
Identification and Genome-Wide Prediction of DNA Binding Specificities for the ApiAP2 Family of Regulators from the Malaria Parasite
The molecular mechanisms underlying transcriptional regulation in apicomplexan parasites remain poorly understood. Recently, the Apicomplexan AP2 (ApiAP2) family of DNA binding proteins was identified as a major class of transcriptional regulators that are found across all Apicomplexa. To gain insight into the regulatory role of these proteins in the malaria parasite, we have comprehensively surveyed the DNA-binding specificities of all 27 members of the ApiAP2 protein family from Plasmodium falciparum revealing unique binding preferences for the majority of these DNA binding proteins. In addition to high affinity primary motif interactions, we also observe interactions with secondary motifs. The ability of a number of ApiAP2 proteins to bind multiple, distinct motifs significantly increases the potential complexity of the transcriptional regulatory networks governed by the ApiAP2 family. Using these newly identified sequence motifs, we infer the trans-factors associated with previously reported plasmodial cis-elements and provide evidence that ApiAP2 proteins modulate key regulatory decisions at all stages of parasite development. Our results offer a detailed view of ApiAP2 DNA binding specificity and take the first step toward inferring comprehensive gene regulatory networks for P. falciparum.
Published in the journal:
. PLoS Pathog 6(10): e32767. doi:10.1371/journal.ppat.1001165
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1001165
Summary
The molecular mechanisms underlying transcriptional regulation in apicomplexan parasites remain poorly understood. Recently, the Apicomplexan AP2 (ApiAP2) family of DNA binding proteins was identified as a major class of transcriptional regulators that are found across all Apicomplexa. To gain insight into the regulatory role of these proteins in the malaria parasite, we have comprehensively surveyed the DNA-binding specificities of all 27 members of the ApiAP2 protein family from Plasmodium falciparum revealing unique binding preferences for the majority of these DNA binding proteins. In addition to high affinity primary motif interactions, we also observe interactions with secondary motifs. The ability of a number of ApiAP2 proteins to bind multiple, distinct motifs significantly increases the potential complexity of the transcriptional regulatory networks governed by the ApiAP2 family. Using these newly identified sequence motifs, we infer the trans-factors associated with previously reported plasmodial cis-elements and provide evidence that ApiAP2 proteins modulate key regulatory decisions at all stages of parasite development. Our results offer a detailed view of ApiAP2 DNA binding specificity and take the first step toward inferring comprehensive gene regulatory networks for P. falciparum.
Introduction
Plasmodium falciparum is responsible for the majority of human malaria cases and causes approximately 1 million deaths every year [1]. The complete lifecycle of P. falciparum includes three developmental stages, which occur in its mosquito vector, the human liver, and human blood. Within each developmental stage the parasite undergoes major morphological changes that are accompanied by precisely timed transcription of genes that are necessary for parasite growth, differentiation, and replication. Detailed transcriptome and proteome studies have been conducted across the different stages of the life cycle [2]–[12]. Despite these advances in our understanding of messenger RNA transcript dynamics in P. falciparum, very little is known regarding the mechanism of transcriptional regulation, including transcription factor binding and sequence specificity.
Basic transcriptional control in P. falciparum appears to resemble that of other eukaryotic organisms, with general transcription factors coordinating the recruitment of RNA polymerase II to core promoter elements [13]–[15]. Experiments aimed at identifying cis-acting sequences required for gene expression have successfully identified specific enhancer and repressor sequences upstream of the core promoter elements [16]–[26]. In the asexual blood stage, regulatory sequence elements have been identified for the gene encoding the knob-associated histidine-rich protein (kahrp) [16], glycophorin binding protein 130 (gbp130) [18], cytidine diphosphate-diacylglycerol synthase (pfcds) [19], the DNA polymerase delta gene [20], a subset of the heat shock protein (hsp) family [22], the rif genes [23] and the falcipains [24]. Additionally, three sequence motifs have been identified upstream of the var genes: the SPE1, CPE, and SPE2 motifs, of which the SPE2 motif has been hypothesized to be involved in silencing of var gene expression [25], [26]. In sexual blood stage parasites three distinct short sequence elements have been found to regulate expression of the gametocyte genes pfs16, pfs25 [17], and pgs28 [21]. In addition to these experimentally derived motifs, bioinformatic analyses of the P. falciparum genome have identified a number of potential cis-elements that may play a role in gene regulation [27]–[36]. However, attempts to identify trans-factors have been largely unsuccessful [13], [15], [37], [38], with the exceptions of Myb1 [39], [40] and the high mobility group box (HMGB) proteins [41], [42].
Recently, a large protein family was identified in P. falciparum, containing Apetala2 (AP2) domains [43]. AP2 domains were originally described in plants as DNA binding domains approximately 60 amino acids in length [44]. In plants, the AP2 family of transcription factors is one of the largest, playing key roles in developmental regulation [44] and stress responses [45]. The Apicomplexan AP2 (ApiAP2) proteins represent a lineage-specific expansion, and are highly conserved across all Plasmodium spp. and in other Apicomplexans including Theileria, Cryptosporidium [43] and Toxoplasma [46]. P. falciparum was initially predicted to contain 26 ApiAP2 factors, each containing one to three AP2 domains [43], while in Toxoplasma the family is expanded to over 50 ApiAP2 proteins [46]. We have noted a 27th highly conserved ApiAP2 protein (PF13_0267), which agrees with recent Pfam predictions for this protein [47]. Although other DNA binding proteins have been reported in the literature, ApiAP2 proteins represent the largest family of transcriptional regulators identified in P. falciparum, where they are expressed throughout the entire developmental lifecycle [43].
Previously, we established that two ApiAP2 proteins, PF14_0633 and PFF0200c, bind DNA with high sequence selectivity [48]. Subsequent work demonstrated that the P. berghei orthologue of PF14_0633 (PBANKA_132980) is essential for the formation of sporozoites [49], and specifically regulates sporozoite target genes by binding to the same GCATGCA motif that we identified [48]. More recently, PFF0200c was shown to function as a DNA tethering protein involved in heterochromatin formation and integrity [50] via binding to the previously identified SPE2 motif [25]. Importantly, PFF0200c does not appear to act as a transcriptional regulator in the blood stage. A third study identified a P. berghei protein, AP2-O [PBANKA_090590 (PF11_0442)], as an activator of genes required for invasion of the mosquito midgut during the mosquito stage of the life cycle [51]. Together, these studies highlight the importance of the ApiAP2 DNA binding proteins in modulating stage-specific gene regulation and chromatin integrity. Despite these recent advances, the regulatory function of the majority of ApiAP2 proteins remains unknown. The DNA sequences recognized by the members of this protein family are largely uncharacterized, and the target genes that these ApiAP2 factors bind are undefined.
Here we biochemically and computationally characterize the global DNA binding specificities for the entire ApiAP2 protein family from P. falciparum. Our results reveal a complex array of DNA sequence elements, with the majority of proteins binding to unique sequences. We demonstrate several cases where multiple AP2 domains within the same ApiAP2 protein are capable of binding distinct DNA sequences. The identification of these unique sequence motifs sheds light on the molecular mechanisms of transcriptional regulation by assigning correlations between putative cis-acting sequences in Plasmodium and the trans-factors that will bind at those sequences genome-wide. Our data reveal the likely identity of trans-acting ApiAP2 factors that specifically bind to previously described cis-motifs, illuminating some of the previously unknown effectors of plasmodial gene expression. For the many new motifs we identified, we predict putative targets for each of the ApiAP2 proteins. This work represents the first comprehensive analysis of the ApiAP2 DNA binding proteins in P. falciparum and provides a crucial missing link toward understanding their role in the regulation of parasite development.
Results
P. falciparum ApiAP2 Domains Bind Diverse Sequence Elements
The 27 plasmodial ApiAP2 proteins vary drastically in size (Figure S1), however, the predicted 60 amino acid AP2 domains, are well-defined and highly conserved. To determine the DNA binding specificity for the P. falciparum AP2 domains, we used protein binding microarrays (PBMs), which enable simultaneous screening of all possible DNA sequences up to ten nucleotides in length without sequence bias [52], [53]. Seminal studies from the Bulyk lab have used PBMs to comprehensively characterize individual transcription factors from a diverse array of organisms including yeast, worm, mouse and human [52]–[54]; and we have previously demonstrated its utility for Apicomplexan AP2 DNA binding proteins [48].
We created 50 constructs for PBM screening (Supplemental Text S1, Figure S2), including individual domains, full length proteins, and tandem domain arrangements (two AP2 domains separated by a short conserved linker sequence of 12 to 79 amino acids; designated DLD). Our analysis by PBM of these P. falciparum AP2 domains revealed motifs for 20 out of the 27 ApiAP2 proteins (Figure 1), including a motif for the recently identified ApiAP2 protein, PF13_0267, helping to confirm the new annotation for this protein. Results from at least two PBM experiments for each AP2 domain were used to generate position weight matrices (PWMs), which represent the DNA binding affinity for a given domain (Dataset S1, Figure 1). Replicate experiments had excellent correlation coefficients illustrating the robustness of the PBM methodology (see Supplemental Text S1). Enrichment scores (E-scores) were assigned for each 8-mer (allowing up to two gaps) [53], with a significance cut-off of 0.45 (E-scores range from -0.5 to +0.5) for specific 8-mers enriched above background. The E-score is a rank-based, nonparametric score that is robust to differences in protein concentration and reflects the relative preference for each 8-mer [55]. In total we identified sequence motifs for 24 AP2 domains found in a variety of protein architectures (Figure 1). While Figure 1 illustrates which motifs are linked to the blood stage of Plasmodium development, several motifs are also associated with ApiAP2 proteins during non-blood stages as well (see Supplemental Text S1). It is noteworthy that different AP2 domains from the same ApiAP2 protein bind distinct DNA sequence elements. However, we do find several motifs that are recognized by multiple ApiAP2 factors (see Supplemental Text S1 and Table S1). This complexity may allow for multifaceted transcriptional regulation using a smaller number of individual factors.

P. falciparum AP2 Domains Can Recognize Multiple Distinct Sequences
Protein-DNA interaction specificities are determined by the chemical interactions of amino acids and DNA bases [56]. Side chain flexibility and DNA distortions allow one DNA binding domain to interact with multiple distinct DNA sequences. For several of the AP2 domains there were significant differences among the top scoring 8-mer sequences that were bound, suggesting multi-motif recognition. Using the Seed and Wobble algorithm [57] we identified alternative motifs associated with 8-mers of high signal intensity that could not be explained by the primary motif for 14 AP2 domains (representing 13 ApiAP2 proteins) (Figure S3, Dataset S2). Some AP2 domains only had a single secondary motif, whereas others had up to four. The secondary motifs can be described based on their relationship with the corresponding primary motifs and fall into the broad categories of end modifications, core changes, variable spacer distances or alternate recognition interfaces [57] (see Supplemental Text S1). The ability of an individual domain to bind anywhere from one to five different DNA sequences would significantly increase the number of target genes that could be regulated by one factor.
We selected two ApiAP2 proteins for confirmation of secondary motif binding by electrophoretic mobility shift assays (EMSAs). Domain 2 of PFD0985w has three predicted secondary motifs in addition to the primary motif. A plot of the E-scores for all ungapped 8-mers reveals that the top 100 matches to both the primary motif and one of the secondary motifs (Figure 2A) are relatively equal in E-score. Therefore, PFD0985w_D2 should bind equally well to these two motifs. To test this hypothesis we generated 60 bp oligonucleotides with the specific motif sequence in the center flanked by random sequences. EMSAs with purified PFD0985w_D2 demonstrate that both oligonucleotides are bound equally well, and that the primary motif is capable of out-competing the secondary motif and vice versa (Figure 2B). No binding is observed with an unrelated non-specific oligonucleotide, indicating specificity for the predicted motifs (data not shown). The second ApiAP2 factor that we selected for confirmation of secondary motifs was PFL1900w_DLD. The highest scoring 8-mers for this tandem domain were represented by completely distinct sequences and a plot of all 8-mers and their E-scores revealed preferential binding with a primary, secondary, and tertiary motif (Figure 2C). PFL1900w_DLD was able to shift all three motifs, but with varying affinities (data not shown for the primary and tertiary motifs), and competition between the secondary and tertiary motifs revealed a clear preference for the secondary motif over the tertiary motif (Figure 2D). These results suggest that the secondary motifs detected represent bona fide sequences bound by the AP2 domains and the E-score distributions accurately reflect binding affinities.

Identification of Putative Trans-Factors for Previously Predicted Plasmodium Regulatory Motifs
Both computational predictions and experimental data have identified a number of DNA sequence motifs upstream of genes in Plasmodium [17]–[22], [27]–[32], but the specific trans-factors that bind to these motifs have mostly remained elusive. For three cases, we now establish plausible links between the newly identified AP2 DNA sequence motifs and these previous reports. Militello et al. identified a specific motif, (A/G)NGGGG(C/A) (called the G-box), upstream of 8 out of 18 Plasmodium heat shock genes [22]. The occurrence of this GC-rich motif in the genome is low (Table S2), suggesting that its presence in upstream sequences may be significant for transcriptional regulation. The sequence motif that we have identified for PF13_0235_D1 is nearly identical to the G-box element (Figure 3A). Furthermore, the expression profiles of pf13_0235, hsp86 (pf07_0029), and hsp70 (pf08_0054), two heat shock genes containing one or more G-boxes exhibit a strong positive correlation (r = 0.93) during the asexual blood stage [2] (Figure 3A), suggesting that PF13_0235 may play a role in regulating hsp gene expression. We performed EMSAs using both G-box elements of the hsp86 upstream region and found that PF13_0235_D1 interacts specifically with the G-box and deletion of both G-boxes is required to completely eliminate binding (Figure 3B, C). In the presence of only one G-box, binding is severely reduced (Figure 3B, C) suggesting that PF13_0235_D1 preferentially interacts with both G-boxes, perhaps through dimerization (see below). No binding is observed with an unrelated non-specific oligonucleotide at similar protein concentration (data not shown). This result is in agreement with in vivo data from transient transfections, where elimination of G-box 1 substantially reduced luciferase expression, but did not completely abolish it [22]. We also tested the G-box from the 5′ flanking region of hsp70 for in vitro binding by EMSA, and confirmed that PF13_0235_D1 binds this sequence in vitro. It is interesting to note that the binding of this single G-box motif is similar to that seen for hsp86 after deletion of one G-box, suggesting that higher affinity interactions require two occurrences of this motif.

Likewise, the sequence element bound by PF10_0075_D3, GTGCA, is enriched in the upstream sequences of genes involved in merozoite development and invasion [31], [58]. Using EMSAs we find that PF10_0075_D3 binds to the GTGCA motif upstream of msp1 (pfi1475w), msp10 (pff0995c) and rhopH 3 (pfi0265c) (Figure S4A), and no binding is observed with an unrelated non-specific oligonucleotide, indicating specificity for the predicted motifs (data not shown). Previous expression studies using a rhoptry gene promoter to drive luciferase expression have demonstrated that the GTGCA motif is important for rhoptry gene-like stage-specific expression [31]. Combined with our EMSA results, this suggests that PF10_0075 may play a role in regulating the expression of invasion-related genes in P. falciparum.
Finally, a specific 5 bp motif in the 5′-upstream region of gbp130 (pf10_0159), GTATT, was previously found to be bound by unknown nuclear factors in a sequence-specific manner [18]. The reverse complement of this 5 bp element is nearly identical to the motif we have identified for PF11_0091. EMSAs using the promoter region of gbp130 and the purified AP2 domain from PF11_0091 confirm its ability to interact with this sequence (Figure S4B), while no binding was observed with an unrelated non-specific oligonucleotide (data not shown), suggesting it is a possible regulator of GBP130 function. The PBM-derived motifs are useful to suggest putative targets for the ApiAP2 trans-factors, especially where previous characterization is available. However, in vivo assays will be required in all cases to validate these interactions on a protein-by-protein basis.
Prediction of ApiAP2 Target Genes
To begin to characterize the functional role of ApiAP2 proteins, we searched the P. falciparum genome for sequences in promoters and untranslated regions that may serve as regulatory sites for ApiAP2 binding. As a first analysis, we used our AP2-specific position weight matrices generated from the PBM data to search the 5′ upstream sequence elements of Plasmodium genes using ScanACE [59], which lists all matches to our position weight matrices within the user defined threshold. Although putative transcription start sites have been predicted [60], actual transcription start sites are still poorly defined in P. falciparum [61]. Therefore, we searched 2 kb upstream of the ATG start codon or until an upstream open reading frame was encountered. While this search provides a list of all possible motif occurrences determined from matches to a specific position weight matrix (Datasets S3 and S4), it is undoubtedly an overestimation of putative target genes. In reality, the presence of a regulatory element upstream of a gene does not confirm a regulatory interaction exists, and many motif occurrences may be inactive [62]. Furthermore, for a regulatory element to be functional, it needs to be accessible for binding, which is in part determined by nucleosome occupancy. Nucleosome occupancy has been mapped during the intraerythrocytic developmental cycle (IDC) of P. falciparum [63] and using this data we were able to determine that between 65 and 97% of our ScanACE predicted binding sites are accessible (nucleosome-free) at some point during the IDC (Table S2). This suggests that the majority of our predictions have the potential to be active; however, in vivo binding affinities may differ from in vitro determined affinities, possibly altering the weighting of specific nucleotide positions within the motifs. Ultimately, the actual target sequences of each ApiAP2 protein will need to be individually determined through experimental validation in vivo during the specific lifecycle stage of interest.
As a test of the ability of our ApiAP2 proteins to bind to the ScanACE predicted targets we selected a putative target for the newly annotated ApiAP2 protein PF13_0267; pfc0975c has a match to the CTAGAA motif at 1469 bp upstream of the start codon. EMSAs showed that the putative target sequence was bound by the purified AP2 domain from PF13_0267, while a mutant oligonucleotide lacking the predicted target sequence did not exhibit significant binding (Figure S5). Although these results demonstrate that our ScanACE-predicted target genes provide a good starting point to search for candidate genes for in vivo testing, this does not indicate if pfc0975c is a true target of PF13_0267. Indeed the motif bound by PF13_0267 is found upstream of almost all genes and in vivo validation will be required to identify actual targets. Complete AP2 motif occurrence data for the P. falciparum genome are available to the malaria community at PlasmoDB (www.plasmodb.org) [64].
Target Gene Refinement Using the IDC Transcriptome
While the ScanACE analysis provides a list of all occurrences for each motif, it is unlikely that the ApiAP2 proteins are binding to all possible motif occurrences, and instead that they bind to a smaller subset of promoters. Proteins that are co-localized in the cell or form sub-cellular structures such as the ribosome have been found to be transcriptionally co-regulated in other organisms such as yeast, and often are regulated by the same cis-elements [65]. Genes that are functionally distinct, but are co-expressed can also be regulated by the same cis-elements in their upstream regions. To narrow the ScanACE list to a more informative subset of putative target genes we used relative mRNA abundance profiles to define relationships between co-expressed Plasmodium genes [2]. We used linear regression to determine at each time point the extent to which each AP2 motif contributes to (or recapitulates) the overall expression of the genes that contain a given motif in the upstream regions (see Methods and Supplemental Text S1 for details [66]). Thus each motif at each time point is associated with a score (i.e. the fitted regression coefficient), which is positive if genes that have the motif tend to go up at that time point, or negative if they tend to go down. These scores define the predicted motif activity at each time point, and an activity profile across the entire IDC. Activity profiles reflect the predictive effect of individual AP2 motifs on gene expression of a set of target genes at a given IDC timepoint (Figure 4A), and are therefore independent of the mRNA expression profiles of the AP2 genes themselves. The activity profile for each motif was then used to iteratively identify genes containing the target motif in their 5′ upstream regions that share an expression profile similar to the activity profile. This provided a refined list of putative target genes that are co-expressed with one another (Dataset S5).

To illustrate how this approach improved our target predictions we focus on the G-box motif. The ScanACE predicted target list for this motif includes 522 genes (Dataset S3), and using the activity profile-based approach we refine this list to a set of 124 putative co-expressed target genes (Table 1, Dataset S5). A comparison of the average expression profiles for these genes with the expression of pf13_0235, the gene for the ApiAP2 factor that binds the G-box, shows a strong positive correlation (0.97; Figure 4). Similar comparisons were made for all ApiAP2 factors and their putative targets (Figure S6), and we observe either significant positive (r-values from 0.97 to 0.43) or negative correlations (r-values from −0.94 to −0.56) for many ApiAP2 factors, implying that this protein family may act as both activators and repressors of target gene expression.
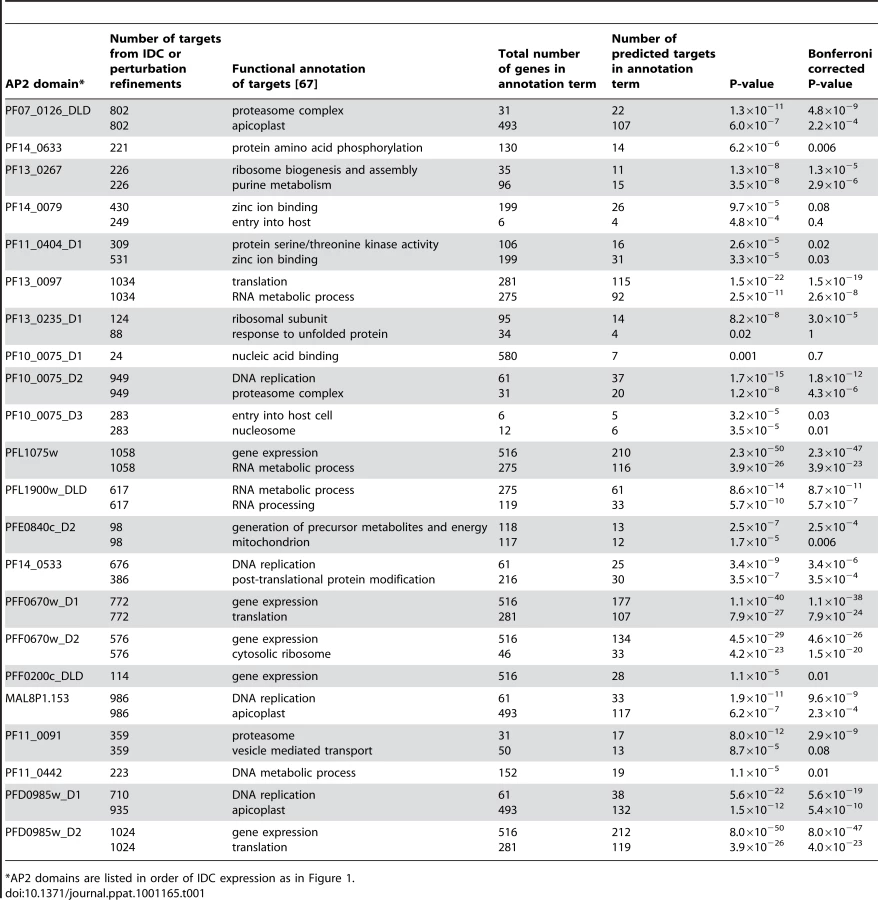
Functional annotation of the predicted targets using the DAVID bioinformatics resource [67] identified enrichment of genes involved in specific cellular processes (Table 1). Targets of PF13_0235_D1, the ApiAP2 factor that binds the G-box motif, include genes involved in ribosome function or translation and heat shock response genes. Genes involved in these functions have previously been suggested to be regulated via the G-box element [22], [33], supporting our target gene predictions. Other notable examples include the enrichment of targets involved in cell invasion and host cell entry for the PF10_0075_D3 motif (GTGCAC) and DNA binding for the MAL8P1.153 motif (ACACA). The involvement of the GTGCAC motif in invasion related processes has been independently predicted by three bioinformatic studies [31]–[33], while the ACACA motif was previously associated with DNA replication [31]. Since the majority of these motifs have not been previously described in P. falciparum, our prediction of target gene functions are novel and warrant further characterization.
Similarly, we used a recently published P. falciparum growth perturbation dataset [68] as an alternative data source to create activity profiles to refine our target gene predictions (Figure S7, Dataset S6, Supplemental Text S1). Genes that respond in a similar manner to a perturbation are more likely to be regulated by the same factor and we observed narrower target gene lists for each motif, many of which overlap with the predictions made using the IDC co-expression data, and others that are novel target gene predictions (Table 1, Figure 4, Figure S8). Further details on the perturbation refinement of target genes can be found in the Supplemental Text S1. Combined, refinement of putative targets using the high resolution temporal gene expression data [2] and the perturbation dataset [68] produce manageable gene sets for further analysis.
Identification of Motifs Enriched Upstream of var Genes
We also looked at motif enrichment in the upstream regions of var genes. There are approximately 60 var genes in P. falciparum that encode the antigenically variant erythrocyte membrane protein 1 (PfEMP1), which is involved in sequestration of infected red blood cells in the vasculature [69]. The var genes have been divided into groups based on their location along the chromosome (internal or at chromosome ends), the direction of transcription, and the sequences of their intron and 5′ and 3′ untranslated regions [70]. Using our ScanACE prediction of binding sites, we observed a striking pattern of ApiAP2 motifs that were clustered in discrete positions of all three types of var promoters (Figure 5). In the upsB promoters we observed repeated motifs for PFF0200c that correspond to the previously identified bipartite SPE2 element [25], located at −2000 to −3000 bp upstream of the ATG start codon. While PF10_0075_D3 binds to a similar sequence as the SPE2 element, recent work has demonstrated that PFF0200c is the primary ApiAP2 factor that binds to this element in vivo [50]. We also predict the SPE1 element of upsB var genes at −1200 bp [25], which matches to the motif we have identified for PF14_0633. In addition to these previously identified sequence elements, we predict binding sites for PF11_0442, which binds to the sequence GCTAGC (Figure 5). Matches to this motif are conserved in all three major types (A, B and C) of var genes. The presence of multiple ApiAP2 binding sites upstream of var genes suggests multiple ApiAP2 factors may be recruited for binding upstream of the var genes. var promoters are silenced by default [71], and it is possible that this silencing is maintained by the co-ordinated action of multiple ApiAP2 factors. Further investigation of these discrete motif-enriched sites will be required to determine the precise role of additional ApiAP2 proteins in var gene regulation.

Target Gene Prediction in P. vivax
The IDC transcriptome of P. vivax [72] suggests that the regulation of development follows a similar cascade of gene expression as that seen for P. falciparum [2], [6]. All P. falciparum ApiAP2 proteins have syntenic homologues in P. vivax and are expressed at a similar stage of development during the IDC with the exception of pf14_0471 (pv118015) which is shifted from trophozoite to late schizont stage [72] (Figure S9A). AP2 domains are highly conserved across all Plasmodium spp. and will likely bind the same motifs. It follows that the ApiAP2 proteins may regulate similar or related target genes in P. vivax compared to P. falciparum. We calculated activity profiles using the P. vivax asexual blood stage transcriptome data [72] (Figure S9B) and as seen for P. falciparum, most of the motifs are associated with activation or repression of target genes at one or more timepoints. However, a comparison of the target gene lists for each motif in P. falciparum and P. vivax shows that the conservation of putative targets is low, ranging from 0 to 53% (Table S3). This implies that although the AP2 DNA binding domains are highly conserved, some of the regulons across these species have diverged and regulation of orthologous genes has evolved independently. It should be noted that a comparison of target genes in non-blood stages, including the mosquito stage, demonstrates substantial statistically significant conservation of motifs among co-regulated genes in P. vivax and P. falciparum [73]. Therefore it will be important to identify the actual target genes bound in vivo for each motif to determine the actual extent of conservation between species.
Target Genes in Non-Blood Stages of Development
While the above analyses are limited to transcriptional regulation during the blood stage of the lifecycle, ApiAP2 function is not limited to the IDC. Accordingly, we analyzed gene expression data from P. falciparum gametocytes, zygotes and sporozoites [6], [10]. Activity profiles for ApiAP2 motifs in gametocyte data revealed activity for a number of the PBM derived motifs during gametocytogenesis (Figure S10A). We found two motifs to be active in zygotes, including the previously identified zygote motif for the P. berghei orthologue of PF11_0442 (PBANKA_090590), as well as the CACACA motif, which is bound by PF13_0026 (Figure S10A, Dataset S7). Activity profiles for AP2 DNA motifs in sporozoites identify PF14_0633, PFD0985w_D2, and PFF0670w_D1 as potentially active AP2 domains during this stage (Figure S10A). Since there is currently no liver stage data available for Plasmodium species that infect humans, we used data from the rodent malaria species P. yoelii [9]. Activity profiles for our PBM derived motifs in the P. yoelii liver stage yielded a number of motifs that are potentially active during this stage (Figure S11). Further discussion of the non-blood stage active motifs can be found in the Supplemental Text S1.
Discussion
Understanding the molecular mechanisms and the regulatory processes that underlie gene expression during the development of P. falciparum is a major challenge in malaria research. The ability to map the recognition sites of DNA binding proteins genome-wide enables an improved understanding of how trans-factors regulate gene expression and has been undertaken for only a few large eukaryotic families of transcription factors [54], [57], [74]–[76]. Here, we have comprehensively characterized the P. falciparum ApiAP2 protein family and established their preferred DNA recognition motifs. A number of findings strongly implicate these factors as major regulators of gene expression in P. falciparum. First, the expression of the ApiAP2 genes at distinct times throughout the IDC suggests they are available to regulate target genes throughout the entire 48-hour blood stage of the parasite. Second, the diversity of sequences bound by these domains is sufficient to account for the full cascade of gene expression observed in the IDC. Furthermore, ApiAP2 genes form an interaction network with themselves during the IDC (Figure S12), suggesting that in addition to regulating target genes they may also regulate their own expression. For all three instances where in vivo data on binding sites for ApiAP2 proteins are available [49]–[51], our PBM derived motifs are excellent matches, emphasizing the quality of these data and the robustness of the PBMs at identifying preferred sequence elements. Finally, half of the motifs that we identified are nearly identical to motifs that were independently predicted computationally as putative cis-elements in the P. falciparum genome [27]–[29], [31]–[33].
Our success rate for identifying DNA binding specificities (20 out of 27 ApiAP2 proteins, 74%) using the PBMs is comparable to results obtained using the same technology with other transcription factor families from yeast and mouse (40–85% successful) [54], [57], [76]. For those AP2 domains that did not yield a result, there are numerous technical reasons why we may fail to detect binding. Although we were able to successfully express all of our GST-fusion constructs, possible reasons for failure to detect binding events include insufficient protein concentration, low protein stability, binding conditions (e.g. ionic strength) used, or low-affinity binding (undetectable by PBM). Another possibility is simply that some predicted AP2 domains lack specific DNA binding activity altogether. As mentioned above, the ApiAP2 proteins vary dramatically in size, including four proteins that are less than 40 kDa (Figure S1). Only one of the four smallest ApiAP2 proteins (<300 amino acids) binds to DNA (PF13_0026). Despite our testing both full-length ApiAP2 proteins and isolated AP2 domains from these small proteins, we could detect no DNA binding by PBM. This is surprising given their lack of any other predicted functional domains.
Both computational prediction of motifs [22], [31]–[33] and experimental data [25], [49]–[51] have identified a number of regulatory elements involving repeated iterations of the same motif. One established way to recruit multiple copies of the same factor to a particular site in the genome is through the formation of dimers or multimers of the same protein. A crystal structure has recently been solved of the AP2 domain from PF14_0633, which reveals that the AP2 domain forms a dimer when bound to DNA [77]. This structure suggests that other ApiAP2 proteins may also form homo - or heterodimers [78], thereby recognizing multiple DNA sequence motifs in concert. Further support for this idea is seen in our EMSA analysis of PF13_0235_D1, which shows that higher affinity binding to the G-boxes upstream of hsp86 (Figure 3) occurs when multiple copies of the motif are present. Similar results were obtained for PF10_0075_D3 and the predicted rhopH 3 target (Figure S4A) suggesting that AP2 domain dimerization may enhance binding to these sequences. Our ScanACE predictions of target genes identify motif repeats in upstream regions (Dataset S3), which may allow for tighter control of target gene expression by the ApiAP2 factors. Similarly, heterodimer formation may facilitate combinatorial regulation of gene expression at co - motifs. Evidence for such interactions can be found in yeast two-hybrid assays that have detected associations between ApiAP2 proteins [79]. Taken together, our genome-wide motif predictions and the ability of plasmodial AP2 domains to form dimers implies that these factors likely work together to regulate target gene expression.
The extremely high level of conservation (>95% identity) for each AP2 domain across all Plasmodium spp. [43] suggests that orthologues from other species will bind to similar DNA sequence motifs. Indeed, motifs for the P. falciparum AP2 domains of PF14_0633 and PF11_0442 are matches to the experimentally determined motifs for their P. berghei orthologues (AP2-Sp and AP2-O, respectively) [49], [51]. However, while the cis-elements bound by individual AP2 domains may be conserved across species, our predicted IDC target gene sets for ApiAP2 proteins appear to differ extensively. This is common in other eukaryotic organisms, where DNA binding domains are highly conserved across species, but downstream target genes are divergent [80]–[85]. Our data suggests that this may be true for Plasmodium spp., as demonstrated by the divergence of IDC target gene sets in P. vivax and P. yoelii compared to P. falciparum, which contrasts with the almost perfect conservation of AP2 domains and the similar temporal expression of ApiAP2 genes. We previously showed that the orthologous AP2 domains from PF14_0633 in P. falciparum and its distant Apicomplexan relative C. parvum (cgd2_3490) bind virtually identical sequence elements [48], but their predicted regulons had virtually no overlap. However, there are some examples of transcription factor binding site conservation among specific groups of target genes. For example, the G-box element has been predicted to regulate heat shock genes in both C. parvum [86] and P. falciparum [22], and both the PF14_0633 (TGCATGCA) and PF10_0075_D3/PFF0200c_DLD (GTGCAC) motifs are conserved among sporozoite and merozoite invasion genes in P. falciparum, P. vivax, P. yoelii, and P. knowlesi [87]. A better assessment of target gene conservation between species will be possible with more accurate target gene lists from chromatin immunoprecipitation experiments for each individual ApiAP2 factor. Indeed, this has recently been demonstrated in the asexual blood stages for PFF0200c [50], and only a subset of predicted targets were actually bound in vivo by this factor. This is also likely to be true for other ApiAP2 factors and further work identifying functional binding sites will clarify the level of conservation of transcription factor binding sites among the different Plasmodium spp.
Combinatorial gene regulation is an important aspect of transcription in many organisms. It controls the level of gene expression, the precise timing of expression, and determines the ability of a regulatory circuit to respond differently to a wide variety of extracellular signals. The finding that there are a relatively small number of specific transcription factors in P. falciparum prompted the hypothesis that combinatorial gene regulation plays an important role in the parasite [29]. Our finding that some ApiAP2 proteins can bind more than one sequence element significantly increases the potential complexity of the regulatory network. We identified secondary DNA binding preferences for 14 AP2 domains. These secondary motifs allow one AP2 domain to regulate a much broader range of targets than initially predicted from our ScanACE analysis using only the primary motifs. Precedent for this has been seen in yeast where the transcriptional activator, HAP1, binds two completely different regulatory sequences, allowing for the regulation of target genes with different promoter elements [88]. A similar observation was made in a recent PBM analysis of 104 mouse transcription factors, where it was found that almost half of the TFs recognized multiple sequence motifs [57]. A re-analysis of previously generated chromatin immunoprecipitation – microarray (ChIP-chip) data illustrated that these newly identified alternate motifs were also bound in vivo [57]. Similar in vivo data will be required to fully elucidate the role of individual ApiAP2 motif occurrences on target gene functions; however it is evident from our data that the ApiAP2 factors have sufficient diversity in sequence recognition to potentially regulate all P. falciparum genes.
While it is clear that the ApiAP2 factors bind DNA and recent work has begun to explore their contribution to transcriptional regulation [49], [51], these factors are also capable of binding DNA as scaffolding and recruitment proteins [50]. This finding opens up new possible functions for this family of DNA binding proteins. The next step in understanding ApiAP2 function will be to address the role of other domains in these proteins. The majority of ApiAP2 proteins are predicted to encode extremely large proteins, yet nothing is known regarding other functional domains outside of the AP2 domain. Yeast two-hybrid assays have identified interactions between ApiAP2 proteins and chromatin modifying proteins [79], which could help recruit ApiAP2 proteins to target sites in the genome. How and when ApiAP2 proteins are targeted to the nucleus and if this is actively regulated also remains unanswered. Our global mapping of ApiAP2 motif preferences represents the first characterization of a Plasmodium family of DNA binding proteins and provides a starting point to investigate transcriptional control in P. falciparum. These results provide an important step toward understanding the role of these proteins as major regulators throughout all stages of parasite development in Plasmodium spp. and other related Apicomplexan species.
Materials and Methods
Cloning, Expression, and Purification of P. falciparum ApiAP2 Domains
Domain boundaries were defined as in [43] and extensions were made based on sequence homology both 5′ and 3′ of the AP2 domains amongst Plasmodium spp. orthologues, as well as using structural predictions from the online secondary structure prediction server Jpred3 [89]. In total 77 different versions of ApiAP2 domains from P. falciparum as well as one from P. berghei were cloned into pGEX-4T1 (GE Life Sciences) to create N-terminal glutathione S-transferase (GST) fusions (Figure S2). Proteins were expressed in BL21-CodonPlus(DE3)-RIL cells (Stratagene) with 0.2 mM IPTG at room temperature and affinity purified using glutathione resin (Clontech). The purity of each protein was estimated by silver stained SDS-PAGE and yields were calculated based on absorbance at 260 nm and specific molar extinction coefficients. All protein concentrations include contaminating products and will be an overestimation of the actual AP2 domain amounts.
Protein-Binding Microarrays (PBMs)
All constructs in Figure S2 were tested at least once on the PBMs, and positive motifs were confirmed at least twice on independent arrays. The PBM experiments were performed as previously described [53]. Briefly, custom designed oligonucleotide arrays are double-stranded using a universal primer, incubated with GST-AP2 fusion proteins, visualized with Alexa-488 conjugated anti-GST antibody, and scanned using an Axon 4200A scanner. Proteins were used at the maximum concentration obtained from purification and represent one-fifth of the total reaction volume used on the PBM. In this study three different universal platforms were used covering all contiguous 8-mers as well as gapped 8-mers spanning up to 10 positions. After data normalization and calculation of enrichment scores [53], [55] the “Seed-and-Wobble” algorithm was applied to combine the data from two separate experiments and create position weight matrices (PWMs) [55]. An enrichment score cut-off of 0.45 was used to distinguish high affinity binding data from low affinity and non-specific binding. The score for each 8-mer reflects the affinity of a DNA binding domain for that sequence, with higher scores representing tighter interactions [55]. Secondary motifs were identified by running the “rerank” program until E-scores below 0.45 were obtained [55]. The PBM analysis suite was downloaded from the Bulyk lab (http://the_brain.bwh.harvard.edu/PBMAnalysisSuite/index.html). For public access, all motifs have been deposited in the UniPROBE database [90].
Electrophoretic Mobility Shift Assays (EMSAs)
N-terminal GST fusions of the ApiAP2 domains were purified as described above. Single-stranded HPLC purified 5′ biotinylated oligonucleotides were purchased from Integrated DNA Technologies (http://www.idtdna.com) and annealed with complementary oligonucleotides to create double-stranded probes (Table S4). EMSAs were performed using the LightShift Chemiluminescent EMSA kit (Pierce). Briefly, purified protein was incubated with 50 ng/µL of poly(dI-dC) and 10 fmol of biotinylated probes. Competitor DNA was added in 50 or 250 fold molar excess. All reactions were incubated in the kit EMSA buffer with 2.5% glycerol, 5 mM MgCl2, 10 mM EDTA, 50 mM KCl, and 0.05% NP-40 at room temperature for 20 minutes. Electrophoresis and transfer to Nylon membrane (Hybond) was performed according to the manufacturer's instructions. The Chemiluminescent Nucleic Acid Detection Module (Pierce) was used according to the manufacturer's instructions to visualize the probes.
Identification of Potential Target Genes Using PBM Derived Position PWMs
2 kb-long upstream regions (or up to the nearest ORF) were first extracted from whole genome sequences and associated gene annotation data (PlasmoDB 6.0). PBM motifs were trimmed down to their 6 most informative consecutive motif positions (motif cores). Then, we determined all core PBM motif occurrences in the 2 kb regions using the ScanACE approach [91]. G+C content was set to 13.1% in P. falciparum, 42.8% in P. vivax and 20.1% in P. yoelii. Score threshold was set to the average score of randomly drawn sequences from the PBM PWMs, minus two standard deviations (this is the default ScanACE setting).
Refinement of Target Gene Lists
In order to identify candidate target genes for each AP2, we reasoned that these target genes should be co-expressed and should share the AP2 binding site we identified using PBMs. Thus, we first identified groups of genes that are co-expressed across multiple experimental conditions or multiple time points (e.g., in the IDC). Then for each AP2 motif, we determined in which of the groups the motif was over-represented. For each of these groups, we extracted the genes associated with one or more motif occurrences. Thus, target genes in this definition can come from multiple co-expression groups (and not all genes in these co-expression groups end up in the target list because not all genes in these groups will have a motif occurrence in their promoter). In order to define co-expressed gene groups, we used the k-means approach together with the Pearson correlation. Motif scanning was performed using the ScanACE approach [91] as described above. In order to determine functional motif score threshold, we used an information-theoretic procedure analogous to that used in FIRE [27]: briefly, we determined the motif score threshold such that the resulting motif occurrences best explain the co-expression clusters obtained by k-means. At a given motif score threshold, motif over-representation in each cluster was assessed using the hypergeometric distribution; to correct for multiple testing (i.e. multiple clusters being evaluated), we used the Benjamini-Hochberg procedure; corrected p-values corresponding to an estimated overall FDR of 0.25 were considered significant and the genes associated with motif occurrences in these clusters were extracted. Because the k-means is dependent on initialization, we repeated the entire procedure 10 times; genes extracted 3 times or more (out of 10) were considered as candidate target genes. Thus, only genes associated with the considered motif and that are consistently found as co-expressed together with other genes sharing that same motif end up as candidate target genes in our analysis. More detailed methods are available in the Supplemental Text S1.
Accession Numbers
PlasmoDB (www.plasmodb.org) accession numbers for genes and proteins discussed in this publication are: hsp86 (PF07_0029); hsp70 (PF08_0054); msp1 (PFI1475w); msp10 (PFF0995c); rhopH 3 (PFI0265c); gbp130 (PF10_0159); AP2-O (PBANKA_090590); AP2-Sp (PBANKA_132980).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. WHO 2009 World Malaria Report 2009
2. BozdechZ
LlinásM
PulliamBL
WongED
ZhuJ
2003 The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLoS Biol 1 E5
3. FlorensL
WashburnMP
RaineJD
AnthonyRM
GraingerM
2002 A proteomic view of the Plasmodium falciparum life cycle. Nature 419 520 526
4. HallN
KarrasM
RaineJD
CarltonJM
KooijTW
2005 A comprehensive survey of the Plasmodium life cycle by genomic, transcriptomic, and proteomic analyses. Science 307 82 86
5. LasonderE
IshihamaY
AndersenJS
VermuntAM
PainA
2002 Analysis of the Plasmodium falciparum proteome by high-accuracy mass spectrometry. Nature 419 537 542
6. Le RochKG
ZhouY
BlairPL
GraingerM
MochJK
2003 Discovery of gene function by expression profiling of the malaria parasite life cycle. Science 301 1503 1508
7. MikolajczakSA
Silva-RiveraH
PengX
TarunAS
CamargoN
2008 Distinct malaria parasite sporozoites reveal transcriptional changes that cause differential tissue infection competence in the mosquito vector and mammalian host. Mol Cell Biol 28 6196 6207
8. SilvestriniF
BozdechZ
LanfrancottiA
Di GiulioE
BultriniE
2005 Genome-wide identification of genes upregulated at the onset of gametocytogenesis in Plasmodium falciparum. Mol Biochem Parasitol 143 100 110
9. TarunAS
PengX
DumpitRF
OgataY
Silva-RiveraH
2008 A combined transcriptome and proteome survey of malaria parasite liver stages. Proc Natl Acad Sci U S A 105 305 310
10. YoungJA
FivelmanQL
BlairPL
de la VegaP
Le RochKG
2005 The Plasmodium falciparum sexual development transcriptome: a microarray analysis using ontology-based pattern identification. Mol Biochem Parasitol 143 67 79
11. Le RochKG
JohnsonJR
FlorensL
ZhouY
SantrosyanA
2004 Global analysis of transcript and protein levels across the Plasmodium falciparum life cycle. Genome Res 14 2308 2318
12. FothBJ
ZhangN
MokS
PreiserPR
BozdechZ
2008 Quantitative protein expression profiling reveals extensive post-transcriptional regulation and post-translational modifications in schizont-stage malaria parasites. Genome Biol 9 R177
13. CoulsonRM
HallN
OuzounisCA
2004 Comparative genomics of transcriptional control in the human malaria parasite Plasmodium falciparum. Genome Res 14 1548 1554
14. CallebautI
PratK
MeuriceE
MornonJP
TomavoS
2005 Prediction of the general transcription factors associated with RNA polymerase II in Plasmodium falciparum: conserved features and differences relative to other eukaryotes. BMC Genomics 6 100
15. BischoffE
VaqueroC
2010 In silico and biological survey of transcription-associated proteins implicated in the transcriptional machinery during the erythrocytic development of Plasmodium falciparum. BMC Genomics 11 34
16. LanzerM
de BruinD
RavetchJV
1992 A sequence element associated with the Plasmodium falciparum KAHRP gene is the site of developmentally regulated protein-DNA interactions. Nucleic Acids Res 20 3051 3056
17. DecheringKJ
KaanAM
MbachamW
WirthDF
ElingW
1999 Isolation and functional characterization of two distinct sexual-stage-specific promoters of the human malaria parasite Plasmodium falciparum. Mol Cell Biol 19 967 978
18. HorrocksP
LanzerM
1999 Mutational analysis identifies a five base pair cis-acting sequence essential for GBP130 promoter activity in Plasmodium falciparum. Mol Biochem Parasitol 99 77 87
19. OstaM
Gannoun-ZakiL
BonnefoyS
RoyC
VialHJ
2002 A 24 bp cis-acting element essential for the transcriptional activity of Plasmodium falciparum CDP-diacylglycerol synthase gene promoter. Mol Biochem Parasitol 121 87 98
20. PorterME
2002 Positive and negative effects of deletions and mutations within the 5′ flanking sequences of Plasmodium falciparum DNA polymerase delta. Mol Biochem Parasitol 122 9 19
21. ChowCS
WirthDF
2003 Linker scanning mutagenesis of the Plasmodium gallinaceum sexual stage specific gene pgs28 reveals a novel downstream cis-control element. Mol Biochem Parasitol 129 199 208
22. MilitelloKT
DodgeM
BethkeL
WirthDF
2004 Identification of regulatory elements in the Plasmodium falciparum genome. Mol Biochem Parasitol 134 75 88
23. ThamWH
PaynePD
BrownGV
RogersonSJ
2007 Identification of basic transcriptional elements required for rif gene transcription. Int J Parasitol 37 605 615
24. SunilS
ChauhanVS
MalhotraP
2008 Distinct and stage specific nuclear factors regulate the expression of falcipains, Plasmodium falciparum cysteine proteases. BMC Mol Biol 9 47
25. VossTS
KaestliM
VogelD
BoppS
BeckHP
2003 Identification of nuclear proteins that interact differentially with Plasmodium falciparum var gene promoters. Mol Microbiol 48 1593 1607
26. VossTS
TonkinCJ
MartyAJ
ThompsonJK
HealerJ
2007 Alterations in local chromatin environment are involved in silencing and activation of subtelomeric var genes in Plasmodium falciparum. Mol Microbiol 66 139 150
27. ElementoO
SlonimN
TavazoieS
2007 A universal framework for regulatory element discovery across all genomes and data types. Mol Cell 28 337 350
28. JurgelenaiteR
DijkstraTM
KockenCH
HeskesT
2009 Gene regulation in the intraerythrocytic cycle of Plasmodium falciparum. Bioinformatics 25 1484 1491
29. van NoortV
HuynenMA
2006 Combinatorial gene regulation in Plasmodium falciparum. Trends Genet 22 73 78
30. WuJ
SieglaffDH
GervinJ
XieXS
2008 Discovering regulatory motifs in the Plasmodium genome using comparative genomics. Bioinformatics 24 1843 1849
31. YoungJA
JohnsonJR
BennerC
YanSF
ChenK
2008 In silico discovery of transcription regulatory elements in Plasmodium falciparum. BMC Genomics 9 70
32. IengarP
JoshiNV
2009 Identification of putative regulatory motifs in the upstream regions of co-expressed functional groups of genes in Plasmodium falciparum. BMC Genomics 10 18
33. EssienK
StoeckertCJJr
2010 Conservation and divergence of known apicomplexan transcriptional regulons. BMC Genomics 11 147
34. GunasekeraAM
MyrickA
Le RochK
WinzelerE
WirthDF
2007 Plasmodium falciparum: genome wide perturbations in transcript profiles among mixed stage cultures after chloroquine treatment. Exp Parasitol 117 87 92
35. GunasekeraAM
MyrickA
MilitelloKT
SimsJS
DongCK
2007 Regulatory motifs uncovered among gene expression clusters in Plasmodium falciparum. Mol Biochem Parasitol 153 19 30
36. MullapudiN
JosephSJ
KissingerJC
2009 Identification and functional characterization of cis-regulatory elements in the apicomplexan parasite Toxoplasma gondii. Genome Biol 10 R34
37. AravindL
IyerLM
WellemsTE
MillerLH
2003 Plasmodium biology: genomic gleanings. Cell 115 771 785
38. TempletonTJ
IyerLM
AnantharamanV
EnomotoS
AbrahanteJE
2004 Comparative analysis of apicomplexa and genomic diversity in eukaryotes. Genome Res 14 1686 1695
39. BoschetC
GissotM
BriquetS
HamidZ
Claudel-RenardC
2004 Characterization of PfMyb1 transcription factor during erythrocytic development of 3D7 and F12 Plasmodium falciparum clones. Mol Biochem Parasitol 138 159 163
40. GissotM
BriquetS
RefourP
BoschetC
VaqueroC
2005 PfMyb1, a Plasmodium falciparum transcription factor, is required for intra-erythrocytic growth and controls key genes for cell cycle regulation. J Mol Biol 346 29 42
41. BriquetS
BoschetC
GissotM
TissandieE
SevillaE
2006 High-mobility-group box nuclear factors of Plasmodium falciparum. Eukaryot Cell 5 672 682
42. GissotM
TingLM
DalyTM
BergmanLW
SinnisP
2008 High mobility group protein HMGB2 is a critical regulator of Plasmodium oocyst development. J Biol Chem 283 17030 17038
43. BalajiS
BabuMM
IyerLM
AravindL
2005 Discovery of the principal specific transcription factors of Apicomplexa and their implication for the evolution of the AP2-integrase DNA binding domains. Nucleic Acids Res 33 3994 4006
44. JofukuKD
den BoerBG
Van MontaguM
OkamuroJK
1994 Control of Arabidopsis flower and seed development by the homeotic gene APETALA2. Plant Cell 6 1211 1225
45. Ohme-TakagiM
ShinshiH
1995 Ethylene-inducible DNA binding proteins that interact with an ethylene-responsive element. Plant Cell 7 173 182
46. IyerLM
AnantharamanV
WolfMY
AravindL
2008 Comparative genomics of transcription factors and chromatin proteins in parasitic protists and other eukaryotes. Int J Parasitol 38 1 31
47. FinnRD
TateJ
MistryJ
CoggillPC
SammutSJ
2008 The Pfam protein families database. Nucleic Acids Res 36 D281 288
48. De SilvaEK
GehrkeAR
OlszewskiK
LeonI
ChahalJS
2008 Specific DNA-binding by apicomplexan AP2 transcription factors. Proc Natl Acad Sci U S A 105 8393 8398
49. YudaM
IwanagaS
ShigenobuS
KatoT
KanekoI
2010 Transcription Factor AP2-Sp and its Target Genes in Malarial Sporozoites. Mol Microbiol 75 854 863
50. FlueckC
BartfaiR
NeiderwieserI
WitmerK
AlakoBTF
2010 A Major Role for the Plasmodium falciparum ApiAP2 Protein PFSIP2 in Chromosome End Biology. PLoS Pathog 6 e1000784
51. YudaM
IwanagaS
ShigenobuS
MairGR
JanseCJ
2009 Identification of a transcription factor in the mosquito-invasive stage of malaria parasites. Mol Microbiol 71 1402 1414
52. BergerMF
BulykML
2006 Protein binding microarrays (PBMs) for rapid, high-throughput characterization of the sequence specificities of DNA binding proteins. Methods Mol Biol 338 245 260
53. BergerMF
PhilippakisAA
QureshiAM
HeFS
EstepPW3rd
2006 Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat Biotechnol 24 1429 1435
54. BergerMF
BadisG
GehrkeAR
TalukderS
PhilippakisAA
2008 Variation in homeodomain DNA binding revealed by high-resolution analysis of sequence preferences. Cell 133 1266 1276
55. BergerMF
BulykML
2009 Universal protein-binding microarrays for the comprehensive characterization of the DNA-binding specificities of transcription factors. Nat Protoc 4 393 411
56. GarvieCW
WolbergerC
2001 Recognition of specific DNA sequences. Mol Cell 8 937 946
57. BadisG
BergerMF
PhilippakisAA
TalukderS
GehrkeAR
2009 Diversity and complexity in DNA recognition by transcription factors. Science 324 1720 1723
58. ZhouY
YoungJA
SantrosyanA
ChenK
YanSF
2005 In silico gene function prediction using ontology-based pattern identification. Bioinformatics 21 1237 1245
59. RothFP
HughesJD
EstepPW
ChurchGM
1998 Finding DNA regulatory motifs within unaligned noncoding sequences clustered by whole-genome mRNA quantitation. Nat Biotechnol 16 939 945
60. BrickK
WatanabeJ
PizziE
2008 Core promoters are predicted by their distinct physicochemical properties in the genome of Plasmodium falciparum. Genome Biol 9 R178
61. WakaguriH
SuzukiY
SasakiM
SuganoS
WatanabeJ
2009 Inconsistencies of genome annotations in apicomplexan parasites revealed by 5′-end-one-pass and full-length sequences of oligo-capped cDNAs. BMC Genomics 10 312
62. HarbisonCT
GordonDB
LeeTI
RinaldiNJ
MacisaacKD
2004 Transcriptional regulatory code of a eukaryotic genome. Nature 431 99 104
63. PontsN
HarrisEY
PrudhommeJ
WickI
Eckhardt-LudkaC
2010 Nucleosome landscape and control of transcription in the human malaria parasite. Genome Res
64. AurrecoecheaC
BrestelliJ
BrunkBP
DommerJ
FischerS
2009 PlasmoDB: a functional genomic database for malaria parasites. Nucleic Acids Res 37 D539 543
65. PlantaRJ
GoncalvesPM
MagerWH
1995 Global regulators of ribosome biosynthesis in yeast. Biochem Cell Biol 73 825 834
66. FoatBC
TepperRG
BussemakerHJ
2008 TransfactomeDB: a resource for exploring the nucleotide sequence specificity and condition-specific regulatory activity of trans-acting factors. Nucleic Acids Res 36 D125 131
67. Huang daW
ShermanBT
LempickiRA
2009 Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4 44 57
68. HuG
CabreraA
KonoM
MokS
ChaalBK
Transcriptional profiling of growth perturbations of the human malaria parasite Plasmodium falciparum. Nat Biotechnol 28 91 98
69. ScherfA
Lopez-RubioJJ
RiviereL
2008 Antigenic variation in Plasmodium falciparum. Annu Rev Microbiol 62 445 470
70. LavstsenT
SalantiA
JensenAT
ArnotDE
TheanderTG
2003 Sub-grouping of Plasmodium falciparum 3D7 var genes based on sequence analysis of coding and non-coding regions. Malar J 2 27
71. VossTS
HealerJ
MartyAJ
DuffyMF
ThompsonJK
2006 A var gene promoter controls allelic exclusion of virulence genes in Plasmodium falciparum malaria. Nature 439 1004 1008
72. BozdechZ
MokS
HuG
ImwongM
JaideeA
2008 The transcriptome of Plasmodium vivax reveals divergence and diversity of transcriptional regulation in malaria parasites. Proc Natl Acad Sci U S A 105 16290 16295
73. WestenbergerSJ
McCleanCM
ChattopadhyayR
DhariaNV
CarltonJM
2010 A systems-based analysis of Plasmodium vivax lifecycle transcription from human to mosquito. PLoS Negl Trop Dis 4 e653
74. GroveCA
De MasiF
BarrasaMI
NewburgerDE
AlkemaMJ
2009 A multiparameter network reveals extensive divergence between C. elegans bHLH transcription factors. Cell 138 314 327
75. NoyesMB
ChristensenRG
WakabayashiA
StormoGD
BrodskyMH
2008 Analysis of homeodomain specificities allows the family-wide prediction of preferred recognition sites. Cell 133 1277 1289
76. ZhuC
ByersKJ
McCordRP
ShiZ
BergerMF
2009 High-resolution DNA-binding specificity analysis of yeast transcription factors. Genome Res 19 556 566
77. LindnerSE
De SilvaEK
KeckJL
LlinásM
2010 Structural determinants of DNA binding by a P. falciparum ApiAP2 transcriptional regulator. J Mol Biol 395 558 567
78. BougdourA
BraunL
CannellaD
HakimiMA
2010 Chromatin modifications: implications in the regulation of gene expression in Toxoplasma gondii. Cell Microbiol 12 413 423
79. LaCountDJ
VignaliM
ChettierR
PhansalkarA
BellR
2005 A protein interaction network of the malaria parasite Plasmodium falciparum. Nature 438 103 107
80. BornemanAR
GianoulisTA
ZhangZD
YuH
RozowskyJ
2007 Divergence of transcription factor binding sites across related yeast species. Science 317 815 819
81. MosesAM
PollardDA
NixDA
IyerVN
LiXY
2006 Large-scale turnover of functional transcription factor binding sites in Drosophila. PLoS Comput Biol 2 e130
82. OdomDT
DowellRD
JacobsenES
GordonW
DanfordTW
2007 Tissue-specific transcriptional regulation has diverged significantly between human and mouse. Nat Genet 39 730 732
83. SchmidtD
WilsonMD
BallesterB
SchwaliePC
BrownGD
2010 Five-Vertebrate ChIP-seq Reveals the Evolutionary Dynamics of Transcription Factor Binding. Science
84. WilsonMD
Barbosa-MoraisNL
SchmidtD
ConboyCM
VanesL
2008 Species-specific transcription in mice carrying human chromosome 21. Science 322 434 438
85. ElementoO
TavazoieS
2005 Fast and systematic genome-wide discovery of conserved regulatory elements using a non-alignment based approach. Genome Biol 6 R18
86. CohnB
ManqueP
LaraAM
SerranoM
ShethN
2010 Putative cis-Regulatory Elements Associated with Heat Shock Genes Activated During Excystation of Cryptosporidium parvum. PLoS One 5 e9512
87. CarltonJM
AdamsJH
SilvaJC
BidwellSL
LorenziH
2008 Comparative genomics of the neglected human malaria parasite Plasmodium vivax. Nature 455 757 763
88. PfeiferK
PrezantT
GuarenteL
1987 Yeast HAP1 activator binds to two upstream activation sites of different sequence. Cell 49 19 27
89. ColeC
BarberJD
BartonGJ
2008 The Jpred 3 secondary structure prediction server. Nucleic Acids Res 36 W197 201
90. NewburgerDE
BulykML
2009 UniPROBE: an online database of protein binding microarray data on protein-DNA interactions. Nucleic Acids Res 37 D77 82
91. HughesJD
EstepPW
TavazoieS
ChurchGM
2000 Computational identification of cis-regulatory elements associated with groups of functionally related genes in Saccharomyces cerevisiae. J Mol Biol 296 1205 1214
92. WorkmanCT
YinY
CorcoranDL
IdekerT
StormoGD
2005 enoLOGOS: a versatile web tool for energy normalized sequence logos. Nucleic Acids Res 33 W389 392
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 10
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Retroviral RNA Dimerization and Packaging: The What, How, When, Where, and Why
- Viral Replication Rate Regulates Clinical Outcome and CD8 T Cell Responses during Highly Pathogenic H5N1 Influenza Virus Infection in Mice
- Antimicrobial Peptides: Primeval Molecules or Future Drugs?
- Crystal Structure of DotD: Insights into the Relationship between Type IVB and Type II/III Secretion Systems
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy