
Autoimmunity in Arabidopsis Is Mediated by Epigenetic Regulation of an Immune Receptor
Certain pathogens deliver effectors into plant cells to modify host protein targets and thereby suppress immunity. These target modifications can be detected by intracellular immune receptors, or Resistance (R) proteins, that trigger strong immune responses including localized host cell death. The accelerated cell death 11 (acd11) “lesion mimic” mutant of Arabidopsis thaliana exhibits autoimmune phenotypes such as constitutive defense responses and cell death without pathogen perception. ACD11 encodes a putative sphingosine transfer protein, but its precise role during these processes is unknown. In a screen for lazarus (laz) mutants that suppress acd11 death we identified two genes, LAZ2 and LAZ5. LAZ2 encodes the histone lysine methyltransferase SDG8, previously shown to epigenetically regulate flowering time via modification of histone 3 (H3). LAZ5 encodes an RPS4-like R-protein, defined by several dominant negative alleles. Microarray and chromatin immunoprecipitation analyses showed that LAZ2/SDG8 is required for LAZ5 expression and H3 lysine 36 trimethylation at LAZ5 chromatin to maintain a transcriptionally active state. We hypothesize that LAZ5 triggers cell death in the absence of ACD11, and that cell death in other lesion mimic mutants may also be caused by inappropriate activation of R genes. Moreover, SDG8 is required for basal and R protein-mediated pathogen resistance in Arabidopsis, revealing the importance of chromatin remodeling as a key process in plant innate immunity.
Published in the journal:
. PLoS Pathog 6(10): e32767. doi:10.1371/journal.ppat.1001137
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1001137
Summary
Certain pathogens deliver effectors into plant cells to modify host protein targets and thereby suppress immunity. These target modifications can be detected by intracellular immune receptors, or Resistance (R) proteins, that trigger strong immune responses including localized host cell death. The accelerated cell death 11 (acd11) “lesion mimic” mutant of Arabidopsis thaliana exhibits autoimmune phenotypes such as constitutive defense responses and cell death without pathogen perception. ACD11 encodes a putative sphingosine transfer protein, but its precise role during these processes is unknown. In a screen for lazarus (laz) mutants that suppress acd11 death we identified two genes, LAZ2 and LAZ5. LAZ2 encodes the histone lysine methyltransferase SDG8, previously shown to epigenetically regulate flowering time via modification of histone 3 (H3). LAZ5 encodes an RPS4-like R-protein, defined by several dominant negative alleles. Microarray and chromatin immunoprecipitation analyses showed that LAZ2/SDG8 is required for LAZ5 expression and H3 lysine 36 trimethylation at LAZ5 chromatin to maintain a transcriptionally active state. We hypothesize that LAZ5 triggers cell death in the absence of ACD11, and that cell death in other lesion mimic mutants may also be caused by inappropriate activation of R genes. Moreover, SDG8 is required for basal and R protein-mediated pathogen resistance in Arabidopsis, revealing the importance of chromatin remodeling as a key process in plant innate immunity.
Introduction
Unlike vertebrates, plants lack a somatic, adaptive immune system and immunological memory [1]. Therefore, plants rely on a large repertoire of pre-existing immune receptors, encoded by hypervariable Resistance (R) genes, which recognize specific pathogens and activate strong defense responses. These responses include the programmed cell death (PCD) of host cells at infection sites to restrict pathogen access in a process called the hypersensitive response (HR). R proteins are triggered by pathogen-specific effector proteins that have evolved to perturb or disrupt host processes to facilitate infection. While some pathogen effectors are recognized extracellularly, the majority are targeted to various intracellular compartments of the plant host and identified there. In most cases, R proteins are activated by detecting modifications to host proteins targeted by pathogen effectors. This model, known as the “guard hypothesis” [2], [3], has been supported in numerous instances. For example RIN4, a host protein with key roles in basal defense, is under surveillance by multiple R proteins, and at the same time is the target of multiple pathogen effectors [4]. Most R proteins have been classified as NB-LRRs, named after their central nucleotide-binding (NB) and C-terminal leucine-rich repeat (LRR) domains, although various exceptions exist [5]. The N-terminal domains of NB-LRR R proteins fall into two broad categories: those with homology to Drosophila Toll and mammalian Interleukin-1 Receptor (TIR), and those with predicted coiled-coil (CC) regions [6]. Members of the animal NOD-like receptor (NLR) family exhibit similar domain architecture to plant NB-LRRs, and NLRs are likewise involved in immunity [7], [8]. Like NB-LRR proteins, NLRs have several types of amino-termini including protein–protein interaction domains associated with proteins involved in programmed cell death and inflammation. Several autoimmune diseases in humans have been associated with mutations in NLRs [9].
In plants, there are numerous examples of mutants with autoimmunity-related phenotypes. These so-called “lesion-mimics” are, in many cases, caused by mutations in genes hypothesized to be negative regulators of the HR [10]. Other examples include point mutations in NB-LRR R proteins [11], [12]. Since R proteins have the potential to trigger host PCD, their activity is tightly regulated. R genes are typically constitutively expressed at low levels and some are up-regulated in response to pathogen-derived peptides or to the accumulation of the phytohormone salicylic acid (SA) [13], [14]. Little is known about the transcriptional control of R genes. Intriguingly, members of a cluster of related Arabidopsis R genes are endogenously suppressed at the post-transcriptional level by RNA silencing, suggesting that pathogens that interfere with the silencing machinery unwittingly up-regulate steady-state R protein levels [15]. At the protein level, inappropriate activation is likely prevented by autoinhibition, high rates of turnover, and alternatively spliced products. Recently, it has become clear that hybrid necrosis, a deleterious genetic incompatibility observed in many intra - and interspecific plant hybrids, is associated with autoimmunity [16]. One example of this type of autoimmune response in Arabidopsis was shown to be dependent on an NB-LRR R protein, suggesting that these immune receptors have a broad mandate over PCD that extends beyond pathogen defense [17].
The lethal, recessive accelerated cell death 11 (acd11) mutant of Arabidopsis is characterized by constitutive activation of immune responses and PCD in the absence of pathogen attack [18]. ACD11 encodes a putative sphingosine transfer protein with homology to HET-C2 of the fungus Podospora anserina. Allelic variants of het-c determine compatibility during fusion of hyphae from different strains, causing PCD in combination with specific alleles at other het loci [19]. acd11 mutants develop normally until the 2–4 leaf stage, and PCD involves the phytohormone SA such that expression of a bacterial SA hydroxylase (NahG) strongly suppresses cell death. Application of SA agonists, such as benzothiadiazol-S-methyl ester (BTH), restores autoimmunity in acd11. Interestingly, the genetic requirements for acd11 cell death are similar to those for the HR triggered by TIR-NB-LRR immune receptors [18], [20].
We report here that cell death in acd11 is suppressed by mutations in genes encoding a histone methyltransferase and a TIR-NB-LRR R protein. In addition, the expression of the R gene is dependent on the activity of the histone modifying enzyme. We propose that the TIR-NB-LRR is triggered by the absence of ACD11, implying that ACD11 (or a complex containing ACD11) may be a guarded pathogen effector target. Alternatively, since ACD11 may be involved in production of a lipid signal, the absence of this signal may induce LAZ5 expression in an SA-dependent manner. Our study provides strong evidence that a specific type of histone modification is directly involved in chromatin remodeling and transcriptional control of a subset of R genes including LAZ5.
Results
laz2 suppresses cell death in acd11
To isolate genes required for cell death in acd11, Landsberg erecta (Ler) ecotype acd11-1 plants harboring the NahG transgene were mutagenized with ethyl-methanesulfonate (EMS), diepoxybutane (DEB) or γ-irradiation. ∼200 suppressors of acd11 were subsequently identified as plants that survived following BTH treatment. Genetic analyses of 43 such suppressors grouped them into 12 recessive and 2 dominant loci referred to as lazarus (laz) mutants, after the biblical resurrection. One of the laz mutants found in the suppressor screen, laz2, abolished cell death in response to BTH in the acd11 NahG background, and exhibited similar levels of cellular ion leakage as wild type (Fig. 1, A and B). laz2-1 acd11-1 NahG plants also exhibited abnormal development (e.g. early flowering, increased shoot branching) that, along with acd11 suppression, was inherited recessively (data not shown). Two other laz2 alleles with similar morphology, laz2-2 and laz2-3, were confirmed by complementation tests (Fig. S1A). Global transcript profiles of laz2-1 acd11-1 NahG, Ler wild-type, NahG, and acd11-1 NahG plants were acquired by hybridizing total mRNA, isolated before and 72 h after BTH treatment, to Affymetrix ATH1 GeneChip arrays. laz2-1 exhibited dramatic suppression of the top 500 most significantly regulated genes in acd11-1 after 72 h BTH (Fig. S2A) In addition, a strong negative Pearson correlation of −0.87 was obtained for global expression fold change between laz2-1 acd11-1 and acd11-1, indicating that gene expression in acd11-1 was strongly affected by the laz2-1 mutation (Fig. S2B).

The LAZ2 locus was identified using a map-based approach. Briefly, Ler laz2-1 acd11 NahG was crossed to Columbia ecotype (Col-0) acd11 NahG to generate a segregating F2 mapping population after BTH treatment. Ecotype-specific linkage markers were used to map laz2-1 to a ∼150 kb region at the bottom of chromosome 1 (Fig. S3). Candidate genes were selected and sequenced based on annotated mutant phenotypes at The Arabidopsis Information Resource (TAIR; http://www.arabidopsis.org), revealing an irradiation-induced 28-bp deletion in the third exon of the gene At1g77300 (Fig. 2A). This locus was also sequenced in laz2-2 acd11-1 NahG, revealing an EMS-induced G to A transition converting tryptophan 1536 to a premature stop.
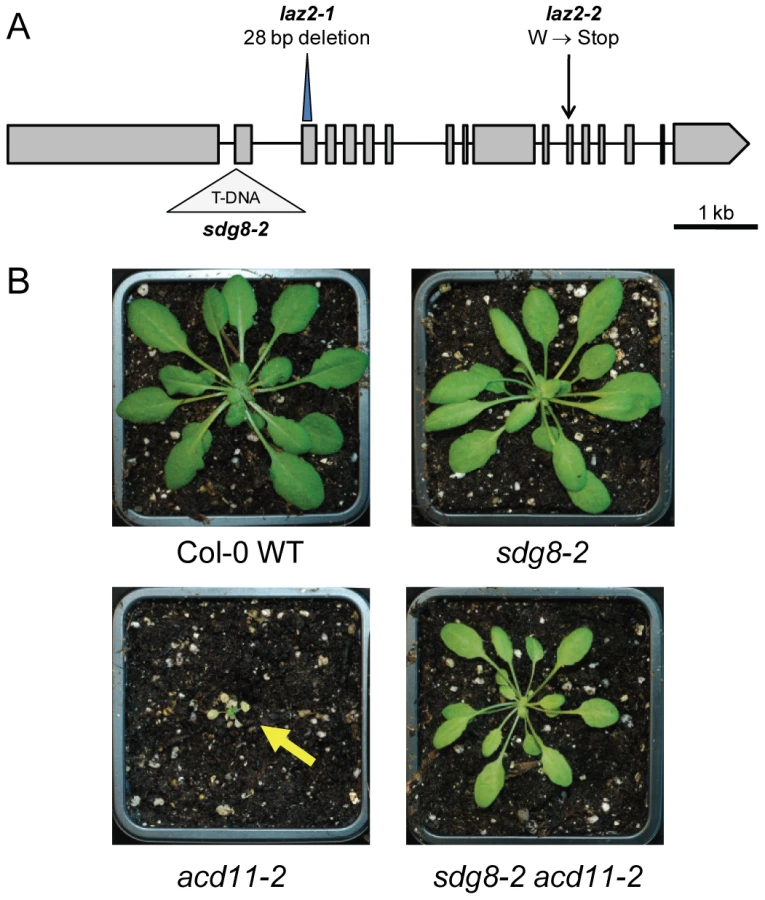
LAZ2 encodes the histone methyltransferase SDG8
Sequence analysis revealed that LAZ2 encodes the histone lysine methyltransferase (HKMT) SET (Su(var)3-9, E(z) and Trithorax-conserved) DOMAIN GROUP 8 (SDG8), otherwise known as EARLY FLOWERING IN SHORT DAYS (EFS) and CAROTENOID CHLOROPLAST REGULATORY 1 (CCR1) [21], [22]. The mutation in laz2-1 causes a frame-shift just upstream of the sequence encoding the conserved SET associated cysteine-rich domains, while that in laz2-2 introduces a stop codon upstream of a motif conserved within the RPB1 subunits of RNA polymerase II [23]. SDG8 is homologous to yeast SET2, which is associated with methylations at histone 3 lysine 36 (H3K36). Another yeast HKMT, SET1, modifies H3K4. Both H3K4 and H3K36 methylation marks are typically associated with active transcription [24]. While Arabidopsis has 43 annotated SDG proteins, SDG8 groups with H3K36-specific HKMTs in fungi and animals along with 4 other Arabidopsis proteins [25]. During transcription in yeast, SET1 and SET2 are recruited to active chromatin by the RNA polymerase II-associated PAF1 complex, where they promote gene expression by facilitating chromatin opening, thus enhancing transcription initiation and elongation, respectively [26]. A similar mechanism seems to be conserved in Arabidopsis based on studies of sdg mutants. SDG8 was first identified as a gene that controlled flowering time via its activity on the transcription of the key floral repressor FLOWERING LOCUS C (FLC), an epigenetically regulated MADS box transcription factor (TF) [27], [28]. Expression of the FLC paralog MADS AFFECTING FLOWERING 1 (MAF1) is also dependent on SDG8, which is required for di - and trimethylation of H3K36 [25]. In addition to flowering time, SDG8 regulates carotenoid composition and shoot branching via modification of chromatin at specific loci [22], [29]. Our microarray expression analysis revealed that MAF1 and CRTISO, both recently confirmed as direct targets of SDG8 [22], [25], exhibited very low expression levels in the absence of LAZ2 (Fig. S4). Deficient expression of these and similar genes likely contributes to the developmental phenotypes observed in laz2. Furthermore, the loss-of-function mutant sdg8-2 (SALK_026642) shared laz2 morphology (Fig. S1B) and suppressed acd11-2, an ACD11 knockout in the Col-0 ecotype (Fig. 2B).
Cell death in acd11 is dependent on the R gene LAZARUS 5
Transcriptome analysis of genes normally induced in acd11-1 NahG after BTH treatment showed that one of the most affected genes in laz2-1 was At5g44870, annotated as an NB-LRR R gene (Fig. 3A). This agrees with data from a previous study showing that At5g44870 is severely down-regulated in ccr1-1 (sdg8) leaf tissues [22]. A number of acd11 suppressors found in the same screen as laz2 were dominant. One of these, laz5 Dominant 1 (laz5-D1), was mapped to a region close to this R gene (Fig. S5). Sequencing of At5g44870 in laz5-D1 revealed a G to A transition at the splice donor site (+1 position) of intron 4 likely resulting in deletion of exon 5 (Fig. 3B). To confirm that this mutation resulted in suppression of acd11, two allelic dominant suppressors, laz5-D2 and laz5-D3, were sequenced: both had lesions in At5g44870 (below), hereafter referred to as LAZ5.

LAZ5 encodes a TIR-class NB-LRR of unknown pathogen specificity with sequence similarity to RPS4 (Fig. S6), an R protein conferring resistance to Pseudomonas syringae expressing the effector AvrRPS4 [30]. The DEB-induced laz5-D2 mutation is a T to A transversion changing isoleucine 287 to asparagine (I287N). This mutation is within the P-loop motif of the NB domain essential for coordination of bound nucleoside triphosphates [5]. The EMS-induced point mutation in laz5-D3 (G811E) lies in the LRR domain, which provides pathogen recognition specificity and has been implicated in R protein activation [31]. Accelerated cell death in acd11-1 was suppressed by laz5-D1 and laz5-D2 (Fig. 3C), and laz5-D alleles suppressed acd11 cell death irrespective of BTH induction or the presence of NahG (Fig. 3D). Furthermore, over-expression of laz5-D2 or laz5-D3 (35S:laz5-D2 or 3) suppressed acd11 death after induction, confirming that dominant negative mutations in LAZ5 are responsible for suppression of the acd11-dependent autoimmune response (Fig. S7).
Transgenic plants over-expressing R-genes can exhibit spontaneous cell death and/or constitutive defense responses [32]. In agreement with these observations and the phenotype associated with deletion of ACD11, over-expression of wild-type LAZ5 (35S:LAZ5) in the Col-0 background resulted in 30 out of 38 transgenic plants exhibiting acd11-like cell death which did not survive to set seed (Fig. S8). Since LAZ5 transcription is likely dependent on SDG8 HKMT activity, and the suppression of acd11 by laz2/sdg8 is recessive, we predicted that a loss-of-function mutation in LAZ5 would suppress acd11 in a recessive manner. As expected, a null T-DNA insertion mutant of At5g44870 (SALK_087262; here termed laz5-1) suppressed acd11-2 cell death recessively in plants without NahG (Fig. 4A). A second T-DNA insertion mutant allele of LAZ5 (SAIL_874-D10) also suppressed cell death in acd11-2 (data not shown). Expression of LAZ5 was assayed by real-time PCR in wild-type, laz5-1, and sdg8-2 plants 24 hours after syringe inoculation with the virulent bacterial pathogen Pseudomonas syringae tomato (P.s.t.) DC3000 or with 10 mM MgCl2 (mock control). While pathogen treatment induced LAZ5 expression in wild type, transcript levels in sdg8-2 were comparable to that in the laz5-1 null mutant (Fig. S9A). This confirms the microarray expression data shown in Fig. 3A. The apparent lack of LAZ5 expression in sdg8-2 was seen in several independent experiments with plants at different stages and/or treated with other pathogen strains (data not shown). Moreover, ACD11 expression was unaffected in laz5-1 and sdg8-2 (Fig. S9B), and transcript accumulation of several TIR-NB-LRR-encoding genes homologous to LAZ5 was seemingly unaffected in 3-week old sdg8-2 plants compared to wild-type control with the possible exception of At5g45230 (Fig. S10).

An important question is whether LAZ5 is the relevant target of SDG8 required for acd11 cell death. To help answer this question, we transformed laz2-1 acd11-1 NahG plants with a genomic construct of LAZ5 under control of a constitutive promoter and monitored cell death by ion leakage after BTH treatment compared to relevant controls (Fig. S11). LAZ5 over-expression restored cell death in leaf discs between 3 and 8 days after induction, indicating that lack of LAZ5 expression in sdg8 is a major cause of the suppression of acd11 cell death. However, it cannot be excluded that other targets of SDG8 histone methyltransferase activity also contribute to BTH-induced cell death in acd11.
SDG8 directly modifies chromatin at the LAZ5 locus
To test whether laz2 directly affects histone methylation at the LAZ5 locus, chromatin immunoprecipiation (ChIP) was conducted using antibodies against specifically modified histones. In laz2-1 acd11-1 NahG, trimethylated (me3) H3K36 levels were reduced in chromatin associated with the 5′ coding regions of MAF1 (control) and LAZ5, when compared to the acd11-1 NahG control (Fig. 4B). Enrichment of H3K36me3 in LAZ5 chromatin was not influenced by BTH treatment or acd11 homozygosity (Fig. S12A). This suggests that activation of cell death in acd11 does not result in hyper-trimethylation at H3K36, but rather that this histone modification is required for proper LAZ5 expression. There was no effect of genotype on levels of total H3 (Fig. 4C). H3K36me3 is not a general mark for genes up-regulated in acd11, such as FMO1 [18], since we found no enrichment at FMO1 chromatin 72 h after BTH induction (Fig. S12B, C). Moreover, absence of LAZ2/SDG8 had no effect on H3K36me3 levels at the constitutively expressed ACTIN locus (Fig. 4C) or the MAP KINASE KINASE 4 (MKK4) locus (Fig. S12D).
To elucidate H3K36 methylation status irrespective of acd11 and NahG, we also conducted ChIP assays on sdg8-2 single mutant and Col-0 wild-type seedlings. It was previously shown that loss of SDG8 resulted in both a decrease in global H3K36me3 levels and a coincident increase in global monomethylated (me1) H3K36, a mark associated with transcriptional repression in Arabidopsis [25]. In wild-type plants, MAF1 and LAZ5 chromatin was enriched for H3K36me3, whereas the level of H3K36me3 was diminished in sdg8-2 (Fig. 4D). Conversely, H3K36me1 levels at these loci were higher in sdg8-2 and reduced in wild type. Treatment of seedlings for 3 hours with an HR-inducing bacterial pathogen had no effect on the methylation status of H3K36 (data not shown). Also, H3 trimethylation of LAZ5 chromatin at other lysine residues (K4, K9, K27), was not affected by loss of SDG8 (Fig. S12E).
SDG8 is required for pathogen resistance in Arabidopsis
To determine whether SDG8 and/or LAZ5 are required for basal resistance to virulent pathogens, leaves of 4-week old sdg8-2, laz5-1, wild-type and an allele of enhanced disease susceptibility 1 (eds1-2 introgressed into Col-0) mutants were syringe-inoculated with P.s.t. DC3000 and growth was assayed after 4 days. Bacteria grew to ∼9-fold higher titers in sdg8-2 than in wild-type or laz5-1, while titers in eds1 were yet another order of magnitude higher (Fig. 5A). Growth of another strain of bacterial pathogen, Pseudomonas syringae maculicola (P.s.m.) ES4326, was tested on sdg8-2, laz5-1, wild-type and eds1 with similar results (Fig. 5B). We did not observe elevated bacterial growth in sdg8-2 when we used P.s.t. DC3000 HrcC - (Fig. S13A), a non-pathogenic mutant defective in delivery of effectors to host cells [33]. These data indicate that SDG8, but not LAZ5, is required for full resistance to virulent pathogens. Furthermore, we found that SDG8 is involved in resistance to avirulent pathogens mediated by other R proteins, for example RPM1. Plants were syringe-inoculated with P.s.t. DC3000 expressing HR-inducing AvrRpm1, AvrRpt2, AvrRps4 or AvrPphB and growth was assayed after 3 or 4 days. Bacterial titers were ∼15-fold higher in sdg8-2 than in wild-type or laz5-1 for P.s.t. expressing AvrRpm1 (Fig. 5C). This suggested that RPM1-mediated resistance is defective in sdg8-2. To confirm this, growth of P.s.m. ES4326 expressing AvrB was assessed after 3 days: AvrB is also recognized by RPM1, and resistance to this avirulent pathogen was affected in sdg8-2 to a similar level as P.s.t. with AvrRpm1 (Fig. 5D). In both cases, bacterial titers were comparable to the rpm1-3 null mutant [34]. Defects in SDG8 had a consistent, yet statistically insignificant effect on growth of P.s.t. DC3000 expressing AvrPphB, (Fig. S13B) resistance to which is dependent on the R gene RPS5 [35]. In addition, sdg8-2 did not affect RPS2 - or RPS4-mediated resistance to AvrRpt2 [36], [37] (Fig. 5E) and AvrRps4 [30] (Fig. 5F). Corroborating the pathogen growth assay, transcript levels of RPM1 and RPS5 were low or absent in 4-week old sdg8-2 compared to wild-type, whereas expression of RPS2 and RPS4 in sdg8-2 was similar to that in wild-type (Fig. 5G and S13C). Defects in LAZ5 did not have a detectable effect on transcript accumulation of RPM1, RPS5, RPS2 or RPS4 (data not shown). As with LAZ5, we conducted ChIP assays at the RPM1 locus in untreated seedling tissue from laz2-1 acd11-1 NahG versus acd11-1 NahG (in Ler) and sdg8-2 versus wild-type (in Col-0). We observed lower H3K36me3 and higher H3K36me1 levels at RPM1 chromatin in the absence of functional LAZ2/SDG8, indicating that RPM1 is an example of another R gene that is regulated by histone methylation (Fig. S14). These results indicate that SDG8 targets a subset of R genes and other genes involved in more general aspects of basal defense.

Discussion
Chromatin remodeling has emerged as a complex regulator of transcription and an epigenetic mechanism to maintain lasting changes in gene activity states. Dynamic post-translational modifications of various residues of histones tails, including methylation, phosphorylation, acetylation, and ubiquitination, play important roles in both promoting and repressing gene expression by recruiting histone binding proteins and chromatin remodeling enzymes [38]. The combinatorial nature of histone modifications results in a complex “histone code” that adds an important level of control to fine-tune gene-specific responses to broader transcriptional inputs [39]. Changes in chromatin state may therefore modulate gene expression in a context-dependent manner to maintain a flexible response to pathogen attack. In plants, this process has been proposed as a mechanism for priming SA-responsive loci during systemic acquired resistance to pathogens [40].
So far, relatively few studies directly associate epigenetic processes related to chromatin modification to plant innate immunity and/or PCD. Defects in HISTONE DEACETYLASE 19 (HDAC19) and HISTONE MONOUBIQUITINATION 1 (HUB1) increase susceptibility to necrotrophic fungal pathogens in Arabidopsis [41], [42]. Furthermore, defects in genes involved in histone variant replacement, and the variant H2A.Z itself, result in increased resistance to virulent bacterial pathogens, some spontaneous cell death, and up-regulation of defense genes [43]. More commonly, the “memory” of chromatin remodeling activity is observed as increased levels of open chromatin marks (H3Ac, H3K4me2, etc) at the promoters of many SA-responsive genes, such as PATHOGENESIS-RELATED 1 (PR-1) and WRKY TFs [40], [44], [45]. The clearest example of immune response at the level of chromatin comes from Alvarez-Venegas and colleagues, who showed that the HKMT ARABIDOPSIS TRITHORAX 1 (ATX1, also known as SDG27) controls expression of WRKY70, a TF involved in pathogen response [46]. ATX1-dependent H3K4me3 signatures at the promoter of WRKY70 correlated with WRKY70 transcriptional up-regulation. Intriguingly, although ATX1 regulates expression of a large set of genes, a high proportion of immunity-related genes exhibited reduced expression in the knockout mutant, including various TIR-NB-LRR R genes [47]. Numerous examples exist of microbes and viruses manipulating host chromatin remodeling machinery or histones directly in animals [48], [49]. Strikingly, toxins from unrelated bacterial pathogens of animals have evolved to modify host histones, reducing transcriptional activity of key immunity genes [50]. The only clear instance of related phenomena identified among plant pathogens is the case of the Crown Gall disease-causing bacterium Agrobacterium tumefaciens which selectively modulates the expression of host variant histone genes to allow genomic integration of its T-DNA [51], [52].
There is conflicting data on whether loss of sdg8 influences H3K4 methylation, H3K36 methylation, or both [22], [23], [25], [28]. We detected a dramatic effect of laz2/sdg8 on H3K36 methylation status of chromatin at various loci and no difference in H3K4me3 levels at LAZ5, although the H3K4 methylation status of chromatin at other loci in laz2 backgrounds remains to be investigated. In addition, our data suggest that monomethylation of H3K36 at MAF1 and LAZ5 chromatin relies on HKMTs other than SDG8. One of these, SDG26, was previously shown to act antagonistically to SDG8 by repressing FLC expression, although global H3K36me1 levels were unaffected in the sdg26 mutant [25]. The significance of H3K36me1 enrichment in sdg8-2 remains unknown. One hypothesis is that H3K36 methylation proceeds in a stepwise fashion, with the accumulation of H3K36me1 (due to activity of an unknown HKMT) being a consequence of a block in further di - and trimethylation at this residue normally mediated by SDG8. Alternatively, monomethylation of H3K36 may represent a transcriptionally repressive mark that accumulates only in the absence of di - and trimethylation due to disruption of the balance between antagonistic chromatin modifiers. For example, the SET-domain containing Arabidopsis proteins TRITHORAX-RELATED PROTEIN 5 (ATXR5, also known as SDG15) and ATXR6/SDG34 are H3K27-specific monomethyltransferases essential for transcriptional repression in heterochromatin [53]. Further studies should examine if other predicted H3K36-specific HKMTs, namely SDG4, SDG7, SDG24 and SDG26, have any role in H3K36 monomethylation, trimethylation and/or antagonistic control of expression of LAZ5 and other genes with roles in immunity or are required for cell death in acd11. Moreover, further work is required to determine the mechanisms by which SDG8-dependent changes in H3 methylation regulates the expression of specific genes.
A clue to the function of LAZ5 activation comes from the isolation in our screen of dominant alleles. This indicates that the mutant form (laz5-D) of the R protein likely interferes with activity of the wild-type copy since plants heterozygous for the laz5 null mutation do not suppress acd11, indicating haplosufficiency of LAZ5. Dominant negative activity has been described for mutations in the R gene N from tobacco, and indeed for a point mutation (G216E) in the P-loop motif of N [54]. N was later found to oligomerize in the presence of a Tobacco mosaic virus elicitor, likely through interaction of TIR domains [55]. This oligomerization was an early event in pathogen perception and was independent of mutations that have an effect on HR induction. Therefore, it is possible that laz5-D mutants form inactive oligomers with wild-type LAZ5 and/or accessory proteins. An example of this scenario from animal innate immunity comes from NOD2, an NLR involved in recognition of bacterial cell wall components: an endogenously truncated form, NOD2-S, interacts with full-length NOD2 to potentiate signaling [56]. In plants, there are examples of truncated R proteins, generated by alternative splicing, playing a key role in signaling [57], [58]. At present, it is an open question whether LAZ5 oligomerizes and how this relates to cell death activation. It should be noted that, while all the laz5 alleles isolated thus far in the acd11 suppressor screen were dominant negative, only 43 of the ∼200 unknown recessive mutants were placed into complementation groups, and even fewer were mapped. Therefore, a recessive laz5 knockout allele may exist among our unmapped suppressors.
In this study we have identified the chromatin modifying enzyme SDG8, and its specific target LAZ5, as regulators of autoimmune cell death in acd11. Furthermore, sdg8 mutants exhibit enhanced susceptibility to virulent and avirulent pathogens, whereas laz5 mutants do not, suggesting that other targets of SDG8 are important for general resistance. We also show that transcription of a subset of R genes, including LAZ5 and RPM1, is likely to be directly or indirectly dependent on LAZ2 activity. One scenario that may account for the enhanced susceptibility of sdg8 mutants to virulent pathogens could be the consequence of SDG8 action on multiple NB-LRR loci. If the suite of effectors delivered by Pseudomonas triggers a weak R gene response, in sdg8 a subset of these do not accumulate and thus are no longer available to signal for defense against the invading pathogen. Intriguingly, SDG8 is not expressed until 8 days after germination [28], a stage preceding the initiation of cell death in acd11. SDG8 may therefore developmentally regulate targets such as LAZ5, and may exemplify a key difference in the programmed defenses required during seed maturation and the inducible defenses used during plant growth.
Lesion mimic mutants such as acd11 are useful tools in the genetic dissection of innate immunity in plants [10]. Whereas several of these mutants have putative roles in ceramide signaling or synthesis [59], [60] or auto-activate R proteins [11], the majority of lesion mimic mutants represent proteins with no straightforward connection to PCD. Milder autoimmunity, associated with constitutive activation of defense responses and dwarf morphology without coincident HR, can similarly be the result of point mutations in immune receptors (Zhang et al., 2003), or deletion of signaling intermediates such as MAP kinases [61]. Knockout mutants that eliminate host guardees mimic the effects of pathogen effectors, and have been found to exhibit R-gene-dependent lethality [62]. Therefore, it is possible that many lesion mimic/autoimmune mutants may correspond to gene functions that are guarded by NB-LRRs. If so, the diverse functions of these genes may be “red herrings” not directly related to PCD but only implicated in this process due to their targeting by pathogen effectors. Such may be the case for acd11, although we have been unable to detect any interaction between full-length or truncated LAZ5 and ACD11 in yeast or in planta (data not shown). Previously, we reported the identification of ACD11-interacting proteins [63], which we are testing for interaction with LAZ5. Two predictions about wild-type products of autoimmune mutants emerge from this model. First, suppressor screens should identify R genes. Second, pathogen effectors should target them either directly or indirectly via interacting partners or products of their activities. We currently have no evidence that ACD11 is targeted by pathogen effectors, or that ACD11 contributes to disease resistance in the absence of LAZ5. While future work may strengthen this hypothesis, an alternative model is that ACD11 is involved in negatively regulating SA-dependent expression of LAZ5 (or a subset of R genes) perhaps via some lipid signal.
Materials and Methods
Plant material and growth conditions
Arabidopsis plants were grown on soil or MS-agar plates at 21°C with an 8 h or 12 h photoperiod. sdg8-2 (SALK_026642) and laz5-1 (SALK_087262) T-DNA insertion lines, both previously described as null mutants [23], [64], were generated by SIGnAL [65] and obtained from the Nottingham Arabidopsis Stock Centre (NASC; Nottingham, UK). Homozygous genotyping primers were 5′-TAAAGAGGGTCTGCATCATGG-3′ with 5′-CACTGTCCAGTAAAAGCTGGC-3′ for sdg8-2 and 5′-TATGTTTTTCCCAGATGCCAG-3′ with 5′-ATCATGCATCTCAACTCGACC-3′ for laz5-1. Sequences of primers used to detect acd11-1, acd11-2, and NahG are available upon request.
Suppressor screen
Three lots of 920–950 mg Ler acd11-1 NahG seeds were incubated for 4 hr in either 0.74% (w/v) EMS (Sigma-Aldrich, St Louis, MO, USA) prepared in 0.1M sodium phosphate buffer, pH 5, with 5% DMSO, or 10 mM DEB (Sigma-Aldrich) in water, followed by rinsing. γ-irradiation of 300 mg acd11-1 NahG seeds was performed at the Risø Reference Laboratory (Denmark) with 500 Gy from a Cobalt-80 source. M1 plants were grown in families of 125 individuals, 3500 M2 plants per family were screened for BTH-resistant suppressors. ∼3 million M2 plants from 845 M1 pools or ∼100.000 M1 plants were scored. Putative mutants were genotyped to be homozygous for acd11-1 by PCR.
Ion leakage assay
Conductivity assays were conducted essentially as previously described [66].
Microarray hybridization
Total RNA was isolated from three independent biological replicates of relevant genotypes at 0 and 72 hr after BTH treatment. RNA was labeled and amplified according to the MessageAmp Biotin-enhanced kit (Ambion) protocol and hybridized to 51 ATH1 GeneChips after Affymetrix protocols.
Chromatin immunoprecipitation and real-time PCR
ChIP antibodies purchased from Abcam (Cambridge, UK) included anti-H3 (ab1791), anti-H3K36me1 (ab9048), anti-H3K36me3 (ab9050) and anti-H3K27me3 (ab6002). ChIP antibodies against H3K4me3 (pAb-056-050) and H3K9me3 (pAb-003-050) were purchased from Diagenode (Liège, Belgium). Quantitative PCR primers for ChIP analysis were LAZ5: 5′-GAGTCGTGGCAAGTGTTCATC-3′ with 5′ - GAAGATGGACAGTGCGATTTC-3′; FMO1: 5′-CTCAGATGGCTTCTAACTATG-3′ with 5′-CTATTATTGGGCCATGGAAAG-3′; MAF1: 5′-CCCTTATCGGAGATTTGAAGC-3′ with 5′-GGAGGATTCACAGAGAATCG-3′; ACTIN: 5′-GGAAACATCGTTCTCAGTGG-3′ with 5′-ACCAGATAAGACAAGACACAC-3′. ChIP was performed essentially as described [67], using 1µg of each antibody. Real-time PCR to quantify the immunoprecipitated DNA was performed using Brilliant II SYBR Green qPCR kit (Stratagene), and reactions were run on an iCycler IQ (Bio-Rad, Hercules, CA, USA). In all cases, ChIP values were calculated using the Delta-Delta-Ct (ddCt) algorithm to determine relative gene expression utilizing the ‘percent input method’. Briefly, signals obtained from the ChIP were divided by signals obtained from an input sample representing the amount of chromatin used in the ChIP. The ‘% input’ value shows what proportion of this starting material is found in the eluate after IP with appropriate Ab.
For expression analyses, RNA was extracted from relevant genotypes using the Qiagen RNeasy RNA extraction kit followed by DNase treatment as per the manufacturer's instructions. Equal amounts of RNA were subjected to one-step real-time PCR using the same kit as described for ChIP except with reverse transcriptase included. For all sample/primer combinations, a control without reverse transcriptase was included to exclude genomic DNA contamination.
Cloning and generation of transgenic plants
3.9-kb fragments of laz5-D alleles were amplified from genomic DNA (laz5-D1 acd11-2 NahG, laz5-D2 acd11-2 NahG, laz5-D3 acd11-2 NahG) and cloned into modified pCAMBIA-3300 as described [68], using a uracil-excision based cloning technique (USER, New England Biolabs). Cloning primers were 5′-ggcttaaUATGGCAGCATCTTCCGAAATAC-3′ and 5′-ggtttaaUTTACAATAAACCCAAGTATAATTTAG-3′. A 3.9-kb fragment of LAZ5 was amplified from genomic DNA (wild type Ler), cloned into pENTR/D-TOPO (Invitrogen) and transferred to Gateway-compatible constitutive expression vectors pGWB502Ω or pGWB521 [69] by LR recombination reaction (Invitrogen). Cloning primers used were 5′-CACCATGGCAGCATCTTCCGAAATAC-3′ and 5′-TTACAATAAACCCAAGTATAATTTAG-3′. The final constructs were verified by sequencing, electroporated into Agrobacterium tumefaciens strain GV3101 and used to transform acd11-1 NahG or wild type plants by floral dip method [70]. Transgenic plants were selected on soil with glufosinate (35S:laz5-D alleles) or on MS-agar plate with (20mg/L) hygromycin B followed by transplanting to soil (35S:LAZ5).
Accession numbers
At2g34690 (ACD11): NP_181016. At1g77300 (LAZ2/SDG8): NP_177854. At5g44870 (LAZ5): NP_199300. At1g77080 (MAF1): NM_180648. At5g10140 (FLC): NM_121052. At1g19250 (FMO1) NP_173359. At5g09810 (ACTIN): NP_196543. At1g06820 (CRTISO): NP_172167. At3g48090 (EDS1) NM_114678. At3g20600 (NDR1): NP_188696. At3g07040 (RPM1): NP_187360. At4g26090 (RPS2): NP_194339. At5g45250 (RPS4): NP_199338. At1g12220 (RPS5): NP_172686. At5g17880 (CSA1): NP_197290. At4g36150: NP_195338. At5g45200: NP_199333. At5g45230: NP_199336.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. JonesJD
DanglJL
2006 The plant immune system. Nature 444 323 329
2. Van der BiezenEA
JonesJD
1998 Plant disease-resistance proteins and the gene-for-gene concept. Trends Biochem Sci 23 454 456
3. DanglJL
JonesJD
2001 Plant pathogens and integrated defence responses to infection. Nature 411 826 833
4. MaratheR
Dinesh-KumarSP
2003 Plant defense: one post, multiple guards?! Mol Cell 11 284 286
5. CaplanJ
PadmanabhanM
Dinesh-KumarSP
2008 Plant NB-LRR immune receptors: from recognition to transcriptional reprogramming. Cell Host Microbe 3 126 135
6. CollierSM
MoffettP
2009 NB-LRRs work a “bait and switch” on pathogens. Trends Plant Sci 14 521 529
7. AusubelFM
2005 Are innate immune signaling pathways in plants and animals conserved? Nat Immunol 6 973 979
8. FritzJH
FerreroRL
PhilpottDJ
GirardinSE
2006 Nod-like proteins in immunity, inflammation and disease. Nat Immunol 7 1250 1257
9. EckmannL
KarinM
2005 NOD2 and Crohn's disease: loss or gain of function? Immunity 22 661 667
10. LorrainS
VailleauF
BalagueC
RobyD
2003 Lesion mimic mutants: keys for deciphering cell death and defense pathways in plants? Trends Plant Sci 8 263 271
11. ShiranoY
KachrooP
ShahJ
KlessigDF
2002 A gain-of-function mutation in an Arabidopsis Toll Interleukin1 receptor-nucleotide binding site-leucine-rich repeat type R gene triggers defense responses and results in enhanced disease resistance. Plant Cell 14 3149 3162
12. BelkhadirY
NimchukZ
HubertDA
MackeyD
DanglJL
2004 Arabidopsis RIN4 negatively regulates disease resistance mediated by RPS2 and RPM1 downstream or independent of the NDR1 signal modulator and is not required for the virulence functions of bacterial type III effectors AvrRpt2 or AvrRpm1. Plant Cell 16 2822 2835
13. XiaoS
BrownS
PatrickE
BrearleyC
TurnerJG
2003 Enhanced transcription of the Arabidopsis disease resistance genes RPW8.1 and RPW8.2 via a salicylic acid-dependent amplification circuit is required for hypersensitive cell death. Plant Cell 15 33 45
14. ZipfelC
RobatzekS
NavarroL
OakeleyEJ
JonesJD
2004 Bacterial disease resistance in Arabidopsis through flagellin perception. Nature 428 764 767
15. YiH
RichardsEJ
2007 A cluster of disease resistance genes in Arabidopsis is coordinately regulated by transcriptional activation and RNA silencing. Plant Cell 19 2929 2939
16. BombliesK
WeigelD
2007 Hybrid necrosis: autoimmunity as a potential gene-flow barrier in plant species. Nat Rev Genet 8 382 393
17. BombliesK
LempeJ
EppleP
WarthmannN
LanzC
2007 Autoimmune response as a mechanism for a Dobzhansky-Muller-type incompatibility syndrome in plants. PLoS Biol 5 e236
18. BrodersenP
PetersenM
PikeHM
OlszakB
SkovS
2002 Knockout of Arabidopsis accelerated-cell-death11 encoding a sphingosine transfer protein causes activation of programmed cell death and defense. Genes Dev 16 490 502
19. FedorovaND
BadgerJH
RobsonGD
WortmanJR
NiermanWC
2005 Comparative analysis of programmed cell death pathways in filamentous fungi. BMC Genomics 6 177
20. AartsN
MetzM
HolubE
StaskawiczBJ
DanielsMJ
1998 Different requirements for EDS1 and NDR1 by disease resistance genes define at least two R gene-mediated signaling pathways in Arabidopsis. Proc Natl Acad Sci U S A 95 10306 10311
21. MichaelsSD
AmasinoRM
1999 FLOWERING LOCUS C encodes a novel MADS domain protein that acts as a repressor of flowering. Plant Cell 11 949 956
22. CazzonelliCI
CuttrissAJ
CossettoSB
PyeW
CrispP
2009 Regulation of carotenoid composition and shoot branching in Arabidopsis by a chromatin modifying histone methyltransferase, SDG8. Plant Cell 21 39 53
23. ZhaoZ
YuY
MeyerD
WuC
ShenWH
2005 Prevention of early flowering by expression of FLOWERING LOCUS C requires methylation of histone H3 K36. Nat Cell Biol 7 1256 1260
24. MartinC
ZhangY
2005 The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol 6 838 849
25. XuL
ZhaoZ
DongA
Soubigou-TaconnatL
RenouJP
2008 Di - and tri - but not monomethylation on histone H3 lysine 36 marks active transcription of genes involved in flowering time regulation and other processes in Arabidopsis thaliana. Mol Cell Biol 28 1348 1360
26. KroganNJ
KimM
TongA
GolshaniA
CagneyG
2003 Methylation of histone H3 by Set2 in Saccharomyces cerevisiae is linked to transcriptional elongation by RNA polymerase II. Mol Cell Biol 23 4207 4218
27. BastowR
MylneJS
ListerC
LippmanZ
MartienssenRA
2004 Vernalization requires epigenetic silencing of FLC by histone methylation. Nature 427 164 167
28. KimSY
HeY
JacobY
NohYS
MichaelsS
2005 Establishment of the vernalization-responsive, winter-annual habit in Arabidopsis requires a putative histone H3 methyl transferase. Plant Cell 17 3301 3310
29. DongG
MaDP
LiJ
2008 The histone methyltransferase SDG8 regulates shoot branching in Arabidopsis. Biochem Biophys Res Commun 373 659 664
30. GassmannW
HinschME
StaskawiczBJ
1999 The Arabidopsis RPS4 bacterial-resistance gene is a member of the TIR-NBS-LRR family of disease-resistance genes. Plant J 20 265 277
31. TakkenFL
TamelingWI
2009 To nibble at plant resistance proteins. Science 324 744 746
32. TianD
TrawMB
ChenJQ
KreitmanM
BergelsonJ
2003 Fitness costs of R-gene-mediated resistance in Arabidopsis thaliana. Nature 423 74 77
33. YuanJ
HeSY
1996 The Pseudomonas syringae Hrp regulation and secretion system controls the production and secretion of multiple extracellular proteins. J Bacteriol 178 6399 6402
34. GrantMR
GodiardL
StraubeE
AshfieldT
LewaldJ
1995 Structure of the Arabidopsis RPM1 gene enabling dual specificity disease resistance. Science 269 843 846
35. ShaoF
GolsteinC
AdeJ
StoutemyerM
DixonJE
2003 Cleavage of Arabidopsis PBS1 by a bacterial type III effector. Science 301 1230 1233
36. MindrinosM
KatagiriF
YuGL
AusubelFM
1994 The A. thaliana disease resistance gene RPS2 encodes a protein containing a nucleotide-binding site and leucine-rich repeats. Cell 78 1089 1099
37. BentAF
KunkelBN
DahlbeckD
BrownKL
SchmidtR
1994 RPS2 of Arabidopsis thaliana: a leucine-rich repeat class of plant disease resistance genes. Science 265 1856 1860
38. PflugerJ
WagnerD
2007 Histone modifications and dynamic regulation of genome accessibility in plants. Curr Opin Plant Biol 10 645 652
39. StrahlBD
AllisCD
2000 The language of covalent histone modifications. Nature 403 41 45
40. van den BurgHA
TakkenFL
2009 Does chromatin remodeling mark systemic acquired resistance? Trends Plant Sci 14 286 294
41. ZhouC
ZhangL
DuanJ
MikiB
WuK
2005 HISTONE DEACETYLASE19 is involved in jasmonic acid and ethylene signaling of pathogen response in Arabidopsis. Plant Cell 17 1196 1204
42. DhawanR
LuoH
FoersterAM
AbuqamarS
DuHN
2009 HISTONE MONOUBIQUITINATION1 interacts with a subunit of the mediator complex and regulates defense against necrotrophic fungal pathogens in Arabidopsis. Plant Cell 21 1000 1019
43. March-DiazR
Garcia-DominguezM
Lozano-JusteJ
LeonJ
FlorencioFJ
2008 Histone H2A.Z and homologues of components of the SWR1 complex are required to control immunity in Arabidopsis. Plant J 53 475 487
44. MosherRA
DurrantWE
WangD
SongJ
DongX
2006 A comprehensive structure-function analysis of Arabidopsis SNI1 defines essential regions and transcriptional repressor activity. Plant Cell 18 1750 1765
45. AyN
IrmlerK
FischerA
UhlemannR
ReuterG
2009 Epigenetic programming via histone methylation at WRKY53 controls leaf senescence in Arabidopsis thaliana. Plant J 58 333 346
46. Alvarez-VenegasR
AbdallatAA
GuoM
AlfanoJR
AvramovaZ
2007 Epigenetic control of a transcription factor at the cross section of two antagonistic pathways. Epigenetics 2 106 113
47. Alvarez-VenegasR
SadderM
HlavackaA
BaluskaF
XiaY
2006 The Arabidopsis homolog of trithorax, ATX1, binds phosphatidylinositol 5-phosphate, and the two regulate a common set of target genes. Proc Natl Acad Sci U S A 103 6049 6054
48. ArbibeL
2008 Immune subversion by chromatin manipulation: a ‘new face’ of host-bacterial pathogen interaction. Cell Microbiol 10 1582 1590
49. LiebermanPM
2006 Chromatin regulation of virus infection. Trends Microbiol 14 132 140
50. HamonMA
BatscheE
RegnaultB
ThamTN
SeveauS
2007 Histone modifications induced by a family of bacterial toxins. Proc Natl Acad Sci U S A 104 13467 13472
51. LiJ
KrichevskyA
VaidyaM
TzfiraT
CitovskyV
2005 Uncoupling of the functions of the Arabidopsis VIP1 protein in transient and stable plant genetic transformation by Agrobacterium. Proc Natl Acad Sci U S A 102 5733 5738
52. YiH
SardesaiN
FujinumaT
ChanCW
Veena
2006 Constitutive expression exposes functional redundancy between the Arabidopsis histone H2A gene HTA1 and other H2A gene family members. Plant Cell 18 1575 1589
53. JacobY
FengS
LeBlancCA
BernatavichuteYV
StroudH
2009 ATXR5 and ATXR6 are H3K27 monomethyltransferases required for chromatin structure and gene silencing. Nat Struct Mol Biol 16 763 768
54. Dinesh-KumarSP
ThamWH
BakerBJ
2000 Structure-function analysis of the tobacco mosaic virus resistance gene N. Proc Natl Acad Sci U S A 97 14789 14794
55. MestreP
BaulcombeDC
2006 Elicitor-mediated oligomerization of the tobacco N disease resistance protein. Plant Cell 18 491 501
56. RosenstielP
HuseK
TillA
HampeJ
HellmigS
2006 A short isoform of NOD2/CARD15, NOD2-S, is an endogenous inhibitor of NOD2/receptor-interacting protein kinase 2-induced signaling pathways. Proc Natl Acad Sci U S A 103 3280 3285
57. Dinesh-KumarSP
BakerBJ
2000 Alternatively spliced N resistance gene transcripts: their possible role in tobacco mosaic virus resistance. Proc Natl Acad Sci U S A 97 1908 1913
58. ZhangXC
GassmannW
2007 Alternative splicing and mRNA levels of the disease resistance gene RPS4 are induced during defense responses. Plant Physiol 145 1577 1587
59. LiangH
YaoN
SongJT
LuoS
LuH
2003 Ceramides modulate programmed cell death in plants. Genes Dev 17 2636 2641
60. WangW
YangX
TangchaiburanaS
NdehR
MarkhamJE
2008 An inositolphosphorylceramide synthase is involved in regulation of plant programmed cell death associated with defense in Arabidopsis. Plant Cell 20 3163 3179
61. PetersenM
BrodersenP
NaestedH
AndreassonE
LindhartU
2000 Arabidopsis map kinase 4 negatively regulates systemic acquired resistance. Cell 103 1111 1120
62. YangS
HuaJ
2004 A haplotype-specific Resistance gene regulated by BONZAI1 mediates temperature-dependent growth control in Arabidopsis. Plant Cell 16 1060 1071
63. PetersenNH
JoensenJ
McKinneyLV
BrodersenP
PetersenM
2009 Identification of proteins interacting with Arabidopsis ACD11. J Plant Physiol 166 661 666
64. Faigon-SovernaA
HarmonFG
StoraniL
KarayekovE
StaneloniRJ
2006 A constitutive shade-avoidance mutant implicates TIR-NBS-LRR proteins in Arabidopsis photomorphogenic development. Plant Cell 18 2919 2928
65. AlonsoJM
StepanovaAN
LeisseTJ
KimCJ
ChenH
2003 Genome-wide insertional mutagenesis of Arabidopsis thaliana. Science 301 653 657
66. AvivDH
RusterucciC
HoltBF3rd
DietrichRA
ParkerJE
2002 Runaway cell death, but not basal disease resistance, in lsd1 is SA - and NIM1/NPR1-dependent. Plant J 29 381 391
67. QiuJL
FiilBK
PetersenK
NielsenHB
BotangaCJ
2008 Arabidopsis MAP kinase 4 regulates gene expression through transcription factor release in the nucleus. Embo J 27 2214 2221
68. Nour-EldinHH
HansenBG
NorholmMH
JensenJK
HalkierBA
2006 Advancing uracil-excision based cloning towards an ideal technique for cloning PCR fragments. Nucleic Acids Res 34 e122
69. NakagawaT
SuzukiT
MurataS
NakamuraS
HinoT
2007 Improved Gateway binary vectors: high-performance vectors for creation of fusion constructs in transgenic analysis of plants. Biosci Biotechnol Biochem 71 2095 2100
70. CloughSJ
BentAF
1998 Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J 16 735 743
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 10
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Retroviral RNA Dimerization and Packaging: The What, How, When, Where, and Why
- Viral Replication Rate Regulates Clinical Outcome and CD8 T Cell Responses during Highly Pathogenic H5N1 Influenza Virus Infection in Mice
- Antimicrobial Peptides: Primeval Molecules or Future Drugs?
- Crystal Structure of DotD: Insights into the Relationship between Type IVB and Type II/III Secretion Systems
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy