
-Induced Inactivation of the Macrophage Transcription Factor AP-1 Is Mediated by the Parasite Metalloprotease GP63
Leishmania parasites have evolved sophisticated mechanisms to subvert macrophage immune responses by altering the host cell signal transduction machinery, including inhibition of JAK/STAT signalling and other transcription factors such as AP-1, CREB and NF-κB. AP-1 regulates pro-inflammatory cytokines, chemokines and nitric oxide production. Herein we show that upon Leishmania infection, AP-1 activity within host cells is abolished and correlates with lower expression of 5 of the 7 AP-1 subunits. Of interest, c-Jun, the central component of AP-1, is cleaved by Leishmania. Furthermore, the cleavage of c-Jun is dependent on the expression and activity of the major Leishmania surface protease GP63. Immunoprecipitation of c-Jun from nuclear extracts showed that GP63 interacts, and cleaves c-Jun at the perinuclear area shortly after infection. Phagocytosis inhibition by cytochalasin D did not block c-Jun down-regulation, suggesting that internalization of the parasite might not be necessary to deliver GP63 molecules inside the host cell. This observation was corroborated by the maintenance of c-Jun cleavage upon incubation with L. mexicana culture supernatant, suggesting that secreted, soluble GP63 could use a phagocytosis-independent mechanism to enter the host cell. In support of this, disruption of macrophage lipid raft microdomains by Methyl β-Cyclodextrin (MβCD) partially inhibits the degradation of full length c-Jun. Together our results indicate a novel role of the surface protease GP63 in the Leishmania-mediated subversion of host AP-1 activity.
Published in the journal:
. PLoS Pathog 6(10): e32767. doi:10.1371/journal.ppat.1001148
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1001148
Summary
Leishmania parasites have evolved sophisticated mechanisms to subvert macrophage immune responses by altering the host cell signal transduction machinery, including inhibition of JAK/STAT signalling and other transcription factors such as AP-1, CREB and NF-κB. AP-1 regulates pro-inflammatory cytokines, chemokines and nitric oxide production. Herein we show that upon Leishmania infection, AP-1 activity within host cells is abolished and correlates with lower expression of 5 of the 7 AP-1 subunits. Of interest, c-Jun, the central component of AP-1, is cleaved by Leishmania. Furthermore, the cleavage of c-Jun is dependent on the expression and activity of the major Leishmania surface protease GP63. Immunoprecipitation of c-Jun from nuclear extracts showed that GP63 interacts, and cleaves c-Jun at the perinuclear area shortly after infection. Phagocytosis inhibition by cytochalasin D did not block c-Jun down-regulation, suggesting that internalization of the parasite might not be necessary to deliver GP63 molecules inside the host cell. This observation was corroborated by the maintenance of c-Jun cleavage upon incubation with L. mexicana culture supernatant, suggesting that secreted, soluble GP63 could use a phagocytosis-independent mechanism to enter the host cell. In support of this, disruption of macrophage lipid raft microdomains by Methyl β-Cyclodextrin (MβCD) partially inhibits the degradation of full length c-Jun. Together our results indicate a novel role of the surface protease GP63 in the Leishmania-mediated subversion of host AP-1 activity.
Introduction
Parasites of the Leishmania genus are the causative agent of leishmaniasis; a disease distributed worldwide affecting more than 12 million people in 88 countries [1]. Leishmaniasis is a complex of diseases ranging from self-healing cutaneous lesions to lethal visceral afflictions [2]. In its mammalian host, Leishmania is an obligate intracellular pathogen infecting hematopoietic cells of the monocyte/macrophage lineage. Macrophages are specialized for the destruction of invading pathogens and priming the immune response. In order to survive within these cells, Leishmania has evolved sophisticated mechanisms to subvert macrophage microbicidal functions such as inhibition of nitric oxide (NO) production and cytokine-inducible macrophage functions [3]. This occurs as the direct consequence of parasite-mediated activation of protein tyrosine phosphatases, alteration of signal transduction and inhibition of nuclear translocation and activity of transcription factors such as NF-κB, STAT, CREB and AP-1[4], [5]. Activated Protein-1 (AP-1) is an important transcription factor that mediates gene regulation in response to physiological and pathological stimuli, including cytokines, growth factors, stress signals, bacterial and viral infections, apoptosis, as well oncogenic responses [6], [7]. AP-1 is formed by homodimers of Jun family members (c-Jun, Jun B and Jun D), or heterodimers of Jun and Fos family members (c-Fos, Fos B, Fra 1 and Fra 2). Homodimers within the Fos family do not occur due to conformational repulsion [8].
Previous studies have reported that the AP-1 transcription factor is inactivated by Leishmania infection. For instance, activation of macrophage AP-1 and NF-κB is inhibited by L. donovani promastigotes through an increase in intracellular ceramide concentration, which leads to the down-regulation of classical PKC activity, up-regulation of calcium independent atypical PKC-ζ and dephosphorylation of Extracellular Signal-Regulated Kinases (ERK) [9], [10]. Other studies have shown that Leishmania alters signal transduction upstream of c-Fos and c-Jun by inhibiting ERK, JNK and p38 MAP Kinases, resulting in a reduction of AP-1 nuclear translocation [11], [12]. However, little is known about the molecular mechanism (s) by which Leishmania parasites are able to inactivate this important transcription factor.
Many Leishmania-specific factors such as lipophosphoglycan (LPG), A2 proteins, cysteine peptidases (CPs) and the protease GP63, contribute to Leishmania virulence and pathogenicity. LPG has been implicated in altering phagosome maturation in L. donovani infection [13].The A2 proteins of L. donovani are involved in intracellular amastigote survival [14].The cysteine peptidases of L. mexicana are implicated in facilitating the survival and growth of the parasite [15]. Furthermore GP63, also known as the major surface protease (MSP), has been related to resistance to complement-mediated lysis, among others [16], [17]. GP63 is a metalloprotease which belongs to the metzincin class. It is the most abundant surface glycoprotein of the parasite and accounts for 1% of the total protein content of L. mexicana promastigotes [18]. GP63 of different Leishmania species encode similar amino acid sequences, although slight substrate specificity variations have been reported [19]. Specific characteristics of this class of metalloproteases include a conserved signature motif HEXXHXXGXXH and an N-terminal pro-peptide that serves to maintain the pro-enzyme inactive during translation, which is removed upon protein maturation and activation [20]. The mature GP63 contains 3 domains: 1) N-terminal (bases ∼101-273) which comprises a structure corresponding to the catalytic module of metzincin class zinc protease, 2) central domain (bases ∼274–391) and 3) C-terminal domain containing the site of glycosylphosphatidylinositol (GPI) anchor addition (bases ∼392–577) [17], [18], [20], [21]. We have previously shown that this protease actively participates in the cleavage of NF-κB [5], protein tyrosine phosphatases (PTP) [22] and actin cytoskeleton regulators [23]. In this study we have investigated how GP63 contributes to the inactivation of AP-1 and the degradation of its subunits. Herein, we report that GP63 enters the host cell via lipid raft microdomains, independently of parasite internalization, and for the first time show that it is able to reach the nuclear compartment shortly after infection where it degrades and cleaves c-Jun and other AP-1 subunits.
Results
Alteration of steady state level of AP-1 upon Leishmania infection involves down-regulation/degradation of selected Jun/Fos family members
We have previously studied the effect of Leishmania promastigote infection on the activity of various macrophage transcription factors: STAT-1α degradation is proteasome and receptor-dependent and is mediated through a mechanism involving PKC-α [4], and cleavage of NF-κB subunits upon Leishmania infection is in part dependent on GP63 [5]. As AP-1 is an important transcription factor regulating the expression of many genes involved in the activation of macrophage functions (TNFα, iNOS, and IL-12) [24], [25], [26] critical for the adequate innate immune response against Leishmania infection, we investigated the mechanisms underlying AP-1 inactivation upon Leishmania infection.
To evaluate nuclear translocation and DNA binding activity of macrophage AP-1 upon infection with Leishmania promastigotes, Electrophoretic Mobility Shift Assays (EMSA) were performed. As shown in Figure 1A, AP-1 nuclear translocation was inhibited as early as 30 min post-infection in L. donovani-infected macrophages. Furthermore, we observed that other mammalian pathogenic Leishmania species (L. mexicana and L. major) were able to alter AP-1 DNA binding (Figure 1B). Of interest, we did not observe any effect on macrophages infected with L. tarentolae, whose pathogenicity is limited to reptilian hosts.

In order to better understand the observed decrease in AP-1 activity, we performed Western Blot (WB) analysis in the total cell extracts to evaluate the various AP-1 subunits during infection. Five out of the seven AP-1 subunits (c-Fos, Fra 1, Fra 2, c-Jun and Jun B) showed decreased expression after infection with L. donovani, whereas Jun D presented a slight reduction and Fos B was maintained intact (Figure 2A). To further confirm the presence of these subunits in the AP-1 complex we used super shift analysis. This approach uses the incorporation of specific antibodies to nuclear protein extracts, allowing the visualization of the antibody: protein: DNA complexes by retarding the migration of the specific bands in the gel. As shown in Figure 2B, inclusion of antibodies specific for Fos B, c-Fos, Fra 1, Fra 2, c-Jun, Jun B and Jun D, demonstrated presence of Fra 1, Fra 2, c-Jun, Jun B and Jun D, but not c-Fos or Fos B, within the macrophage nuclear AP-1 complex. Importantly, L. donovani infection clearly affected the AP-1 complex as the bands observed for Fra-1, Fra-2, c-Jun, Jun B and Jun D in the super shift assay was greatly reduced. Whereas the c-Fos protein was not detectable by super shift assay, this protein was still affected by Leishmania infection since less expression was observed by WB (see Figures 2A and 3), suggesting that the amount of c-Fos might not be enough to be detected by super shift assay.
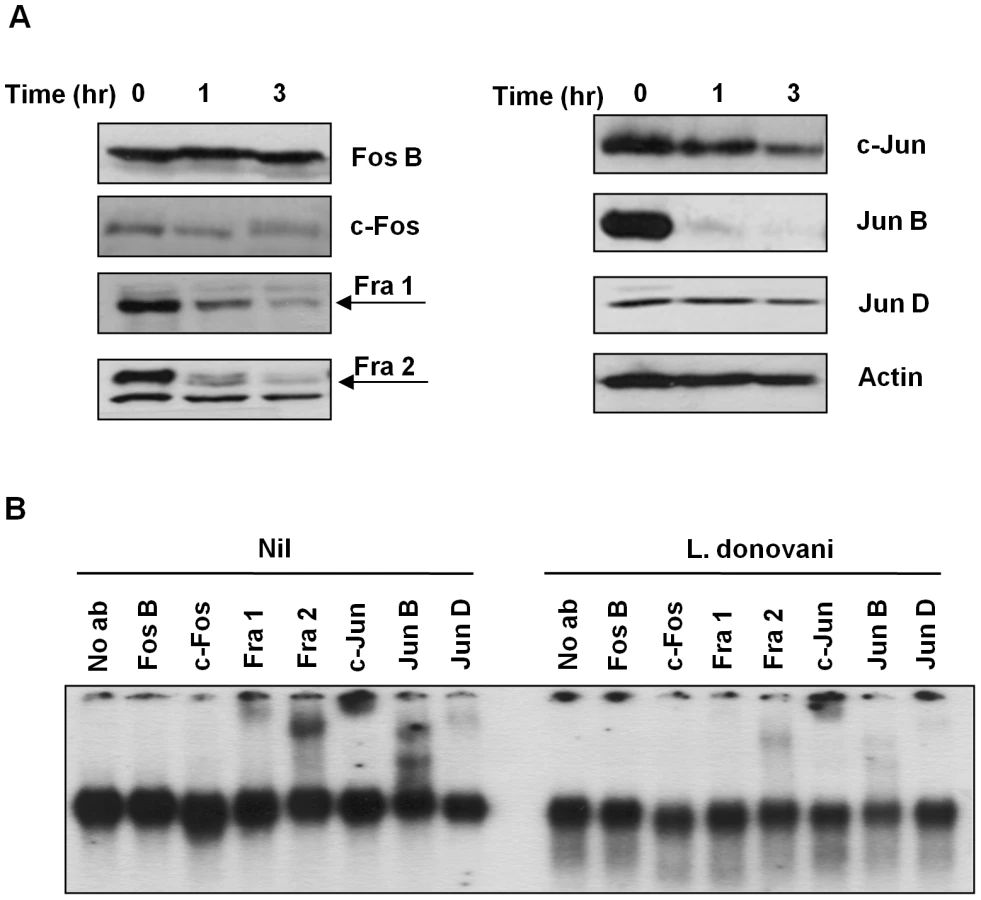

After phosphorylation AP-1 subunits are translocated into the nucleus where they dimerize with another subunit to form an active AP-1 complex [6], [7], [8], [27]. To determine the level of expression of each AP-1 subunit in the different cellular compartments (cytoplasm vs nucleus), we performed WB analysis on separated nuclear and cytoplasmic fractions. As shown in Figure 3, different phenomena can be observed. c-Fos and Fra-1 expression in the cytoplasmic fraction are not altered with Leishmania infection, but their expression in the nuclear fraction is decreased in infected macrophages; Fra-2 and c-Jun have decreased expression in both cytoplasmic and nuclear fractions, and Fra-2 in the nuclear fraction presents a band with less migration than the band observed in the cytoplasmic fractions, possible due to post-nuclear translocation modifications. On the other hand, Jun-B and Jun-D were detected only in the nuclear fraction; however, only Jun-B expression is affected by Leishmania infection. The lower expression of the different subunits in the nucleus could be due to decreased complex formation and/or cleavage and further degradation of the subunits, as it is possible to detect smaller bands (c-Jun and Jun-B).
Leishmania major surface protease GP63 is involved in AP-1 inactivation
Leishmania surface molecules such as LPG and GP63, among others, play important roles as virulence factors and modulators of host cell signalling. LPG, for instance, has been implicated in the interference of phagolysosome maturation and inactivation of PKC signalling [13], [28]. GP63 has been related to resistance to complement-mediated lysis, migration of Leishmania parasites through the extracellular matrix by degradation of casein, fibrinogen and collagen [16], [21] and inhibition of JAK/STAT signalling by modulation of PTP activities [22]. To address the role of LPG and GP63 in AP-1 inactivation we performed EMSA with extracts from cells infected with Leishmania mutants for these two surface molecules. As shown in Figure 4A, LPG is not involved in the AP-1 degradation induced by Leishmania infection since DNA binding in macrophages infected with either L. donovani or L. donovani LPG−/− promastigotes was similarly altered. Importantly, however, we observed that cells infected with an L. major strain lacking GP63 (L. major GP63−/−) [29] showed normal AP-1 DNA binding capacity, compared to uninfected controls. This suggests that GP63 but not LPG is highly involved in the mechanism responsible for the inactivation of AP-1 transcription factor.

To further elucidate the role of GP63, we performed WB analysis of all the AP-1 subunits of macrophages infected with L. major, L. major GP63−/− and L. major GP63 Rescued. Results obtained further revealed that in the absence of GP63, no degradation or cleavage of any AP-1 subunit was evident (Figure 4B); supporting the finding that AP-1 activity is unaffected in L. major GP63−/−-infected macrophages.
In addition, to validate the role of GP63 on AP-1 activity we verified the expression of IL-12 transcripts, as it is known that AP-1 regulates its transcription [24]. As shown in the Figure S1, LPS-induced IL-12 expression is fully blocked by all infectious Leishmania species but not by L. major GP63−/− and L. tarentolae. As expected, the JNK/c-Jun inhibitor has completely inhibited LPS-induced IL-12 transcripts.
GP63 action requires macrophage lipid raft and is not dependent on parasite phagocytosis
Leishmania GP63 can be found in three different forms: 1) Intracellular GP63, 2) Surface GPI-anchored GP63 and 3) secreted or released GP63 [21], [30]. For GP63 to target its intracellular macrophage substrates, it needs to gain access to or be internalized by the macrophage. To explore whether the internalization of the parasite is necessary to deliver GP63 inside the cell, murine macrophages were pre-treated with the phagocytosis inhibitor cytochalasin D which inhibits actin polymerization, therefore blocking internalization by phagocytosis (Figure S2A). We used c-Jun as a model protein to evaluate the cleavage and degradation of the AP-1 subunits. WB analysis showed that parasite phagocytosis was not necessary for c-Jun cleavage and less expression (Figure 5A). To confirm this, we incubated macrophages with the culture supernatant of L. mexicana promastigotes, which is rich in soluble GP63 [18], [31]. WB showed that even in the absence of the parasite, c-Jun degradation was observed (Figure 5B).

Since phagocytosis seems not to be completely required in the internalization of GP63 we addressed whether GP63 internalization could be dependent on lipid raft-mediated endocytosis, given the fact that GP63 is an excreted and membrane-GPI anchored protein. On the other hand, lipid raft microdomains are highly dynamic membrane domains rich in cholesterol and sphingolipids, and present high affinity for proteins containing GPI anchors [32], [33], [34]. In order to examine the possible role of host lipid raft microdomains in GP63 internalization, we pre-treated cells with a non-cytotoxic dose (Figure S2B) of the cholesterol chelator and inhibitor of lipid raft integrity methyl-β-clyclodextrin (MβCD) prior to infection. As shown in Figure 5C, full length c-Jun was not degraded in cells infected under these conditions, although interestingly, a cleavage fragment was still observed. Pre-treatment of macrophages with MβCD and subsequent incubation with L. mexicana supernatant showed that lipid raft disruption altered internalization of parasite-free soluble GP63 and also impaired c-Jun degradation (Figure 5D). As shown in Figure S3, confocal microscopy confirmed an interaction between lipid raft microdomains and GP63, since in macrophages infected with L. major, GP63 (green) partially co-localized with the lipid raft marker Choleratoxin B (red). Furthermore, we have previously shown that, pre-treatment with MβCD before infection abrogates GP63 internalization [22]. To determine if MβCD had any effect over the c-Jun expression we performed a time course analysis of macrophages stimulated with MβCD. As shown in Figure S4A, there was no alteration in the expression of c-Jun after 2 hr of incubation of the macrophages with the drug. Together these data strengthen the hypothesis that GP63 uses lipid raft microdomains for internalization independent of parasite entry.
To evaluate the role of the GPI anchor in mediating GP63 internalization via lipid raft microdomains, macrophages were incubated with a GPI-deficient recombinant GP63 (rGP63) and c-Jun degradation was monitored. WB analysis evidenced that neither degradation nor cleavage of c-Jun occurred (Figure 5E) in the presence or absence of MβCD, similarly to what we have previously shown for GP63-mediated PTP cleavage. Moreover, although rGP63 is still internalized in macrophages to a limited extent, perinuclear localization was never detected [22].
To demonstrate that the less expression and cleavage of c-Jun observed in this set of experiments were occurring inside the cells and not as an effect of proteolysis during the preparation of the lysates, we included two experiments as controls; first, we lysed the cells using sample loading buffer 1× and the samples were boiled right after, to stop the proteolysis; second, we added 1 mM of phenanthroline (a Zn chelator [35]) to the lysis buffer to abrogate post-infection GP63 activity. In both experiments, we observed that cleavage of c-Jun under these conditions still occurs, suggesting that the cleavage of c-Jun occurs inside the cell and not during the sample preparation (Figure S4B and S4C). In addition, to establish whether GP63 proteolytic activity is critical for c-Jun cleavage in the macrophage, L. mexicana culture supernatant was treated with the GP63 inhibitor phenanthroline prior to its incubation with macrophages. As shown in the Figure S4D, phenanthroline fully inhibited GP63-mediated c-Jun degradation.
GP63-mediated c-Jun cleavage occurs at perinuclear compartment
One of the most surprising elements of the evidence presented above was the fact that GP63 is able to act on its substrate proteins within the nucleus of its host cell. In order to further demonstrate that GP63 reaches the nucleus, we separated cytoplasmic and nuclear proteins from macrophages infected with L. major, L. major GP63−/− and L. major GP63 Rescued. WB analysis using an anti-GP63 antibody revealed that this protease is present in both fractions of Leishmania-infected cell extracts. As expected, there was no GP63 in macrophages infected with L. major GP63 −/− or the uninfected control (Figure 6A). Confocal microscopy of Leishmania-infected macrophages confirmed that GP63 reaches the nuclear membrane as early as 1 hr post-infection (Figure 6B). In order to demonstrate the purity of our fractions, we performed WB of the cytoplasmic and nuclear proteins against the lysosomal marker LAMP-1, the ER specific marker (the KDEL protein - Lys-Asp-Glu-Leu endoplasmic reticulum protein retention receptor), histone 2B (nuclear marker), and actin (cytoplasm marker). Figure S5 shows that actin, LAMP-1 and KDEL are only present in the cytoplasmic fraction, in contrast, histone is only detected in the nuclear fraction, and this way we are confident to say that GP63 was present in both protein fractions.

In order to confirm nuclear interaction of GP63 with c-Jun we performed a Co-Immunoprecipitation (IP) assay. c-Jun was immunoprecipitated from nuclear extracts of Leishmania-infected macrophages and subjected to WB analysis of GP63. This result revealed a band around 65 kDa, confirming the interaction between nuclear c-Jun and GP63 in the macrophages infected with L. major and L. major GP63 Rescue, but not with L. major GP63 −/− as is shown in Figure 7.

To further support that degradation of c-Jun could occurs in the nucleus we performed confocal microscopy. As shown in Figure 8A (upper panel), c-Jun (red) is localized inside the nucleus in uninfected cells. However, after 1 hr of infection GP63 was detected in the perinuclear area and the fluorescence intensity of c-Jun was considerably diminished (Figure 8A, lower panel), such reduction in the fluorescence was not observed in macrophages infected either with L. major GP63−/− or L. tarentolae (Figure 8C upper and lower panels, respectively). The upper panel of Figure 8B shows partial co-localization between the nuclear stain (blue) and GP63 (green) in the periphery of the nucleus, giving a light blue signal. Of utmost importance, the panel representing c-Jun (red) versus nucleus (blue) co-localization, clearly reveals that c-Jun is absent from perinuclear area as this one is solely stained in blue (Figure 8B, lower panel). To discard possible unspecific signals in the confocal micrographs we included specific isotype and secondary antibody controls (Figure S6B). Collectively, our results suggest that GP63 reaches the perinuclear area of the cell shortly after macrophage-parasite contact occurred leading to degradation and cleavage of various members of AP-1 subunits, leading to its inability to dimerize and bind DNA and therefore, altering AP-1 transcriptional activity on genes under its regulation.

GP63 can directly cleave c-Jun
To further understand the direct effect of GP63 on c-Jun, we used a purified GST tagged-c-Jun protein and incubated it with Leishmania promastigotes of different species (including L. donovani, L. mexicana, L. major, L. major GP63−/− and L. major GP63 Rescued). WB analysis showed that direct contact of parasites expressing GP63 and c-Jun protein is sufficient to induce c-Jun degradation (Figure 9A). This was corroborated by the reduction of c-Jun degradation when incubated with the GP63−/− strain. Figure S7 shows that L. tarentolae has no effect on the degradation of GST-c-Jun.

GP63 recognizes a four amino acid motif in its target protein substrates based on amino acid characteristics: polar/hydrophobic/basic/basic amino acids (P1 - P′1-P′2-P′3) [19]. Sequence analysis of AP-1 subunits revealed putative cleavage sites within c-Jun, Jun B, Jun D and c-Fos (Figure 9B). Of interest, one of the sequence-identified cleavage sites of c-Jun was found between the leucine zipper and the DNA binding domain as shown in Figure 9C. In addition, this motif is found at amino acids 271-275, which will generate cleaved fragments with molecular weight similar to the one detected by WB of lysates from infected cells (∼30 kDa).
Collectively, these data indicate that the Leishmania protease GP63 actively participates in altering the DNA binding capacity of AP-1 as a consequence of the diminished expression and cleavage of its subunits. These data further corroborate a mechanism whereby GP63 can enter the cell using lipid raft microdomains, and show for the first time that GP63 reaches the perinuclear area where it proteolytically degrades AP-1 subunits.
Discussion
Leishmania parasites have evolved many mechanisms to undermine macrophage signalling pathways in order to survive and replicate inside these cells. For instance, parasite-mediated activation of macrophage PTPs leads to protein dephosphorylation resulting in the inactivation of transcription factors controlling the expression of many genes required for the effective activation of the innate immune response [36], and macrophage effector functions such as NO production [37]. We have previously reported that Leishmania promastigote infection induces degradation and inactivation of some transcription factors. For example, STAT 1 is inactivated by a proteasome mediated mechanism [4], and NF-kB activity is altered in a cleavage-dependent fashion [5]. We show that cleavage of p65 generates an active fragment, p35, which is able to translocate into the nucleus, where it dimerizes with p50 to induce specific chemokine gene expression. Interestingly this cleavage event was found to occur in the macrophage cytoplasm in a GP63-dependent mechanism [5].
Along with STAT and NF-κB, AP-1 is responsible for the transcription of iNOS [38]. NO is a by-product of iNOS-mediated conversion of L-arginine to L-citruline and is essential for the control of Leishmania infection [3], [36]. Among other genes regulated by AP-1 in macrophages and known to be affected by Leishmania infection are TNFα, IL-1β and IL-12 [24], [25], [26], [39]. The breadth and importance of the immunological functions of AP-1 highlights how detrimental its degradation is to host defence against Leishmania infection. Previously, Descoteaux and Matlashewski (1989) demonstrated that the c-fos gene, one of the main activators of AP-1, was down-regulated due to abnormal PKC signalling [40]. More recently Ghosh and colleagues (2002) reported Leishmania-dependent inactivation of both AP-1 and NF-κB in a ceramide dependent mechanism, where increased levels of intracellular ceramide conducted to the down-regulation of classical PKC activity and impartment of the phosphorylation of ERK, which results in decreased AP-1 activation [10]. These previous reports have given some indication of AP-1 inactivation by Leishmania. Here we further demonstrated the molecular mechanisms involved in the AP-1 inactivation by Leishmania parasites and its impact on IL-12 expression.
We have found that infection with several Leishmania species alters the DNA binding capacity of AP-1. In particular, we have shown that the parasite metalloprotease GP63 is responsible for this DNA binding alteration and is able to induce the degradation/down-regulation and cleavage of c-Jun, the central component of the AP-1 transcription factor [41], as well of other components including c-Fos, Fra-1, Fra-2 and Jun B. We provide evidence that GP63 exerts its effect by its internalization into the host cell, in a mechanism that is independent of parasite internalization, and induces AP-1 proteolysis within the nucleus or in the nuclear membrane.
The present study corroborates along with previous studies (Ghosh et. al) that AP-1 is down-regulated by Leishmania parasites. Alteration of AP-1 activity varies according to the pathogen, for instance it has been shown, that the hepatitis C virus alters MAP kinases and AP-1 to accelerate the cell cycle progression, helping the development of hepatocellular carcinoma and HCV development [42]. Another example is the Edema toxin produced by Bacillus anthracis; this toxin is able to up-regulate macrophage gene expression, among them genes that are known to be involved in inflammatory responses, regulation of apoptosis, adhesion, immune cell activation, and transcription regulation. Interestingly this up-regulation was found to correlate with induced activation of AP-1 and CAAAT/enhancer-binding protein-beta [43]. In contrast with these reports where different pathogens up-regulate AP-1 to survive inside the host cell, herein we have shown how this transcription factor is down-regulated after Leishmania infection in a cleavage-dependent manner. Whether AP-1 down-regulation is a general mechanism used by different intracellular protozoa requires further investigation.
The AP-1 transcription factor is formed by dimers of Jun and Fos family members. In addition, the Jun proteins can dimerize with other proteins that share the leucine zipper region such as ATF-1 and ATF-2 [8], [27]. Although we did not test other non-classical AP-1 subunits, we demonstrated that at least 5 of the classical subunits belonging the Jun and Fos families are degraded by the parasite within 1 hr of infection. Of interest, c-Jun subunit, one of the main activators of AP-1 along with c-Fos, is cleaved generating a GP63-mediated 30 kDa fragment. The cleaved product would be unable to dimerize and bind DNA, as it has been demonstrated that truncated c-Jun deprived of either the leucine zipper or the DNA binding domain results in only marginal AP-1 transactivation [41], [44], [45]. The generation of c-Jun fragment by GP63 can explain the lower AP-1 binding activity observed in the EMSA experiment. Furthermore, Fos B, which is not cleaved or degraded and also apparently absent in AP-1 complexes (Figure 2B), lacks putative GP63 cleavage sites. Surprisingly Jun D presents two putative sites of cleavage by GP63. However, we did not detect either complete degradation/down-regulation or cleavage products. One possible explanation is that the structural conformation of this protein renders these sites unavailable for GP63-Jun D interaction.
GP63 is known to interact with various substrates. For instance we have recently reported that Leishmania GP63 impacts the stability of cortactin and caspase-3, and negatively regulates p38 kinase activity [23]. Furthermore, we have shown that GP63 cleaves host PTPs resulting in enzymatic activation and leading to JAK 2 dephosphorylation, and inhibition of NO production in IFN-γ primed and infected macrophages [22]. Our current study further supports the important role of GP63 as a negative regulator of host cell functions, actively participating in the pleiotropic effects excreted by Leishmania parasite to suppress the immune response, our results showed that internalization of GP63 by cells from innate immune response is independent of parasite internalization, as our data revealed GP63 proteolytic activity was not affected by inhibition of phagocytosis, but clearly abolished by a lipid raft disruptor, strongly suggesting that lipid rafts microdomains are important for internalization of GP63. Proteins that have a GPI anchor have affinity for lipid rafts, and it has been reported that these rafts recognize these GPI anchors allowing the entrance of GPI-bearing proteins in endocytic vesicles [33], [34]. In addition, Brittingham and collaborators showed interaction of GP63 with the fibronectin receptor (α4β1), that also translocate into the lipid rafts microdomains [46], suggesting that GP63 could have two different ways to get access into the cell: 1) GPI-anchor (native and excreted) and 2) receptor mediated (RGP63). Additionally, we have shown that the GPI anchor is important for the internalization of GP63 since recombinant GP63 (rGP63) lacking the GPI anchor is less internalized [22]. Most importantly, GPI anchor seems to be required for the cellular localization of GP63 since rGP63 is localized inside intracellular compartments whereas GPI-GP63 (native protein) is found within nuclear membrane (see Figure 6). Despite this evidence we have not excluded the possibility that GP63 could use other mechanisms to enter the cells, such as micropinocytosis or classical endocytosis pathways.
One of the main finding of this research is the macrophage nuclear localization of GP63. One plausible mechanism is by the recognition of its GPI domain by the recently described lipid microdomains rich in cholesterol and sphingolipids in the nuclear membrane [47]. Another possible mechanism for the internalization of GP63 inside the nucleus is the presence of a nuclear localization signal (NLS)-like motif (Figure S8) in the GP63 sequence. Nuclear proteins are usually transported inside the nucleus by recognition of a NLS, which usually consist of short chains of basic amino acids with the signature motif K-K/R-X-K/R [48]. These NSLs are recognized by the adaptor molecule importin α, which forms a hetorodimer with the transporter receptor importin β. The importin α/β-NSL-cargo complex is then translocated through the nuclear pore complex [48], [49]. The exact mechanisms of how the nuclear translocation of GP63 occurs are currently under investigation.
In summary, GP63 seems to preferentially target AP-1 subunits within the nuclear membrane, altering its DNA binding capacity. Given the critical role of this transcription factor in the transcription of several genes involved in the innate immune response, alterations in AP-1 activity can dramatically contribute to the down-regulation of innate immune functions observed during the early stages of Leishmania infection. Therefore, this novel mechanism of evasion by Leishmania further demonstrates the complex negative regulatory mechanisms developed by the parasite, which has permitted its adaptation to the harsh intracellular environment leading to its survival and propagation within its mammalian host.
Materials and Methods
Cell culture, macrophage infection and reagents
Immortalised murine bone marrow derived macrophages B10R cell line were maintained at 37°C in 5% CO2 in Dulbecco's Modified Eagle medium (DMEM) supplemented with 10% heat inactivated FBS (Invitrogen, Burlington, ON, Canada) and 100 U/ml penicillin 100 µg/ml streptomycin and 2 mM of L-glutamine (Wisent, St-Bruno, QC, Canada). Leishmania promastigotes (L. donovani infantum, L. donovani 1S2D, L. donovani R2D (LPG −/−), L. mexicana, L. major A2 (WT), L. major GP63 −/−, L. major GP63 Rescued [29] and L. tarentolae) were grown and maintained at 25°C in SDM-79 culture medium supplemented with 10% FBS by bi-weekly passage. Macrophages were infected at parasite to macrophage ratio 20∶1 with stationary phase promastigotes for the times specified in each Figure legend. Using this ratio of infection we normally observe around 30% and 60% of infected cells in 1 or 2 hr, respectively. When chemical inhibitors were used, 2 µM cytochalasin D (Sigma-Aldrich, St-Louis MO, USA) and 20 mM Methylβ-cyclodextrin (MβCD) (Sigma-Aldrich, St-Louis MO, USA), cells were treated 1 hr prior to infection and the inhibitor remained throughout the infection time.
Electrophoresis Mobility Shift Assay (EMSA) and supershift assays
B10R macrophages (2×106) were infected, washed three times with Phosphate Buffered Saline (PBS) to remove non-internalized parasites, and processed for nuclear extraction as previously described [4], [50]. Briefly, macrophages were collected in 1 ml of cold PBS, centrifuged and pellets were resuspended in 400 µl of ice-cold buffer A (10 mM HEPES, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT and 0.5 mM of PMSF) and incubated 15 min on ice. Twenty five µl of IGEPAL 10% (Sigma-Aldrich, St-Louis MO, USA) were added, and samples vortexed for 30 sec. Nuclear proteins were pelleted by centrifugation and resuspended in 50 µl of cold buffer C (20 mM HEPES, 400 mM NaCl 1 mM EDTA, 1 mM EGTA 1 mM DTT and 0.5 mM PMSF).
Protein concentrations were determined by Bradford assay (Bio-Rad, Hercules CA, USA). 6 µg of nuclear proteins were incubated for 20 min at room temperature with 1 µl of binding buffer (100 nM Hepes pH 7.9, 8% v/v glycerol, 1% w/v Ficoll, 25 mM KCl, 1 mM DTT, 0.5 mM EDTA, 25 mM NaCl, and 1 µg/µl BSA) and 200 ng/µl of poly (dI-dC), 0.02% bromophenol blue and 1 µl of γ-P32labeled oligonucleotide containing a consensus sequence for AP-1 binding complexes (5′-CGTTTGATGACTCAGCCGGAA-3′) (Santa Cruz Biotechnology Inc, Sta Cruz CA, USA). After incubation, DNA-protein complexes were resolved by electrophoresis in non-denaturing polyacrylamide gel 5% (w/v). Subsequently gels were dried and autoradiographed. Competition assays were conducted by adding a 100-fold molar excess of homologous unlabeled AP-1 oligonucleotide, or the non-specific competitor sequence for SP-1 binding (5′-ATTCGAATCGGGGCGGGGCGAGC-3′).
For supershift assay, 2 µg of nuclear protein extract were incubated for 1 hr at room temperature with binding buffer, poly (dI-dC), 0.02% bromophenol blue, labeled oligonucleotide and 4 µg of individual specific antibodies (α-c-Jun, Jun B, Jun D, c-Fos, Fos B, Fra 1 or Fra 2; Santa Cruz Biotech Inc, Sta Cruz CA, USA). Complexes were resolved on standard non-denaturing polyacrilamide gel 5% (w/v).
Western blot
Infected and non infected cells (1×106) were washed 3 times with PBS and lysed with cold buffer (50 mM Tris (pH 7), 0.1 mM, 0.1 mM EGTA, 0.1% 2-mercaptoethanol, 1% NP-40, 40 µg/ml aproptinin and 20 µg/ml of leupeptin). Proteins were dosed by Bradford (Bio-Rad, Hercules CA, USA), and 30 µg of proteins were separated by SDS-PAGE, and transferred onto PVDF membranes (GE healthcare, Piskataway NJ, USA). Membranes were blocked in 5% non-fat dry milk, washed and incubated for 1 hr with α-c-Jun (BD Biosciences, San Jose, CA, USA), α-Jun B, α-Jun D, α-c-Fos, α-Fos B, α-Fra 1 and α-Fra 2 (Santa Cruz Biotech Inc. Sta Cruz CA, USA). After washing, membranes were incubated 1 hr with α-mouse or α-rabbit HRP-conjugated antibody (GE healthcare, Piskataway NJ, USA), and developed by autoradiography.
Imunoprecipitation
B10R macrophages (10×106) were infected with either L. major A2, or L. major GP63 −/− or L. major GP63 Rescued for 1 hr, and nuclear proteins were extracted as previously described in [5]. c-Jun was immunoprecipitated from pre-cleared nuclear extracts with 2 µg antibody, followed by addition of 12.5 µl (packed volume) of protein A/G PLUS agarose beads (Santa Cruz Biotech Inc. Sta Cruz CA USA). Beads were washed three times and bound proteins were analyzed by WB as described above.
Confocal microscopy
B10R macrophages (0.5×106) were plated ON in glass cover slips. After infection for 30, 60 and 180 min cells were gently washed 3 times with PBS, and then fixed with 4% p-formaldehyde for 30 min at 4°C. Slides were permeabilized for 5 minutes with PBS containing 1% BSA and 0.05% NP-40 and blocked with 5% non-fat dry milk for 1 hr. Incubation with primary antibody α-c Jun or α-c-Fos or α-GP63 mouse monoclonal antibody clone #96 [51] was conducted in humid dark chamber for 1 hr at room temperature. After three washes with PBS, cells were incubated with secondary antibody (Alexa Fluor 488 or 594, from Molecular probes, Burlington ON, Canada) for 1 hr. Nuclei were stained with propidium iodide or DAPI for 10 min and slides were mounted in permaflour medium (Thermo Co. Waltham MA, USA). Images were taken using an Olympus FV1000 confocal microscope and a Zeiss LCS 500.
IL-12 mRNA expression analysis
B10R macrophages (10×106) were infected with either L. major A2, L. major GP63−/−, L. major GP63 Rescued or L. tarentolae for 18 hr or treated with 20 µM of JNK inhibitor SP600125 for 1 hr. Thereafter cells were stimulated with 100 ng/ml of LPS for 6 hr and RNA extracted using TRIzol reagent according to the manufacturer's instruction (Invitrogen Canada). Reverse transcriptase was performed using oligo(dT) primers. Quantitative relative PCR was performed using Invitrogen Platinum qPCR Super-Mixes and 0.4 µM primer in 25 µl and the following parameters: 50°C for 2 min and 95°C for 3 min (95°C for 20 s, 60°C for 30 s, 72°C for 20 s, 80°C (reading step) for 20 s) for 40 cycles followed by a melting curve. Annealing temperature was 60°C. IL-12 primer sequences: 5′-GGA AGCACG GCA GCA GAA TA-3′ and 3′-AAC TTG AGG GAG AAG TAGGAA TGG-5′.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. DesjeuxP
2004 Leishmaniasis: current situation and new perspectives. Comp Immunol Microbiol Infect Dis 27 305 318
2. PiscopoTV
MalliaAC
2006 Leishmaniasis. Postgrad Med J 82 649 657
3. OlivierM
GregoryDJ
ForgetG
2005 Subversion mechanisms by which Leishmania parasites can escape the host immune response: a signaling point of view. Clin Microbiol Rev 18 293 305
4. ForgetG
GregoryDJ
OlivierM
2005 Proteasome-mediated degradation of STAT1α following infection of macrophages with Leishmania donovani. J Biol Chem 280 30542 30549
5. GregoryDJ
ContrerasI
ForgetG
OlivierM
2008 A novel form of NF-κB is induced by Leishmania infection: Involvement in macrophage gene expression. Eur J Immunol 38 1071 1081
6. HessJ
AngelP
Schorpp-KistnerM
2004 AP-1 subunits: quarrel and harmony among siblings. J Cell Sci 117 5965 5973
7. WisdomR
1999 AP-1: One switch for many signals. Exp Cell Res 253 180 185
8. KarinM
LiuZ-g
ZandiE
1997 AP-1 function and regulation. Curr Opin Cell Biol 9 240 246
9. GhoshS
BhattacharyyaS
DasS
RahaS
MaulikN
2001 Generation of ceramide in murine macrophages infected with Leishmania donovani alters macrophage signaling events and aids intracellular parasitic survival. Molecular and Cellular Biochemistry 223 47 60
10. GhoshS
BhattacharyyaS
SirkarM
SaGS
DasT
2002 Leishmania donovani suppresses Activated Protein 1 and NF-κB activation in host macrophages via ceramide generation: involvement of Extracellular Signal-Regulated Kinase. Infect Immun 70 6828 6838
11. PrivéC
DescoteauxA
2000 Leishmania donovani promastigotes evade the activation of mitogen-activated protein kinases p38, c-Jun N-terminal kinase, and extracellular signal-regulated kinase-1/2 during infection of naive macrophages. Eur J Immunol 30 2235 2244
12. NandanD
LoR
ReinerNE
1999 Activation of phosphotyrosine phosphatase activity attenuates Mitogen-Activated Protein kinase signaling and inhibits c-FOS and nitric oxide synthase expression in macrophages infected with Leishmania donovani. Infect Immun 67 4055 4063
13. DescoteauxA
TurcoSJ
1999 Glycoconjugates in Leishmania infectivity. Biochim Biophys Acta (BBA) - Molecular Basis of Disease 1455 341 352
14. ZhangW-W
MatlashewskiG
1997 Loss of virulence in Leishmania donovani deficient in an amastigote-specific protein, A2. Proc Natl Acad Sci U S A 94 8807 8811
15. MottramJC
CoombsGH
AlexanderJ
2004 Cysteine peptidases as virulence factors of Leishmania. Curr Opin Microbiol 7 375 381
16. McGwireBS
ChangK-P
EngmanDM
2003 Migration through the extracellular matrix by the parasitic protozoan Leishmania is enhanced by surface metalloprotease gp63. Infect Immun 71 1008 1010
17. BrittinghamA
MorrisonCJ
McMasterWR
McGwireBS
ChangKP
1995 Role of the Leishmania surface protease gp63 in complement fixation, cell adhesion, and resistance to complement-mediated lysis. J Immunol 155 3102 3111
18. YaoC
DonelsonJE
WilsonME
2003 The major surface protease (MSP or GP63) of Leishmania sp. Biosynthesis, regulation of expression, and function. Mol Biochem Parasitol 132 1 16
19. BouvierJ
SchneiderP
EtgesR
BordierC
1990 Peptide substrate specificity of the membrane-bound metalloprotease of Leishmania. Biochemistry 29 10113 10119
20. Bianchini Gianluca ABPAEGFMnA 2006 Molecular dynamics simulation of Leishmania major surface metalloprotease GP63 (leishmanolysin). Proteins: Structure, Function, and Bioinformatics 64 385 390
21. McGwireBS
O'ConnellWA
ChangK-P
EngmanDM
2002 Extracellular release of the Glycosylphosphatidylinositol (GPI)-linked Leishmania surface metalloprotease, gp63, is independent of GPI phospholipolysis. Implications for parasite. J Biol Chem 277 8802 8809
22. GomezMA
ContrerasI
HalleM
TremblayML
McMasterRW
2009 Leishmania GP63 alters host signaling through cleavage-activated protein tyrosine phosphatases. Sci Signal 2 ra58
23. HalleM
GomezMA
StuibleM
ShimizuH
McMasterWR
2009 The Leishmania surface protease GP63 cleaves multiple intracellular proteins and actively participates in p38 mitogen-activated protein kinase Inactivation. J Biol Chem 284 6893 6908
24. FolettaVC
SegalDH
CohenDR
1998 Transcriptional regulation in the immune system: all roads lead to AP-1. J Leukoc Biol 63 139 152
25. LeeK-Y
ItoK
HayashiR
JazrawiEPI
BarnesPJ
2006 NF-κB and Activator Protein 1 Response Elements and the Role of Histone Modifications in IL-1β-Induced TGFβ1 Gene Transcription. J Immunol 176 603 615
26. NewellC
DeisserothAB
Lopez-BeresteinG
1994 Interaction of nuclear proteins with an AP-1/CRE-like promoter sequence in the human TNF-alpha gene. J Leukoc Biol 56 27 35
27. KarinM
HawkinsPT
1996 The regulation of AP-1 activity by mitogen-activated protein kinases. Philos Trans R Soc Lond B Biol Sci 351 127 134
28. DescoteauxA
TurcoSJ
2002 Functional aspects of the Leishmania donovani lipophosphoglycan during macrophage infection. Microbes and Infection 4 975 981
29. JoshiPB
KellyBL
KamhawiS
SacksDL
McMasterWR
2002 Targeted gene deletion in Leishmania major identifies leishmanolysin (GP63) as a virulence factor. Mol Biochem Parasitol 120 33 40
30. YaoC
DonelsonJE
WilsonME
2007 Internal and surface-localized major surface proteases of Leishmania spp. and their differential release from promastigotes. Eukaryotic Cell 6 1905 1912
31. EllisM
SharmaDK
HilleyJD
CoombsGH
MottramJC
2002 Processing and trafficking of Leishmania mexicana GP63. Analysis using gp18 mutants deficient in glycosylphosphatidylinositol protein anchoring. J Biol Chem 277 27968 27974
32. AllenJA
Halverson-TamboliRA
RasenickMM
2007 Lipid raft microdomains and neurotransmitter signalling. Nat Rev Neurosci 8 128 140
33. SimonsK
ToomreD
2000 Lipid rafts and signal transduction. Nat Rev Mol Cell Biol 1 31 39
34. LajoiePNR
2007 Regulation of raft-dependent endocytosis. J Cell Mol Med 11 644 653
35. ChaudhuriG
ChaudhuriM
PanA
ChangKP
1989 Surface acid proteinase (gp63) of Leishmania mexicana. A metalloenzyme capable of protecting liposome-encapsulated proteins from phagolysosomal degradation by macrophages. J. Biol Chem 264 7483 7489
36. ForgetG
GregoryDJ
WhitcombeLA
OlivierM
2006 Role of host protein tyrosine phosphatase SHP-1 in Leishmania donovani-induced inhibition of nitric oxide production. Infect Immun 74 6272 6279
37. LiewFY
MillottS
ParkinsonC
PalmerRM
MoncadaS
1990 Macrophage killing of Leishmania parasite in vivo is mediated by nitric oxide from L-arginine. J Immunol 144 4794 4797
38. KristofAS
Marks-KonczalikJ
MossJ
2001 Mitogen-activated protein kinases mediate activator protein-1-dependent human inducible nitric-oxide synthase promoter activation. J Biol Chem 276 8445 8452
39. Abu-DayyehI
ShioMT
SatoS
AkiraS
CousineauB
2008 Leishmania-Induced IRAK-1 Inactivation Is Mediated by SHP-1 Interacting with an Evolutionarily Conserved KTIM Motif. PLoS Negl Trop Dis 2 e305
40. DescoteauxA
MatlashewskiG
1989 c-fos and tumor necrosis factor gene expression in Leishmania donovani-infected macrophages. Mol Cell Biol 9 5223 5227
41. AngelP
KarinM
1991 The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta (BBA) - Reviews on Cancer 1072 129 157
42. KoikeK
2007 Pathogenesis of HCV-associated HCC: Dual-pass carcinogenesis through activation of oxidative stress and intracellular signaling. Hepatol Res 37 S-115 120
43. ComerJE
GalindoCL
ZhangF
WenglikowskiAM
BushKL
2006 Murine macrophage transcriptional and functional responses to Bacillus anthracis edema toxin. Microb Pathog 41 96 110
44. AlaniR
BrownP
BinétruyB
DosakaH
RosenbergRK
1991 The transactivating domain of the c-Jun proto-oncoprotein is required for co-transformation of rat embryo cells. Mol Cell Biol 11 6286 6295
45. Tiliang DengMK
1992 Construction and expression of a monomeric c-Jun protein that binds and activates transcription of AP-1-responsive genes. Proc Natl Acad Sci USA 89 8572 8576
46. BrittinghamA
ChenG
McGwireBS
ChangK-P
MosserDM
1999 Interaction of Leishmania gp63 with Cellular Receptors for Fibronectin. Infect Immun 67 4477 4484
47. CascianelliG
VillaniM
TostiM
MariniF
BartocciniE
2008 Lipid Microdomains in Cell Nucleus. Mol Biol Cell 19 5289 5295
48. PembertonLF
PaschalBM
2005 Mechanisms of receptor-mediated nuclear import and nuclear export. Traffic 6 187 198
49. ArnoldM
NathA
WohlwendD
KehlenbachRH
2006 Transportin is a major nuclear import receptor for c-Fos: a novel mode of cargo interaction. J Biol Chem 281 5492 5499
50. JaramilloM
OlivierM
2002 Hydrogen peroxide induces murine macrophage chemokine gene transcription via extracellular signal-regulated kinase - and cyclic adenosine 5′-monophosphate (cAMP)-dependent pathways: involvement of NF-kappa B, activator protein 1, and cAMP response element binding protein. J Immunol 169 7026 7038
51. MacdonaldMH
MorrisonC
McMasterRW
1995 Analysis of the active site and activation mechanism of the Leishmania surface metalloproteinase GP63. Biochim Biophys Acta (BBA) - Molecular Basis of Disease 1253 199 207
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 10
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Retroviral RNA Dimerization and Packaging: The What, How, When, Where, and Why
- Viral Replication Rate Regulates Clinical Outcome and CD8 T Cell Responses during Highly Pathogenic H5N1 Influenza Virus Infection in Mice
- Antimicrobial Peptides: Primeval Molecules or Future Drugs?
- Crystal Structure of DotD: Insights into the Relationship between Type IVB and Type II/III Secretion Systems
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy