
SWAN-1 Binds to EGL-9 and Regulates HIF-1-Mediated Resistance to the Bacterial Pathogen PAO1
Pseudomonas aeruginosa is a nearly ubiquitous human pathogen, and infections can be lethal to patients with impaired respiratory and immune systems. Prior studies have established that strong loss-of-function mutations in the egl-9 gene protect the nematode C. elegans from P. aeruginosa PAO1 fast killing. EGL-9 inhibits the HIF-1 transcription factor via two pathways. First, EGL-9 is the enzyme that targets HIF-1 for oxygen-dependent degradation via the VHL-1 E3 ligase. Second, EGL-9 inhibits HIF-1-mediated gene expression through a VHL-1-independent mechanism. Here, we show that a loss-of-function mutation in hif-1 suppresses P. aeruginosa PAO1 resistance in egl-9 mutants. Importantly, we find stabilization of HIF-1 protein is not sufficient to protect C. elegans from P. aeruginosa PAO1 fast killing. However, mutations that inhibit both EGL-9 pathways result in higher levels of HIF-1 activity and confer resistance to the pathogen. Using forward genetic screens, we identify additional mutations that confer resistance to P. aeruginosa. In genetic backgrounds that stabilize C. elegans HIF-1 protein, loss-of-function mutations in swan-1 increase the expression of hypoxia response genes and protect C. elegans from P. aeruginosa fast killing. SWAN-1 is an evolutionarily conserved WD-repeat protein belonging to the AN11 family. Yeast two-hybrid and co-immunoprecipitation assays show that EGL-9 forms a complex with SWAN-1. Additionally, we present genetic evidence that the DYRK kinase MBK-1 acts downstream of SWAN-1 to promote HIF-1-mediated transcription and to increase resistance to P. aeruginosa. These data support a model in which SWAN-1, MBK-1 and EGL-9 regulate HIF-1 transcriptional activity and modulate resistance to P. aeruginosa PAO1 fast killing.
Published in the journal:
. PLoS Pathog 6(8): e32767. doi:10.1371/journal.ppat.1001075
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1001075
Summary
Pseudomonas aeruginosa is a nearly ubiquitous human pathogen, and infections can be lethal to patients with impaired respiratory and immune systems. Prior studies have established that strong loss-of-function mutations in the egl-9 gene protect the nematode C. elegans from P. aeruginosa PAO1 fast killing. EGL-9 inhibits the HIF-1 transcription factor via two pathways. First, EGL-9 is the enzyme that targets HIF-1 for oxygen-dependent degradation via the VHL-1 E3 ligase. Second, EGL-9 inhibits HIF-1-mediated gene expression through a VHL-1-independent mechanism. Here, we show that a loss-of-function mutation in hif-1 suppresses P. aeruginosa PAO1 resistance in egl-9 mutants. Importantly, we find stabilization of HIF-1 protein is not sufficient to protect C. elegans from P. aeruginosa PAO1 fast killing. However, mutations that inhibit both EGL-9 pathways result in higher levels of HIF-1 activity and confer resistance to the pathogen. Using forward genetic screens, we identify additional mutations that confer resistance to P. aeruginosa. In genetic backgrounds that stabilize C. elegans HIF-1 protein, loss-of-function mutations in swan-1 increase the expression of hypoxia response genes and protect C. elegans from P. aeruginosa fast killing. SWAN-1 is an evolutionarily conserved WD-repeat protein belonging to the AN11 family. Yeast two-hybrid and co-immunoprecipitation assays show that EGL-9 forms a complex with SWAN-1. Additionally, we present genetic evidence that the DYRK kinase MBK-1 acts downstream of SWAN-1 to promote HIF-1-mediated transcription and to increase resistance to P. aeruginosa. These data support a model in which SWAN-1, MBK-1 and EGL-9 regulate HIF-1 transcriptional activity and modulate resistance to P. aeruginosa PAO1 fast killing.
Introduction
Pseudomonas aeruginosa is a ubiquitous bacterial pathogen that can infect a wide range of animals and plants, and hospital-acquired P. aeruginosa infections are often lethal to patients with respiratory ailments or immune system dysfunction [1], [2]. The cyanide produced by P. aeruginosa is thought to contribute to the potentially devastating effects of P. aeruginosa respiratory infections in cystic fibrosis patients [3]. Antibiotic-resistant strains of P. aeruginosa are becoming more prevalent, and it is increasingly important to understand the pathogenicity of this microbe and the mechanisms that enable resistance [4], [5].
During infection and inflammation, multicellular tissues must adapt to changing levels of oxygen. The hypoxia-inducible factor (HIF) transcription complex mediates most of the transcriptional responses to hypoxia (low oxygen) [6], [7]. While HIF transcription complexes have been shown to play key roles in mammalian innate immunity, the mechanisms by which HIF regulatory networks influence pathogenicity and disease progression are not yet fully understood [8], [9], [10], [11], [12].
In recent years, the nematode Caenorhabditis elegans has emerged as a powerful genetic system to study innate immunity and resistance to bacterial pathogens [13], [14], [15], [16], [17], [18]. Many of the genes that contribute to C. elegans pathogen resistance are evolutionarily conserved [19], [20], [21]. Interestingly, there is a strong correlation between C. elegans genes that mediate resistance to bacterial pathogens and genes that protect C. elegans from stresses and extend lifespan [22], [23], [24], [25], [26].
Loss-of-function mutations in the C. elegans egl-9 gene enable the animals to survive fast killing by P. aeruginosa PAO1 [27], [28]. While some Pseudomonas strains (such as PA14 on NGM growth media) kill C. elegans slowly through colonization in the gut, logarithmically growing P. aeruginosa PAO1 emits cyanide and kills C. elegans within hours [14], [27], [28], [29]. C. elegans egl-9 mutants are also resistant to Crystal or Vibrio cholerae pore-forming toxins [30]. The egl-9 gene encodes a 2-oxoglutarate-dependent dioxygenase that hydroxylates the HIF-1 transcription factor. Once HIF-1 is hydroxylated, it interacts with the VHL-1 E3 ligase and is targeted for proteasomal degradation [31]. EGL-9 has also been shown to inhibit HIF-1 transcriptional activity via a vhl-1-independent pathway that has little or no requirement for EGL-9 hydroxylase activity [32], [33]. Moderate over-expression of HIF-1 has been shown to increase resistance to heat and to increase adult longevity in C. elegans [34], [35], [36], [37].
In this study, we directly test the hypothesis that increased expression and activation of the HIF-1 transcription factor in egl-9 mutants protect C. elegans from P. aeruginosa PAO1 fast killing. We show that resistance to P. aeruginosa fast killing requires both stabilization of HIF-1 protein and derepression of HIF-1-mediated gene expression. Using forward genetic screens, we identify additional mutations that confer resistance to P. aeruginosa PAO1 fast killing. This leads to the discovery that SWAN-1 inhibits HIF-1 transcriptional activity and modulates resistance to P. earuginosa PAO1 fast killing. SWAN-1 is an evolutionarily conserved protein with WD40 repeats [38]. Further, we demonstrate that SWAN-1 interacts with EGL-9 protein in yeast two-hybrid and co-immunoprecipitation studies.
Results
hif-1 is required for the egl-9-mediated resistance to PAO1 fast killing
The egl-9(sa307) strong loss-of-function mutation has been shown to protect C. elegans from P. aeruginosa PAO1 fast killing [27], [28] As shown in Figure 1A, wild-type animals are paralyzed when placed on P. aeruginosa PAO1, while egl-9 mutant animals remain motile for several hours. We tested the hypothesis that egl-9-mediated resistance to fast killing required hif-1 function. As shown in Figure 1A, the hif-1(ia04) loss-of-function allele totally suppressed the egl-9-mediated resistance phenotype. The rate at which the egl-9, hif-1 double mutant was killed by the pathogen was very similar to the killing curves for wild-type or hif-1(ia04) animals (Figure 1A, Text S1). A prior study had shown that the fast killing of C. elegans by P. aeruginosa required cyanide synthesis [27]. Consistent with this, we found that while P. aeruginosa PAO1 killed wild-type or hif-1-deficient C. elegans within 2 hours, the hydrogen cyanide synthase mutant P. aeruginosa MP507 did not kill C. elegans in this time interval (Text S1).

We next investigated which EGL-9 functions were most critical to the P. aeruginosa PAO1 fast killing phenotype. EGL-9 regulates HIF-1 via at least two pathways: EGL-9 is the oxygen-sensitive enzyme that targets HIF-1 protein for degradation through the VHL-1 pathway, and EGL-9 inhibits HIF-1-mediated transcriptional activity by a vhl-1-independent mechanism [32], [33], [39]. We first tested the hypothesis that stabilization of HIF-1 protein was sufficient to increase resistance to P. aeruginosa PAO1 fast killing. The HIF-1(P621G) mutation precludes hydroxylation of HIF-1 by EGL-9 and stabilizes HIF-1 protein [31], [32], [34]. We assayed four transgenic strains, each expressing either wild-type HIF-1 or the HIF-1(P621G) stabilized protein. Remarkably, none of the transgenic strains were resistant to P. aeruginosa PAO1, as they died at rates similar to wild-type animals (Figure 1B, Text S1). Consistent with this result, the vhl-1(ok161) mutation did not protect C. elegans from fast killing (Text S1).
Genetic screens identify mutations in swan-1 and rhy-1 that increase HIF-1-mediated transcription and protect C. elegans from P. aeruginosa PAO1 fast killing
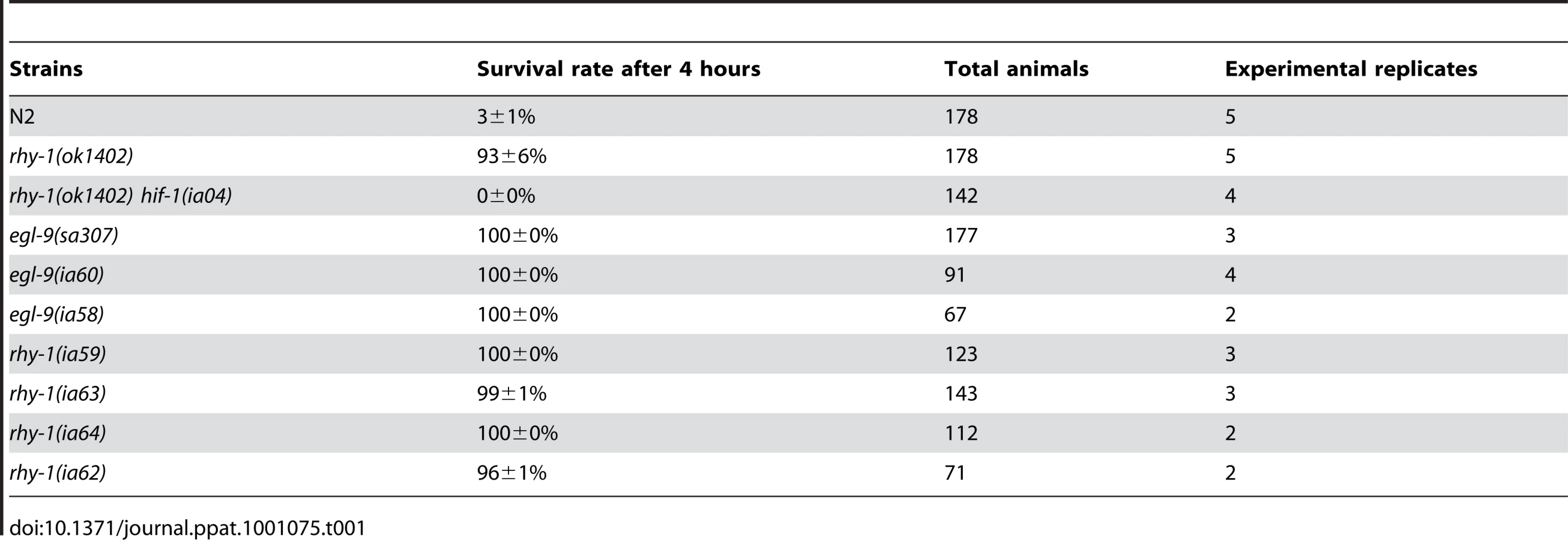
The results thus far suggested that resistance to fast killing required multiple EGL-9 functions. To gain insight to the mechanisms by which EGL-9 repressed HIF-1 transcriptional activity and to better understand P. aeruginosa PAO1 pathogenicity, we conducted forward genetic screens. Using chemical or transposon-mediated mutagenesis, we screened for mutations in C. elegans that caused dramatic over-expression of HIF-1 target genes. As a primary screen, we assayed for the increased expression of Pnhr-57::GFP, a reporter that is expressed at very low levels in wild-type animals and is expressed at high levels in egl-9 mutants [32], [33]. These screens identified novel loss-of-function mutations in rhy-1 (Text S1). Prior studies had shown that rhy-1 encoded a multipass transmembrane protein, and loss-of-function mutations in rhy-1 had been shown to elevate hif-1 mRNA levels slightly and to increase HIF-1 transcriptional activity [33]. As shown in Table 1, animals that lacked rhy-1 function were resistant to P. aeruginosa PAO1 fast killing, and rhy-1-mediated resistance was completely suppressed by the hif-1(ia04) strong loss-of-function mutation.
Reasoning that the effects of some mutations that derepressed HIF-1 activity might only be evident if HIF-1 protein were stable, we crossed a vhl-1 loss-of-function mutation into the parental strain and screened for mutations that increased expression of the reporter. One such screen identified ia50, a mutation that enhanced expression of the Pnhr-57::GFP reporter in vhl-1(ok161) mutants (compare Figure 2C to 2B). While vhl-1(ok161) animals over-expressed the reporter in the intestine, the ia50 mutation expanded expression of Pnhr-57::GFP to other tissues, including the hypodermis and the excretory cell (Figure 2C). The enhanced GFP expression phenotype of ia50 was completely recessive. The ia50, vhl-1(ok161) double mutants had morphological defects that were similar to those seen in egl-9 loss-of-function mutants, including egg-laying defects (data not shown). Additionally, both strains exhibited reduced fertility (Figure 2G).

Genetic mapping with single-nucleotide polymorphisms placed ia50 near +5.67 map units on chromosome five of the C. elegans genome (Figure 3A). Cosmid rescue experiments further delimited a genomic region that could restore a wild-type Pnhr-57::GFP expression pattern to ia50 mutant animals (Figure 3A). We sequenced the genes in this region and found that ia50 mutants carried a single nucleotide mutation in the splice acceptor site for swan-1 (F53C11.8) exon 3 (Figure 3A, 3B). Full-length cDNA sequencing confirmed that ia50 mutants did not splice intron 2 from the swan-1 mRNA, and this introduced an early stop codon. The gene name swan-1 means “seven WD repeats, AN11 family”, and this family of genes includes Petunia AN11, Arabidopsis TTG1, zebrafish Wdr68, and human HAN11 [40], [41], [42], [43]. WD repeat proteins have beta propeller tertiary structures and often serve as platforms for the assembly of larger protein complexes. The swan-1(ia50) mutant allele is predicted to encode a truncated protein including only 2 of the WD repeats, and this suggests that it is a loss-of-function mutation.

To further test the hypothesis that the ia50 mutant phenotype was due to defects in the swan-1 gene, we used bacterially mediated RNAi to deplete swan-1 mRNA. swan-1 RNAi increased the expression of Pnhr-57::GFP, as assayed by protein blots. In control experiments, RNAi for a neighboring gene, swan-2, did not change the expression of the reporter (compare to the empty vector control in Figure 3C).
Prior studies had characterized the swan-1(ok267) deletion mutation as a strong loss-of-function allele [38] (illustrated in Figure 3A). When HIF-1 protein was stabilized by the vhl-1(ok161) mutation, the swan-1(ok267) allele increased expression of the Pnhr-57::GFP reporter (Figure 3D). This phenotype was suppressed by a loss-of-function mutation in hif-1. The swan-1 deletion allele also reduced fertility in a vhl-1 mutant background (Figure 2G). These similarities between the swan-1(ia50) and swan-1(ok267) phenotypes provided additional support for the conclusion that ia50 was a loss-of-function mutation in the swan-1 gene. For more in-depth analyses of swan-1 function, we used the swan-1(ok267) deletion allele, as it had been characterized in prior studies [38].
Having established that swan-1 negatively regulated Pnhr-57::GFP expression, we next asked whether a strong loss-of-function mutation in swan-1 also increased the expression of other HIF-1 target genes. Prior studies had demonstrated that K10H10.2 and F22B5.4 were induced by hypoxia in a hif-1-dependent manner and that they were over-expressed in vhl-1, egl-9, or rhy-1 loss-of-function mutants [31], [32], [33], [39]. As shown in Figure 4A and 4B, mRNA levels for both K10H10.2 and F22B5.4 increased in swan-1(ok267), vhl-1(ok161) double mutants, relative to the vhl-1(ok161) single mutants. These data represent at least three biological replicates of realtime RT-PCR experiments. As shown in Figure 4C, the swan-1 deletion mutation did not have a significant effect on HIF-1 protein levels.

In the presence of stabilized HIF-1, a swan-1 loss-of-function mutation protected C. elegans from P. aeruginosa fast killing
We hypothesized that the combination of HIF-1 stabilization and deletion of swan-1 might result in a P. aeruginosa resistance phenotype similar to that of egl-9 or rhy-1 mutants. Stabilization of HIF-1, through either the stabilizing P621G mutation in HIF-1 transgenes or by mutation of the vhl-1 E3 ligase, was not sufficient to protect C. elegans from P. aeruginosa PAO1 fast killing (Figure 1B and 5A). However, swan-1(ok267), vhl-1(ok161) double mutants were much more resistant than wild-type animals (>40% survived after two hours) (Figure 5A and Text S1). As expected, this resistance was suppressed by a hif-1 loss-of-function mutation (Text S1). Similarly, in transgenic animals expressing HIF-1(P621G), the swan-1(ok267) mutation enabled almost 100% survival after two hours on a P. aeruginosa PAO1 lawn (Figure 5B). Similar results were obtained using an independent hif-1(P621G) transgenic line (Text S1).

Interaction between the EGL-9 and SWAN-1 proteins
Since the genetic data suggested that SWAN-1 and EGL-9 acted in concert to inhibit HIF-1 activity, we next asked whether the two proteins interacted directly. To address this, we performed yeast-two-hybrid assays. In these assays, the EGL-9 catalytic domain was fused to the GAL4 DNA binding domain. In control experiments, this protein fusion by itself did not activate expression of reporter genes that were positively regulated by GAL4 upstream activating sequences. When the EGL-9 protein fusion was combined with a protein containing SWAN-1 fused to the GAL4 activation domain, the two proteins interacted to allow yeast growth on nutrient deficient plates (-Ade/-His/-Leu/-Trp) and to activate α-galactosidase expression (Figure 6A). To further define the regions of SWAN-1 that interacted with EGL-9, we assayed five SWAN-1 deletions, and these results are summarized in Figure 6B. A construct containing only the first three WD repeats of SWAN-1 was able to interact strongly with EGL-9 in yeast two-hybrid assays, whereas a construct that lacked the first four WD repeats did not interact with EGL-9.

To further test the hypothesis that EGL-9 and SWAN-1 could interact in a common complex, we conducted co-immunoprecipitation studies. In these experiments, the EGL-9 catalytic domain was fused to maltose binding protein and expressed in E. coli. A SWAN-1::GFP fusion protein was expressed in C. elegans and purified with a GFP-specific monoclonal antibody coupled to Sepharose beads. In control experiments, GFP alone was purified from worms. To assess interactions, the SWAN-1::GFP and MBP::EGL-9 proteins were co-incubated. Then, GFP-interacting proteins were isolated, and unbound proteins were washed away. As shown in Figure 6C, MBP::EGL-9 was coimmunoprecipitated with SWAN-1::GFP.
Prior studies had demonstrated that SWAN-1 interacted with Rac GTPases and the Rac effector UNC-115, and swan-1 had been shown to repress Rac GTPase activity in neurons [38]. Thus, we considered models in which swan-1 repressed HIF-1 activity by inhibiting Rac GTPases. However, depletion of unc-115, rac-2, ced-10 or mig-2 by mutation or RNAi did not abolish the induction of Pnhr-57::GFP expression in swan-1(ok267), vhl-1(ok161) animals (Text S1). This suggested that SWAN-1 had at least two functions: it interacted with Rac GTPases in neurons to regulate cell migration, and it inhibited HIF-1 transcriptional activity, probably through interaction with EGL-9.
Mutation of mbk-1/DYRK suppresses the swan-1 mutant phenotype
Homologs of SWAN-1 have been shown to interact with DYRK dual-specificity tyrosine-phosphorylation regulated kinases in yeast, zebrafish, and mammalian systems [42], [44]. We hypothesized that swan-1 could interact with a DYRK homolog to regulate HIF-1 transcriptional activity. To test this, we used bacterially-mediated RNAi to knock down the expression of mbk-1, hpk-1 and E02H4.3, three C. elegans genes homologous to mammalian DYRK genes. As shown in Figure 7A, mbk-1 RNAi suppressed Pnhr-57::GFP expression in swan-1(ok267), vhl-1(ok161) double mutants, while the other two RNAi treatments did not. Interestingly, mbk-1 RNAi did not inhibit expression of the reporter in egl-9 mutant animals (Figure 7B). We obtained similar results using the mbk-1(pk1389) loss-of-function mutation (Figure 7C). The mbk-1 deletion allele also inhibited Pnhr-57::GFP expression in swan-1(ok267) animals expressing the hif-1(P621G) transgene (Figure 7D).

We next asked whether mbk-1 contributed to swan-1-mediated P. aeruginosa PAO1 resistance. As shown in Figure 7E and Text S1, the mbk-1(pk1389) mutation completely suppressed the PAO1 fast killing resistance phenotype in swan-1(ok267), vhl-1(ok161) double mutant animals. Further, mbk-1(pk1389) reduced PAO1 resistance in the swan-1(ok267), hif-1(P621G) genetic background, as assayed in two independently isolated hif-1(P621G) transgenic lines (Figure 7F and Text S1). Interestingly, egl-9 mbk-1 double mutants are highly resistant to fast killing (Text S1). Thus, in assays of Pnhr-57::GFP or P. aeruginosa PAO1 fast killing, the mbk-1 mutation suppresses the swan-1 vhl-1 double mutant phenotype, but not the egl-9 loss-of-function phenotype.
Discussion
HIF-1 protects C. elegans from P. aeruginosa PAO1 fast killing
Mutations, alone or in combination, that dramatically increase HIF-1-mediated gene expression can protect C. elegans from P. aeruginosa PAO1 fast killing. Prior studies had discovered that loss-of-function mutations in egl-9 conferred resistance to P. aeruginosa PAO1 fast killing and to cyanide poisoning, but the role of hif-1 had not been investigated [27], [28]. Here, we establish that the resistance of egl-9 mutants to this pathogen is dependent upon hif-1 function (Figure 1A). EGL-9 is a bifunctional protein, and it regulates both HIF-1 protein stability and HIF-1 transcriptional activity [32], [33] (Figure 8). Interestingly, we find that stabilization of HIF-1 protein is not sufficient to protect C. elegans from P. aeruginosa fast killing, but mutations that disable both EGL-9 pathways confer resistance. We propose that over-expression of HIF-1 targets beyond a threshold level protects C. elegans from the cyanide produced by P. aeruginosa PAO1. It is also possible that mutation of egl-9 allows HIF-1-mediated transcription in specific cells or tissues that are especially important to this resistance phenotype.

Hydrogen cyanide is an inhibitor of cytochrome c oxidase, and it is a potent toxin. The cyanide produced by P. aeruginosa in cystic fibrosis patients is recognized as a clinically important virulence factor [3]. Cyanide inhibits cytochrome c oxidase, severely disabling ATP synthesis through oxidative phosphorylation. Interestingly, egl-9 mutant animals are also resistant to hydrogen sulfide, and H2S is also a cytochrome c oxidase inhibitor [45], [46]. A parsimonious explanation is that persistent over-expression of HIF-1 targets protects C. elegans from cyanide or hydrogen sulfide treatments that disable oxidative phosphorylation. These findings introduce an important question: which HIF-1 target genes protect C. elegans from P. aeruginosa PAO1 fast killing, cyanide exposure, and/or hydrogen sulfide? Prior studies have investigated the genes induced by short-term moderate hypoxia at 0.1% oxygen for 4 hours at room temperature [6]. Future studies will examine the changes in gene expression that are common to mutants or mutant combinations that activate HIF-1 and confer resistance to P. aeruginosa.
Increased expression of HIF-1 targets has been shown to protect C. elegans from diverse pathogens or stresses [30], [36], [37], [45], [47]. Bellier et al. isolated a loss-of-function mutation in egl-9 in a screen for mutations that protected C. elegans from pore-forming toxins [30]. egl-9 mutants have also been shown to be resistant to enteropathogenic E. coli E2348/69 [47]. Additionally, mutations that stabilize HIF-1 or increase expression of HIF-1 targets have been shown to increase C. elegans resistance to polyglutamine or beta-amyloid toxicity and heat stress [30], [34], [36], [37]. It is not yet known whether the same HIF-1 targets mediate all of these resistance phenotypes, but we anticipate that each of these functions may require multiple direct and indirect HIF-1 targets.
SWAN-1, a novel regulator of HIF-1
SWAN-1 is an evolutionarily conserved WD-repeat protein of the AN11 family [38]. The data presented here show that SWAN-1 represses HIF-1-mediated gene expression, but does not control HIF-1 protein levels. While swan-1 RNAi does increase expression of the Pnhr-57:GFP reporter in an otherwise wild-type background (Figure 3C), loss of swan-1 function alone is not sufficient to confer resistance to cyanide released by P. aeruginosa PAO1 (Figures 5A and 5B). Resistance to fast killing requires a second mutation that protects HIF-1 protein from oxygen-dependent degradation. Double mutants that carry a loss-of-function mutation in swan-1 and a mutation that stabilizes HIF-1 protein are phenotypically similar to egl-9 loss-of-function mutants, as assayed by fertility, egg laying defects, over-expression of HIF-1 targets, and resistance to P. aeruginosa fast killing (Figures 1A, 2G, 4A, 4B, 5A). swan-1, vhl-1 double mutants also exhibit increased resistance to hydrogen cyanide (unpublished data). Importantly, we show that SWAN-1 forms a complex with EGL-9 (Figure 6A, 6C). Collectively, these data suggest that SWAN-1 and EGL-9 interact directly to repress HIF-1 transcriptional activity.
The AN11 family is evolutionarily conserved, and comparative studies may provide important insights to the roles of these proteins in stress resistance and transcriptional regulation. Petunia AN11 interacts with a MYB family transcription factor, and the human HAN11 gene has been shown to partially rescue the Petunia an11 mutant phenotype [43]. In human cells, zebrafish, and in yeast, AN11 homologs have been shown to form complexes with DYRK kinases [42], [44], [48]. There are five DYRK members in mammals: DYRK1A, DYRK1B, DYRK2, DYRK3 and DYRK4 [49]. Of these, DYRK1A has been characterized most extensively, and it is associated with Down Syndrome [50], [51], [52]. DYRK1A is a multifunctional protein and has more than two dozen targets or interacting proteins, including GLI1, STAT3, and eIF2Bε [49]. HAN11 was shown to decrease DYRK1A-mediated phosphorylation of GLI1 in a HEK293T cell line [53].
The genetic analyses presented here suggest that in the absence of SWAN-1, MBK-1/DYRK activates HIF-1 (illustrated in Figure 8). Specifically, a loss-of-function mutation in mbk-1 suppresses the swan-1, vhl-1 double mutant phenotypes, as assayed by expression of Pnhr-57::GFP expression and by P. aeruginosa PAO1 fast killing (Figure 7). These genetic data suggest that mbk-1 acts downstream of swan-1. Interestingly, mutation of mbk-1 does not suppress the egl-9 mutant phenotype (Text S1). There are at least two models that could explain these findings. First, EGL-9-mediated repression of HIF-1 transcriptional activity may be modulated by SWAN-1 and MBK-1 without being totally dependent upon these regulators. An alternative model is that MBK-1 and SWAN-1 act in parallel to EGL-9 to repress HIF-1 activity. While we favor the first model, we recognize both possibilities in Figure 8. A goal for future studies will be to identify the targets of MBK-1/DYRK to better understand how MBK-1 promotes HIF-1 activity.
A long-term goal will be to understand how hypoxia-induced gene expression influences the progression of P. aeruginosa infections. P. aeruginosa can survive in anaerobic environments, and the formation of biofilms likely restricts oxygen availability to infected tissues in human patients. Our findings in C. elegans suggest that pharmacological inhibitors of the HIF prolyl hydroxylases might contribute to combinatorial therapies to protect cells from the cyanide produced by P. aeruginosa PAO1.
Materials and Methods
Alleles and worm culture
C. elegans were grown at 20°C using standard methods, unless other culture conditions are specified [54]. The loss-of-function alleles and transgenic lines used in this study are listed in Text S1. The swan-1(ok267) mutant allele was backcrossed to wild-type animals three times prior to phenotypic analyses [38].
Mutagenesis
The EMS forward genetic screen was performed as described previously [33]. Briefly, the parental strain carrying Pnhr-57::GFP and vhl-1(ok161) was mutagenized with EMS, and the F2 progeny were screened for increased expression of the Pnhr-57::GFP reporter using fluorescent stereomicroscopy. We generated the rhy-1 loss-of-function alleles ia59, ia62, ia63, ia64 in a screen for Mos1 transposon-mediated mutations that caused Pnhr-57::GFP overexpression. The methods for Mos1 mobilization have been described previously [32], [55].
Mapping ia50 to the swan-1 locus
The ia50 mutant allele was out-crossed twice to the parental strain prior to any further mapping or characterization. Chromosome and interval mapping were performed as described previously using single-nucleotide polymorphisms (SNPs) between the Bristol N2 and Hawaiian strains [56]. Briefly, the iaIs07 (Pnhr-57::GFP) transgene and the vhl-1(ok161) mutation were crossed extensively into the Hawaiian genetic background. The resulting males were crossed to ia50 vhl-1(ok161) double mutants carrying the iaIs07(Pnhr-57::GFP) marker. Fifty F2 animals exhibiting the vhl-1, ia50 double mutant phenotype and fifty animals exhibiting the vhl-1 (ok161) single mutant phenotype (intestinal GFP expression) were picked into separate tubes, and genomic DNA was prepared. Analyses of the divergent SNPs showed enrichment of Bristol bands in mutant lanes and an enrichment of Hawaiian bands in non-mutant lanes for SNPs lying between -5 and +13 mu on chromosome V. For interval mapping, individual self-progeny of F1 hermaphrodites (described above) with the double mutant phenotype were picked into a 96-well plate to prepare the genomic DNA, and four SNPs were analyzed (−5, +1, +6, +13) [56]. We then used two SNPs, R10D6 (+5.83 mu) and pkP5086 (+6.42 mu), to do three point mapping of the ia50 mutation.
RNA interference
RNAi was performed as previously described [34]. Bacterial strains containing RNAi constructs were purchased from Geneservice Ltd., and inserts were validated by sequencing.
Fertility assays
Individual L4-stage worms were placed on NGM plates with fresh OP50 bacterial food. The worms were transferred onto fresh plates every 12 hours, and the total progeny laid on each plate was counted and recorded at each time point. This procedure was continued until the worms reached the end of their reproductive capacity.
MBP::EGL-9 protein expression
To build pSZ18, the bacterial expression vector for EGL-9::MBP, egl-9 cDNA was amplified with two primers: 5′GGTGGATCCAAACCAACGGTATCCAGAAC and 5′GGGATCCGATGTAATACTCTGGGTTTGTGG. The PCR products were cut with BamHI and PstI and ligated into the pMal-p2x vector (from New England Biolabs). The plasmid expressing EGL-9 fused to maltose binding protein was transformed into BL21 (DE3) bacteria, and a single colony was inoculated into liquid media and cultured for 16 hours at 37°C. The bacterial culture was diluted by 1 : 200 to 50 ml and cultured for 2.5 hours. IPTG was added to a final concentration of 1 mM. After 4 hours, the bacteria were pelleted and frozen in liquid nitrogen. The bacteria were frozen at −80°C overnight before adding bacterial lysis buffer (50 mM Tris PH 7.5, NaCl 150 mM, NP-40 0.1%), 0.25 mg/ml lysozyme, 0.01 mg/ml DNase, and 1× protease inhibitor cocktail (from Roche). After four hours at 4°C, the lysate was centrifuged at 12000 g for 20 min and the supernatant was kept for further use.
P. aeruginosa fast killing assays
The fast killing assay follows the approach described previously [27], [28]. Briefly, a single P. aeruginosa PAO1 colony was inoculated and cultured at 37°C for 16 hours in 3–5 ml brain heart infusion (BHI) broth. The culture was then diluted 100-fold. 300 µl of diluted bacteria was evenly spread on 60 mm diameter Petri dishes with 7 ml BHI agar. To minimize the possibility of animals escaping from the bacterial lawn, bacteria were spread to cover the whole plate. The plates were incubated at 37°C for 24 hours and then cooled to room temperature for 30 minutes. Thirty to fifty developmentally synchronized L4-stage C. elegans were put on the lawn and incubated at room temperature (22°C). Lids to the petri plates remained closed during this time to keep hydrogen cyanide from evaporating. C. elegans were scored as paralyzed or dead if they did not move when the plate was tapped against the microscope stage. Each data point represents at least three independent replicate experiments.
Protein blots
The methods for protein blots have been described previously [32]. At least three biological replicates were analyzed for each experiment. Transgenic strains expressing epitope-tagged HIF-1 were used to assay HIF-1 protein levels and each lane of a protein gel included lysate from 40–100 L4-stage animals. For the Pnhr-57::GFP reporter, we used 5–50 L4 animals. The statistical significance of differences was assessed by two-sample paired t-tests or one way ANOVA with Bonferroni post test.
Real time PCR
The methods for real time PCR were as described previously [32]. At least three biological replicates were analyzed for each experiment, and each PCR reaction was performed in duplicate. The statistical significance of differences was assessed by two-sample paired t-tests or one way ANOVA with Bonferroni post test.
Yeast two-hybrid assays
For yeast two-hybrid experiments, C. elegans cDNA sequences were inserted in to the pGBKT7 and pGADT7 vectors from Clonetech. The sequences encoding the EGL-9 catalytic domain were amplified with two primers, egl-9-F: CCGGAATTCGGTCTCGCACTAAGCATTCACC and egl-9-R: CGCGGATCCCCGTGGT CTCAAAAGTGATCCAAT, and cloned into pGBKT7 (GAL4 DNA-binding domain vector). This construct did not result in detectable autoactivation. Dr. Erik Lundquist kindly provided the plasmid which expressed SWAN-1 fused to the GAL4 transcriptional activation domain. The deletion vectors (shown in Figure 6B) were amplified from this swan-1 prey plasmid [38]. The primer sets are listed in Text S1.
The AH109 yeast strain was used. Transformations of plasmids into yeast were performed as described previously [57], [58]. The negative controls included the co-transformation of pGADT7-swan-1 plasmids with pGBKT7 (empty vector); the co-transformation of pGBKT7-egl-9 and pGADT7; and the co-transformation of pGBKT and pGADT7. To test for interactions, yeast colonies carrying both plasmids (selected on Leu−, Trp− plates) were cultured in 2 ml Leu−/Trp− liquid SD-medium for 24 hours at 30°C. Twenty individual colonies were then assayed for growth on Ade−/His−/Leu−/Trp− X-α-gal SD-medium plates. Additionally, six Leu+/Trp+ colonies were cultured for quantification of the β-galactosidase activity using O-nitrophenyl-B-D-galactopyranoside as a substrate.
Co-immunoprecipitation experiments
The C. elegans strain expressing SWAN-1::GFP [transgenic array lqEx19 (Pswan-1:: swan-1::gfp)] was generously provided by Erik Lundquist. C. elegans were grown on 100 mm enriched media plates with NA22 bacteria, and 0.4 ml mixed-stage worms were harvested. Animals were washed with cold M9 buffer three times and were washed once with worm lysis buffer (50 mM Tris-HCl PH 7.4, 150 mM NaCl, 0.5% Triton-X-100, 10% glycerol, 1 mM DTT, and 1X protease inhibitor cocktail from Roche). The worm pellet was resuspended in 1.2 ml worm lysis buffer, and the animals were lysed in a french press (Thermo Electron Corporation) three times at 1000 psi. To perform immunoprecipitations, 80 µl G-Sepharose beads were washed with worm lysis buffer three times for 10 minutes each, and they were then incubated with 80 µl 0.4 mg/ml GFP antibody (Roche) overnight. Beads were washed with worm lysis buffer three times for 5 minutes and divided into two 40 µl aliquots, each of which was incubated with 600 µl of SWAN-1::GFP or GFP worm lysate at 4°C for 4 hours. Beads with either control GFP [Pnhr-57::GFP] or SWAN-1::GFP were washed (three times 10 minutes in cold lysis buffer) and incubated with MBP::EGL-9. After four hours incubation at 4°C, beads were washed with worm lysis buffer and boiled in 40 µl 1xSDS buffer. The proteins were fractionated by 10% SDS polyacrylamide gel electrophoresis, and blots were probed with maltose binding protein rabbit antiserum (from NEB at 1∶3000 dilution) or mouse GFP monoclonal antibody (clones 7.1 and 13.1 from Roche at 1∶1000 dilution).
Supporting Information
Zdroje
1. LeeSC
HuaCC
YuTJ
ShiehWB
SeeLC
2005 Risk factors of mortality for nosocomial pneumonia: importance of initial anti-microbial therapy. Int J Clin Pract 59 39 45
2. GomezMI
PrinceA
2007 Opportunistic infections in lung disease: Pseudomonas infections in cystic fibrosis. Curr Opin Pharmacol 7 244 251
3. AndersonRD
RoddamLF
BettiolS
SandersonK
ReidDW
2010 Biosignificance of bacterial cyanogenesis in the CF lung. J Cyst Fibros 9 158 164
4. PageMG
HeimJ
2009 Prospects for the next anti-Pseudomonas drug. Curr Opin Pharmacol 9 558 565
5. PageMG
HeimJ
2009 New molecules from old classes: revisiting the development of beta-lactams. IDrugs 12 561 565
6. ShenC
NettletonD
JiangM
KimSK
Powell-CoffmanJA
2005 Roles of the HIF-1 hypoxia-inducible factor during hypoxia response in Caenorhabditis elegans. J Biol Chem 280 20580 20588
7. SemenzaGL
2001 Hypoxia-inducible factor 1: control of oxygen homeostasis in health and disease. Pediatr Res 49 614 617
8. NizetV
JohnsonRS
2009 Interdependence of hypoxic and innate immune responses. Nat Rev Immunol 9 609 617
9. HongSW
YooJW
KangHS
KimS
LeeDK
2009 HIF-1alpha-dependent gene expression program during the nucleic acid-triggered antiviral innate immune responses. Mol Cells 27 243 250
10. WalmsleySR
McGovernNN
WhyteMK
ChilversER
2008 The HIF/VHL pathway: from oxygen sensing to innate immunity. Am J Respir Cell Mol Biol 38 251 255
11. RiusJ
GumaM
SchachtrupC
AkassoglouK
ZinkernagelAS
2008 NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 453 807 811
12. ZinkernagelAS
JohnsonRS
NizetV
2007 Hypoxia inducible factor (HIF) function in innate immunity and infection. J Mol Med 85 1339 1346
13. KurzCL
EwbankJJ
2000 Caenorhabditis elegans for the study of host-pathogen interactions. Trends Microbiol 8 142 144
14. AballayA
AusubelFM
2002 Caenorhabditis elegans as a host for the study of host-pathogen interactions. Curr Opin Microbiol 5 97 101
15. MylonakisE
AusubelFM
TangRJ
CalderwoodSB
2003 The art of serendipity: killing of Caenorhabditis elegans by human pathogens as a model of bacterial and fungal pathogenesis. Expert Rev Anti Infect Ther 1 167 173
16. EwbankJ
2003 The nematode Caenorhabditis elegans as a model for the study of host-pathogen interactions. J Soc Biol 197 375 378
17. AlegadoRA
CampbellMC
ChenWC
SlutzSS
TanMW
2003 Characterization of mediators of microbial virulence and innate immunity using the Caenorhabditis elegans host-pathogen model. Cell Microbiol 5 435 444
18. TanMW
2002 Identification of host and pathogen factors involved in virulence using Caenorhabditis elegans. Methods Enzymol 358 13 28
19. SchulenburgH
KurzCL
EwbankJJ
2004 Evolution of the innate immune system: the worm perspective. Immunol Rev 198 36 58
20. KimDH
AusubelFM
2005 Evolutionary perspectives on innate immunity from the study of Caenorhabditis elegans. Curr Opin Immunol 17 4 10
21. NicholasHR
HodgkinJ
2004 Responses to infection and possible recognition strategies in the innate immune system of Caenorhabditis elegans. Mol Immunol 41 479 493
22. EvansEA
KawliT
TanMW
2008 Pseudomonas aeruginosa suppresses host immunity by activating the DAF-2 insulin-like signaling pathway in Caenorhabditis elegans. PLoS Pathog 4 e1000175
23. TroemelER
ChuSW
ReinkeV
LeeSS
AusubelFM
2006 p38 MAPK regulates expression of immune response genes and contributes to longevity in C. elegans. PLoS Genet 2 e183
24. SinghV
AballayA
2006 Heat-shock transcription factor (HSF)-1 pathway required for Caenorhabditis elegans immunity. Proc Natl Acad Sci U S A 103 13092 13097
25. LawsTR
HardingSV
SmithMP
AtkinsTP
TitballRW
2004 Age influences resistance of Caenorhabditis elegans to killing by pathogenic bacteria. FEMS Microbiol Lett 234 281 287
26. KurzCL
TanMW
2004 Regulation of aging and innate immunity in C. elegans. Aging Cell 3 185 193
27. GallagherLA
ManoilC
2001 Pseudomonas aeruginosa PAO1 kills Caenorhabditis elegans by cyanide poisoning. J Bacteriol 183 6207 6214
28. DarbyC
CosmaCL
ThomasJH
ManoilC
1999 Lethal paralysis of Caenorhabditis elegans by Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 96 15202 15207
29. TanMW
Mahajan-MiklosS
AusubelFM
1999 Killing of Caenorhabditis elegans by Pseudomonas aeruginosa used to model mammalian bacterial pathogenesis. Proc Natl Acad Sci U S A 96 715 720
30. BellierA
ChenCS
KaoCY
CinarHN
AroianRV
2009 Hypoxia and the hypoxic response pathway protect against pore-forming toxins in C. elegans. PLoS Pathog 5 e1000689
31. EpsteinAC
GleadleJM
McNeillLA
HewitsonKS
O'RourkeJ
2001 C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107 43 54
32. ShaoZ
ZhangY
Powell-CoffmanJA
2009 Two distinct roles for EGL-9 in the regulation of HIF-1-mediated gene expression in Caenorhabditis elegans. Genetics 183 821 829
33. ShenC
ShaoZ
Powell-CoffmanJA
2006 The Caenorhabditis elegans rhy-1 gene inhibits HIF-1 hypoxia-inducible factor activity in a negative feedback loop that does not include vhl-1. Genetics 174 1205 1214
34. ZhangY
ShaoZ
ZhaiZ
ShenC
Powell-CoffmanJA
2009 The HIF-1 hypoxia-inducible factor modulates lifespan in C. elegans. PLoS One 4 e6348
35. ChenD
ThomasEL
KapahiP
2009 HIF-1 modulates dietary restriction-mediated lifespan extension via IRE-1 in Caenorhabditis elegans. PLoS Genet 5 e1000486
36. MehtaR
SteinkrausKA
SutphinGL
RamosFJ
ShamiehLS
2009 Proteasomal regulation of the hypoxic response modulates aging in C. elegans. Science 324 1196 1198
37. TreininM
ShliarJ
JiangH
Powell-CoffmanJA
BrombergZ
2003 HIF-1 is required for heat acclimation in the nematode Caenorhabditis elegans. Physiol Genomics 14 17 24
38. YangY
LuJ
RovnakJ
QuackenbushSL
LundquistEA
2006 SWAN-1, a Caenorhabditis elegans WD repeat protein of the AN11 family, is a negative regulator of Rac GTPase function. Genetics 174 1917 1932
39. BishopT
LauKW
EpsteinAC
KimSK
JiangM
2004 Genetic analysis of pathways regulated by the von Hippel-Lindau tumor suppressor in Caenorhabditis elegans. PLoS Biol 2 e289
40. DresselA
HemlebenV
2009 Transparent Testa Glabra 1 (TTG1) and TTG1-like genes in Matthiola incana R. Br. and related Brassicaceae and mutation in the WD-40 motif. Plant Biol (Stuttg) 11 204 212
41. NissenRM
AmsterdamA
HopkinsN
2006 A zebrafish screen for craniofacial mutants identifies wdr68 as a highly conserved gene required for endothelin-1 expression. BMC Dev Biol 6 28
42. SkuratAV
DietrichAD
2004 Phosphorylation of Ser640 in muscle glycogen synthase by DYRK family protein kinases. J Biol Chem 279 2490 2498
43. de VettenN
QuattrocchioF
MolJ
KoesR
1997 The an11 locus controlling flower pigmentation in petunia encodes a novel WD-repeat protein conserved in yeast, plants, and animals. Genes Dev 11 1422 1434
44. HoY
GruhlerA
HeilbutA
BaderGD
MooreL
2002 Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 415 180 183
45. BuddeMW
RothMB
2010 Hydrogen sulfide increases hypoxia-inducible factor-1 activity independently of von Hippel-Lindau tumor suppressor-1 in C. elegans. Mol Biol Cell 21 212 217
46. CooperCE
BrownGC
2008 The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: chemical mechanism and physiological significance. J Bioenerg Biomembr 40 533 539
47. AnyanfulA
Dolan-LivengoodJM
LewisT
ShethS
DezaliaMN
2005 Paralysis and killing of Caenorhabditis elegans by enteropathogenic Escherichia coli requires the bacterial tryptophanase gene. Mol Microbiol 57 988 1007
48. MazmanianG
KovshilovskyM
YenD
MohantyA
MohantyS
2010 The zebrafish dyrk1b gene is important for endoderm formation. Genesis 48 20 30
49. ParkJ
SongWJ
ChungKC
2009 Function and regulation of Dyrk1A: towards understanding Down syndrome. Cell Mol Life Sci 66 3235 3240
50. SmithDJ
StevensME
SudanaguntaSP
BronsonRT
MakhinsonM
1997 Functional screening of 2 Mb of human chromosome 21q22.2 in transgenic mice implicates minibrain in learning defects associated with Down syndrome. Nat Genet 16 28 36
51. ShindohN
KudohJ
MaedaH
YamakiA
MinoshimaS
1996 Cloning of a human homolog of the Drosophila minibrain/rat Dyrk gene from “the Down syndrome critical region” of chromosome 21. Biochem Biophys Res Commun 225 92 99
52. AltafajX
DierssenM
BaamondeC
MartiE
VisaJ
2001 Neurodevelopmental delay, motor abnormalities and cognitive deficits in transgenic mice overexpressing Dyrk1A (minibrain), a murine model of Down′s syndrome. Hum Mol Genet 10 1915 1923
53. MoritaK
Lo CelsoC
Spencer-DeneB
ZouboulisCC
WattFM
2006 HAN11 binds mDia1 and controls GLI1 transcriptional activity. J Dermatol Sci 44 11 20
54. BrennerS
1974 The genetics of Caenorhabditis elegans. Genetics 77 71 94
55. GrangerL
MartinE
SegalatL
2004 Mos as a tool for genome-wide insertional mutagenesis in Caenorhabditis elegans: results of a pilot study. Nucleic Acids Res 32 e117
56. DavisMW
HammarlundM
HarrachT
HullettP
OlsenS
2005 Rapid single nucleotide polymorphism mapping in C. elegans. BMC Genomics 6 118
57. GietzD
St JeanA
WoodsRA
SchiestlRH
1992 Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res 20 1425
58. SchiestlRH
GietzRD
1989 High efficiency transformation of intact yeast cells using single stranded nucleic acids as a carrier. Curr Genet 16 339 346
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 8
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- The Transcription Factor Rbf1 Is the Master Regulator for -Mating Type Controlled Pathogenic Development in
- PKC Signaling Regulates Drug Resistance of the Fungal Pathogen via Circuitry Comprised of Mkc1, Calcineurin, and Hsp90
- Contribution of Coagulases towards Disease and Protective Immunity
- Early Severe Inflammatory Responses to Uropathogenic Predispose to Chronic and Recurrent Urinary Tract Infection
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy