
Parvovirus Minute Virus of Mice Induces a DNA Damage Response That Facilitates Viral Replication
Infection by DNA viruses can elicit DNA damage responses (DDRs) in host cells. In some cases the DDR presents a block to viral replication that must be overcome, and in other cases the infecting agent exploits the DDR to facilitate replication. We find that low multiplicity infection with the autonomous parvovirus minute virus of mice (MVM) results in the activation of a DDR, characterized by the phosphorylation of H2AX, Nbs1, RPA32, Chk2 and p53. These proteins are recruited to MVM replication centers, where they co-localize with the main viral replication protein, NS1. The response is seen in both human and murine cell lines following infection with either the MVMp or MVMi strains. Replication of the virus is required for DNA damage signaling. Damage response proteins, including the ATM kinase, accumulate in viral-induced replication centers. Using mutant cell lines and specific kinase inhibitors, we show that ATM is the main transducer of the signaling events in the normal murine host. ATM inhibitors restrict MVM replication and ameliorate virus-induced cell cycle arrest, suggesting that DNA damage signaling facilitates virus replication, perhaps in part by promoting cell cycle arrest. Thus it appears that MVM exploits the cellular DNA damage response machinery early in infection to enhance its replication in host cells.
Published in the journal:
. PLoS Pathog 6(10): e32767. doi:10.1371/journal.ppat.1001141
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1001141
Summary
Infection by DNA viruses can elicit DNA damage responses (DDRs) in host cells. In some cases the DDR presents a block to viral replication that must be overcome, and in other cases the infecting agent exploits the DDR to facilitate replication. We find that low multiplicity infection with the autonomous parvovirus minute virus of mice (MVM) results in the activation of a DDR, characterized by the phosphorylation of H2AX, Nbs1, RPA32, Chk2 and p53. These proteins are recruited to MVM replication centers, where they co-localize with the main viral replication protein, NS1. The response is seen in both human and murine cell lines following infection with either the MVMp or MVMi strains. Replication of the virus is required for DNA damage signaling. Damage response proteins, including the ATM kinase, accumulate in viral-induced replication centers. Using mutant cell lines and specific kinase inhibitors, we show that ATM is the main transducer of the signaling events in the normal murine host. ATM inhibitors restrict MVM replication and ameliorate virus-induced cell cycle arrest, suggesting that DNA damage signaling facilitates virus replication, perhaps in part by promoting cell cycle arrest. Thus it appears that MVM exploits the cellular DNA damage response machinery early in infection to enhance its replication in host cells.
Introduction
It has become increasingly clear that viruses, especially DNA viruses, can provoke DNA damage responses (DDRs) in infected cells, either in response to virally encoded proteins or to the large amount of foreign DNA produced during viral replication. These cellular responses are varied, and have the potential to impede or facilitate virus replication (reviewed in [1], [2]). In the case of adenovirus (Ad), the DDR constitutes a barrier that must be overcome in order for viral replication to proceed [3], [4], [5], [6], [7], [8]. In contrast, the small DNA tumor viruses, polyomavirus and simian virus type 40 (SV40), activate a DDR that facilitates their replication [9], [10], [11]. Herpesviruses show complex interactions with the DDR pathway: while there is reduced replication of herpes simplex virus (HSV) in the absence of some DNA damage proteins [12], the viral immediate early protein ICP0 also inhibits accumulation of certain repair factors at sites of DNA damage [13].
Parvoviruses are small non-enveloped icosahedral viruses that are important pathogens in many animal species including humans [14], [15]. They are the only known viruses of vertebrates that contain genomes of single-stranded linear DNA [16]. Minute virus of mice (MVM) is an autonomously replicating parvovirus which is lytic in murine cells and transformed human cells. The viral genome is approximately 5 kb and possesses inverted terminal repeats at each end which form different hairpin structures and serve as origins of replication [17]. MVM encodes two non-structural proteins: the larger non-structural phosphoprotein NS1 is required for viral replication, while NS2 plays important roles in the normal murine host but is dispensable for replication in many permissive transformed human cell lines [18].
In contrast to the DNA tumor viruses, parvoviruses cannot induce entry of cells into S-phase, and must wait for cells to cycle into S-phase to begin replication [17]. Other than the non-structural NS1 protein, they do not encode their own replicative machinery, and depend on cellular replication factors. Following initiation of parvovirus replication, there is a reorganization of the nucleus, leading to formation of distinct nuclear foci termed “autonomous parvovirus-associated replication” (APAR) bodies [19], [20], [21]. NS1 co-localizes with replicating viral DNA in APAR bodies, which also accumulate host replication proteins such as PCNA, RPA and DNA polymerases α and δ [19], [20], [21].
Several studies have suggested links between MVM, cell cycle and apoptosis [22], [23], [24]. MVM has been shown to cause cell cycle arrest in S and G2 phases, and this has been suggested to be mediated at least in part by stabilization of p53 [22], [23], [24]. Following MVM infection, there is a reduction in cellular DNA replication [25], and the endonuclease activity of NS1 has been suggested to cause nicks in cellular chromatin [23]. The related parvovirus H1 has been shown to cause apoptosis [26], [27], and in a recent report to induce phosphorylation of the histone variant H2AX [28]. In addition to the effects of the non-structural proteins, single-stranded parvoviruses present cells with distinct forms of foreign DNA during the infectious cycle. The related helper-dependent parvovirus adeno-associated virus (AAV) can induce a DDR. UV-inactivated AAV genomes delivered in very high amounts induce a robust cellular response [29], [30], and recent studies have suggested that AAV replication, in the context of helper Ad co-infection or Ad helper proteins, resulted in a unique DNA damage response which was not seen by infection with virus alone [31], [32]. Detailed investigation and interpretation of AAV induction of cellular DDRs during replication is complicated, however, by the potent effects of its helper viruses (Ad and HSV).
The cellular MRN complex consists of the Mre11, Rad50 and Nbs1 proteins, and acts as a sensor of double strand breaks (reviewed in [33], [34], [35], [36]). This complex is redistributed to DNA damage sites and recruits the ataxia telangiectasia mutated (ATM) kinase. In addition to ATM, two other phosphatidylinositol 3-kinase-like kinases (PIKKs) act as transducer kinases that communicate DNA damage signals to downstream targets in response to various stimuli: ATM-related (ATR), and DNA-dependent protein kinase (DNA-PK) [37], [38]. ATR is mainly activated following replicative stress [39], [40] while DNA-PK is mainly involved in DNA repair via the non-homologous end joining pathway (NHEJ) [41]. Once at the site of damage, these kinases phosphorylate histone H2AX [42], [43] and activate additional cell cycle checkpoint kinases [44], [45]. This leads to cell cycle arrest or induction of apoptosis in cases of severe DNA damage [46], [47].
In this report we have investigated the interaction between MVM and the cellular DNA damage response pathway. We show that MVM induces a robust DDR in infected murine, hamster, and transformed human cells. The cellular response requires replicating viral DNA. DDR sensor and response proteins accumulate in MVM replication centers, but at late times there is a proteasome-dependent loss of Mre11. We show that ATM is the primary kinase required for DDR signaling in response to MVM infection of rodent cells. The response observed with this autonomous parvovirus is distinct from that described for AAV in the presence of helper virus [31], [32], and appears to be more similar to that observed with papovaviruses. Chemical inhibitors of ATM restrict MVM replication and ameliorate virus-induced cell cycle arrest. MVM therefore appears to exploit the cellular DNA damage response to enhance its replication in host cells.
Results
MVM infection induces a DNA damage response in both murine and human cells
To examine whether MVM induced a DDR, para-synchronized murine A9 cells were infected at low multiplicity infection (MOI) with the MVM prototype strain MVMp. Phosphorylation of damage response proteins was detected by Western blotting and visualized by immunofluorescence (Fig. 1). Expression of the viral replicator protein NS1, a marker for viral replication, was detected by 12 hours post-infection (h.p.i.), and by 18 h.p.i. robust expression of both NS1 and NS2 could be observed (Fig. 1A, lanes 4–6). Co-incident with viral gene expression, we observed activation of a cellular DNA damage response that persisted throughout the time course monitored (Fig. 1A, lanes 4–6). We detected the appearance of phosphorylated H2AX (γH2AX), as well as p53 phosphorylated at S18 (S15 in human cells), and phosphorylated RPA32 (as identified with an antibody detecting phosphorylated S4/8 and by the concomitant decrease in electrophoretic mobility using antibodies to total RPA32). In addition, there was an increase in total p53 levels following MVM infection, consistent with a previous report [24]. The appearance of these DDR markers was similar to that seen following treatment with hydroxyurea (Fig. 1A, lane 7), a known inducer of the DDR [48], and absent from para-synchronized, mock-infected cells (Fig. 1A, lanes 1 and 8).

When examining downstream DNA damage effector kinases during MVMp infection, we observed phosphorylation of Chk2, a downstream target of ATM [49], [50], [51], as evident by an decrease in electrophoretic mobility (Fig. 1A, lanes 4–6), and confirmed by both reactivity with anti-Chk2-P-T68 antibody and phosphatase susceptibility (data not shown). In contrast, we did not detect phosphorylation of Chk1, a kinase typically phosphorylated at serine 345 in response to ATR activation [52], [53], [54], [55]. A significant DDR was also detected when re-infection was blocked by added neutralizing antibody in the media (data not shown), suggesting that a single round of infection was sufficient to induce the DDR. The DDR was also observed during infection of asynchronous A9 cells (data not shown).
We confirmed that activation of the DDR by MVM was not limited to the A9 cell line or MVMp strain. Additional permissive cell lines murine NIH3T6 and human NB324K were infected with MVMp and analyzed for DDR signaling. Infection of these cell lines resulted in the phosphorylation of H2AX, RPA32, p53 and Chk2 (Fig. 1B). Similar results were also seen following infection with the lymphotropic variant of MVM, MVMi, in the murine lymphocyte lines S49 and EL4 (Fig. 1C). Differences in the relative levels of NS1∶NS2 by MVMp and MVMi have been previously reported [56]. Taken together, these results demonstrated that autonomous parvoviruses induced a significant DDR in permissive murine and human cells.
Components of the DDR localize to sites of MVM replication
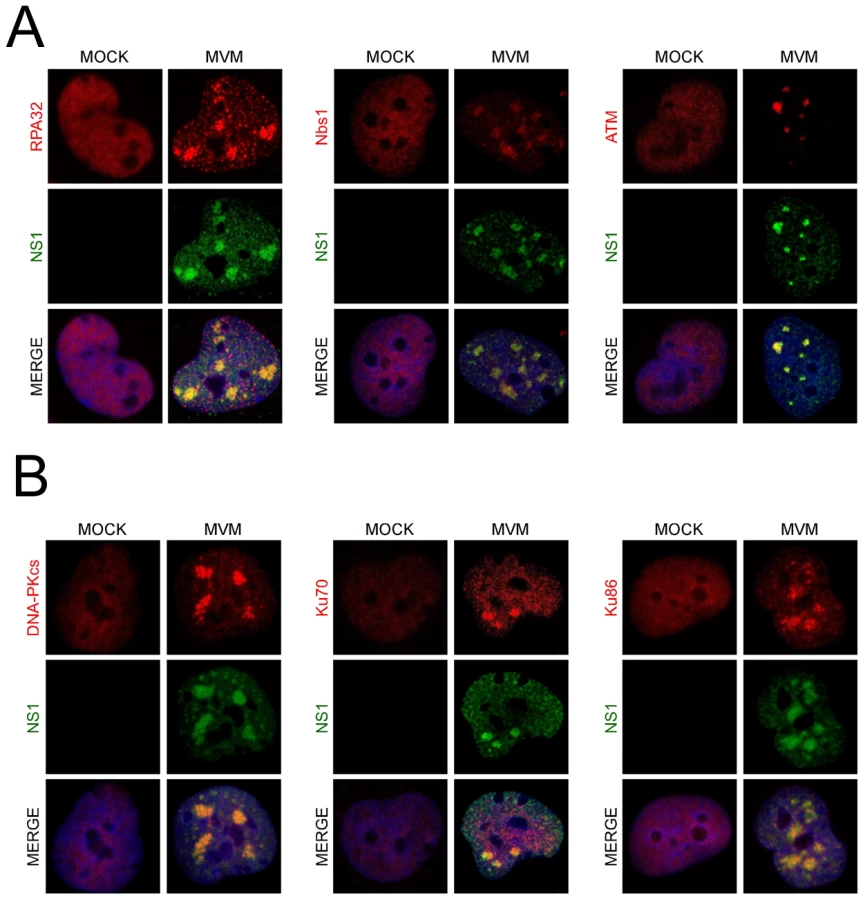
MVM replication takes place in viral-induced APAR bodies, which contain RPA, DNA pol-α, pol-δ, and cyclin A, in addition to the viral replicator protein NS1 and replicating genomes [19], [20], [21]. The MVM-induced phosphorylated DDR proteins could also be detected by immunofluorescence at APAR bodies (Fig. 1D). After 24 h of MVMp infection in murine A9 cells, γH2AX staining was detected and primarily co-localized with NS1 at distinct APAR bodies that represent early stages of infection (Fig. 1D, left panel, second column). In cells with more diffuse NS1 patterns, that represent later stages of the infection cycle, we observed pan-nuclear staining of γH2AX (Fig. 1D, left panel, third column). The majority of infected cells also showed co-localization of NS1 with staining patterns for S4/8-phosphorylated RPA32, and the S343–phosphorylated form of the MRN complex component Nbs1 (Fig. 1D, center and right panels, respectively). Similar results were obtained in NB324K cells (data not shown).
We also found that MVM dramatically redistributed bulk DDR-associated proteins within the nucleus of infected NB324K cells. In addition to the previously reported redistribution of RPA into APAR bodies [19] (Fig. 2A, left panel), we also found that MVM infection led to accumulation of Nbs1 into NS1-containing APAR bodies, in contrast to the diffuse nuclear staining pattern observed in mock cells (Fig. 2A, center panel). Nbs1 has been shown to recruit ATM to sites of DNA damage via its C-terminus [57], [58]. We examined ATM localization using the rabbit monoclonal antibody Y170, which has previously been validated for ATM immunostaining [59]. We detected endogenous ATM redistributed to APAR bodies in MVM-infected cells (Fig. 2A, right panel). In addition, we also observed redistribution of DNA-PK and its components Ku70 and Ku86 to MVM replication compartments (Fig. 2B). Taken together these results show that MVM replication leads to a reorganization and redistribution of DDR proteins to MVM sites of virus replication.
MVM-induced DDR is associated with reduction in Mre11 levels
We observed that levels of the Mre11 component of the MRN complex, which is associated with ATM activation [33], [34], were reduced approximately 3-fold by 24 hrs following MVM infection of A9 cells (Fig. 3A). Loss of Mre11 was also reported following infection by SV40 [11], and adenovirus [8], [60]. This reduction, which was not seen during the DDR induced by hydroxyurea (Fig. 3A), was partially reversed in the presence of MG132, suggesting its loss was due to proteasome-mediated degradation (Fig. 3B). Surprisingly, and in contrast to the case for SV40 and adenovirus, no decrease was observed for other MRN components, such as Nbs1 (Fig. 1B). In addition, we did not observe any reduction in the levels of ATM or DNA-PK following MVM infection (data not shown). Immunofluorescence analysis demonstrated that Mre11 was redistributed to co-localize with NS1 in cells with distinct MVM APAR bodies, but staining was completely undetectable in cells that represent the late stages of MVM infection, with robust and diffusely nuclear NS1 expression (Fig. 3C, left panel). These assays also demonstrated the sustained presence of Nbs1 throughout infection (Fig. 3C, right panel). These results suggest the specific down-regulation of Mre11 at the late stages of MVM infection.

MVM-induced DDR requires viral replication
The MVM-induced DDR correlated with the onset of viral DNA replication (Fig. 1A). We thus attempted to determine which viral elements or functions induced the cellular response. As expected, infection with UV-inactivated MVM (calculated prior to treatment to infect at an MOI of 10) did not generate the viral non-structural replication proteins (Fig. 4A). When UV-MVM was compared to a parallel infection done at the same multiplicity with untreated virus, activation of the DDR proteins was not detectable (Fig. 4A, compare lane 3 to lane 2). These results suggested that neither the infecting virion itself, nor the incoming viral genomes, were sufficient to bring about a DDR at low MOI. This implicated either expression of the viral genes and/or viral replication as responsible for the induction of the cellular DDR.

To assess the contribution of MVM proteins to activation of the DDR, we transfected expression vectors for non-structural NS1 and NS2 proteins into A9 cells. Ectopic expression of NS1, NS2 or NS1+NS2 did not generate significant levels of DDR signaling (Fig. 4B), although expression of NS1 alone led to a slight increase in phosphorylated H2AX above background levels. The requirement for NS2 in activation of the DDR was further analyzed using the NS2-null host-range mutant MVM1989, which is restricted in murine hosts but grows to wild-type levels in human NB324K cells [18]. Infection with MVM1989 generated a DDR to levels similar to wild-type virus in the permissive cells, confirming NS2 was unnecessary to generate this response (data not shown).
These results suggest that viral replication is necessary for induction of the DDR, which cannot be accomplished efficiently merely by expression of MVM RNA or proteins. Since MVM replication is restricted to S-phase, it also follows that the DDR response that it induced is initiated during that phase of the cell cycle.
ATM is activated during MVM infection and predominately mediates the DDR
To address which cellular kinase(s) might be responsible for signaling activated during MVM infection, we analyzed the DDR in the presence of chemical inhibitors and utilized mutant cell lines (Fig. 5). Treatment of MVM-infected A9 cells with caffeine, which inhibits both ATM and ATR [61], dramatically reduced the DDR, while the DNA-PK inhibitor NU7026 [62], [63] reduced the DDR only minimally (data not shown). More specifically, the ATM inhibitor KU55933 [64] could inhibit DDR signaling as monitored by γH2AX and phosphorylated RPA32 (Fig. 5A, compare lane 2 to 3). The ATM inhibitor KU55933 and caffeine produced similar levels of inhibition of DDR signaling when compared in parallel (data not shown), suggesting that ATM was the primary mediator of this effect, and that ATR likely played only a minor or auxiliary role. Whether the reduction in the observed levels of the highly labile NS2 was a direct effect of ATMi, or merely reflective of less transcription template due to the inhibition of virus replication, is not known. When infected with MVM (as confirmed by expression of NS1 and NS2, data not shown) a CHO cell line deficient in DNA-PK (CHO V3) [65] responded with an efficient DDR, as evidenced by phosphorylation of H2AX, RPA32 and p53 (Fig. 5B), although the magnitude of the response was somewhat lower than the parent cell line CHO AA8 (Fig. 5B, compare lanes 2 and 4). Signaling in the DNA-PK-deficient cells was blocked by the ATM inhibitor, confirming that the DDR is ATM-dependent. Consistent with a role for ATM in the mediation of the DDR, phosphorylated ATM was detected in MVM-infected NB324K cells by Western blot analysis using an antibody generated to the S1981 residue auto-phosphorylated in ATM (Fig. 5C). Similar results were seen for A9 cells (data not shown). We used immunofluorescence to assess MVM-infected human NB324K cells for reactivity to this antibody against phosphorylated ATM. Staining with this antibody co-localized with MVM NS1 in viral replication APAR bodies (Fig. 5D). Taken together, these results demonstrated that ATM is the primary mediator of the DDR to MVM infection.

ATM kinase activity is required for efficient MVM replication
We tested the importance of the DDR for MVM replication by assessing accumulation of viral DNA in the presence and absence of kinase inhibitors using Southern blotting of DNA extracted from infected cells (Fig. 6A). Pre-treatment of murine A9 cells with the ATM inhibitor KU55933 significantly reduced MVM replication, compared to untreated cells and cells treated with the DNA-PK inhibitor NU7026 (Fig. 6A, compare lane 3 to lanes 2 and 4). The reduction achieved by the ATM inhibitor was similar to that obtained by pre-treatment with caffeine, or the combination of NU7026 and KU55933 (Fig. 6A, lanes 5 and 6). These results demonstrate that the ATM kinase activity, which mediates the DDR response to MVM infection, facilitates efficient virus replication.

We assessed the effect of the DDR on the cell cycle arrest induced by MVM infection (Fig. 6). Parvovirus replication is dependent on cells cycling through S-phase, and is known to induce a cell cycle block past this point, either in late S-phase or in G2/M [22], [24]. Cells were pre-treated with the inhibitors to ATM and DNA-PK and then infected with MVM. As expected, treatment with the ATM inhibitor alone did not induce a cell cycle block that might inhibit MVM infection (Fig. 6B). Treatment of infected cells with the ATM inhibitor, however, did overcome the replication-dependent MVM-induced block at G2/M (Fig. 6B). In contrast, the DNA-PK inhibitor NU7026 had a negligible effect. These results suggested that the MVM-induced DNA damage response facilitates viral replication, perhaps in part by promoting cell cycle arrest.
Discussion
In this study we investigated interaction of the autonomous parvovirus MVM with the cellular pathways that respond to DNA damage. We have shown via Western blots and immunofluorescence that MVM replication induced a strong DNA damage response, characterized by phosphorylation of H2AX, RPA32, p53, Nbs1, Chk2 and ATM. The majority of these phosphoproteins accumulated at MVM-induced APAR bodies in the nuclei of infected cells. The DDR was observed following MVM infections of permissive cells that were transformed and untransformed, and in cells of murine, hamster and human origin. In addition, the lymphotrophic strain of MVM also led to robust signaling events following infection of murine lymphocyte lines. Together these results demonstrate that infection with the autonomous parvovirus MVM leads to activation of the cellular signaling pathways that form the DDR. Our study is the first to report induction of a DDR by an autonomous parvovirus. Similar results have recently been observed for the autonomous parvovirus minute virus of canine (MVC) (J. Qiu, personal communication).
Comparing cellular responses to different viruses is often revealing about virus-host interactions in general, and can provide insights into fundamental cellular processes. Replication of the related helper-dependent parvovirus AAV, which also consists of a single-stranded linear DNA genome, can occur in the absence of helper following UV-treatment of host cells [66]. AAV has recently been shown to induce a unique DDR [31], [32], and, in contrast to what we report here for MVM infection, the DDR signaling events activated in response to AAV replication appear to be predominantly mediated by the DNA-PK kinase [31], [32]. Interpretation of the DDR induced during AAV replication is, however, complicated by the confounding effects of its helper virus. For example, the MRN complex which plays important roles in ATM-mediated DNA damage responses is degraded in the context of Ad infection [8], and has been shown to be inhibitory to AAV transduction and replication [67]. It is possible that loss of MRN during AAV/Ad co-infection limits the ATM response during AAV infection. The primary role played by ATM following MVM infection is similar to that reported following infections with other viruses such as human papillomavirus HPV [68], SV40 [10], [11] and HSV [12], [69]. It will be interesting to determine if MVM-induced signaling is altered by murine adenovirus co-infection.
DNA damage response signaling is initiated by sensor proteins which recruit transducer kinases and mediators to damage sites [36], [38]. In MVM-infected cells we observed redistribution of MRN complex proteins from their diffuse nuclear localization seen in mock-infected cells into APAR bodies, the sites of ongoing viral replication. Similar accumulation of MRN constituents at sites of viral replication has also been observed during replication of SV40 and AAV [11], [67]. Redistribution of DNA-PKcs, Ku86 and Ku70 has also been observed during AAV replication [31], [32]. These DNA damage proteins may be re-localized as a result of interactions with parvovirus replication proteins [70], [71] or through binding to elements in the virus genome [67]. Although we observed accumulation of Mre11 at the distinct APAR bodies early in MVM infection, we also observed that Mre11 levels were significantly diminished at late time points. The loss of Mre11 was likely due to proteasome-mediated degradation, since it was partially reversed with the proteasome inhibitor MG132. Mre11 has been shown to be inhibitory to the replication of the parvovirus AAV [67], and one of the helper functions provided by Ad is the targeting of the MRN complex for degradation [8], [67]. Proteasome-mediated Mre11 loss has also been observed at late time points during infection with the small DNA tumor virus, SV40 [11]. Surprisingly, the levels of Nbs1 protein, another member of the MRN complex, remained unchanged during MVM infection. The discriminatory loss of a single component of the MRN complex has been previously reported to occur following HSV infection [72], however, in that case Mre11 loss was not proteasome-mediated. Since MRN is required for robust ATM signaling [73], it is possible that it plays important roles early in MVM infection to activate signaling but must be degraded at late times due to an inhibitory function.
Our data suggests that the full spectrum of DNA damage responses during MVM infection required ongoing viral DNA replication. It will be of interest to determine which specific replication intermediates provoke the DDR. Supplying non-replicating input MVM genomes at low MOIs was not sufficient to activate damage signaling. Ectopic expression of NS1 by itself was found to result in a slight but reproducible inductioin of signaling events, including phosphorylation of H2AX. NS1 expression is known to be cytotoxic to cells [74]. The NS1 protein has nickase activities and has been suggested to cause nicks in cellular chromatin [23]. In a recent report the NS1 protein of the closely related parvovirus H1 was shown to cause apoptosis via induction of reactive oxygen species [28]. Any of these NS1 functions could mediate induction of a DDR upon high level expression. Whether these effects are involved in the DDR seen during viral infection is not yet known.
Inhibition of the ATM kinase led to reduced accumulation of MVM replication products and intermediates, suggesting that signaling events enhance viral replication and are exploited by the virus to its benefit. Similar observations have been made with the papovaviruses SV40 [10], [11], JCV [75], polyomavirus [9], and HPV 16 [68], as well as HSV [12]. It is not known if enhancement of MVM replication by ATM signaling events is direct or indirect. For the polyomaviruses, ATM kinase activity contributes directly to increased viral replication, at least in part via phosphorylation of large T antigen on S120 [10]. Since we observed accumulation of ATM in MVM APAR bodies, it is possible that ATM and NS1 interact at these sites. ATM phosphorylates several proteins on well characterized S/TQ sites [76], and four of these motifs are present in NS1. The analogous AAV Rep78 protein seems to be phosphorylated by DNA-PK [32]. It will be interesting to determine whether ATM phosphorylates NS1 and whether this contributes to MVM replication.
MVM infection has been reported to bring about cell cycle arrest at G2/M [24]. Here we extend those findings by showing that ATM catalytic activity is required to allow MVM-induced cell cycle arrest. Inhibition of virus replication by the ATM inhibitor could in turn lead to an abrogation of the virally-induced cell cycle block. Alternatively, or in addition to a potential direct effect of ATM on the main MVM replication protein, ATM might contribute to MVM infection by providing an environment suitable for prolonged viral replication [77]. MVM replication does not commence until cells enter S-phase, yet within infected cells MVM replication continues longer than the duration of normal S-phase (Tullis and Pintel, data not shown). Induction of the DDR results in a halt in cellular DNA replication, yet MVM replication persists. It may be that the ATM kinase activity contributes to MVM replication, at least in part, by governing an extended G2 arrest that facilitates prolonged MVM replication. Thus, MVM induction of the DDR would simultaneously inhibit cellular replication and provide an environment suitable for its own replication in host cells. Similar conclusions have recently been suggested for JCV infection [75]. It is perhaps not surprising that smaller DNA viruses, which encode fewer accessory proteins that might counteract a deleterious DDR, have evolved to exploit the DNA damage response for their replication.
Materials and Methods
Cell lines
Murine A9, EL4, S49 and human 324K cells were propagated as previously described [78]. NIH3T6 cells were kindly supplied by Kathy Spindler (University of Michigan) and were maintained in DMEM with 5% heat-inactivated bovine serum. Chinese hamster ovary (CHO), AA8 and DNA-PK null V3 cell lines were kindly supplied by Dr David Chen (University of Texas) and were maintained in DMEM with 10% bovine serum.
Cell synchronization and drug treatments
A9 cells were para-synchronized in G0 by isoleucine deprivation (described in [79]). Hydroxyurea (Sigma) was used at a final concentration of 2 mM, caffeine (Sigma) was used at a final concentration of 2.5 mM, InSolution ATM kinase inhibitor (KU 55933) was obtained from Calbiochem and used at a final concentration of 7.5 µM, DNA-PK inhibitor (NU 7026) was obtained from Sigma and used at a final concentration of 10 µM, MG132 was obtained from Calbiochem and used at a final concentration of 10 µM, and controls were treated with vehicle (DMSO). Cells were pretreated with the inhibitors for 1 hr before infection and for the duration of infection unless otherwise indicated.
Plasmids and transfections
The NS1 plasmid, NS2 plasmid, and the NS1/2 plasmid which generates a single, slower migrating isoform of NS2 have all been previously described [80]. Transfection of A9 cells was performed using Lipofectamine (Invitrogen) as previously described [78].
Viruses and infections
Wild-type MVMp, MVMi, and MVM1989 were propagated in 324K cells and titred by plaque assay as previously described [81]. Infections were carried out at an MOI of 10 unless otherwise indicated. Where indicated, re-infection was blocked by addition of neutralizing antibody to the media. UV-inactivation of wild-type MVMp was carried out by exposure of virus to 900 mJ of UV radiation using a GS gene linker UV chamber (BioRad).
Antibodies
Commercially available antibodies used in this study were obtained from Cell signaling (Mre11, cat # 4895; Nbs1, cat # 3002; p53-P-S15, cat # 9284; Chk1-P-S345, cat # 2348; Chk2-P-T68, cat # 2662; ATM, cat # 2873), Millipore (γH2AX, cat # 05-636; Chk2, cat # 05-649), Santa Cruz (p53, cat # sc-6243), Thermo Fisher (DNA-PKcs, cat # MS-423-P1), Rockland (ATM-P-S1981, cat # 200-301-400), GeneTex (RPA32, cat # GTX 70258), Bethyl (RPA32-P-S4/8, cat # A300-245A), Sigma (Tubulin, cat # T4026) and Abcam (Actin, cat # ab8226). Additional antibodies used for immunofluorescence were obtained from Novus (Nbs1, cat # NB100-143; Nbs1-P-S343, cat # NB100-284A3), Epitomics (ATM, cat # 1549-1; ATM-P-S1981, cat # 2152-1), R&D systems (γH2AX, cat # AF2288), Neomarkers (DNA-PKcs, cat # MS-370-P1), Santa Cruz (Ku70, cat # SC-9033; Ku86 cat # SC-5280) and Genetex (Mre11, cat # GTX70212). The mouse monoclonal antibody against RPA32 was a gift from Tom Melendy (SUNY-Buffalo). All secondary antibodies were from Invitrogen. Other antibodies used include: a polyclonal rabbit antibody raised to the NH2-terminus of NS1/2, a polyclonal rabbit antibody to NS1 (91W12), a polyclonal rabbit antibody to NS2 and a mouse monoclonal antibody to NS1 (CE10) kindly provided by Carol Astell (University of British Columbia).
Immunoblot analysis
Cells grown and infected in 60 mm dishes were harvested and lysed in modified RIPA buffer containing 20 mM Tris HCL pH 7.5, 150 mM NaCL, 10% glycerol, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, 1 mM EDTA, 10 mM trisodium pyrophosphate, 20 mM sodium fluoride, 2 mM sodium orthovanadate and 1× protease inhibitor cocktail (Sigma). Alternatively, cells were lysed in 2% SDS lysis buffer as previously described [78]. Protein concentrations were quantified by Bradford assay and equal amounts of lysates were loaded in wells and used for western blot analyses as previously described [78].
Immunofluorescence
For immunofluorescence, NB324K cells or para-synchronized A9 cells were grown on glass coverslips in 24-well plates and infected with MVMp using an MOI of 10. After 16–24 hr, cells were washed with PBS, fixed with 4% paraformaldehyde for 15 min and extracted with 0.5% Triton X-100 in PBS for 10 min. Nuclei were visualized by staining with DAPI (4′,6′-diaminido-2-phenylindole). The coverslips were mounted in Fluoromount-G (Southern Biotech) and images were acquired using a Leica TCS SP2 confocal microscope. All images were captured using an objective of 63×.
Analysis of viral DNA
Cell pellets from 60 mm dishes were split in two, with one half used for western blot analysis and the other half for Southern blot analysis. Southern blots were carried out as previously described [56], using whole MVM genome probes.
Cell cycle analysis
Cells were harvested and fixed in 4% formaldehyde for 15 min at room temperature. Alternatively, cells were fixed in 70% ethanol for 15 min on ice. Cells were then pelleted, washed in PBS and resuspended in 50 µg/ml propidium iodide solution containing 0.1 mg/ml RNAase A as well as 0.05% Trition X-100 for 40 min at 37°C. Cells were resuspended in PBS and flow cytometry was performed using FACScan (BD biosciences). Data were analyzed using Flowjo software (Tree Star, OR).
Zdroje
1. LilleyCE
SchwartzRA
WeitzmanMD
2007 Using or abusing: viruses and the cellular DNA damage response. Trends Microbiol 15 119 126
2. WeitzmanMD
CarsonCT
SchwartzRA
LilleyCE
2004 Interactions of viruses with the cellular DNA repair machinery. DNA Repair (Amst) 3 1165 1173
3. EvansJD
HearingP
2005 Relocalization of the Mre11-Rad50-Nbs1 complex by the adenovirus E4 ORF3 protein is required for viral replication. J Virol 79 6207 6215
4. CarsonCT
OrazioNI
LeeDV
SuhJ
Bekker-JensenS
2009 Mislocalization of the MRN complex prevents ATR signaling during adenovirus infection. Embo J 28 652 662
5. LakdawalaSS
SchwartzRA
FerenchakK
CarsonCT
McSharryBP
2008 Differential requirements of the C terminus of Nbs1 in suppressing adenovirus DNA replication and promoting concatemer formation. J Virol 82 8362 8372
6. MathewSS
BridgeE
2007 The cellular Mre11 protein interferes with adenovirus E4 mutant DNA replication. Virology 365 346 355
7. MathewSS
BridgeE
2008 Nbs1-dependent binding of Mre11 to adenovirus E4 mutant viral DNA is important for inhibiting DNA replication. Virology 374 11 22
8. StrackerTH
CarsonCT
WeitzmanMD
2002 Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature 418 348 352
9. DahlJ
YouJ
BenjaminTL
2005 Induction and utilization of an ATM signaling pathway by polyomavirus. J Virol 79 13007 13017
10. ShiY
DodsonGE
ShaikhS
RundellK
TibbettsRS
2005 Ataxia-telangiectasia-mutated (ATM) is a T-antigen kinase that controls SV40 viral replication in vivo. J Biol Chem 280 40195 40200
11. ZhaoX
Madden-FuentesRJ
LouBX
PipasJM
GerhardtJ
2008 Ataxia telangiectasia-mutated damage-signaling kinase - and proteasome-dependent destruction of Mre11-Rad50-Nbs1 subunits in Simian virus 40-infected primate cells. J Virol 82 5316 5328
12. LilleyCE
CarsonCT
MuotriAR
GageFH
WeitzmanMD
2005 DNA repair proteins affect the lifecycle of herpes simplex virus 1. Proc Natl Acad Sci U S A 102 5844 5849
13. LilleyCE
ChaurushiyaMS
BoutellC
LandryS
SuhJ
2010 A viral E3 ligase targets RNF8 and RNF168 to control histone ubiquitination and DNA damage responses. Embo J 29 943 955
14. TattersallP
2006 The evolution of Parvovirus taxonomy.
KerrJR
CotmoreS
BloomME
Parvoviruses London, UK Hodder Arnold 5 14
15. WeitzmanMD
2006 The parvovirus life cycle: an introduction to molecular interactions important for infection.
KerrJR
CotmoreS
BloomME
Parvoviruses London, UK Hodder Arnold 143 156
16. CotmoreSF
TattersallP
2006 Structure and organization of the viral genome.
KerrJR
CotmoreS
BloomME
Parvoviruses London, UK Hodder Arnold 73 94
17. CotmoreSF
TattersallP
2006 A rolling-hairpin strategy: basic mechanisms of DNA replication in the parvoviruses.
KerrJR
CotmoreS
BloomME
Parvoviruses London, UK Hodder Arnold 171 188
18. NaegerLK
CaterJ
PintelDJ
1990 The small nonstructural protein (NS2) of the parvovirus minute virus of mice is required for efficient DNA replication and infectious virus production in a cell-type-specific manner. J Virol 64 6166 6175
19. BashirT
RommelaereJ
CziepluchC
2001 In vivo accumulation of cyclin A and cellular replication factors in autonomous parvovirus minute virus of mice-associated replication bodies. J Virol 75 4394 4398
20. CziepluchC
LampelS
GrewenigA
GrundC
LichterP
2000 H-1 parvovirus-associated replication bodies: a distinct virus-induced nuclear structure. J Virol 74 4807 4815
21. YoungPJ
JensenKT
BurgerLR
PintelDJ
LorsonCL
2002 Minute virus of mice NS1 interacts with the SMN protein, and they colocalize in novel nuclear bodies induced by parvovirus infection. J Virol 76 3892 3904
22. Op De BeeckA
AnoujaF
MoussetS
RommelaereJ
Caillet-FauquetP
1995 The nonstructural proteins of the autonomous parvovirus minute virus of mice interfere with the cell cycle, inducing accumulation in G2. Cell Growth Differ 6 781 787
23. Op De BeeckA
Caillet-FauquetP
1997 The NS1 protein of the autonomous parvovirus minute virus of mice blocks cellular DNA replication: a consequence of lesions to the chromatin? J Virol 71 5323 5329
24. Op De BeeckA
Sobczak-ThepotJ
SirmaH
BourgainF
BrechotC
2001 NS1 - and minute virus of mice-induced cell cycle arrest: involvement of p53 and p21(cip1). J Virol 75 11071 11078
25. CotmoreSF
TattersallP
1987 The autonomously replicating parvoviruses of vertebrates. Adv Virus Res 33 91 174
26. MoehlerM
BlechaczB
WeiskopfN
ZeidlerM
StremmelW
2001 Effective infection, apoptotic cell killing and gene transfer of human hepatoma cells but not primary hepatocytes by parvovirus H1 and derived vectors. Cancer Gene Ther 8 158 167
27. RayetB
Lopez-GuerreroJA
RommelaereJ
DinsartC
1998 Induction of programmed cell death by parvovirus H-1 in U937 cells: connection with the tumor necrosis factor alpha signalling pathway. J Virol 72 8893 8903
28. HristovG
KramerM
LiJ
El-AndaloussiN
MoraR
2010 Through its nonstructural protein NS1, parvovirus H-1 induces apoptosis via accumulation of reactive oxygen species. J Virol 84 5909 5922
29. JurvansuuJ
RajK
StasiakA
BeardP
2005 Viral transport of DNA damage that mimics a stalled replication fork. J Virol 79 569 580
30. RajK
OgstonP
BeardP
2001 Virus-mediated killing of cells that lack p53 activity. Nature 412 914 917
31. CollacoRF
BevingtonJM
BhriguV
Kalman-MalteseV
TrempeJP
2009 Adeno-associated virus and adenovirus coinfection induces a cellular DNA damage and repair response via redundant phosphatidylinositol 3-like kinase pathways. Virology 392 24 33
32. SchwartzRA
CarsonCT
SchuberthC
WeitzmanMD
2009 Adeno-associated virus replication induces a DNA damage response coordinated by DNA-dependent protein kinase. J Virol 83 6269 6278
33. LavinMF
2007 ATM and the Mre11 complex combine to recognize and signal DNA double-strand breaks. Oncogene 26 7749 7758
34. PaullTT
LeeJH
2005 The Mre11/Rad50/Nbs1 complex and its role as a DNA double-strand break sensor for ATM. Cell Cycle 4 737 740
35. WilliamsRS
WilliamsJS
TainerJA
2007 Mre11-Rad50-Nbs1 is a keystone complex connecting DNA repair machinery, double-strand break signaling, and the chromatin template. Biochem Cell Biol 85 509 520
36. PetriniJH
StrackerTH
2003 The cellular response to DNA double-strand breaks: defining the sensors and mediators. Trends Cell Biol 13 458 462
37. ShrivastavM
De HaroLP
NickoloffJA
2008 Regulation of DNA double-strand break repair pathway choice. Cell Res 18 134 147
38. LovejoyCA
CortezD
2009 Common mechanisms of PIKK regulation. DNA Repair (Amst) 8 1004 1008
39. WardIM
MinnK
ChenJ
2004 UV-induced ataxia-telangiectasia-mutated and Rad3-related (ATR) activation requires replication stress. J Biol Chem 279 9677 9680
40. CimprichKA
CortezD
2008 ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol 9 616 627
41. WeteringsE
ChenDJ
2007 DNA-dependent protein kinase in nonhomologous end joining: a lock with multiple keys? J Cell Biol 179 183 186
42. StiffT
O'DriscollM
RiefN
IwabuchiK
LobrichM
2004 ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res 64 2390 2396
43. DickeyJS
RedonCE
NakamuraAJ
BairdBJ
SedelnikovaOA
2009 H2AX: functional roles and potential applications. Chromosoma 118 683 692
44. AbrahamRT
2001 Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev 15 2177 2196
45. HarperJW
ElledgeSJ
2007 The DNA damage response: ten years after. Mol Cell 28 739 745
46. RoosWP
KainaB
2006 DNA damage-induced cell death by apoptosis. Trends Mol Med 12 440 450
47. JacksonSP
BartekJ
2009 The DNA-damage response in human biology and disease. Nature 461 1071 1078
48. KumarS
DodsonGE
TrinhA
PuchalskiJR
TibbettsRS
2005 ATR activation necessary but not sufficient for p53 induction and apoptosis in hydroxyurea-hypersensitive myeloid leukemia cells. Cell Cycle 4 1667 1674
49. ChaturvediP
EngWK
ZhuY
MatternMR
MishraR
1999 Mammalian Chk2 is a downstream effector of the ATM-dependent DNA damage checkpoint pathway. Oncogene 18 4047 4054
50. MatsuokaS
HuangM
ElledgeSJ
1998 Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 282 1893 1897
51. MatsuokaS
RotmanG
OgawaA
ShilohY
TamaiK
2000 Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc Natl Acad Sci U S A 97 10389 10394
52. GuoZ
KumagaiA
WangSX
DunphyWG
2000 Requirement for Atr in phosphorylation of Chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in Xenopus egg extracts. Genes Dev 14 2745 2756
53. LiuQ
GuntukuS
CuiXS
MatsuokaS
CortezD
2000 Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev 14 1448 1459
54. Lopez-GironaA
TanakaK
ChenXB
BaberBA
McGowanCH
2001 Serine-345 is required for Rad3-dependent phosphorylation and function of checkpoint kinase Chk1 in fission yeast. Proc Natl Acad Sci U S A 98 11289 11294
55. ZhaoH
Piwnica-WormsH
2001 ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol 21 4129 4139
56. ChoiEY
NewmanAE
BurgerL
PintelD
2005 Replication of minute virus of mice DNA is critically dependent on accumulated levels of NS2. J Virol 79 12375 12381
57. FalckJ
CoatesJ
JacksonSP
2005 Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 434 605 611
58. YouZ
ChahwanC
BailisJ
HunterT
RussellP
2005 ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol Cell Biol 25 5363 5379
59. BitonS
DarI
MittelmanL
PeregY
BarzilaiA
2006 Nuclear ataxia-telangiectasia mutated (ATM) mediates the cellular response to DNA double strand breaks in human neuron-like cells. J Biol Chem 281 17482 17491
60. CarsonCT
SchwartzRA
StrackerTH
LilleyCE
LeeDV
2003 The Mre11 complex is required for ATM activation and the G2/M checkpoint. Embo J 22 6610 6620
61. SarkariaJN
BusbyEC
TibbettsRS
RoosP
TayaY
1999 Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res 59 4375 4382
62. HollickJJ
GoldingBT
HardcastleIR
MartinN
RichardsonC
2003 2,6-disubstituted pyran-4-one and thiopyran-4-one inhibitors of DNA-Dependent protein kinase (DNA-PK). Bioorg Med Chem Lett 13 3083 3086
63. VeugerSJ
CurtinNJ
RichardsonCJ
SmithGC
DurkaczBW
2003 Radiosensitization and DNA repair inhibition by the combined use of novel inhibitors of DNA-dependent protein kinase and poly(ADP-ribose) polymerase-1. Cancer Res 63 6008 6015
64. HicksonI
ZhaoY
RichardsonCJ
GreenSJ
MartinNM
2004 Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res 64 9152 9159
65. BluntT
FinnieNJ
TaccioliGE
SmithGC
DemengeotJ
1995 Defective DNA-dependent protein kinase activity is linked to V(D)J recombination and DNA repair defects associated with the murine scid mutation. Cell 80 813 823
66. YakobsonB
HrynkoTA
PeakMJ
WinocourE
1989 Replication of adeno-associated virus in cells irradiated with UV light at 254 nm. J Virol 63 1023 1030
67. SchwartzRA
PalaciosJA
CassellGD
AdamS
GiaccaM
2007 The Mre11/Rad50/Nbs1 complex limits adeno-associated virus transduction and replication. J Virol 81 12936 12945
68. MoodyCA
LaiminsLA
2009 Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog 5 e1000605
69. ShirataN
KudohA
DaikokuT
TatsumiY
FujitaM
2005 Activation of ataxia telangiectasia-mutated DNA damage checkpoint signal transduction elicited by herpes simplex virus infection. J Biol Chem 280 30336 30341
70. NashK
ChenW
SalganikM
MuzyczkaN
2009 Identification of cellular proteins that interact with the adeno-associated virus rep protein. J Virol 83 454 469
71. NicolasA
Alazard-DanyN
BiollayC
ArataL
JolinonN
2010 Identification of rep-associated factors in herpes simplex virus type 1-induced adeno-associated virus type 2 replication compartments. J Virol 84 8871 8887
72. GregoryDA
BachenheimerSL
2008 Characterization of mre11 loss following HSV-1 infection. Virology 373 124 136
73. UzielT
LerenthalY
MoyalL
AndegekoY
MittelmanL
2003 Requirement of the MRN complex for ATM activation by DNA damage. Embo J 22 5612 5621
74. AnoujaF
WattiezR
MoussetS
Caillet-FauquetP
1997 The cytotoxicity of the parvovirus minute virus of mice nonstructural protein NS1 is related to changes in the synthesis and phosphorylation of cell proteins. J Virol 71 4671 4678
75. OrbaY
SuzukiT
MakinoY
KubotaK
TanakaS
2010 Large T antigen promotes JC virus replication in G2-arrested cells by inducing ATM - and ATR-mediated G2 checkpoint signaling. J Biol Chem 285 1544 1554
76. TravenA
HeierhorstJ
2005 SQ/TQ cluster domains: concentrated ATM/ATR kinase phosphorylation site regions in DNA-damage-response proteins. Bioessays 27 397 407
77. KudohA
FujitaM
ZhangL
ShirataN
DaikokuT
2005 Epstein-Barr virus lytic replication elicits ATM checkpoint signal transduction while providing an S-phase-like cellular environment. J Biol Chem 280 8156 8163
78. MillerCL
PintelDJ
2002 Interaction between parvovirus NS2 protein and nuclear export factor Crm1 is important for viral egress from the nucleus of murine cells. J Virol 76 3257 3266
79. SchoborgRV
PintelDJ
1991 Accumulation of MVM gene products is differentially regulated by transcription initiation, RNA processing and protein stability. Virology 181 22 34
80. MillerCL
PintelDJ
2001 The NS2 protein generated by the parvovirus minute virus of mice is degraded by the proteasome in a manner independent of ubiquitin chain elongation or activation. Virology 285 346 355
81. NaegerLK
SalomeN
PintelDJ
1993 NS2 is required for efficient translation of viral mRNA in minute virus of mice-infected murine cells. J Virol 67 1034 1043
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 10
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Retroviral RNA Dimerization and Packaging: The What, How, When, Where, and Why
- Viral Replication Rate Regulates Clinical Outcome and CD8 T Cell Responses during Highly Pathogenic H5N1 Influenza Virus Infection in Mice
- Antimicrobial Peptides: Primeval Molecules or Future Drugs?
- Crystal Structure of DotD: Insights into the Relationship between Type IVB and Type II/III Secretion Systems
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy