
The Proteasome Active Site Threonine Is Essential for Persistence Yet Dispensable for Replication and Resistance to Nitric Oxide
Previous work revealed that conditional depletion of the core proteasome subunits PrcB and PrcA impaired growth of Mycobacterium tuberculosis in vitro and in mouse lungs, caused hypersusceptibility to nitric oxide (NO) and impaired persistence of the bacilli during chronic mouse infections. Here, we show that genetic deletion of prcBA led to similar phenotypes. Surprisingly, however, an active site mutant proteasome complemented the in vitro and in vivo growth defects of the prcBA knockout (ΔprcBA) as well as its NO hypersensitivity. In contrast, long-term survival of M. tuberculosis in stationary phase and during starvation in vitro and in the chronic phase of mouse infection required a proteolytically active proteasome. Inhibition of inducible nitric oxide synthase did not rescue survival of ΔprcBA, revealing a function beyond NO defense, by which the proteasome contributes to M. tuberculosis fitness during chronic mouse infections. These findings suggest that proteasomal proteolysis facilitates mycobacterial persistence, that M. tuberculosis faces starvation during chronic mouse infections and that the proteasome serves a proteolysis-independent function.
Published in the journal:
. PLoS Pathog 6(8): e32767. doi:10.1371/journal.ppat.1001040
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1001040
Summary
Previous work revealed that conditional depletion of the core proteasome subunits PrcB and PrcA impaired growth of Mycobacterium tuberculosis in vitro and in mouse lungs, caused hypersusceptibility to nitric oxide (NO) and impaired persistence of the bacilli during chronic mouse infections. Here, we show that genetic deletion of prcBA led to similar phenotypes. Surprisingly, however, an active site mutant proteasome complemented the in vitro and in vivo growth defects of the prcBA knockout (ΔprcBA) as well as its NO hypersensitivity. In contrast, long-term survival of M. tuberculosis in stationary phase and during starvation in vitro and in the chronic phase of mouse infection required a proteolytically active proteasome. Inhibition of inducible nitric oxide synthase did not rescue survival of ΔprcBA, revealing a function beyond NO defense, by which the proteasome contributes to M. tuberculosis fitness during chronic mouse infections. These findings suggest that proteasomal proteolysis facilitates mycobacterial persistence, that M. tuberculosis faces starvation during chronic mouse infections and that the proteasome serves a proteolysis-independent function.
Introduction
Most cells continuously synthesize and degrade proteins in a regulated manner. Protein degradation is highly selective and this is achieved in part by localization of protease active sites within a barrel-shaped complex. This self-compartmentalization was first discovered for the proteasome [1], [2]. In all genera, the proteasome consists of a 20S cylindrical core particle, which contains two heptameric outer rings composed of α subunits, and two heptameric inner rings composed of the proteolytically active β subunits. The 20S proteasome belongs to the class of N-terminal nucleophile (Ntn) hydrolases, with a hydroxyl group of the amino-(N) terminal threonine functioning as catalytic nucleophile that reacts with peptide bonds of substrates or the electrophilic functional groups of proteasome inhibitors [3].
Bacterial proteasomes are only found in Actinomycetes [4], while other chambered proteases such as ClpAP, ClpXP, Lon, HslUV and FtsH are common in most bacteria [5], [6]. Mycobacterium tuberculosis encodes a proteasome and two CLP proteases, but lacks homologs of Lon and HslUV [7]. The proteasome accessory factors, Mycobacterium proteasomal ATPase (Mpa) and proteasome accessory factor A (PafA), are important for defense against reactive nitrogen intermediates (RNI) and for virulence of M. tuberculosis in the mouse [8]. Mpa assembles into a hexameric ATPase similar to the archeal proteasome associating nucleotidase (PAN) and the eukaryotic regulatory 19S cap [9], [10]. The M. tuberculosis 20S proteasome harbors electron dense plugs at the barrel ends created by the N-termini of its α subunits [11]. Removal of the N-terminal eight amino acids resulted in enhanced peptidolytic activity, suggesting that the M. tuberculosis proteasome has a gated structure and implying a role for accessory factors including Mpa in “gate opening” [9], [12], [13]. A direct interaction of purified Mpa with the 20S open gate mutant proteasome was demonstrated by electron microscopy [14].
In eukaryotic cells a covalently attached polymeric chain of ubiquitin targets proteins for degradation by the proteasome [15]. In M. tuberculosis, Pup, a prokaryotic ubiquitin-like protein, is ligated by PafA to proteasomal substrate proteins and serves as degradation signal [16], [17], [18]. Pup must be deamidated by Dop (deamidase of Pup) to activate it for conjugation to a substrate [16], [17], [18]. In vitro reconstitution assays with purified Dop, PafA, Pup, ATP and substrate proteins FabD (malonyl acyltransferase) or PanB (ketopantoate hydroxymethyltranferase) revealed that Dop and PafA are necessary and sufficient for in vitro pupylation of proteasome target proteins. Accordingly pupylation was severely impaired and PanB and FabD accumulated in an M. smegmatis dop deletion mutant [19]. Recently, the Mpa-proteasome complex has been reconstituted in vitro and shown to unfold and degrade Pup-tagged substrates via interaction of Mpa with Pup [20]. Interestingly Pup is degraded together with the substrate, in contrast to ubiquitin, which is recycled.
Numerous pupylated proteins of diverse cellular functions have been identified in M. smegmatis and M. tuberculosis [21], [22]. The overlap between nitrosylated and pupylated proteins suggests that the proteasome is important for turnover of nitrosylated proteins [22], [23]. This hypothesis is substantiated by hypersusceptibility to reactive nitrogen intermediates (RNI) of M. tuberculosis lacking proteasome associated factors or depleted for the proteasome core subunits PrcBA [8], [24]. However, it is unclear if accumulation of nitrosylated proteins or any other proteasome substrate(s) caused the growth and persistence defects of proteasome deficient M. tuberculosis in mouse lungs. To gain more insight into proteasome core function, we constructed a prcBA deletion mutant (ΔprcBA) and complemented it with either an active wild type core proteasome or a proteolytically defective, active site mutant proteasome. Our data suggest that proteasomal proteolysis is dispensable for in vitro and in vivo replication of M. tuberculosis and for resistance to RNI. Inhibition of inducible nitric oxide synthase (iNOS) did not affect killing of ΔprcBA, indicating that defense against NO is likely not a major activity by which the proteasome facilitates mycobacterial persistence. However, M. tuberculosis expressing the proteolysis defective proteasome was severely impaired in stationary phase survival, died in response to carbon starvation and failed to persist during chronic mouse infections. Thus, the M. tuberculosis proteasome may promote survival in vivo by opposing starvation.
Results
The 20S core proteasome is not essential but is required for optimal growth and resistance to nitric oxide
The genes encoding the M. tuberculosis proteasome core subunits PrcB and PrcA were predicted to be essential or required for optimal growth in vitro [25]. Conditional depletion of the proteasome core subunits via transcriptional silencing of prcBA resulted in impaired growth on agar plates and in liquid culture [24]. Here, we genetically deleted prcBA (Figure S1) resulting in loss of proteasome activity (Figure 1A). Expression of prcBA from a constitutive promoter on an episomal plasmid restored PrcB expression and proteasome activity in the complemented mutant (ΔprcBA+PrcBA) (Figure 1A, S1C). The prcBA knockout (ΔprcBA) was viable, yet impaired for growth on agar plates and had a small but reproducible growth defect in liquid culture, confirming previous observations (Figure 1B, C). The growth defects were restored in the complemented mutant. These data demonstrate that while the core proteasome is required for optimal growth of M. tuberculosis in vitro, it is not essential. The growth defect of ΔprcBA was more evident on agar plates than in liquid medium, similar to what we previously observed after prcBA silencing [24].

M. tuberculosis mutants that lack the mycobacterial proteasome ATPase Mpa or the Pup ligase PafA or are depleted for PrcBA are hypersusceptible to RNI [8], [24]. Similarly, viability of ΔprcBA was almost ten-fold reduced compared to wt M. tuberculosis after exposure to acidified sodium nitrite (Figure 1D). This increased killing was complemented when PrcBA were expressed from a plasmid. Thus, the 20 S proteasome core is required for resistance against RNI in vitro.
The core proteasome is required for virulence in immune competent and immune compromised mice
The mouse model of tuberculosis is characterized by an acute phase, in which the bacteria replicate actively for approximately three weeks and a chronic phase, during which the bacteria persist at stable numbers. Silencing of prcBA reduced replication of M. tuberculosis during the acute phase and persistence during the chronic phase of infection in mouse lungs [24]. Genetic deletion of prcBA similarly affected in vivo growth and persistence of M. tuberculosis (Figure 2A, B). At three weeks post infection CFU in lungs were 1.5 log10 lower in ΔprcBA infected mice than in mice infected with wt M. tuberculosis and at 16 weeks post infection this difference increased to 2 log10 (P = 0.001 and P = 0.009). The virulence defects were fully restored in the complemented mutant. Nitric oxide generated by inducible nitric oxide synthase (iNOS) is required to control mycobacterial replication in mice [26] and lack of proteasome activity resulted in increased susceptibility of M. tuberculosis to RNI (Figure 1D). To determine if NO produced by iNOS was responsible for the decline in viability of ΔprcBA during the chronic phase of the infection, infected mice were treated with an iNOS-specific inhibitor L-N6-iminoethyl-lysine (L-NIL) [27], [28] starting at day 25 post infection (Figure 2C). In mice infected with wt M. tuberculosis, L-NIL treatment resulted in a failure to control bacterial replication, so that bacillary loads were increased by two orders of magnitude in lungs at 25 days post treatment (day 50 post infection) compared to the control group treated with the inactive enantiomer (D-NIL) (Figure 2C). The remaining L-NIL-treated mice infected with wt M. tuberculosis succumbed between day 50 and day 75 post-infection. In contrast, only a 2-fold increase in bacillary burden of ΔprcBA was observed in lungs upon L-NIL treatment compared to D-NIL treatment at 25 days post treatment. L-NIL treated mice infected with ΔprcBA survived until the end of the experiment (day 200), and bacterial numbers in the lungs of both L-NIL and D-NIL treated mice declined by 20-fold (Figure 2C). There was only a slight increase in the number of nodular lesions at day 200 in mice infected with ΔprcBA and treated with L - Nil compared to D-Nil treated mice (not shown). Altogether, these data suggest that iNOS is not required to control ΔprcBA during chronic infection in mice.

Mutation of the PrcB active site residue threonine dramatically impairs proteolytic activity
The 20S proteasome is a multimeric protein complex and we hypothesized that lack of expression of the proteasome core subunits PrcB and PrcA could have different physiological consequences than lack of PrcB-mediated proteolytic activity. To test this, we expressed a proteasome active site mutant in ΔprcBA. In this mutant, the N-terminal threonine residue of the mature PrcB subunit was mutated to alanine (T1A). This mutation abolished proteolytic activity of the proteasome from Thermoplasma acidophilum [3], [29], [30]. To allow assembly of this mutant proteasome subunit into a 20S complex, we also deleted the pro-peptide of the PrcB subunit, which in an active proteasome is autocatalytically removed by the active site Thr, thereby exposing the amino group of Thr for nucleophilic attack on the target peptide bond [12], [29], [31]. Similar mutagenesis of the T. acidophilum proteasome allowed assembly of a mature 20S proteasome core [29], [31], [32]. The mutated prcB gene (pcrBT1A) was cloned including a C-terminal histidine tag in an operon with prcA (prcABT1A) and expressed in ΔprcBA. Immunoprecipitation of the PrcB subunit from lysates of this M. tuberculosis strain co-purified PrcA as determined by liquid chromatography-tandem mass spectrometry (Figure S2) indicating that a complex containing both subunits formed in vivo despite the PrcBT1A mutation. As expected, the mutant proteasome failed to complement proteolytic activity of ΔprcBA, measured by cleavage of the peptide substrate Suc-LLVY-AMC (Figure 3A), although the expression level of the PrcB subunit containing the T1A mutation was similar to that of wt PrcB (Figure 3B). To determine whether the T1A mutation affected proteolytic activity within the bacteria, GFP was fused to the proteasome substrate PanB (ketopantoate hydroxymethyltransferase). PanB has been shown to accumulate in M. tuberculosis lacking Mpa and in wt M. tuberculosis treated with the proteasome inhibitor epoxomicin [33]. We confirmed by 2-D SDS page analysis that PanB also accumulated in ΔprcBA (not shown). Similarly, the PanB-GFP fusion protein accumulated in ΔprcBA compared to wt M. tuberculosis as shown by GFP activity (Figure 3C) and GFP protein levels (Figure 3D). This was complemented when the intact core proteasome (PrcBA) was expressed in ΔprcBA. In contrast, the active site mutant proteasome (PrcAB-T1A) did not revert the accumulation of PanB-GFP (Figure 3C, D). Thus, the active site mutation T1A not only abolished proteasome activity against a peptide substrate in vitro, but also disrupted proteolytic activity of the proteasome within the bacteria.

The active site mutant proteasome enables optimal growth in vitro and in vivo, confers RNI resistance but is not sufficient for persistence in mice
Surprisingly, expression of PrcAB-T1A in ΔprcBA complemented its growth defect both on solid and in liquid media similar to expression of wt PrcBA (Figure 4A, B). The RNI hypersusceptibility of ΔprcBA was also complemented to a large degree by the active site mutant proteasome (Figure 4C). We next asked if the catalytic activity of the proteasome is required for M. tuberculosis to grow and persist in mice. The mutant proteasome complemented the in vivo growth defect of ΔprcBA similar to the wt proteasome (Figure 5A), suggesting that a proteolysis-independent activity of the 20S core is required for optimal growth of M. tuberculosis in mouse lungs. However, the persistence defect of ΔprcBA during the chronic phase of infection was not complemented by the T1A mutant proteasome and bacterial numbers in the lungs declined by 3 log10 between day 28 and day 200. Similarly, lung pathology, which was easily detectable on day 56 post-infection, decreased from day 56 to day 200 in mice infected with the T1A mutant proteasome complemented ΔprcBA (Figure 5B). Of note, ΔprcBA complemented with the mutant proteasome lost viability faster than ΔprcBA in mouse lungs (Figure 5A). The higher bacterial burden reached by the strain expressing the mutant proteasome at three weeks post infection likely resulted in a more efficient activation of the immune system resulting in faster killing of the bacilli. This hypothesis is supported by the increased kinetics of killing of ΔprcBA in the face of a higher bacterial burden when mice were infected with a mixture of equal numbers of wt M. tuberculosis and ΔprcBA (Figure 5C).

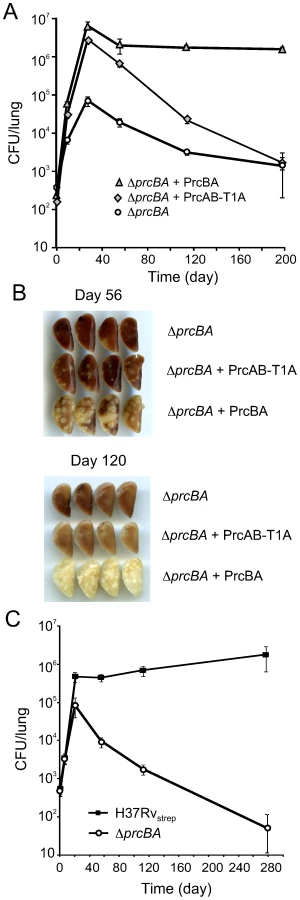
Altogether, these data suggest that the proteolytic activity of the proteasome is dispensable for growth of M. tuberculosis in vitro and in mice and for RNI resistance, yet proteasome mediated proteolysis is required for persistence of M. tuberculosis in mouse lungs.
A proteolytically active proteasome is required for survival in stationary phase and during starvation
Proteasomal proteolysis may be required during bacteriostasis or periods of slow replication to counter the effects of accumulating protein damage as well as to provide amino acids for energy metabolism during starvation. Proteolysis mediated by ClpP and Lon is important for the ability of E. coli to sustain starvation [34], [35]. We monitored survival during stationary phase and during complete starvation of wt M. tuberculosis, ΔprcBA and ΔprcBA complemented with the wt proteasome and the T1A active site mutant proteasome. In normal growth medium, M. tuberculosis grew exponentially for about 10 days, after which bacterial numbers remained almost constant over the next 170 days (Figure 6A). ΔprcBA was, as expected from earlier growth characterizations, impaired for growth. Upon entering stationary phase, viability of the ΔprcBA declined steadily. At 180 days post inoculation there was an approximately 3 log10 difference in viable counts between wt and ΔprcBA (Figure 6A). Both the growth and persistence defects were largely complemented by expression of the wt proteasome. The T1A mutant proteasome complemented the initial growth defect but failed to complement the persistence defect. Wt M. tuberculosis and ΔprcBA transformed with the wt proteasome also survived conditions of complete starvation without a significant decline in viability (Figure 6B). In contrast, viability of ΔprcBA and ΔprcBA transformed with the T1A mutant proteasome declined steadily during starvation (Figure 6B). Collectively, these data indicate that the proteasomal proteolysis is important for the ability of M. tuberculosis to survive conditions of starvation and stationary phase in vitro.

Discussion
The eukaryotic proteasome is ubiquitous and essential for many basic cellular processes including differentiation, proliferation, transcription, signal transduction, metabolic regulation, immune surveillance and others [36], [37]. In prokaryotes, proteasome deletion mutants have been generated in Thermoplasma acipophilum, Streptomyces lividans, S. coelicolor and M. smegmatis. T. acidophilum proteasome mutants were impaired for survival post heat shock but not under normal growth conditions [38]. Deletion of the proteasome in S. lividans, S. coelicolor and M. smegmatis did not reveal phenotypic defects [39], [40], [41]. The current work proves that the 20S proteasome is not essential for growth of M. tuberculosis. However, consistent with previous studies [24], [25] lack of the core proteasome resulted in a growth defect on plates and in liquid culture, suggesting that proteasome-mediated proteolysis is important for optimal in vitro growth of M. tuberculosis.
Surprisingly, however, a proteolytically defective proteasome fully complemented the growth defects and partially restored the RNI hypersusceptibility of ΔprcBA. Thus, these phenotpyes of ΔprcBA are likely not due to lack of proteasomal proteolysis. We cannot exclude that mutation of the active site threonine to alanine failed to completely abolish proteolysis. However, peptidolytic activity of the proteasome was undetectable in lysates expressing the T1A mutant proteasome and the proteasome substrate PanB tagged with GFP accumulated similarly in ΔprcBA and ΔprcBA expressing the T1A mutant proteasome. Thus, proteasome-mediated proteolysis was drastically impaired when the active site threonine was mutated to alanine in PrcB. The 20S core requires accessory factors for protein targeting to the proteolytic chamber [42]. In eukaryotes, proteasome accessory factors are not only found as part of the proteasome complex; they also form subcomplexes that regulate transcription and DNA repair and have chaperone function [43], [44], [45]. The stoichiometry of proteasome accessory factors that are free or in complex with the 20S core might be regulated. In the absence of the 20S core an excess of free accessory factors might affect growth. The T1A active-site mutant proteasome is likely to interact with Mpa and potentially other proteasome accessory factors despite its catalytic defect and thereby prevent phenotypes caused by an imbalance of free and complexed accessory factors.
Depletion of the 20S proteasome in M. tuberculosis caused hypersusceptibility to RNI and impaired persistence in the mouse [24]. Deletion of Mpa and PafA sensitized M. tuberculosis to RNI in vitro and resulted in impaired growth in the mouse [8]. However, the proteolytically defective active site mutant proteasome complemented the RNI hypersusceptibility of ΔprcBA to a large degree, suggesting that proteasomal proteolysis is not essential for conferring RNI resistance. Moreover, inhibition of iNOS, the major producer of nitric oxide in macrophages, had no impact on survival of ΔprcBA in mouse lungs. Thus other factors of the adaptive immune response appear responsible for killing ΔprcBA during the chronic phase of the infection. Of note, the attenuated growth of the M. tuberculosis Mpa mutant in the acute phase of infection was not rescued in iNOS-deficient mice [8]. Phagocyte oxidase activity may have compensated for inhibited iNOS activity; however, proteasome-depleted M. tuberculosis and the Mpa mutant were hyperresistant to oxidative stress in vitro [8], [24].
We propose that nutrient limitation might be responsible for the killing of ΔprcBA in mice. M. tuberculosis lacking the 20S core failed to survive prolonged stationary phase and nutrient starvation in vitro and was unable to persist in vivo. Proteasome-mediated proteolytic turnover seems essential for in vitro and in vivo persistence of M. tuberculosis, because the active site mutant proteasome expressing strain phenocopied the inability of ΔprcBA to persist both in vitro and in vivo. Nutritionally starved and clinically persistent M. tuberculosis share phenotypic similarities, including reduced acid-fastness and drug tolerance [46], [47], [48]. Starvation of M. tuberculosis reduced respiration to minimal levels, indicating a low metabolic activity, but the bacilli remained viable and were recoverable when returned to rich medium [49], [50]. Nutrient starvation-induced transcripts can be detected in human tuberculous granulomas [46], [51]. Moreover, the stringent response, required for long-term survival in culture, was also required for persistence of M. tuberculosis in mice [52], [53], [54]. In E. coli amino acid starvation is followed by increased ribosomal protein degradation via Lon protease to provide amino acids for the synthesis of new enzymes important for adaptation to starvation [35], [55]. Moreover, starvation and growth arrest are linked to the production of misfolded and aberrant proteins isoforms that need to be degraded to prevent toxicity [56]. In mammalian cells proteasomal protein degradation is crucial in supplying amino acids for the synthesis of new proteins during amino acid deprivation [57]. Similarly, M. tuberculosis might require the proteasome for amino acid supply and turnover of damaged proteins during long term persistence within its host.
In summary, this work demonstrates the essential role of the 20S proteasome proteolytic activity for M. tuberculosis to persist in vivo and reveals a mechanism beyond nitric oxide defense by which the proteasome contributes to mycobacterial fitness.
Materials and Methods
Ethics statement
All mouse procedures performed in this study were conducted following the National Institutes of Health guidelines for housing and care of laboratory animals and performed in accordance with institutional regulations after protocol review and approval by the Institutional Animal Care and Use Committee of Weill Cornell Medical College.
Strains, media and culture conditions
Wild-type M. tuberculosis (H37Rv) was obtained from Dr. Robert North, Trudeau Institute. Mycobacteria were grown at 37°C in Middlebrook 7H9 medium (Difco) containing 0.2% glycerol, 0.5% bovine serum albumin, 0.2% dextrose, 0.085% NaCl, and 0.05% Tween 80. Hygromycin B (50 mg/ml), kanamycin (15 mg/ml) and streptomycin (20 mg/ml) were included when required for selection.
Construction of ΔprcBA
PrcBA genes were deleted from the chromosome via homologous recombination following transduction with temperature-sensitive mycobacteriophage phAE87 [58]. 768 bp upstream of the start codon of prcB and 524 bp downstream of the stop codon of prcA were amplified by PCR from H37Rv genomic DNA and cloned into pJSC284 to flank the hygromycin resistance gene. pJSC284 is a derivative of pYUB854 containing a lambda cos site, and a unique PacI site. The resulting plasmid was ligated with the temperature-sensitive phage phAE87 and the resulting phage was used to infect M. tuberculosis. Hygromycin-resistant transductants were selected on 7H11 agar plates with 50 µg/ml hygromycin for 3 weeks and analyzed by Southern blot.
Complementation of ΔprcBA
PrcBA were PCR amplified from H37Rv genomic DNA with a forward primer specific to prcB (5′ - CGTCCGCGCATGCGTCCAGGAGGGCGGACAG-3′) and a reverse primer specific to prcA (5′-GACACGCGTCGGACGTTTAAACTCAGCCCG-3′). The resulting fragment was cloned into an episomal mycobacterial plasmid containing the mycobacterial promoter Pmyc1tetO [59] and a kanamycin resistance gene.
Construction of T1A proteasome mutant
PrcA was PCR amplified using primers 5′ - CGGGTGCGCATGCTTTCGGCTCCGAAGGAGGTGAG-3′ and 5′-ACTCAGCCCGACGATTCGCCGTCAGACTGC-3′ resulting in introduction of an SphI site at the 5′ end of prcA followed by a synthetic ribosome binding site (RBS). prcB was PCR amplified using primers 5′-GGCCACCATTGTCGCGCTGAAATACCCC-3′ and 5′ - CGCCTGCTCTGCAGTCAATGATGATGATGATGATGCTTCTCACCGCCATCGGAGCCGAAAGTATCC-3′ from the H37Rv genome resulting in deletion of the 5′ end encoding its pro-peptide, mutation of threonine 1 to alanine and addition of a C terminal hexahistidine tag, followed by a PstI site 3′ of prcB-T1AHis6 (encoded protein referred to as PrcAB-T1A). For expression of the active proteasome, prcBA was amplified from the H37Rv genome using primers 5′ - CGTCCGCGCATGCGTCCAGGAGGGCGGACAG-3′ and 5′-GGGGGCCCATCGATCTCTTAATTAAGGTAGAC-3′. The amplified fragments were cloned into an episomal mycobacterial plasmid containing the mycobacterial promoter Pmyc1tetO [59] and a kanamycin resistance gene.
Construction of PanB-GFP expression vector
The panB-gfp fusion was generated by PCR and cloned using the Gateway Cloning Technology (Invitrogen) behind a constitutive promoter into an integrative mycobacterial plasmid containing a streptomycin resistance gene.
Immunoblots
Cell lysates were prepared by bead-beating the cell pellets in PBS containing protease inhibitor cocktail (Complete Mini, Roche). Clarified cell lysates were filter sterilized by passage through a 0.2 µm filter. 15 µg cell lysates were subjected to SDS-PAGE, followed by transfer to a nitrocellulose membrane. Blots were probed with PrcB-specific and DlaT-specific rabbit sera at 1∶15,000 and 1∶10,000 dilutions in 5% skimmed-milk containing Tris-buffered saline with 0.05% Tween 20 (TBST). To assess PanB-GFP accumulation, blots were probed with anti-GFP antibody (Invitrogen). Secondary antibodies, donkey anti-rabbit (horse radish peroxidase coupled) or LI-COR 800 goat anti rabbit were used at 1∶30,000 dilution in 2% skimmed-milk containing TBST and at 1∶15,000 dilution in Odyssey blocking buffer, respectively. Blots were developed using Immobilon Western Chemiluminescent HRP substrate (Millipore) or using the Odyssey Infrared Imaging System (LI-COR Biosciences).
Proteasome activity assay
Bacteria were grown to OD580nm 1.0, washed as described above and cell pellets were lysed in 450 µl PBS containing protease inhibitor cocktail (Complete Mini, Roche) using a bead beater. Clarified cell lysates were filter sterilized by passage through a 0.2 µm filter and then adjusted to a final glycerol concentration of 10%. Proteasome activity was assessed as previously described [12]. Briefly, 50 µg of lysate were incubated with 100 µM Succinyl-Leu-Leu-Val-Tyr-aminomethyl coumarin (Suc-LLVY-AMC) in 20 mM HEPES, 0.5 mM EDTA buffer and fluorescence was monitored at excitation of 370 nm and emission of 430 nm at 37°C over 60 min in a 96 well-plate fluorimeter (Molecular Devices).
Determination of in vivo GFP and PanB-GFP accumulation
Cultures were grown to OD580nm 0.4–0.9 (mid log) and 1.0–1.5 ml of cultures were harvested, resuspended in 100 ml PBS and aliquoted into a black 96 well plate. Fluorescence was measured using excitation at 485 nm and emission at 515 nm. Relative fluorescence units were normalized to OD580nm.
In vitro stress susceptibility assays
Cultures were grown to log phase (OD580nm 0.6), washed in growth medium and single cell suspensions prepared in assay medium by centrifugation at 800 rpm for 12 minutes. Single cell suspensions were subsequently diluted to OD580nm 0.01. To test susceptibility to RNI, diluted cultures were incubated at pH 5.5 with or without 3mM or 5mM NaNO2 for 3 days at 37°C. To determine viability, serial dilutions of cultures were plated on 7H11 plates.
Stationary phase survival and starvation assays
For long-term survival experiments M. tuberculosis strains were grown in 7H9 medium to mid log phase. Single cell suspensions were prepared in 7H9 as described above and diluted into fresh medium to OD580n 0.01, in 10 ml triplicate cultures. For starvation conditions, single cell suspensions were prepared in PBS with 0.02% Tween 80 and diluted into PBS-Tween to OD580n 0.01 in triplicate 10 ml cultures. Cultures were incubated at 37°C under constant shaking (50 rpm). To determine growth and viability, serial dilutions of cultures were plated on 7H11 plates.
Animal infections
Eight week old, female C57BL/6 mice (Jackson Laboratory) were infected with M. tuberculosis strains by aerosol as described [24]. Bacterial numbers in organs were enumerated by plating organ homogenates for colony forming units (CFU) at indicated times. N6-(1-Iminoethyl)lysine (NIL; L - and D-enantiomers) (custom synthesized by DeCODE Chemicals [60], and a kind gift from Dr. C. Nathan) were given in acidified (pH 2.7) drinking water (4 mM) beginning on day 21 post infection and freshly prepared every 48 hr until the end of the experiment [26].
Supporting Information
{kind=link}
Zdroje
1. ArrigoAP
TanakaK
GoldbergAL
WelchWJ
1988 Identity of the 19S ‘prosome’ particle with the large multifunctional protease complex of mammalian cells (the proteasome). Nature 331 192 194
2. BaumeisterW
DahlmannB
HegerlR
KoppF
KuehnL
1988 Electron microscopy and image analysis of the multicatalytic proteinase. FEBS Lett 241 239 245
3. KisselevAF
SongyangZ
GoldbergAL
2000 Why does threonine, and not serine, function as the active site nucleophile in proteasomes? J Biol Chem 275 14831 14837
4. LupasA
ZuhlF
TamuraT
WolfS
NagyI
1997 Eubacterial proteasomes. Mol Biol Rep 24 125 131
5. De MotR
NagyI
WalzJ
BaumeisterW
1999 Proteasomes and other self-compartmentalizing proteases in prokaryotes. Trends Microbiol 7 88 92
6. GottesmanS
2003 Proteolysis in bacterial regulatory circuits. Annu Rev Cell Dev Biol 19 565 587
7. ColeST
BroschR
ParkhillJ
GarnierT
ChurcherC
1998 Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393 537 544
8. DarwinKH
EhrtS
Gutierrez-RamosJC
WeichN
NathanCF
2003 The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science 302 1963 1966
9. DarwinKH
LinG
ChenZ
LiH
NathanCF
2005 Characterization of a Mycobacterium tuberculosis proteasomal ATPase homologue. Mol Microbiol 55 561 571
10. NeuwaldAF
AravindL
SpougeJL
KooninEV
1999 AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res 9 27 43
11. HuG
LinG
WangM
DickL
XuRM
2006 Structure of the Mycobacterium tuberculosis proteasome and mechanism of inhibition by a peptidyl boronate. Mol Microbiol 59 1417 1428
12. LinG
HuG
TsuC
KunesYZ
LiH
2006 Mycobacterium tuberculosis prcBA genes encode a gated proteasome with broad oligopeptide specificity. Mol Microbiol 59 1405 1416
13. Cerda-MairaF
DarwinKH
2009 The Mycobacterium tuberculosis proteasome: more than just a barrel-shaped protease. Microbes Infect 11 1150 1155
14. WangT
LiH
LinG
TangC
LiD
2009 Structural insights on the Mycobacterium tuberculosis proteasomal ATPase Mpa. Structure 17 1377 1385
15. KerscherO
FelberbaumR
HochstrasserM
2006 Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu Rev Cell Dev Biol 22 159 180
16. PearceMJ
MintserisJ
FerreyraJ
GygiSP
DarwinKH
2008 Ubiquitin-like protein involved in the proteasome pathway of Mycobacterium tuberculosis. Science 322 1104 1107
17. BurnsKE
LiuWT
BoshoffHI
DorresteinPC
BarryCE3rd
2009 Proteasomal protein degradation in Mycobacteria is dependent upon a prokaryotic ubiquitin-like protein. J Biol Chem 284 3069 3075
18. StriebelF
ImkampF
SutterM
SteinerM
MamedovA
2009 Bacterial ubiquitin-like modifier Pup is deamidated and conjugated to substrates by distinct but homologous enzymes. Nat Struct Mol Biol 16 647 651
19. ImkampF
RosenbergerT
StriebelF
KellerPM
AmstutzB
2009 Deletion of dop in Mycobacterium smegmatis abolishes pupylation of protein substrates in vivo. Mol Microbiol 75 744 754
20. StriebelF
HunkelerM
SummerH
Weber-BanE
2010 The mycobacterial Mpa-proteasome unfolds and degrades pupylated substrates by engaging Pup's N-terminus. EMBO J
21. FestaRA
McAllisterF
PearceMJ
MintserisJ
BurnsKE
2010 Prokayrotic ubiquitin-like protein (Pup) proteome of Mycobacterium tuberculosis. PLoS One 5 e8589
22. WatrousJ
BurnsK
LiuWT
PatelA
HookV
2010 Expansion of the mycobacterial “PUPylome”. Mol Biosyst 6 376 385
23. RheeKY
Erdjument-BromageH
TempstP
NathanCF
2005 S-nitroso proteome of Mycobacterium tuberculosis: Enzymes of intermediary metabolism and antioxidant defense. Proc Natl Acad Sci U S A 102 467 472
24. GandotraS
SchnappingerD
MonteleoneM
HillenW
EhrtS
2007 In vivo gene silencing identifies the Mycobacterium tuberculosis proteasome as essential for the bacteria to persist in mice. Nature Medicine 13 1515 1520
25. SassettiCM
BoydDH
RubinEJ
2003 Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol 48 77 84
26. MacMickingJD
NorthRJ
LaCourseR
MudgettJS
ShahSK
1997 Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc Natl Acad Sci U S A 94 5243 5248
27. MooreWM
WebberRK
JeromeGM
TjoengFS
MiskoTP
1994 L-N6-(1-iminoethyl)lysine: a selective inhibitor of inducible nitric oxide synthase. J Med Chem 37 3886 3888
28. StengerS
ThuringH
RollinghoffM
ManningP
BogdanC
1995 L-N6-(1-iminoethyl)-lysine potently inhibits inducible nitric oxide synthase and is superior to NG-monomethyl-arginine in vitro and in vivo. Eur J Pharmacol 294 703 712
29. SeemullerE
LupasA
BaumeisterW
1996 Autocatalytic processing of the 20S proteasome. Nature 382 468 471
30. SeemullerE
LupasA
StockD
LoweJ
HuberR
1995 Proteasome from Thermoplasma acidophilum: a threonine protease. Science 268 579 582
31. ZuhlF
SeemullerE
GolbikR
BaumeisterW
1997 Dissecting the assembly pathway of the 20S proteasome. FEBS Lett 418 189 194
32. ZwicklP
KleinzJ
BaumeisterW
1994 Critical elements in proteasome assembly. Nat Struct Biol 1 765 770
33. PearceMJ
AroraP
FestaRA
Butler-WuSM
GokhaleRS
2006 Identification of substrates of the Mycobacterium tuberculosis proteasome. EMBO J 25 5423 5432
34. DamerauK
St JohnAC
1993 Role of Clp protease subunits in degradation of carbon starvation proteins in Escherichia coli. J Bacteriol 175 53 63
35. KurodaA
NomuraK
OhtomoR
KatoJ
IkedaT
2001 Role of inorganic polyphosphate in promoting ribosomal protein degradation by the Lon protease in E. coli. Science 293 705 708
36. DahlmannB
2007 Role of proteasomes in disease. BMC Biochem 8 Suppl 1 S3
37. JungT
CatalgolB
GruneT
2009 The proteasomal system. Mol Aspects Med 30 191 296
38. RueppA
EckerskornC
BogyoM
BaumeisterW
1998 Proteasome function is dispensable under normal but not under heat shock conditions in Thermoplasma acidophilum. FEBS Lett 425 87 90
39. HongB
WangL
LammertynE
GeukensN
Van MellaertL
2005 Inactivation of the 20S proteasome in Streptomyces lividans and its influence on the production of heterologous proteins. Microbiology 151 3137 3145
40. KnipferN
ShraderTE
1997 Inactivation of the 20S proteasome in Mycobacterium smegmatis. Mol Microbiol 25 375 383
41. NagyI
BanerjeeT
TamuraT
SchoofsG
GilsA
2003 Characterization of a novel intracellular endopeptidase of the alpha/beta hydrolase family from Streptomyces coelicolor A3(2). J Bacteriol 185 496 503
42. DarwinKH
2009 Prokaryotic ubiquitin-like protein (Pup), proteasomes and pathogenesis. Nat Rev Microbiol 7 485 491
43. ChenZJ
SunLJ
2009 Nonproteolytic functions of ubiquitin in cell signaling. Mol Cell 33 275 286
44. FerdousA
KodadekT
JohnstonSA
2002 A nonproteolytic function of the 19S regulatory subunit of the 26S proteasome is required for efficient activated transcription by human RNA polymerase II. Biochemistry 41 12798 12805
45. NishiyamaA
TachibanaK
IgarashiY
YasudaH
TanahashiN
2000 A nonproteolytic function of the proteasome is required for the dissociation of Cdc2 and cyclin B at the end of M phase. Genes Dev 14 2344 2357
46. BettsJC
LukeyPT
RobbLC
McAdamRA
DuncanK
2002 Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol Microbiol 43 717 731
47. NykaW
1974 Studies on the effect of starvation on mycobacteria. Infect Immun 9 843 850
48. WayneLG
HayesLG
1996 An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect Immun 64 2062 2069
49. LoebelRO
ShorrE
RichardsonHB
1933 The Influence of Adverse Conditions upon the Respiratory Metabolism and Growth of Human Tubercle Bacilli. J Bacteriol 26 167 200
50. LoebelRO
ShorrE
RichardsonHB
1933 The Influence of Foodstuffs upon the Respiratory Metabolism and Growth of Human Tubercle Bacilli. J Bacteriol 26 139 166
51. FenhallsG
StevensL
MosesL
BezuidenhoutJ
BettsJC
2002 In situ detection of Mycobacterium tuberculosis transcripts in human lung granulomas reveals differential gene expression in necrotic lesions. Infect Immun 70 6330 6338
52. DahlJL
KrausCN
BoshoffHI
DoanB
FoleyK
2003 The role of Rel Mtb-mediated adaptation to stationary phase in long-term persistence of Mycobacterium tuberculosis in mice. Proc Natl Acad Sci U S A 100 10026 10031
53. PrimmTP
AndersenSJ
MizrahiV
AvarbockD
RubinH
2000 The stringent response of Mycobacterium tuberculosis is required for long-term survival. J Bacteriol 182 4889 4898
54. StallingsCL
StephanouNC
ChuL
HochschildA
NickelsBE
2009 CarD is an essential regulator of rRNA transcription required for Mycobacterium tuberculosis persistence. Cell 138 146 159
55. KurodaA
TanakaS
IkedaT
KatoJ
TakiguchiN
1999 Inorganic polyphosphate kinase is required to stimulate protein degradation and for adaptation to amino acid starvation in Escherichia coli. Proc Natl Acad Sci U S A 96 14264 14269
56. NystromT
2002 Translational fidelity, protein oxidation, and senescence: lessons from bacteria. Ageing Res Rev 1 693 703
57. VabulasRM
HartlFU
2005 Protein synthesis upon acute nutrient restriction relies on proteasome function. Science 310 1960 1963
58. BardarovS
KriakovJ
CarriereC
YuSW
VaamondeC
1997 Conditionally replicating mycobacteriophages: A system for transposon delivery to Mycobacterium tuberculosis. Proceedings Of The National Academy Of Sciences Of The United States Of America 94 10961 10966
59. EhrtS
GuoXV
HickeyCM
RyouM
MonteleoneM
2005 Controlling gene expression in mycobacteria with anhydrotetracycline and Tet repressor. Nucleic Acids Res 33 e21
60. DumontM
WilleE
CalingasanNY
NathanC
Flint BealM
2010 N-iminoethyl-L-lysine improves memory and reduces amyloid pathology in a transgenic mouse model of amyloid deposition. Neurochem Int 56 345 351
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 8
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- The Transcription Factor Rbf1 Is the Master Regulator for -Mating Type Controlled Pathogenic Development in
- PKC Signaling Regulates Drug Resistance of the Fungal Pathogen via Circuitry Comprised of Mkc1, Calcineurin, and Hsp90
- Contribution of Coagulases towards Disease and Protective Immunity
- Early Severe Inflammatory Responses to Uropathogenic Predispose to Chronic and Recurrent Urinary Tract Infection
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy