higher doses of TcdBHIST or for longer periods of time increased toxicity but did not alter the physiological damage from this toxin. These findings indicate that TcdBHV impacts a broader number of cell types in vivo compared to TcdBHIST. However, corresponding to our previous report TcdBHIST preferentially targets cardiac cells in the zebrafish embryo system.
Recent studies determined the relative cytotoxicity of TcdBHV and TcdBHIST on eight different cell types [17]. Because this analysis did not include cells of cardiac lineage, we compared the two toxins on HL-1 cells, which are derived from mouse cardiac tissue [38]. We also examined the effects of the two toxins on CHO cells for a relative comparison to the cardiomyocytes. As shown in Fig. 3, similar to previous observations, TcdBHV was more cytotoxic to CHO cells (TCD50 2.37×10−13 M) than was TcdBHIST (TCD50 2.53×10−11 M). In contrast, TcdBHV was not more cytotoxic on cardiomyocytes and displayed a very similar activity to TcdBHIST. Upon further investigation of the cardiomyocytes, the cytotoxicity of TcdBHV was found to be slightly lower than TcdBHIST (p<0.05) with a TCD50 approximately 10-fold higher (3.37×10−10 M) than TcdBHIST (TCD50 2.80×10−11 M). These data indicate that while TcdBHV has a broader cell tropism and is most likely more cytotoxic overall, TcdBHIST cardiotropism is more pronounced between the two forms of this toxin.

Comparison of intracellular effects of TcdBHV and TcdBHIST
We next determined if the variation in cytotoxicity was due to differences in the cytosolic activities of the two forms of TcdB. As an approach to this problem we took advantage of a previously described system used for heterologous delivery of proteins and protein fragments into the cytosol of target cells [39], [40]. This system is composed of the cell entry components of anthrax lethal toxin. Briefly, protective antigen (PA) delivers lethal factor (LF) into the cytosol of mammalian cells. The heterologous delivery system is derived from the amino-terminus of LF (LFn), which interacts with PA and can be delivered into cells, but lacks enzymatic activity. In our experiments, the DNA fragment encoding the enzymatic domain of TcdB was genetically fused to lfn, yielding a DNA construct that expresses the cell entry portion of LF with the enzymatic component of TcdB. This heterologous delivery system allowed us to regulate the cell entry of the enzymatic component of TcdBHV and TcdBHIST so that these domains were identical in the way in which they entered the cell. We predicted that if the differences in cytotoxicity were due to factors other than intracellular activity of these forms of TcdB, then the fusions should exhibit identical cytotoxic effects.
The results of the PA, LFn-TcdB fusion experiments are shown in Fig. 4. CHO cells were treated with a fixed amount of PA (500 nM) plus a range of concentrations of LFnTcdBHV(enz) or LFnTcdBHIST(enz) in order to generate a standard killing curve for this assay. As controls, CHO cells were treated with PA, LFnTcdBHV(enz), or LFnTcdBHIST(enz) separately. Following 24 h of treatment the cells were assayed for viability using WST-8 colorimetric assay and the percent survival was plotted versus concentration of the fusion protein. Treatment with each of the components alone had no effect on cell viability in this assay (data not shown). Treatments with PA plus LFnTcdBHV(enz) or PA plus LFnTcdBHIST(enz) resulted in similar (p<0.05) cytotoxicity at each of the concentrations tested (Fig. 4). To confirm that PA was not limiting in these experiments, cytotoxicity of the fusions was tested with 10-fold higher amounts of PA, and this additional amount of PA did not change the level of cytotoxicity for either fusion (data not shown). The results from this experiment suggested that the differences in the cytotoxicity of LFnTcdBHV(enz) and LFnTcdBHIST(enz) were not due to variations in intracellular activities of the enzymatic domains.

Flow-cytometry analysis of TcdBHIST and TcdBHV interaction with CHO cells and cardiomyocytes
The results from the experiment using an identical method of cell entry, suggested the differences in cytotoxicity might be associated with early steps in cell binding and cell entry. To address this hypothesis, we compared the interaction of TcdBHV and TcdBHIST with cultured cells. Cultured cells were incubated with Alexa-647-labeled TcdBHV or Alexa-647-labeled TcdBHIST and the extent of toxin binding was determined by flow cytometry. This analysis was performed on CHO cell and HL-1 cardiomyocytes. As shown in Fig. 5, CHO cells and HL-1 cells exhibited a higher degree of fluorescence when incubated with labeled TcdBHIST than when incubated with labeled TcdBHV. A biphasic profile was detected in CHO cells with a smaller population of cells exhibiting a distinct, reduced, toxin-binding pattern. In contrast, binding to cardiomyocytes was uniform and revealed a profile expected for a single population of cells.

Experiments were next performed to determine the apparent Kd for binding of TcdBHIST and TcdBHV. Interestingly, within the constraints of these experimental conditions we were not able to achieve saturable binding of either form of the toxin to target cells. Fig. 5C shows a nearly linear correlation between the increase in toxin concentration and the mean fluorescence intensity (MFI) of HL-1 cells despite reaching toxin concentrations of over 300 nM. Additionally, Fig. 5C further emphasizes the extremely low level of interaction of TcdBHV with target cells in comparison to the high MFI achieved with TcdBHIST. These data suggest that cell binding involves a higher order and more complex process than expected for a single receptor-ligand interaction.
Rates of cell entry differ between TcdBHIST and TcdBHV
Experiments were next performed to assess the difference in the rates of cell entry between the two toxins. In previous work on historical TcdB, we found that lysosomotropic inhibitors could completely block cytopathic effects of the toxin for up to 16 h, even if added up to 20 min following exposure of the cells to the toxin [41]. These findings indicate interaction with the cell, uptake, and then translocation into the cytosol requires at least 20 min and acidification of endosomes is necessary. To determine if TcdBHV differed from TcdBHIST in rates of cell entry, cultured CHO cells were treated with the two forms of the toxin and a lysosomotropic agent was added to the cells at time-points ranging from 5 to 60 min following treatment with toxin. The lysosomotropic agent was also added prior to or at the same time cells were treated with the toxins. The effect of the lysosomotropic agent was then assessed by determining the level of cytopathic effects (CPE) either 2 h or 12 h after treatment with the toxin. For this experiment CPE was determined rather than cytotoxicity due to toxicity of ammonium chloride at the later time points necessary for cytotoxicity assays. As shown in Fig. 6, based on the extent of cell rounding, there appeared to be a clear difference in the rates of translocation between TcdBHV and TcdBHIST. Unlike our earlier findings on TcdBHIST, the cytotoxic effects of TcdBHV could not be prevented when the lysosomotropic agent was added as soon as 10 min following treatment with the toxin (Fig. 6A). Furthermore, addition of the lysosomotropic agent within 10 min of treatment of TcdBHV only provided a slight delay in CPE, as all inhibitor treated cells showed complete rounding by 12 h (Fig. 6B). In contrast, the CPE of TcdBHIST could be prevented by adding the inhibitor up to 30 min following treatment with the toxin. These findings indicate TcdBHV translocates to the cytosol more rapidly than TcdBHIST.

Hydrophobic transitions occur at a higher pH in TcdBHV
Previous studies from our group found that acidic pH triggers hydrophobic transitions in TcdBHIST [41]. Studies by Barth et al. found that this hydrophobic transition in TcdB correlated with membrane insertion by the toxin [42]. These conformational changes corresponded to the decrease in endosome pH that led to translocation of the toxin into the cytosol. Thus, it was reasonable to suspect that TcdBHV translocates more quickly into the cytosol because the hydrophobic transition was induced at a higher pH and thus at an earlier stage of endocytosis. To address this possibility, in the next series of experiments we identified the pH dependent conformational transitions of TcdBHV by observing changes in TNS fluorescence when the toxin was incubated at various pHs. To identify whether TcdBHV exhibits differential transitions compared to TcdBHIST, the proteins were preincubated with 150 µM TNS at pH 4.0, 5.0, 6.0, and 7.0, and then analyzed for changes in TNS fluorescence. As shown in Fig. 7, TcdBHV exhibited a significant increase in hydrophobicity at pH 5.0, while TcdBHIST did not undergo this transition until pH 4.0. Further examination of a narrower pH range revealed that a significant shift occurred between pH 5.4 and 5.6 in TcdBHV (Fig. 7D). In comparison, TNS fluorescence of TcdBHIST at these pHs was just above background levels.

These pH transitions were also studied using the inherent fluorescence of TcdBHIST and TcdBHV from the emission of tryptophan residues. Unfolding of the hydrophobic region should expose portions of the protein to a more aqueous environment, quenching tryptophan fluorescence. Environmental changes surrounding the tryptophan residues over a broad range of pH are shown in Fig. 8A and 8B. A gradual quenching of fluorescence was detected in TcdBHIST from pH 7 to pH 4, while the tryptophan emission spectra of TcdBHV indicated a sudden shift between pH 5 and pH 6. Fig. 8D reveals that this shift took place between pH 5.4 and 5.2, similar to the increase in TNS fluorescence seen at pH 5.4.

Discussion
In the current study we compared the sequences and activities of TcdB from hypervirulent and historical strains of C. difficile. Because TcdB has been shown to be the major virulence factor of C. difficile [43], we reasoned that changes in the activity of this toxin could have a profound impact on the severity of disease. The findings support this notion, as TcdBHV exhibited a broader tropism and higher potency than TcdBHIST. Among the possible explanations for this increased toxicity are the observations that TcdBHV enters cells more rapidly than TcdBHIST, and TcdBHV undergoes conformational changes at a higher pH than TcdBHIST.
Based on the sequence comparisons and the results of the experiments using the heterologous delivery system (Figs. 1 and 3), it appears that the differences in tropism and cytotoxicity are due to changes in regions outside of the enzymatic domain. Rapid cell entry could lead to more efficient cell killing by providing the toxin an endocytic condition in which the toxin is not subject to possible destruction by lysosomal proteases. The data from the lysosomotropic inhibitor assays (Fig. 6) support the idea that TcdBHV does not reside within the endosome as long as TcdBHIST. Among the possible reasons for more rapid cell entry is a differential sensitivity to levels of IP6 that trigger autoproteolytic processing associated with translocation. We also noted a difference in the sequence of the hydrophobic region of TcdB, and if, as has been proposed [41], [42], this region mediates membrane insertion, such differences could allow TcdBHV to insert into the membrane at an earlier stage of cell entry. We reasoned that if this possibility were true, there should be a difference in the pH-induced transitions of the two forms of TcdB, with the hydrophobic regions of TcdBHV becoming exposed at a pH higher than the pH necessary for triggering this transition in TcdBHIST. The results from the TNS experiments (Fig. 7) indicate that TcdBHV is able to undergo the hydrophobic transition at a higher pH than TcdBHIST, providing further evidence that TcdBHV has higher translocation efficiency than TcdBHIST. Studies looking at the environment surrounding tryptophan residues of TcdBHIST and TcdBHV at lower pH (Fig. 8) support the idea that TcdBHV undergoes a structural change at higher pH than TcdBHIST. Additionally, these experiments revealed that the transition of TcdBHIST occurs gradually, while TcdBHV demonstrates sudden shifts upon lowering the pH. This could be indicative of a more efficient unfolding of TcdBHV, which may contribute to an enhanced ability to traverse the endosomal membrane. Our current working model is that TcdBHV is able to translocate at an earlier point in endocytosis and this contributes, at least in part, to a more efficient intoxication.
We also recognize that the expanded tropism, along with more efficient cell entry could combine to enhance the in vivo toxicity of TcdBHV. The results from the zebrafish experiments (Fig. 2) indicate TcdBHV targets a broader array of cells in vivo than does TcdBHIST. Defining the specific tropism in the murine model or an infection model is more difficult, but it is reasonable to consider the possibility that TcdBHV is more lethal because the toxin targets an extensive variety of cell types systemically. Unfortunately, the TcdB receptor has been difficult to identify. Several attempts by our group to identify the TcdB receptor using standard techniques that have been successful with other toxins have failed. The results from the flow analyses in the current study suggest that the interaction of TcdB with the cell surface does not fit a single ligand-receptor model; this observation may explain why it has been so difficult to identify a receptor for this toxin. We were not able to achieve saturable binding, and interestingly TcdBHV interacted less efficiently than TcdBHIST, despite the fact that TcdBHV is clearly more cytotoxic than TcdBHIST. Undoubtedly, future studies on characterizing this complex interaction with target cells will provide important insight into a novel mechanism of TcdB intoxication.
Previous work by Razaq et al. found that C. difficile BI/NAP1/027 strains were more lethal than historical strains of C. difficile [44]. As mentioned in the introduction of this paper, there are several differences in the phenotypes of the hypervirulent and historical strains of C. difficile. NAP1 strains sporulate at a higher efficiency and are resistant to fluoroquinolones. Both of these characteristics may make the NAP1 strains more difficult to manage in the hospital setting and increase the frequency of disease, but are unlikely to increase virulence. Likewise, the binary toxin has been shown to enhance colonization [13], but clinical data have revealed little correlation between the increase in disease severity and production of this toxin [45], [46]. In addition, previous work found binary toxin to be enterotoxic, but strains producing binary toxin alone did not cause disease in hamsters [47]. Clearly, an increase in toxin production such as that reported for NAP1 strains could enhance virulence, but a recent report suggests that the tcdC mutation in epidemic strains does not always correlate with the overexpression of TcdA and TcdB [48]. Based on the findings from the current study, we suggest that variations in TcdB sequence and activity could be an important determining factor in the hypervirulence of NAP1 strains.
The recent work of Lyras et al. [43] found that TcdB is critical to C. difficile virulence in a hamster model of CDAD. Thus, variations in the antigenic region (e.g. carboxy terminus) of TcdB could allow repeated C. difficile infections of the same host by strains with antigenic variants of this toxin. In a recent publication by He and colleagues it was estimated that C. difficile diverged into a distinct species between 1.1 and 85 million years ago, and has gone through remarkable genetic variation over time [49]. The authors also posited that immune selection could have influenced the genetic variation, and they examined candidate immunogenic proteins that might fit this profile and 12 such proteins were identified. TcdB was not among these candidate proteins. It is unclear whether TcdB fits the criteria established for a positively selected core gene of C. difficile in this study, but it is reasonable to suspect the gene may have varied to avoid immune responses and this hypervariability enriched for a more potent form of the toxin. It is worth noting that while the protein identity was around 92%, the DNA homology was 93%. Nearly all of the residue changes occur as a single nucleotide substitution that result in amino acid substitutions. This further suggests a possible change in the sequence of TcdB that has been selected through an enhancement in virulence and perhaps by immune evasion.
Materials and Methods
Reagents and cell culture
Chinese hamster ovary-K1 (CHO) cells were maintained in F-12K medium (American Tissue and Culture Collection; ATCC) along with 10% fetal bovine serum (ATCC). HL-1 cardiomyocytes were obtained from the Claycomb laboratory [38] and maintained in Claycomb medium (Sigma) supplemented with 10% fetal bovine serum (ATCC), 0.1 mM Norepinephrine (Sigma), and 2 mM L-glutamine (Invitrogen). Cultures were grown at 37°C in the presence of 6% CO2. C. difficile VPI 10463 (produces TcdB with identical sequence to the 630 strain) and C. difficile BI17 6493 (a gift from Dr. Dale Gerding), were used in this study for the purification of TcdBHIST and TcdBHV. The tcdB gene was sequenced from both of these strains and the sequence was confirmed as exact matches to Genbank deposited sequences of strain 630 and R20291 (Genbank numbers AM180355 and FN545816). Cultures were grown as previously described [41], and TcdB was isolated by consecutive steps of anion-exchange (Q-Sepharose) and high-resolution anion-exchange (Mono-Q) chromatography in 20 mM Tris-HCl, 20 mM CaCl2, pH 8.0. Purification steps were followed by protein determination using the Bradford method, visualization of a single band by SDS-PAGE, and LC/MS/MS analysis (University of Oklahoma Health Science Center) to confirm protein identity. Cytotoxicity was determined using a WST-8 [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodiumsalt] (Dojindo Laboratories) according to manufacturer's instructions.
Zebrafish husbandry and experiments
Zebrafish maintenance and experiments were performed in accordance with the PHS Principles for The Utilization and Care of Vertebrate Animals Used in Testing, Research, and Training, and followed the recommendations in the Guide for the Care and Use of Laboratory Animals under the approval of The University of Oklahoma Health Sciences Center Campus IACUC (OUHSC #06-126). Zebrafish were obtained from Aquatic Eco-System (Apopka, FL). Zebrafish were maintained at 28.5°C on a 14 h light/10 h dark cycle in 10 gallon tanks equipped with pumps for mechanical and chemical filtration. Matings were performed in false bottom tanks, and embryos were washed briefly with 0.5% bleach after collection. Embryos were incubated in embryo water (60 mM NaCl, 1.2 mM NaHCO3, 0.9 mM CaCl2, 0.7 mM KCl) in petri dishes at 28.5°C, and water was changed daily. For TcdB treatment experiments, embryos were used between 48 and 72 h post fertilization, with chorions removed. Embryos were placed (5 embryos per well) into 48-well plates and treated with TcdBHIST or TcdBHV in embryo water at concentrations ranging from 50 nM to 0.01 nM. The embryos were observed for 72 h after treatment for morphological changes by using a SZX-7 microscope with a DP70 camera (Olympus). All images were captured and processed by using DP controller and DP manager software (Olympus).
Construction of LFn-fusions and related assays
The region encoding the enzymatic domain of TcdBHV was amplified from C. difficile NAP1 genomic DNA by PCR using the forward primer 5′-ACGTCCCGGGATGAGTTTAGTTAATA-3′ and the reverse primer 5′-ACTGGATCCTCATTATACTGTATTTTG-3′ to generate the tcdB gene fragment encoding residues 1 to 1668 of tcdB (tcdB1–1668) with a 3′ XmaI/SmaI and a 5′ BamHI site. The restricted gene fragment was fused to lfn by overnight ligation at 16°C with a Xma1/BamHI-restricted pET15b derivative containing lfn. The resulting plasmid was cloned into Escherichia coli XL-1 blue (Novagen) and candidate clones were screened for the correct insert and orientation by restriction analysis and DNA sequencing. LFnTcdBHIST(enz) which had been previously cloned and described [40] and the newly synthesized LFnTcdBHV(enz) were expressed using E. coli BL-21 Star (Invitrogen). Both fusions were purified by Ni2+ affinity chromatography (His-Trap, GE Life Sciences) and the purified protein migrated within the predicted size range of ∼94 kDa on SDS-PAGE. Protective antigen was expressed and purified as previously described [50].
Cell binding analyses
TcdBHIST or TcdBHV were labeled with Alexa Fluor 647 C5 maleimide (Invitrogen) according to manufacturer's instructions. Briefly, a 10 M excess of dye was added to TcdB in 20 mM Tris-HCl, pH 8.0, and incubated overnight at 4°C. Conjugated protein was separated from unincorporated dye using Sephadex G-25, and efficiency of labeling was confirmed to be between 80% and 100%. The activity of labeled TcdB was confirmed by cytotoxicity on CHO and HL-1 cells and was not reduced by >10%. Binding of each toxin to CHO and HL-1 cells was examined as follows. Cells were dissociated from flasks using 1 mM EDTA in PBS, centrifuged at 500× g, and washed once with PBS. One hundred thousand cells were incubated with a range from 10 nM to 320 nM of labeled toxin in 1 mL of PBS on ice for 1 h, washed twice, and the pellets were resuspended in 1 mL of PBS. The samples were analyzed using a FACSCalibur flow cytometer (University of Oklahoma Health Sciences Center) and FLOWJO software (Tree Star, San Carlos, CA). The emission wavelength was set to 665 nm, and the excitation was set at 633 nm with a bandpass of 30 nm.
Lysosomotropic inhibitor assays
CHO cells were plated at 5×104 cells/well in a 96-well plate and incubated overnight. The following day, TcdBHIST or TcdBHV was added to the cells at a final concentration of 0.1 µg/mL. At the indicated time points, the cells were washed to remove unbound toxin, and ammonium chloride (Sigma) was added to the cells at final concentration of 100 mM. Each sample was monitored for 24 h, and CPE (cytopathic effect) was determined by visualization. Percent CPE was calculated by counting a minimum of 100 cells in 3 different fields for each sample. Cells scored positive for CPE only when fully rounded, and the percent CPE was calculated as % rounded cellstest - % rounded cellscontrol, where control refers to cells treated with media alone.
TNS assays and tryptophan analysis
2-(p-Toluidinyl) naphthalene-6-sulfonic acid, sodium salt (TNS; Invitrogen) solutions were prepared to a final concentration of 150 µM in pH specific buffers. For pHs ranging from 4.0 to 6.0, 100 mM NaCl-100 mM ammonium acetate-1 mM EDTA was used. For pH 6.0 to 7.0, 100 mM NaCl-100 mM MES-1 mM EDTA was used. For pH 7.0 to 8.0, 100 mM NaCl - 100 mM HEPES-1mM EDTA was used. 40 pmol of TcdBHIST or TcdBHV was added to the buffer/TNS mixture in a final volume of 2.5 mL and allowed to incubate for 20 min and 37°C. Each sample was analyzed on a Fluorolog R928P PMT fluorometer (HORIBA Jobin Yvon) with an excitation of 366 nm and an emission scan of 380 to 500 nm with a slit width of 2.0. Tryptophan fluorescence of TcdBHIST and TcdBHV was also compared in the same manner, using an excitation of 270 nm and an emission scan of 310 nm to 400 nm.
Statistical analyses
Data are expressed as the means ± S.E.M. Statistical analyses were performed using two-tailed unpaired Student's t-test in GraphPad Prism (La Jolla, CA). Statistical significance is indicated as * p<0.05; ** p<0.01; *** p<0.001.
Zdroje
1. HallIC
O'TooleE
1935 Intestinal flora in new-born infants: with a description of a new pathogenic anaerobe, Bacillus difficilis. Am J Dis Child 49 390 402
2. GeorgeRH
SymondsJM
DimockF
BrownJD
ArabiY
1978 Identification of Clostridium difficile as a cause of pseudomembranous colitis. Br Med J 1 695
3. ChangTW
BartlettJG
GorbachSL
OnderdonkAB
1978 Clindamycin-induced enterocolitis in hamsters as a model of pseudomembranous colitis in patients. Infect Immun 20 526 529
4. BartlettJG
1992 Antibiotic-associated diarrhea. Clin Infect Dis 15 9
5. MutoCA
PokrywkaM
ShuttK
MendelsohnAB
NouriK
2005 A Large Outbreak of Clostridium difficile-Associated Disease With an Unexpected Proportion of Deaths and Colectomies at a Teaching Hospital Following Increased Fluoroquinolone Use. Infection Control and Hospital Epidemiology 26 273 280
6. LooVG
PoirierL
MillerMA
OughtonM
LibmanMD
2005 A Predominantly Clonal Multi-Institutional Outbreak of Clostridium difficile-Associated Diarrhea with High Morbidity and Mortality. N Engl J Med 353 2442 2449
7. McDonaldLC
KillgoreGE
ThompsonA
OwensRCJr
KazakovaSV
2005 An Epidemic, Toxin Gene-Variant Strain of Clostridium difficile. N Engl J Med 353 2433 2441
8. WarnyM
PepinJ
FangA
KillgoreG
ThompsonA
2005 Toxin production by an emerging strain of Clostridium difficile associated with outbreaks of severe disease in North America and Europe. The Lancet 366 1079 1084
9. AkerlundT
PerssonI
UnemoM
NorenT
SvenungssonB
2008 Increased Sporulation Rate of Epidemic Clostridium difficile Type 027/NAP1. J Clin Microbiol 46 1530 1533
10. MacCannellDR
LouieTJ
GregsonDB
LaverdiereM
LabbeA-C
2006 Molecular Analysis of Clostridium difficile PCR Ribotype 027 Isolates from Eastern and Western Canada. J Clin Microbiol 44 2147 2152
11. GericB
RupnikM
GerdingDN
GrabnarM
JohnsonS
2004 Distribution of Clostridium difficile variant toxinotypes and strains with binary toxin genes among clinical isolates in an American hospital. J Med Microbiol 53 887 894
12. GoncalvesC
DecreD
BarbutF
BurghofferB
PetitJ-C
2004 Prevalence and Characterization of a Binary Toxin (Actin-Specific ADP-Ribosyltransferase) from Clostridium difficile. J Clin Microbiol 42 1933 1939
13. SchwanC
StecherBr
TzivelekidisT
van HamM
RohdeM
2009 Clostridium difficile Toxin CDT Induces Formation of Microtubule-Based Protrusions and Increases Adherence of Bacteria. PLoS Pathog 5 e1000626
14. BourgaultA-M
LamotheF
LooVG
PoirierL
the CDAD-CSI Study Group 2006 In Vitro Susceptibility of Clostridium difficile Clinical Isolates from a Multi-Institutional Outbreak in Southern Quebec, Canada. Antimicrob Agents Chemother 50 3473 3475
15. DrudyD
QuinnT
O'MahonyR
KyneL
O'GaoraP
2006 High-level resistance to moxifloxacin and gatifloxacin associated with a novel mutation in gyrB in toxin-A-negative, toxin-B-positive Clostridium difficile. J Antimicrob Chemother 58 1264 1267
16. DrudyD
KyneL
ROMS
Fanning
2007 gyrA mutations in fluoroquinolone-resistant Clostridium difficile PCR-027. Emerg Infect Dis 13 2
17. StablerR
HeM
DawsonL
MartinM
ValienteE
2009 Comparative genome and phenotypic analysis of Clostridium difficile 027 strains provides insight into the evolution of a hypervirulent bacterium. Genome Biology 10 R102
18. TanKS
WeeBY
SongKP
2001 Evidence for holin function of tcdE gene in the pathogenicity of Clostridium difficile. J Med Microbiol 50 613 619
19. HammondGA
JohnsonJL
1995 The toxigenic element of Clostridium difficile strain VPI 10463. Microbial Pathogenesis 19 203 213
20. JustI
SelzerJ
WilmM
Eichel-StreiberCv
MannM
1995 Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature 375 500 503
21. Eichel-StreiberCv
Dagmar Meyer zu HeringdorfEH
Sabine Sartingen
1995 Closing in on the toxic domain through analysis of a variant Clostridium difficile cytotoxin B. Mol Microbiol 17 313 321
22. FaustC
YeB
SongK-P
1998 The Enzymatic Domain of Clostridium difficile Toxin A Is Located within Its N-Terminal Region. Biochemical and Biophysical Research Communications 251 100 105
23. HofmannF
BuschC
PrepensU
JustI
AktoriesK
1997 Localization of the Glucosyltransferase Activity of Clostridium difficile Toxin B to the N-terminal Part of the Holotoxin. J Biol Chem 272 11074 11078
24. BuschC
HofmannF
SelzerJr
MunroS
JeckelD
1998 A Common Motif of Eukaryotic Glycosyltransferases Is Essential for the Enzyme Activity of Large Clostridial Cytotoxins. J Biol Chem 273 19566 19572
25. BuschC
HofmannF
GerhardR
AktoriesK
2000 Involvement of a Conserved Tryptophan Residue in the UDP-Glucose Binding of Large Clostridial Cytotoxin Glycosyltransferases. J Biol Chem 275 13228 13234
26. HofmannF
BuschC
AktoriesK
1998 Chimeric Clostridial Cytotoxins: Identification of the N-Terminal Region Involved in Protein Substrate Recognition. Infect Immun 66 1076 1081
27. RupnikM
PabstS
RupnikM
von Eichel-StreiberC
UrlaubH
2005 Characterization of the cleavage site and function of resulting cleavage fragments after limited proteolysis of Clostridium difficile toxin B (TcdB) by host cells. Microbiology 151 199 208
28. ReinekeJ
TenzerS
RupnikM
KoschinskiA
HasselmayerO
2007 Autocatalytic cleavage of Clostridium difficile toxin B. Nature 446 415 419
29. EgererM
GiesemannT
JankT
SatchellKJF
AktoriesK
2007 Auto-catalytic Cleavage of Clostridium difficile Toxins A and B Depends on Cysteine Protease Activity. J Biol Chem 282 25314 25321
30. DingleT
WeeS
MulveyGL
GrecoA
KitovaEN
2008 Functional properties of the carboxy-terminal host cell-binding domains of the two toxins, TcdA and TcdB, expressed by Clostridium difficile. Glycobiology 18 698 706
31. WrenBW
1991 A family of clostridial and streptococcal ligand-binding proteins with conserved C-terminal repeat sequences. Mol Microbiol 5 797 803
32. Eichel-StreiberC
Laufenberg-FeldmannR
SartingenS
SchulzeJ
SauerbornM
1992 Comparative sequence analysis of the Clostridium difficile toxins A and B. Molecular and General Genetics 233 260 268
33. von Eichel-StreiberC
SauerbornM
1990 Clostridium difficile toxin A carries a C-terminal repetitive structure homologous to the carbohydrate binding region of streptococcal glycosyltransferases. Gene 96 107 113
34. StablerRA
GerdingDN
SongerJG
DrudyD
BrazierJS
2006 Comparative phylogenomics of Clostridium difficile reveals clade specificity and microevolution of hypervirulent strains. J Bacteriol 188 7297 7305
35. von Eichel-StreiberC
SauerbornM
KuramitsuHK
1992 Evidence for a modular structure of the homologous repetitive C-terminal carbohydrate-binding sites of Clostridium difficile toxins and Streptococcus mutans glucosyltransferases. J Bacteriol 174 6707 6710
36. Albesa-JovÈD
BertrandT
CarpenterEP
SwainGV
LimJ
Four Distinct Structural Domains in Clostridium difficile Toxin B Visualized Using SAXS. Journal of Molecular Biology 396 1260 1270
37. HammEE
VothDE
BallardJD
2006 Identification of Clostridium difficile toxin B cardiotoxicity using a zebrafish embryo model of intoxication. Proc Natl Acad Sci U S A 103 14176 14181
38. ClaycombWC
LansonNA
StallworthBS
EgelandDB
DelcarpioJB
1998 HL-1 cells: A cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci U S A 95 2979 2984
39. BallardJD
CollierRJ
StarnbachMN
1996 Anthrax toxin-mediated delivery of a cytotoxic T-cell epitope in vivo. Proc Natl Acad Sci U S A 93 12531 12534
40. SpyresLM
Qa'DanM
MeaderA
TomasekJJ
HowardEW
2001 Cytosolic Delivery and Characterization of the TcdB Glucosylating Domain by Using a Heterologous Protein Fusion. Infect Immun 69 599 601
41. Qa'DanM
SpyresLM
BallardJD
2000 pH-Induced Conformational Changes in Clostridium difficile Toxin B. Infect Immun 68 2470 2474
42. BarthH
PfeiferG
HofmannF
MaierE
BenzR
2001 Low pH-induced Formation of Ion Channels by Clostridium difficile Toxin B in Target Cells. J Biol Chem 276 10670 10676
43. LyrasD
O'ConnorJR
HowarthPM
SambolSP
CarterGP
2009 Toxin B is essential for virulence of Clostridium difficile. Nature 458 1176 1179
44. RazaqN
SambolS
NagaroK
ZukowskiW
CheknisA
2007 Infection of Hamsters with Historical and Epidemic BI Types of Clostridium difficile. J Infect Dis 196 1813 1819
45. RupnikM
GrabnarM
GericB
2003 Binary toxin producing Clostridium difficile strains. Anaerobe 9 289 294
46. BarbutF
DecreD
LalandeV
BurghofferB
NoussairL
2005 Clinical features of Clostridium difficile-associated diarrhoea due to binary toxin (actin-specific ADP-ribosyltransferase)-producing strains. J Med Microbiol 54 181 185
47. GericB
CarmanRJ
RupnikM
GenheimerCW
SambolSP
2006 Binary Toxin Producing, Large Clostridial Toxin Negative Clostridium difficile Strains Are Enterotoxic but Do Not Cause Disease in Hamsters. J Infect Dis 193 1143 1150
48. MurrayR
BoydD
LevettP
MulveyM
AlfaM
2009 Truncation in the tcdC region of the Clostridium difficile PathLoc of clinical isolates does not predict increased biological activity of Toxin B or Toxin A. BMC Infectious Diseases 9 103
49. HeM
SebaihiaM
LawleyTD
StablerRA
DawsonLF
2010 Evolutionary dynamics of Clostridium difficile over short and long time scales. Proc Natl Acad Sci U S A: 107 7527 7532
50. VothDE
HammEE
NguyenLG
TuckerAE
SallesII
2005 Bacillus anthracis oedema toxin as a cause of tissue necrosis and cell type-specific cytotoxicity. Cell Microbiol 7 1139 1149
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 8
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- The Transcription Factor Rbf1 Is the Master Regulator for -Mating Type Controlled Pathogenic Development in
- PKC Signaling Regulates Drug Resistance of the Fungal Pathogen via Circuitry Comprised of Mkc1, Calcineurin, and Hsp90
- Contribution of Coagulases towards Disease and Protective Immunity
- Early Severe Inflammatory Responses to Uropathogenic Predispose to Chronic and Recurrent Urinary Tract Infection
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy