
Direct Interaction between Two Viral Proteins, the Nonstructural Protein 2C and the Capsid Protein VP3, Is Required for Enterovirus Morphogenesis
In spite of decades-long studies, the mechanism of morphogenesis of plus-stranded RNA viruses belonging to the genus Enterovirus of Picornaviridae, including poliovirus (PV), is not understood. Numerous attempts to identify an RNA encapsidation signal have failed. Genetic studies, however, have implicated a role of the non-structural protein 2CATPase in the formation of poliovirus particles. Here we report a novel mechanism in which protein-protein interaction is sufficient to explain the specificity in PV encapsidation. Making use of a novel “reporter virus”, we show that a quasi-infectious chimera consisting of the capsid precursor of C-cluster coxsackie virus 20 (C-CAV20) and the nonstructural proteins of the closely related PV translated and replicated its genome with wild type kinetics, whereas encapsidation was blocked. On blind passages, encapsidation of the chimera was rescued by a single mutation either in capsid protein VP3 of CAV20 or in 2CATPase of PV. Whereas each of the single-mutation variants expressed severe proliferation phenotypes, engineering both mutations into the chimera yielded a virus encapsidating with wild type kinetics. Biochemical analyses provided strong evidence for a direct interaction between 2CATPase and VP3 of PV and CAV20. Chimeras of other C-CAVs (CAV20/CAV21 or CAV18/CAV20) were blocked in encapsidation (no virus after blind passages) but could be rescued if the capsid and 2CATPase coding regions originated from the same virus. Our novel mechanism explains the specificity of encapsidation without apparent involvement of an RNA signal by considering that (i) genome replication is known to be stringently linked to translation, (ii) morphogenesis is known to be stringently linked to genome replication, (iii) newly synthesized 2CATPase is an essential component of the replication complex, and (iv) 2CATPase has specific affinity to capsid protein(s). These conditions lead to morphogenesis at the site where newly synthesized genomes emerge from the replication complex.
Published in the journal:
. PLoS Pathog 6(8): e32767. doi:10.1371/journal.ppat.1001066
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1001066
Summary
In spite of decades-long studies, the mechanism of morphogenesis of plus-stranded RNA viruses belonging to the genus Enterovirus of Picornaviridae, including poliovirus (PV), is not understood. Numerous attempts to identify an RNA encapsidation signal have failed. Genetic studies, however, have implicated a role of the non-structural protein 2CATPase in the formation of poliovirus particles. Here we report a novel mechanism in which protein-protein interaction is sufficient to explain the specificity in PV encapsidation. Making use of a novel “reporter virus”, we show that a quasi-infectious chimera consisting of the capsid precursor of C-cluster coxsackie virus 20 (C-CAV20) and the nonstructural proteins of the closely related PV translated and replicated its genome with wild type kinetics, whereas encapsidation was blocked. On blind passages, encapsidation of the chimera was rescued by a single mutation either in capsid protein VP3 of CAV20 or in 2CATPase of PV. Whereas each of the single-mutation variants expressed severe proliferation phenotypes, engineering both mutations into the chimera yielded a virus encapsidating with wild type kinetics. Biochemical analyses provided strong evidence for a direct interaction between 2CATPase and VP3 of PV and CAV20. Chimeras of other C-CAVs (CAV20/CAV21 or CAV18/CAV20) were blocked in encapsidation (no virus after blind passages) but could be rescued if the capsid and 2CATPase coding regions originated from the same virus. Our novel mechanism explains the specificity of encapsidation without apparent involvement of an RNA signal by considering that (i) genome replication is known to be stringently linked to translation, (ii) morphogenesis is known to be stringently linked to genome replication, (iii) newly synthesized 2CATPase is an essential component of the replication complex, and (iv) 2CATPase has specific affinity to capsid protein(s). These conditions lead to morphogenesis at the site where newly synthesized genomes emerge from the replication complex.
Introduction
Morphogenesis is a crucial step at the end of the virus' life cycle that provides newly synthesized genomes with a protective shell to survive in the extracellular environment yet assures attachment to and penetration into subsequent host cells. Morphogenesis of viral genomes must be specific because encapsidation of non-progeny nucleic acid is wasteful for the virus, for which reason elaborate mechanisms have evolved to discriminate against nucleic acids other than its own genome.
Here we describe our studies of the morphogenesis of a group of single, plus-stranded RNA viruses that belong to the genus Enterovirus of Picornaviridae, a family of viruses containing a large number of human and animal pathogens. Poliovirus (PV), the prototype enterovirus, has been extensively studied for a century and although much is known about its virion structure, uptake into host cell, genome structure and macromolecular events of replication, the mechanism of particle assembly is only poorly understood [1]. The key requirement of morphogenesis, namely the specific selection of viral genomes, has also remained obscure. We have discovered a novel mechanism for enteroviruses in which the specificity of encapsidation is facilitated by protein-protein interaction. It should be noted that this mechanism is different from the one used by some other RNA viruses such as hepatitis B virus and alphaviruses [2], [3]. The specificity of encapsidation with these viruses is dependent on an RNA encapsidation signal and RNA/protein interactions.
Enteroviruses synthesize only one protein, the polyprotein, which is cleaved by two virus-encoded proteinases, 2Apro and 3Cpro/3CDpro, into intermediates expressing specific functions (e.g. 3CDpro) and into mature proteins (Figure 1A). After its release from the polyprotein by 2Apro, the precursor of the structural proteins (P1) interacts with cellular chaperone Hsp90 [4], a requirement for its subsequent processing by 3CDpro into capsid proteins VP0, VP3 and VP1 (Fig. 1A) [5]. These cleavage products will spontaneously form a 5S protomer (VP0, VP3, VP1) that can oligomerize to the 14S pentamer (VP0, VP3, VP1)5; twelve pentamers, subsequently, assemble into a 75S empty capsid [(VP0, VP3, VP1)5]12, also called procapsid [6], [7]. It is not known at what stage progeny genomes interact with the capsid precursors. They may be inserted into the procapsid or, alternatively, pentamers may condense around RNA emerging from the replication complex. Either process will yield provirions {[(VP0, VP3, VP1)5]12RNA} [8], [9], [10] that mature to virions when VP0 is cleaved to VP4 and VP2 by a mechanism possibly involving an RNA-dependent autocatalytic process [6], [7].

The encapsidation process in PV morphogenesis is highly restricted to newly synthesized plus strand progeny RNA [11], [12]. Under normal conditions of replication in HeLa cells, cellular RNAs, PV mRNA lacking VPg or viral VPg-linked minus strand RNA are excluded from mature viral particles [6]. Numerous studies aimed at determining the specificity of encapsidation by searching for an RNA packaging signal have been unsuccessful. The very long 5′NTR of PV can be replaced with that of the distantly related coxsackie B3 virus (CVB3) [13] or CVB4 [14] yielding virions containing chimeric genomes that proliferate with PV wild type (wt) kinetics. Similarly, the cloverleaf of PV can be changed to that of HRV2 [15], or the PV IRES has been exchanged with IRESes from other picornaviruses [16], [17], [18] and even with that of HCV [19] without yielding impaired encapsidation phenotypes. The 3′NTR of PV, in turn, has been exchanged with that of HRV14, a single stem-loop structure with no apparent similarity in structure and sequence to that of PV. This chimera too proliferated with wt kinetics [20]. This makes it highly unlikely that the 5′ - and 3′-NTRs of poliovirus contain packaging determinants. The genomic sequence encoding the capsid P1 precursor cannot harbor an encapsidation signal since the entire P1 encoding region can be deleted [21] or replaced by foreign genes [14], [22], [23]. Such PV replicons, all of which can replicate, can be efficiently encapsidated with PV capsid proteins in trans. Recent experiments from this laboratory, employing a “scrambled” sequence [24] of the P2 coding region (scramble of synonymous codons) have eliminated this region too from carrying an encapsidation signal (Song, Mueller, Ward, Skiena, Futcher, Paul and Wimmer, manuscript in preparation). Finally, genetic modification of the PV VPg coding sequence [25], [26] or engineering PVs carrying VPg sequences of other picornaviruses [25], [27], [28] have also eliminated the VPg coding sequence as providing an encapsidation signal. VPg, however, may still play a role in encapsidation (see below). Currently it seems unlikely that poliovirus or other enteroviruses (including the rhinoviruses that have recently been classified as enteroviruses) harbor an RNA signal that would instruct the capsid components to bind to and enclose the viral genome in a specific manner. Only one member of the extended family of Picornaviridae, Aichi virus (Kobuvirus genus), was reported to contain a 5′-terminal RNA stem loop with a role in particle assembly [29].
Among the nonstructural proteins of PV, 2CATPase and 3CDpro, have been reported to be involved in packaging although no mechanism(s) is known. Studies with an in vitro translation/RNA replication system, which produces viable viruses [11], have suggested that 3CDpro functions at a late step in the assembly process just before or during the maturation cleavage of VP0 to VP2 and VP4 [30]. Protein 2CATPase of PV has been implicated in virion capsid formation through genetic analysis of a cold-sensitive mutant [31] or by determining escape mutants from drug (hydantoin) inhibition [32]. The multifunctional 329 amino acids-long 2CATPase is the most complex and least understood nonstructural proteins of enteroviruses. The functions of this protein that are highly conserved among picornaviruses, include, in addition to encapsidation, host cell membrane rearrangements [33], [34], genome replication [35], [36] and even uncoating of viral particles [37]. Based on sequence analyses the protein has been classified as a member of the superfamily III helicases, which contain 3 conserved motifs (A–C), including two classical ATP binding motifs (A and B) (Fig. 1B) [38]. Purified 2CATPase possesses ATPase activity [39], [40], which is inhibited by guanidine HCl (GnHCl) [41], a known potent inhibitor of PV RNA replication [18]. In vitro the protein forms homo-oligomeric structures required for ATPase activity [42]. The N-terminal part of the protein contains a RNA binding domain and an amphipathic helix, which is involved in membrane binding and oligomerization [36], [42], [43]. Another amphipathic helix, a RNA binding domain and a cysteine rich domain that binds zinc are located near the C-terminus [44], [45]. In infected cells 2CATPase appears to be associated with viral RNA in the replication complexes on the surface of membranous vesicles [46].
Available evidence suggests that genome replication is a precondition of PV encapsidation [11], [12]. Electron-microscopic studies, which showed that RNA replication complexes co-localize with capsid precursors on membranous vesicles during infection [47], supported these observations. Nugent et al. [12] hypothesized that encapsidation specificity may be determined by the spatial arrangement of replication complexes with the capsid precursors. This intriguing hypothesis lacked an essential component: what brings the capsid precursors into the vicinity of the replication complexes since PV replicons lacking the P1 domain altogether can be efficiently encapsidated in trans?
Human enteroviruses have been divided into several clusters based on genotype relationships [48]. PV types 1–3, and eleven C-cluster coxsackie A virus serotypes share the C-cluster, also referred to as C-cluster human enteroviruses (C-HEVs). Their difference in affinity to cellular receptors, PVs using CD155 [49], [50] while C-CAVs using ICAM-1 [51], accounts for significant capsid dissimilarities between the member viruses of this species [52], [53]. In contrast, the differences between the non-structural proteins of PV vs C-CAVs are less pronounced. We have used the similarities and dissimilarities between PV and C-CAVs to separate RNA replication from encapsidation by constructing chimeric viruses in which capsid precursor P1 and/or 2CATPase have been exchanged. All of the chimeric viruses studied here replicated their genomes with wt kinetics in tissue culture cells but were blocked in encapsidation, a phenotype that we examined by genetic analyses. We present genetic evidence suggesting a specific interaction between 2CATPase and VP3, which is essential for genome encapsidation. The genetic evidence of the 2CATPase-VP3 interaction was further substantiated by biochemical assays. We propose that the primary determinant of encapsidation specificity in the enterovirus life cycle is protein-protein interaction.
Materials and Methods
Cells
HeLa H1 cells were maintained in DMEM (Life Technology), supplemented with 10% FCS, 100 units of penicillin, and 100 mg of streptomycin per milliliter.
Viruses
The prototype strain of C-CAVs, CAV20, CAV21 (Kuykendall) and CAV18, propagated in HeLa H1 cells, were obtained from the American Type Culture Collection. Polioviruses (PVM) were derived from cDNA pT7PVM [54] by transfection.
Plasmids
Parental plasmids of PV: pT7PVM contain a full-length infectious cDNA of PVM.
Parental plasmids of C-CAVs: pT7CAV20 contains a full-length infectious cDNA of CAV20 [48]. pGEM-CAV21 and pT7CAV18, which contain a full length infectious cDNA of CAV21 and CAV18 (Kuykendall), respectively, were constructed by Elizabeth Rieder.
Chimeric genomes: Parental plasmids of PVs and C-CAVs were used as the backbone for cloning as described below. All plasmids contain the T7 promoter in front of the 5′ end of the full length genomic cDNA for in vitro RNA transcription by T7 RNA polymerase [54].
Construction of chimeric genomes between C-CAVs
Parental plasmid cDNAs of CAV20, CAV21 and CAV18 were used as the backbone for cloning. Using a three-step overlapping PCR, chimeras between CAV20 and CAV21 (or CAV18) were generated by precise swapping of the genetic segment encoding the P1 region of the polyprotein [48]. The oligonucleotides and the templates for PCR are summarized in Supplementary Table 2 in Text S1. The overlapping PCR fragment and the vectors were digested with the same pair of restriction enzymes and ligated to produce the cDNA clone of the chimeric genome. The vectors and the restriction sites for construction of the chimeric genomes are listed in Supplementary Table 3 in Text S1. The DNA sequence of the final constructs was verified by sequencing analysis using BigDye Kit and ABI Prism DNA sequencer (model 310). Replacement of the original 2CATPase coding region in each of the chimeric genome with that of the same origin as the capsid coding region follows the same strategy described above (Supplementary Table 2 & 4 in Text S1).
Construction of C20PP derivatives
Construction of the chimeric C20PP genome was described before [48]. For construction of the C20PP derivatives, listed in Supplementary Table 3 in Text S1 and Figure 4, a two-step overlapping PCR similar to the one described above was performed using C20PP as the template with the mutation(s) introduced into the internal primers (Supplementary Table 3 & 4 in Text S1).
Construction of Renilla luciferase reporter viruses
To test the RNA replication efficiency of chimeric viruses, we used novel reporter viruses, which contain the Renilla luciferase gene fused to the N-terminal of the P1 coding region of the chimeric genomes. The same strategy (three-step overlapping PCR), described above, was used to introduce the Renilla luciferase gene into the N-terminal of P1 the coding region of the chimeric genomes (Supplementary Table 3 & 4 in Text S1). The luciferase protein is post-translationally cleaved from the remainder of the polyprotein by 3CDpro at a recombinant 3CDpro cleavage site.
Analysis of the growth phenotypes of the parental and chimeric viruses
Parental plasmid cDNAs pT7PVM, pT7CAV20, pGEM-CAV21, pT7CAV18 and the chimeric constructs were linearized at a unique restriction sites downstream the poly(A) tract (Supplementary Table 5 in Text S1) and used as templates for in vitro RNA synthesis using T7 RNA polymerase. RNA transcripts were transfected into HeLa H1 cell monolayers by the DEAE-Dextran method as described before [54]. Following transfection, virus was harvested from the transfected cells when 90–95% of the cells displayed cytopathic effect (CPE). Lysates of transfected cells from the chimeric genomes showing no CPE were inoculated into 35-mm-diameter HeLa H1 cell monolayers for 6–8 subsequent serial passages. The plaque phenotypes and virus titers (PFU/ml) of the parental and chimeric viruses were determined in triplicate by plaque assay [11] using 0.6% tragacanth gum. The identity of the chimeric viruses was confirmed by RT-PCR/sequencing analysis.
In vitro translation
In vitro RNA translations were performed with HeLa cell S10 cytoplasmic extracts at 34 degree Celsius as described previously [11].
RT-PCR and sequencing analysis of viral RNA
HeLa H1 cell monolayers (5×106) were infected with viruses that were purified from plaque assay. At 7-hr post infection, total cytoplasmic RNA was extracted with 1 ml Trizol reagent (Invitrogen) and amplified into DNA using Titan one tube RT-PCR system (Roche). The RT-PCR products were sequenced using the Bigdye Terminator Sequencing Kit (ABI, Applied Biosystems).
Luciferase assay
Dishes (35-mm diameter) of monolayered HeLa H1 cells were transfected with 5 µg of replicon RNAs and were incubated at 37 degree Celsius in standard tissue culture medium in the presence and absence of 2 mM GnHCl. Luciferase activities were determined in lysates of cells harvested 16 hrs after transfection. Cell lysates (10 µl) was mixed with 20 µl of luciferase assay reagent (Promega; luciferase assay system catalog no. E2810) and Renilla luciferase activity was measured in an Optocomp I luminometer (MGM Instruments, Inc.). Cell lysates from transfections were used to re-infect HeLa H1 cells in the presence and absence of 2 mM GnHCl and luciferase activities were determined in lysates of cells harvested 8 hrs after infection. Luciferase activity ratio (−GnHCL/+GnHCl) represents: luciferase activity without GnHCl divided by luciferase activity with GnHCl in either transfection or infection.
Construction of His-tagged VP3
A PCR fragment containing full length PV VP3 was amplified and cloned into the pET21b vector (Novagen) with the restriction enzymes Sac I and Xho I.
Purification of GST-2CATPase and His-tagged VP3 proteins
GST-tagged 2CATPase and His-tagged VP3 recombinant proteins were expressed in E. coli. The GST-2CATPase proteins were expressed from pGEX-2C vector and purified by glutathione sepharose column (GE Healthcare) as described before [41]. The His-VP3 proteins were purified by nickel column chromatography (QIAGEN).
GST pull-down assay
Briefly, 5 µg GST-2CATPase (or 2 µg GST as a control) were incubated with glutathione sepharose beads at 4°C for 3 hr in buffer containing 50 mM Tris-HCl pH7.5, 140 mM NaCl, 0.1% TritonX-100 with protease inhibitor cocktail tablets (Roche). The protein bound GSH beads were washed with PBS 3 times and then 5 µg His-VP3 was added. After 1 hr incubation at 4 degree Celsium, the glutathione beads were washed 3 times and were boiled in 1x SDS sample buffer for 5 min. The samples were analyzed by SDS-polyacrylamide gel (12.5% acrylamide) electrophoresis and followed by western blot analysis using antibodies against PV VP3 (polyclonal, kindly contributed by Dr. Delpeyroux, Pasteur Institute, France).
Co-immunoprecipitation assay using 2CATPase and VP3 proteins translated in an in vitro cell-free translation system
Plasmids used for in vitro translation of CAV20 structural protein VP3 (wt), VP3 (E180G) and CAV20 non-structural protein 2CATPase (wt), PV non-structural proteins 2CATPase (wt) and 2CATPase (N252S) were generated with A2 plasmid [55] and PCR fragments encoding wt and mutant proteins of VP3 and 2CATPase according to methods described previously [55]. 2 µg of each VP3 and 2C RNA transcripts generated in vitro by T7 RNA polymerase were co-translated in HeLa extract and labeled by 35S-labeled methionine. Using anti-PV 2C polyclonal antibody, Co-IP assay was performed with co-translated 35S labeled 2CATPase and VP3 proteins following standard protocols using protein A/G plus-agarose (Santa Cruz Biotechnology) and [56]. The radioactive signals from input proteins and Co-IP reaction products were quantified by a PhosphorImager (Molecular Dynamics, Storm 860) by measuring the amount of 35S incorporated into product. Interactions between 2CATPase and VP3 were represented by percentages of the levels observed in the Co-IP reaction with CAV 2CATPase and CAV VP3 after normalizing the amount of input 2CATPase and VP3. Numbers given for the extents of interactions represented the average of three independent experiments.
Results
Rationale for generating PV and CAV chimeras to study enterovirus encapsidation
The lack of evidence for an RNA packaging signal in enterovirus proliferation has prompted us to study the specificity of encapsidation by searching for possible protein-protein interactions needed for this process. Previous studies have shown that chimeric constructs of the PV polyprotein with exchanges of varying coding regions of closely related picornaviruses can be utilized to analyze determinants of viral macromolecular interactions and replication [57], [58], [59], [60], [61], [62]. PV and C-CAVs share a high degree of amino acid identity in their nonstructural proteins but are not closely related in their capsid sequences probably because they evolved to use different cellular receptors [48]. In a chimera with the capsid of one C-HEV and the nonstructural proteins of another C-HEV, this difference may produce specific morphogenesis phenotypes due to incompatibility or poor interaction between capsid and nonstructural proteins. In our previous work, we have already observed that the replacement of the PV type 1 Mahoney (PVM) capsid with that of its closest relative, CAV20, resulted in a quasi-infectious virus, C20PP (Figure 2A) [48]. In C20PP, the first letter refers to the origin of the P1 region, the 2nd and 3rd letters refer to the origins of the P2 and P3 regions, respectively. The quasi-infectious phenotype means that a step in the life cycle of C20PP is so severely debilitated that only escape variants can be recovered from transfections with RNA transcripts [48]. The molecular basis of the defective phenotype of C20PP was not elucidated but could be proteolytic processing of the capsid precursor, genome replication or encapsidation. The observed quasi-infectious phenotype of C20PP made it possible to subject this chimera to a genetic analysis that may reveal a defect in encapsidation.

C20PP is defective in encapsidation
We first provided evidence that the quasi-infectious phenotype of C20PP was not due to abnormal translation or protein processing resulting from poor compatibility between the heterologous capsid and the 3CDpro polypeptide in C20PP. Translation of RNA transcripts of wt and chimeric constructs in a HeLa cell-free extract [11] showed normal translation and protein processing patterns (Figure 2B).
The search for the block of C20PP proliferation led us to develop a novel reporter virus in which the PV open reading frame (ORF) of the Renilla Luciferase (R-Luc) protein was fused to the N-terminus of the viral polyprotein. In the course of the infection the R-Luc is cleaved off from the viral polyprotein at an engineered 3CDpro proteinase cleavage site. Due to the small size of the inserted R-Luc gene this virus was stable for 1 passage after transfection and, thus, suitable for our experiments. This construct is similar to a previously described recombinant coxsackie B3 virus that stably expressed eGFP in tissue culture [63]. The advantage of using our reporter virus over conventional reporter replicons, in which the P1-coding sequence is replaced by the luciferase gene, is that it can distinguish between a defect in replication and encapsidation. A reporter viral genome (with the R-Luc sequence) that is unable to encapsidate itself will exhibit normal RNA replication levels as evidenced by a wt-like Renilla luciferase signal after RNA transfection. However, it would not generate infectious progeny and, consequently, passage to fresh cells will fail, leading to the loss of the luciferase signal.
We have made such reporter viruses from both the parental wt CAV20 and the chimeric C20PP (Figure 2C). RNA transcripts were transfected into HeLa H1 cells both in the absence and presence of 2 mM GnHCl, a potent inhibitor of PV [18] and CAV20 RNA replication [18], [48]. Luciferase activity was determined at 16-hr post transfection either in the presence of GnHCl (+GnHCl) throughout the incubation period that allows us to measure the translation of the transfecting RNA, or in the absence of GnHCl (−GnHCl) when the luciferase signal is increased because of RNA synthesis. The ratio of the luciferase signals –GnHCl/+GnHCl indicates the extent of genome replication. As shown in Figure 2D there was a 100 fold increase of the luciferase signal at −GnHCl compared to +GnHCl with both wt R-Luc-CAV20 and R-Luc-C20PP viruses, an observation indicating robust RNA synthesis under these conditions. Lysates of transfected cells were then inoculated to fresh HeLa H1 cells as 1st passage either in the presence or absence of GnHCl. Eight hours post infection the luciferase activity was measured (Figure 2D). The high level of luciferase activity obtained after the first passage of the R-Luc-CAV20 virus indicated the formation of virions that had encapsidated the wt genome in the course of transfection. In contrast, no luciferase signal could be detected after passage of the lysate harboring the R-Luc-C20PP chimera (Figure 2D). We conclude that the genome of the C20PP chimera, although competent in RNA replication, cannot form infectious progeny, e.g. it is defective in genome encapsidation. The reason for the defect in encapsidation, however, remains elusive.
The encapsidation defect of chimera C20PP is rescued either by a mutation in capsid protein VP3 or by a mutation in the nonstructural protein 2CATPase
As mentioned above, the C20PP chimera is quasi-infectious. Variants that escaped the block in encapsidation were found only after three blind passages following transfection. This indicated the emergence of mutation(s). To identify the rescuing mutation(s), we plaque purified two viruses that had emerged after three passages from two independent transfections with C20PP transcripts. RT-PCR and sequence analyses of the viral genomes revealed two independent single mutations that mapped to the coding region of either VP3 (E180G) or 2CATPase (N252S). By separately engineering these two mutations back into the cDNA of C20PP we obtained two viable viruses C20PP-VP3E180G and C20PP-2CN252S (Figure 3A). Their titers after transfection were as low as those observed with isolates after three passage of C20PP (Figure 2A) and 1000 fold lower than that observed with wt CAV20. Moreover, C20PP-VP3E180G and C20PP-2CN252S expressed a small plaque phenotype compared to that of wt CAV20 (Figures 2A and 3A).

The double mutation (VP3E180G&2CN252S) fully rescues C20PP to a wt encapsidation phenotype
The phenotypes of C20PP-VP3E180G and C20PP-2CN252S did not change after further passages, e.g. all attempts to isolate variants with improved proliferation phenotypes failed (data not shown). This prompted us to engineer both the VP3E180G and 2CN252S mutations into C20PP (C20PP-DM). Variant C20PP-DM expressed phenotypes (virus titer and plaque size) almost the same as that of wt CAV20 (compare Figures 2A and 3A). Currently, we cannot explain why in our experiments the C20PP-DM-like variant did not evolve during passaging of C20PP.
To confirm the rescue of the encapsidation defect of C20PP by the mutations in VP3 and 2CATPase, we constructed reporter viruses R-Luc-C20PP-VP3E180G, R-Luc-C20PP-2CN252S and R-Luc-C20PP-DM (Figure 3B). All three produced strong luciferase signals after transfection of their genomic RNAs into HeLa H1 cells, as expected (Figure 3C). After a passage into fresh HeLa H1 cells, the luciferase activity was highly impaired with C20PP but was found to be partially rescued if the virions carried the single 2CN252S or VP3E180G mutation or fully rescued by the double mutation (Figure 3C). Thus, although a single mutation can partially rescue the proliferation phenotype of C20PP, the double mutation 2CN252S/VP3E180G in the genome of the chimera is capable of producing a proliferation phenotype similar to that of wt CAV20. This observation, confirms the previous genetic data by Vance et al. [32] that the coding region of 2CATPase is linked to encapsidation. More importantly, the cooperative activity of VP3 with 2CATPase suggests that capsid protein VP3 functions through a direct interaction with 2CATPase in encapsidation. As indicated before, the coding sequence of 2CATPase, however, can be eliminated as carrying an encapsidation signal. The cre, the only known essential RNA structure in coding sequence of 2CATPase, are highly homologous in sequence between PV and CAV20. Moreover, scrambling of the P2 RNA sequence has no influence on PV proliferation if the essential cre is transplanted to the 5′NTR (Song, Mueller, Ward, Skiena, Futcher, Paul, and Wimmer, manuscript in preparation). It is, thus, likely that VP3 and 2CATPase or their precursors cooperate by protein-protein interaction, a novel mechanism for specific genome encapsidation of enteroviruses.
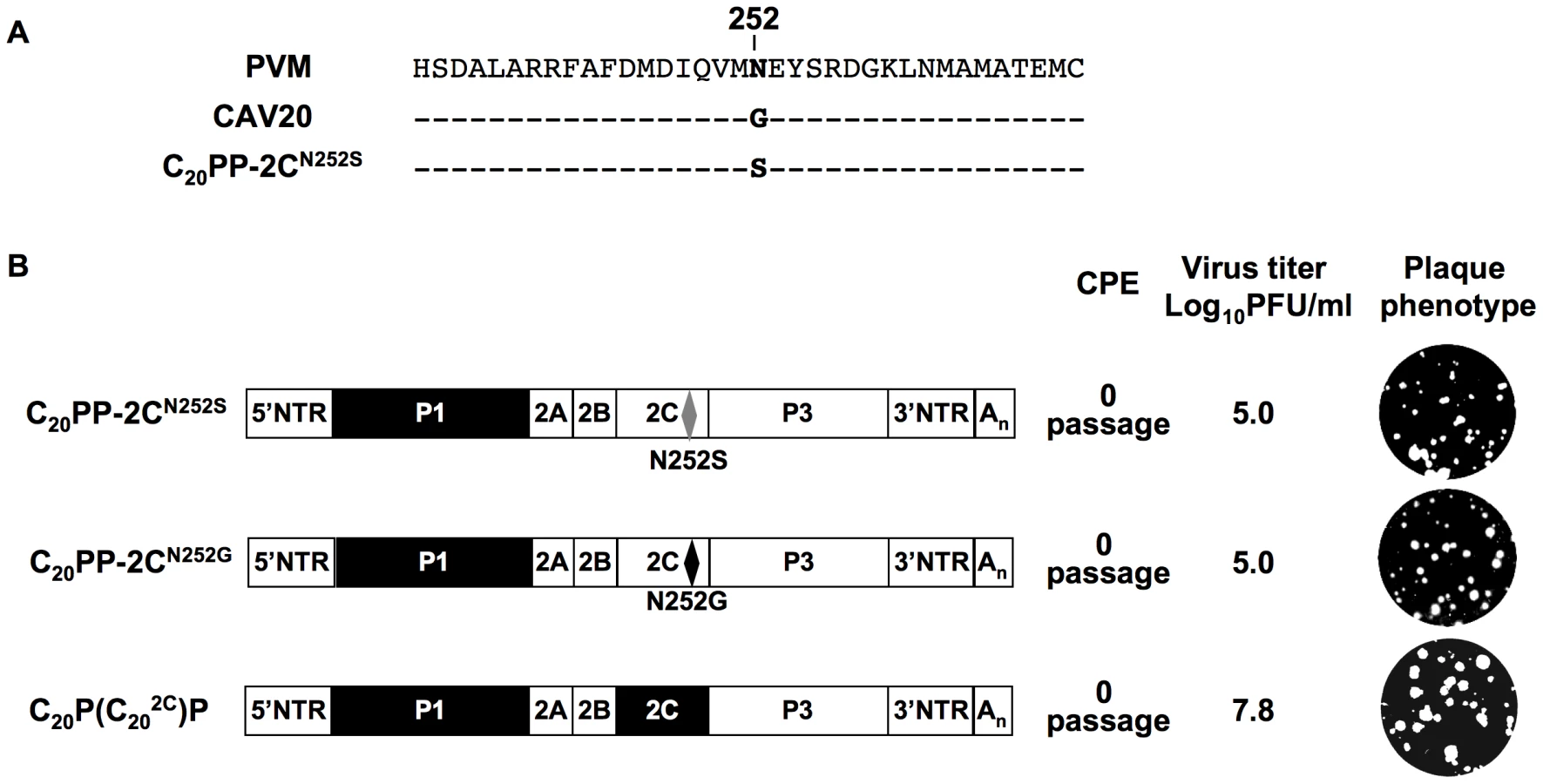
A CAV20-like 2CATPase rescues the encapsidation defect of C20PP
It should be noted that the mutation in 2CATPase, which rescues in part the encapsidation of C20PP-2CN252S, is an N/S change in PV 2CATPase at position 252 (Figure 4A). Amino acid sequence alignment demonstrated that CAV20 has a Gly at this position (Figure 4A), an observation indicating that a CAV-20 like, uncharged residue might be favorable at this position. Thus, since C20PP-2CN252S expressed a severe encapsidation phenotype, it was of interest to determine whether a C20PP-2CN252G mutant would yield a chimera equal or superior in encapsidation to C20PP-2CN252S. Similar to C20PP-2CN252S, the N252G mutation in the PV 2CATPase only partially rescued the encapsidation phenotype of C20PP (Figure 4B). The observed N/S mutation in a naturally selected escape mutant can be explained by the reasoning that the CAV20-like N/G change in codon N252 would have required two nucleotide changes (AAT/GGT) while the N/S mutation entails only a single nucleotide change (AAT/AGT). Overall, the CAV20 like single amino acid change at 252 of PV 2CATPase was favorable but not sufficient to fully rescue the VP3-related function of the PV 2CATPase protein in encapsidation.
Based on this observation, we reasoned that the replacement of the entire 2CATPase coding region in C20PP with its CAV20 counterpart should yield a chimera whose protein/protein interaction required for encapsidation would be sufficient and, thus, yield a virus with proliferation phenotypes similar to that of wt CAV20. Therefore, we generated a chimera designated as C20P(C202C)P that showed CPE after transfection with a virus titer comparable to that of wt CAV20 (compare Figures 2A and 4B). These results provide further support for the hypothesis that the 2CATPase protein is a partner required for encapsidation. It should be noted that the chimera C20P(C202C)P had a growth phenotype more similar to that of the wt CAV20 virus than the chimera C20PP-2CN252S (compare Figures 2A and 4B), an observation indicating that sequences besides residue 252 in 2CATPase are also important for function during viral encapsidation.
The encapsidation of other C-CAV chimeras requires capsid protein(s) and 2CATPase of the same origin
The observation that CAV20 VP3 needs its own 2CATPase to fully rescue the defect in encapsidation suggests that the cooperation or interaction between 2CATPase and capsid might be specific and generally true for C-HEVs. To test this hypothesis we extended our analyses to CAV18 and CAV21, two viruses that are phylogenetically related to CAV20 and PV [48]. Chimeras C20C21C21 and C18C20C20 (Figure 5A) displayed non-viable phenotypes as judged by the lack of virus in plaque assays even after 8 blind passages on fresh HeLa H1 monolayers (Figure 5A). In order to test whether the lethal phenotypes of the two chimeric viruses were due to the same encapsidation defect as that of C20PP, we constructed reporter viruses of CAV18 and of the chimeras C20C21C21 and C18C20C20 (Figure 5B) just as that of CAV20 (Figure 2C). After transfection of RNA transcripts into HeLa H1 cells, R-Luc activity at 16-hr post transfection showed that the parental and chimeric viruses replicated their genomes with nearly wt efficiency (Figure 5C). However, after the first passage on fresh HeLa H1 cells only wt CAV20 and wt CAV18 reporter viruses yielded normal luciferase signals (Figure 5C). The chimeric genomes R-Luc-C20C21C21 and R-Luc-C18C20C20 could not produce an infection, a result demonstrating an encapsidation defect.

To test whether the lethal proliferation phenotypes of the two chimeras (C20C21C21 and C18C20C20) are related to an incompatibility between the capsid and the 2CATPase proteins derived from different parental viruses, we constructed new chimeras [C20C21(C202C)C21, C18C20(C182C)C20] in which the capsid and 2CATPase proteins were derived from the same origin (Figure 6A). The resulting chimeras all showed CPE after transfection (Figure 6B) and the virus titers were comparable to that of the parental viruses (data not shown). The finding that the lethal growth phenotypes of the chimeras were fully rescued when the capsid and 2CATPase were derived from the same origin serves as further support of our hypothesis that 2CATPase and capsid proteins communicate with each other during the process of encapsidation. So far, we have not been able to determine, by continued passage, the necessary amino acid changes in the 2CATPase protein of CAV20 and CAV21 to allow encapsidation of the C20C21C21 and C18C20C20 chimeras. This observation might be explained by the fact that there are many amino acid differences either between CAV20 and CAV21 or between CAV20 and CAV18 flanking residue 252 of the 2CATPase protein (Figure 6C). This may make it difficult for the two chimeras (C20C21C21 and C18C20C20) to generate escape mutants simply by natural selection during passages.

A direct interaction between proteins 2CATPase and VP3 is revealed by biochemical assays
The genetic evidence described above strongly suggested a direct interaction between the capsid proteins and 2CATPase, which is required for encapsidation. To confirm this interaction, we carried out a GST-pull down assay with purified PV proteins GST-2CATPase and His-VP3 (Figure 7A, lane 2). The same assay was performed with purified GST protein as a control (Figure 7A, lane 1). Our results clearly showed that PV GST-2CATPase interacts directly with the PV His-tagged VP3 protein. To provide further proof that direct interaction between VP3 and 2CATPas is required for the encapsidation process, co-immunoprecipitation (Co-IP) assays were performed with VP3 and 2CATPase proteins in three different combinations: CAV20 VP3 & CAV20 2CATPase, CAV20 VP3 & PV 2CATPase, CAV20 VP3 (E180G) & PV 2CATPase (N252S), which correspond to those observed in wt CAV20, nonviable chimera C20PP, and rescued C20PP-DM, respectively. In vitro transcribed RNA transcripts of 2C and VP3 coding sequences in different combinations were co-translated in HeLa cell extracts (Figure 7B, lanes 4–6) [11], [55]. Using PV 2C polyclonal antibody, which recognizes PV 2CATPase and CAV20 2CATPase with the same efficiency (data not shown) [56], CAV20 VP3 was co-immunoprecipitated readily by CAV20 2CATPase (100%, Figure 7B, lane 1) but only weakly by PV 2CATPase (32%, Figure 7B, lane 2). The extent of interaction between 2CATPase and VP3 was quantified using a PhosphorImager and is expressed as percentage of the level observed in the CAV20 2CATPase and CAV20 VP3 Co-IP reaction. These results indicated a strong, direct interaction between 2CATPase and VP3 of the same origin (CAV20) but not between PV 2CATPase and CAV20 VP3, a combination that yielded the nonviable C20PP chimera. In contrast, the interaction between PV 2CATPase (N252S) and CAV20 VP3 (E180G) proteins was restored (78%, Figure 7B, lane 3) when the two mutations were incorporated into PV 2CATPase and CAV20 VP3, respectively, This observation, which correlates with the rescue of the nonviable phenotype of C20PP by the two mutations, strongly support the notion that sufficient protein-protein interaction between 2CATPase and VP3 is essential for the encapsidation process. The same Co-IP assays were also performed with α-actin antibody and empty resin as controls to ensure that the interactions were not due to non-specific binding of the proteins to the antibody or to the resin (data not shown). It should be noted that there was an extra protein band shown below the band of 2CATPase in each of the input lanes (Figure 7B, lane 4–6, indicated by asterisk), which was possibly generated from the internal initiation or premature termination of the translation of RNA transcripts of 2C coding sequences. Apparently these incomplete translation products could not be recognized by the 2C antibody since protein bands disappeared after Co-IP (Figure 7B, lane 1–3). These results confirm the specificity of the Co-IP assay in which the detection of the VP3 protein was due to its co-immunoprecipatation with the 2CATPase protein, recognized by anti-2CATPase antibody.

The data support our hypothesis that 2CATPase is required for viral encapsidation through a direct interaction with capsid protein VP3 and also confirm that 2CATPase interacts with VP3 through protein–protein rather than RNA-protein interaction. Given that 2CATPase and capsid proteins are colocalized on the surface of membranous vesicles in the RNA replication complex [46], [47], it is likely that 2CATPase interacts with VP3 either in the form of the mature protein or in the context of one of the VP3-containing capsid precursors (5S, 14S, 75S and 150S).
Discussion
The mechanism of picornavirus genome encapsidation has been a conundrum for many years. In previous studies on poliovirus morphogenesis, mostly trans encapsidation experiments were performed to determine the specificity of PV morphogenesis. In trans encapsidation experiments, the capsid proteins are offered from a different molecular entity to the parental genome either by coinfecting picornaviruses [14], by the vaccinia system [22], [23] or by expression from co-transfected cDNAs [64]. However, differences in the experimental design also affect the outcome of the experiments. Jia et al., [64] have reported trans-encapsidation of PV replicons into capsids of coxsackie B3, human rhinovirus 14, and coxsackie B24 viruses. These data, however, are in contrast to those of Porter et al., [23] and Barclay et al., [14], who failed to trans encapsidate the reporter PV replicon by super-infecting with CAV21.
In the current study, we have used a novel system to study encapsidation of closely related enteroviruses, the C-cluster enteroviruses (C-HEVs). They consist of two classes of virus serotypes: PV and C-CAVs. We have made use of differences between these viruses to investigate the effect of capsid exchanges on C-HEV morphogenesis. By studying a variety of C-HEV chimeric viruses in which the capsid P1 precursors were exchanged, we now present direct evidence for the involvement of 2CATPase in enterovirus morphogenesis via direct interaction with capsid protein VP3. Different from trans encapsidation assays, in which the capsid proteins are offered from a different molecular entity to the parental genome, our experiments were designed to measure cis encapsidation of genomes, in which the capsid is provided (generated) by the chimeric genome itself. It appears that in these chimeras the non-structural proteins are more discriminatory to capsid proteins because they are required to proteolytically process the heterologous capsid precursor. In addition, the quality and quantity of the heterologous capsid proteins produced might not be ideal for the chimeric genome. As we have shown here, favorable conditions for encapsidation are rarely met in the chimera, even if the viruses are as closely related as PV and CAV20.
In a previous study on genetic recombination between PV and C-CAVs, we observed that capsid chimera C20PP initially could not grow [48], an observation indicating that it harbors defect(s) debilitating the viral replication life cycle. Translation in HeLa cell extracts showed, however, that both translation and proteolytic processing of the CAV20 capsid precursor in C20PP was unimpaired. This is not surprising since the 3CDpro proteins of poliovirus and CAV20 are closely related in amino acid sequence [48] and because the 3CDpro cleavage sites within the P1 precursor of PVM and CAV20/21 are well conserved (Supplementary Table 1 in Text S1). These two facts reduce the likelihood of a processing defect of the foreign capsid precursor prior to packaging. In contrast, a chimera consisting of the PV capsid and the coxsackie virus B3 (CBV3) nonstructural domains was not only dead but it also revealed a processing defect of the PV capsid precursor [65]. CBV3 is an enterovirus belonging to the B-cluster, and its genetic kinship with PV is much more distant than that between PV and CAV20. Thus, CBV3 3CDpro proteinase was apparently unable to properly cleave the PV capsid precursor. Similar results were obtained with a chimera of PV in which the 3C-coding region was derived from HRV14. The foreign proteinase was not capable of recognizing the PV-specific processing sites within the capsid precursor [57].
The robust RNA replication phenotype of C20PP demonstrated here with the use of a new reporter virus construct suggested to us that this chimera might be quasi-infectious with respect to encapsidation. This was proven to be correct since viable viruses were found upon blind passages of C20PP but they harbored mutations. The virus isolates from two independent transfections contained a mutation either in 2CATPase (N252S) or in VP3 (E180G). Either of these single mutations was able to partially rescue the defective encapsidation and growth phenotype of C20PP. Introducing both mutations together into the C20PP genome fully rescued packaging and resulted in normal production of progeny virus. It is noteworthy that we never found both mutations in a single isolate even after eight passages. Perhaps the mutations conferred to the variants too little of an advantage to be selected under the conditions of the experiments. As discussed earlier, an involvement of essential RNA sequences in the 2CATPase and VP3 coding sequences during the process of encapsidation can be excluded. A direct interaction between the 2CATPase and VP3 proteins, suggested by the genetic experiments, was confirmed by biochemical assays using either purified or in vitro translated PV and CAV20 proteins. It should be noted that 2CATPase also functions in the viral life cycle in the form of its precursor 2BCATPase. A requirement of an interaction between 2BCATPase and VP3 for packaging is, however, unlikely since 2B is less homologous between PV and CAV20 in sequence than 2CATPase and the exchange of the mature 2CATPase protein alone is sufficient for full rescue. We are currently investigating whether the interaction between 2CATPase and VP3 involves the mature VP3 polypeptide or one of the capsid intermediates during viral assembly and/or maturation.
The encapsidation defect of C20PP could also be rescued by replacing the entire 2CATPase coding sequence of PV with that of CAV20. Additional experiments indicated that the lethal growth phenotypes of other CAV/CAV capsid chimeras could also be reversed by replacing their 2CATPase coding sequence with that of the capsid donor virus. It is noteworthy that these observations are not contradictory to the scenario of another chimera previously described. PC20C20, which, in contrast to C20PP, possesses a chimeric genome encoding PV capsid and CAV20 nonstructural protein sequences, grows as well as wt PV [48]. We have previously proposed that the evolutionary direction is from C-CAV to PV within C-HEVs resulting in a receptor switch from ICAM-1 to CD155 during the speciation [48]. If so, the newly emerged PV capsid may still be compatible with 2CATPase of the C-CAV ancestors and achieve sufficient interaction required for encapsidation.
Our data also clarify some unanswered questions about previous trans encapsidation experiments using poliovirus replicons containing a reporter gene in the capsid-coding region [14], [23]. Those studies showed that CAV21 was not able to encapsidate a PV replicon even though co-infection of cells with CAV21 resulted in high levels of replication of both CAV21 and the PV replicon. The most likely reason for those results is that the CAV21 capsid, similar to CAV20 capsid, fails to properly interact with PV 2CATPase. From their studies with hydantoin, Vance et al., have suggested that 2CATPase might have a role in encapsidation by an association of the progeny RNA with the capsid [32]. In other experiments with the same drug, however, Oh et al., [66] have recently proposed that hydantoin inhibits the release of the progeny RNA from the replication complex prior to encapsidation. Whether the interaction between 2CATPase and VP3, as we have observed in our studies, is required during or just before the union of the RNA with capsid proteins remains to be determined.
An intriguing phenomenon of poliovirus encapsidation is that only newly replicated RNA molecules are incorporated into virions [11], [12], an observation reported also for some other RNA viruses such as flock house virus [67], [68], [69], and brome mosaic virus [70]. Coupling encapsidation specifically with replication offers an efficient mechanism of discriminating against cellular RNAs or viral mRNA. Since genome replication is coupled with translation [71], [72] the link to encapsidation “can impose a form of late proofreading” for the progeny virus [12]. In Figure 8, we present a model of morphogenesis that is based, admittedly, on much speculation. We currently propose that in the context of the membrane-associated replication complex 2CATPase will directly interact with a 14S pentamer via VP3. The pentamer will then bind the newly emerging, VPg-linked genomic RNA [9] while the assembly of the virion proceeds in close contact with the membranous environment. This model offers a new mechanism for the specificity of enterovirus encapsidation: it is dependent on protein/protein interactions at the site of the active replication complex.

It is likely that our model will be applicable to most, if not all, picornaviruses as well as to other families of plus strand RNA viruses. For example, the requirement for an interaction between a capsid protein and nonstructural proteins for encapsidation has also been observed for members of the Flaviviridae. Murray et al., reported that several assembly-deficient core mutants of HCV genotype 2a (Hepacivirus genus) could be rescued by compensatory mutations in p7 or NS2 [73]. In addition, it was shown that HCV core and NS5A colocalize on the surface of lipid droplets, a process required for particle assembly [74]. Furthermore, recent reports indicate the importance of nonstructural proteins in the maturation of Kunjin virus and Yellow fever virus (Flavivirus genus) [75], [76] and of bovine diarrhea virus (Pestivirus genus) [77].
We have noted before that Aichi virus, a member of the Kobuvirus genus in the Picornaviridae, requires a 5′-terminal RNA element for encapsidation [29]. This signal by itself, however, is not sufficient to confer encapsidation specificity since it can be replaced by a similar stem loop from hepatitis A virus, a member of the genus Hepatovirus of Picornaviridae, and the resulting chimera expresses a severe proliferation phenotype [27]. On the other hand different genera of Picornaviridae may have evolved different strategies of encapsidation. This is not entirely unlikely considering the fundamentally different strategies that different genera of Flaviviridae are using to control genome translation (cap-dependent vs. IRES-dependent initiation of translation [78].
Our model does not explain, however, how VPg-linked minus-stranded poliovirus RNA is discriminated against in encapsidation. Previous studies have reported that several picornaviral 2CATPase proteins bind specifically to the 3′-end of minus strand RNA in vitro [79], [80]. However, the importance of this interaction, if any, for encapsidation is unlikely. Normally, plus strands are produced in great excess over minus strands [81], a phenomenon thought to lead to the depletion of free minus strands by forming the replicative form (RF) or replication intermediates (RI) [18]. If the balance between the plus and the minus strands is disturbed, free minus strands may emerge from the replication complex and they may then be encapsidated. Indeed, encapsidation of both plus and minus strand genomic RNAs was observed in non-cytopathogenic CBV3 that were isolated from persistently infected murine hearts and cardiac myocyte cultures [82]. It is possible that the non-cytopathogenic CBV3 produces minus stranded RNA in excess such that it will emerge from replication complexes where capsid precursors are waiting to encapsidate them. Whether the “cap” of a positively charged VPg on both plus or minus strands plays a role in this process remains to be seen.
Supporting Information
Zdroje
1. SemlerBL
WimmerE
2002 Molecular biology of picornaviruses. Washington D.C. ASM press 502
2. BartenschlagerR
Junker-NiepmannM
SchallerH
1990 The P gene product of hepatitis B virus is required as a structural component for genomic RNA encapsidation. J Virol 64 5324 5332
3. FrolovaE
FrolovI
SchlesingerS
1997 Packaging signals in alphaviruses. J Virol 71 248 258
4. GellerR
VignuzziM
AndinoR
FrydmanJ
2007 Evolutionary constraints on chaperone-mediated folding provide an antiviral approach refractory to development of drug resistance. Gene Dev 21 195 205
5. Ypma-WongMF
DewaltPG
JohnsonVH
LambJG
SemlerBL
1988 Protein 3CD is the major poliovirus proteinase responsible for cleavage of the P1 capsid precursor. Virology 166 265 270
6. HellenCUT
WimmerE
1995 Maturation of Poliovirus capsid proteins.
RotbartHA
Human enterovirus infections Washington, DC ASM Press 155 174
7. RacanielloVR
2007 Picornaviridae: The Viruses and Their Replication.
KnipeDM
Howley
PM
Fundamental virology New York Lippincott Williams & Wilkins 795 838
8. JacobsonMF
BaltimoreD
1968 Morphogenesis of poliovirus. I. Association of the viral RNA with coat protein. J Mol Biol 33 369 378
9. NugentCI
KirkegaardK
1995 RNA binding properties of poliovirus subviral particles. J Virol 69 13 22
10. PfisterT
EggerD
BienzK
1995 Poliovirus subviral particles associated with progeny RNA in the replication complex. J Gen Virol 76 (Pt 1) 63 71
11. MollaA
PaulAV
WimmerE
1991 Cell-free, de novo synthesis of poliovirus. Science 254 1647 1651
12. NugentCI
JohnsonKL
SarnowP
KirkegaardK
1999 Functional coupling between replication and packaging of poliovirus replicon RNA. J Virol 73 427 435
13. JohnsonVH
SemlerBL
1988 Defined recombinants of poliovirus and coxsackievirus: sequence-specific deletions and functional substitutions in the 5′-noncoding regions of viral RNAs. Virology 162 47 57
14. BarclayW
LiQ
HutchinsonG
MoonD
RichardsonA
1998 Encapsidation studies of poliovirus subgenomic replicons. J Gen Virol 79 (Pt 7) 1725 1734
15. XiangW
HarrisKS
AlexanderL
WimmerE
1995 Interaction between the 5′-terminal cloverleaf and 3AB/3CDpro of poliovirus is essential for RNA replication. J Virol 69 3658 3667
16. GromeierM
AlexanderL
WimmerE
1996 Internal ribosomal entry site substitution eliminates neurovirulence in intergeneric poliovirus recombinants. Proc Natl Acad Sci U S A 93 2370 2375
17. AlexanderL
LuHH
WimmerE
1994 Polioviruses containing picornavirus type 1 and/or type 2 internal ribosomal entry site elements: genetic hybrids and the expression of a foreign gene. Proc Natl Acad Sci U S A 91 1406 1410
18. WimmerE
HellenCU
CaoX
1993 Genetics of poliovirus. Annu Rev Genet 27 353 436
19. LuHH
WimmerE
1996 Poliovirus chimeras replicating under the translational control of genetic elements of hepatitis C virus reveal unusual properties of the internal ribosomal entry site of hepatitis C virus. Proc Natl Acad Sci U S A 93 1412 1417
20. RohllJB
MoonDH
EvansDJ
AlmondJW
1995 The 3′ untranslated region of picornavirus RNA: features required for efficient genome replication. J Virol 69 7835 7844
21. KajigayaS
ArakawaH
KugeS
KoiT
ImuraN
1985 Isolation and characterization of defective-interfering particles of poliovirus Sabin 1 strain. Virology 142 307 316
22. PorterDC
AnsardiDC
MorrowCD
1995 Encapsidation of poliovirus replicons encoding the complete human immunodeficiency virus type 1 gag gene by using a complementation system which provides the P1 capsid protein in trans. J Virol 69 1548 1555
23. PorterDC
AnsardiDC
WangJ
McPhersonS
MoldoveanuZ
1998 Demonstration of the specificity of poliovirus encapsidation using a novel replicon which encodes enzymatically active firefly luciferase. Virology 243 1 11
24. MuellerS
PapamichailD
ColemanJR
SkienaS
WimmerE
2006 Reduction of the rate of poliovirus protein synthesis through large-scale codon deoptimization causes attenuation of viral virulence by lowering specific infectivity. J Virol 80 9687 9696
25. ReuerQ
KuhnRJ
WimmerE
1990 Characterization of poliovirus clones containing lethal and nonlethal mutations in the genome-linked protein VPg. J Virol 64 2967 2975
26. CaoX
WimmerE
1996 Genetic variation of the poliovirus genome with two VPg coding units. Embo J 15 23 33
27. CheneyIW
NaimS
ShimJH
ReinhardtM
PaiB
2003 Viability of poliovirus/rhinovirus VPg chimeric viruses and identification of an amino acid residue in the VPg gene critical for viral RNA replication. J Virol 77 7434 7443
28. PaulAV
PetersJ
MugaveroJ
YinJ
van BoomJH
2003 Biochemical and genetic studies of the VPg uridylylation reaction catalyzed by the RNA polymerase of poliovirus. J Virol 77 891 904
29. SasakiJ
TaniguchiK
2003 The 5′-end sequence of the genome of Aichi virus, a picornavirus, contains an element critical for viral RNA encapsidation. J Virol 77 3542 3548
30. FrancoD
PathakHB
CameronCE
RombautB
WimmerE
2005 Stimulation of poliovirus RNA synthesis and virus maturation in a HeLa cell-free in vitro translation-RNA replication system by viral protein 3CDpro. Virol J 2 86
31. LiJP
BaltimoreD
1988 Isolation of poliovirus 2C mutants defective in viral RNA synthesis. J Virol 62 4016 4021
32. VanceLM
MoscufoN
ChowM
HeinzBA
1997 Poliovirus 2C region functions during encapsidation of viral RNA. J Virol 71 8759 8765
33. ChoMW
TeterinaN
EggerD
BienzK
EhrenfeldE
1994 Membrane rearrangement and vesicle induction by recombinant poliovirus 2C and 2BC in human cells. Virology 202 129 145
34. EggerD
GosertR
BienzK
2002 Role of cellular structures in viral RNA replication.
SemlerBL
WimmerE
Molecular Biology of Picornaviruses Washington, D. C ASM Press 247 255
35. PaulAV
BelovGA
EhrenfeldE
WimmerE
2009 Model of picornavirus RNA replication.
CameronCE
GotteM
RaneyKD
Viral Genome Replication: springer 3 24
36. RodriguezPL
CarrascoL
1995 Poliovirus protein 2C contains two regions involved in RNA binding activity. J Biol Chem 270 10105 10112
37. LiJP
BaltimoreD
1990 An intragenic revertant of a poliovirus 2C mutant has an uncoating defect. J Virol 64 1102 1107
38. GorbalenyaAE
KooninEV
1993 Helicases: amino acid sequence comparisons and structure-function relationships. Curr Opin Struct Biol 3 419 429
39. MirzayanC
WimmerE
1994 Biochemical studies on poliovirus polypeptide 2C: evidence for ATPase activity. Virology 199 176 187
40. RodriguezPL
CarrascoL
1993 Poliovirus protein 2C has ATPase and GTPase activities. J Biol Chem 268 8105 8110
41. PfisterT
WimmerE
1999 Characterization of the nucleoside triphosphatase activity of poliovirus protein 2C reveals a mechanism by which guanidine inhibits poliovirus replication. J Biol Chem 274 6992 7001
42. AdamsP
KandiahE
EffantinG
StevenAC
EhrenfeldE
2009 Poliovirus 2C protein forms homo-oligomeric structures required for ATPase activity. J Biol Chem 284 22012 22021
43. PaulAV
MollaA
WimmerE
1994 Studies of a putative amphipathic helix in the N-terminus of poliovirus protein 2C. Virology 199 188 199
44. TeterinaNL
GorbalenyaAE
EggerD
BienzK
EhrenfeldE
1997 Poliovirus 2C protein determinants of membrane binding and rearrangements in mammalian cells. J Virol 71 8962 8972
45. PfisterT
JonesKW
WimmerE
2000 A cysteine-rich motif in poliovirus protein 2C(ATPase) is involved in RNA replication and binds zinc in vitro. J Virol 74 334 343
46. BienzK
EggerD
PasamontesL
1987 Association of polioviral proteins of the P2 genomic region with the viral replication complex and virus-induced membrane synthesis as visualized by electron microscopic immunocytochemistry and autoradiography. Virology 160 220 226
47. PfisterT
PasamontesL
TroxlerM
EggerD
BienzK
1992 Immunocytochemical localization of capsid-related particles in subcellular fractions of poliovirus-infected cells. Virology 188 676 684
48. JiangP
FaaseJA
ToyodaH
PaulA
WimmerE
2007 Evidence for emergence of diverse polioviruses from C-cluster coxsackie A viruses and implications for global poliovirus eradication. Proc Natl Acad Sci U S A 104 9457 9462
49. MendelsohnCL
WimmerE
RacanielloVR
1989 Cellular receptor for poliovirus: molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell 56 855 865
50. KoikeS
HorieH
IseI
OkitsuA
YoshidaM
1990 The poliovirus receptor protein is produced both as membrane-bound and secreted forms. Embo J 9 3217 3224
51. NewcombeNG
AnderssonP
JohanssonES
AuGG
LindbergAM
2003 Cellular receptor interactions of C-cluster human group A coxsackieviruses. J Gen Virol 84 3041 3050
52. HogleJM
ChowM
FilmanDJ
1985 Three-dimensional structure of poliovirus at 2.9 A resolution. Science 229 1358 1365
53. XiaoC
Bator-KellyCM
RiederE
ChipmanPR
CraigA
2005 The crystal structure of coxsackievirus A21 and its interaction with ICAM-1. Structure 13 1019 1033
54. van der WerfS
BradleyJ
WimmerE
StudierFW
DunnJJ
1986 Synthesis of infectious poliovirus RNA by purified T7 RNA polymerase. Proc Natl Acad Sci U S A 83 2330 2334
55. LiuY
FrancoD
PaulAV
WimmerE
2007 Tyrosine 3 of poliovirus terminal peptide VPg(3B) has an essential function in RNA replication in the context of its precursor protein, 3AB. J Virol 81 5669 5684
56. EminiEA
SchleifWA
ColonnoRJ
WimmerE
1985 Antigenic conservation and divergence between the viral-specific proteins of poliovirus type 1 and various picornaviruses. Virology 140 13 20
57. DewaltPG
LawsonMA
ColonnoRJ
SemlerBL
1989 Chimeric picornavirus polyproteins demonstrate a common 3C proteinase substrate specificity. J Virol 63 3444 3452
58. CornellCT
SemlerBL
2002 Subdomain specific functions of the RNA polymerase region of poliovirus 3CD polypeptide. Virology 298 200 213
59. BellYC
SemlerBL
EhrenfeldE
1999 Requirements for RNA replication of a poliovirus replicon by coxsackievirus B3 RNA polymerase. J Virol 73 9413 9421
60. TeterinaNL
GorbalenyaAE
EggerD
BienzK
RinaudoMS
2006 Testing the modularity of the N-terminal amphipathic helix conserved in picornavirus 2C proteins and hepatitis C NS5A protein. Virology 344 453 467
61. LiX
LuHH
MuellerS
WimmerE
2001 The C-terminal residues of poliovirus proteinase 2A(pro) are critical for viral RNA replication but not for cis - or trans-proteolytic cleavage. J Gen Virol 82 397 408
62. LuHH
LiX
CuconatiA
WimmerE
1995 Analysis of picornavirus 2A(pro) proteins: separation of proteinase from translation and replication functions. J Virol 69 7445 7452
63. FeuerR
MenaI
PagariganR
SlifkaMK
WhittonJL
2002 Cell cycle status affects coxsackievirus replication, persistence, and reactivation in vitro. J Virol 76 4430 4440
64. JiaXY
Van EdenM
BuschMG
EhrenfeldE
SummersDF
1998 trans-encapsidation of a poliovirus replicon by different picornavirus capsid proteins. J Virol 72 7972 7977
65. VegaE
PallanschMA
ObersteMS
Interspecies enterovirus recombination;2009 University of British Columbia Vancouver, BC, Canada
66. OhHS
PathakHB
GoodfellowIG
ArnoldJJ
CameronCE
2009 Insight into poliovirus genome replication and encapsidation obtained from studies of 3B-3C cleavage site mutants. J Virol 83 9370 9387
67. VenterPA
KrishnaNK
SchneemannA
2005 Capsid protein synthesis from replicating RNA directs specific packaging of the genome of a multipartite, positive-strand RNA virus. J Virol 79 6239 6248
68. KhromykhAA
VarnavskiAN
SedlakPL
WestawayEG
2001 Coupling between replication and packaging of flavivirus RNA: evidence derived from the use of DNA-based full-length cDNA clones of Kunjin virus. J Virol 75 4633 4640
69. VolkovaE
GorchakovR
FrolovI
2006 The efficient packaging of Venezuelan equine encephalitis virus-specific RNAs into viral particles is determined by nsP1-3 synthesis. Virology 344 315 327
70. AnnamalaiP
RaoAL
2006 Packaging of brome mosaic virus subgenomic RNA is functionally coupled to replication-dependent transcription and translation of coat protein. J Virol 80 10096 10108
71. NovakJE
KirkegaardK
1994 Coupling between genome translation and replication in an RNA virus. Gene Dev 8 1726 1737
72. Hagino-YamagishiK
NomotoA
1989 In vitro construction of poliovirus defective interfering particles. J Virol 63 5386 5392
73. MurrayCL
JonesCT
TasselloJ
RiceCM
2007 Alanine scanning of the hepatitis C virus core protein reveals numerous residues essential for production of infectious virus. J Virol 81 10220 10231
74. AppelN
ZayasM
MillerS
Krijnse-LockerJ
SchallerT
2008 Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathog 4 e1000035
75. KummererBM
RiceCM
2002 Mutations in the yellow fever virus nonstructural protein NS2A selectively block production of infectious particles. J Virol 76 4773 4784
76. KhromykhAA
VarnavskiAN
WestawayEG
1998 Encapsidation of the flavivirus kunjin replicon RNA by using a complementation system providing Kunjin virus structural proteins in trans. J Virol 72 5967 5977
77. AgapovEV
MurrayCL
FrolovI
QuL
MyersTM
2004 Uncleaved NS2-3 is required for production of infectious bovine viral diarrhea virus. J Virol 78 2414 2425
78. LiuY
WimmerE
PaulAV
2009 Cis-acting RNA elements in human and animal plus-strand RNA viruses. Biochim Biophys Acta 1789 495 517
79. BanerjeeR
TsaiW
KimW
DasguptaA
2001 Interaction of poliovirus-encoded 2C/2BC polypeptides with the 3′ terminus negative-strand cloverleaf requires an intact stem-loop b. Virology 280 41 51
80. BanerjeeR
EcheverriA
DasguptaA
1997 Poliovirus-encoded 2C polypeptide specifically binds to the 3′-terminal sequences of viral negative-strand RNA. J Virol 71 9570 9578
81. NovakJE
KirkegaardK
1991 Improved method for detecting poliovirus negative strands used to demonstrate specificity of positive-strand encapsidation and the ratio of positive to negative strands in infected cells. J Virol 65 3384 3387
82. KimKS
TracyS
TapprichW
BaileyJ
LeeCK
2005 5′-Terminal deletions occur in coxsackievirus B3 during replication in murine hearts and cardiac myocyte cultures and correlate with encapsidation of negative-strand viral RNA. J Virol 79 7024 7041
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 8
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- The Transcription Factor Rbf1 Is the Master Regulator for -Mating Type Controlled Pathogenic Development in
- PKC Signaling Regulates Drug Resistance of the Fungal Pathogen via Circuitry Comprised of Mkc1, Calcineurin, and Hsp90
- Contribution of Coagulases towards Disease and Protective Immunity
- Early Severe Inflammatory Responses to Uropathogenic Predispose to Chronic and Recurrent Urinary Tract Infection
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy