
Gene Annotation and Drug Target Discovery in with a Tagged Transposon Mutant Collection
Candida albicans is the most common human fungal pathogen, causing infections that can be lethal in immunocompromised patients. Although Saccharomyces cerevisiae has been used as a model for C. albicans, it lacks C. albicans' diverse morphogenic forms and is primarily non-pathogenic. Comprehensive genetic analyses that have been instrumental for determining gene function in S. cerevisiae are hampered in C. albicans, due in part to limited resources to systematically assay phenotypes of loss-of-function alleles. Here, we constructed and screened a library of 3633 tagged heterozygous transposon disruption mutants, using them in a competitive growth assay to examine nutrient - and drug-dependent haploinsufficiency. We identified 269 genes that were haploinsufficient in four growth conditions, the majority of which were condition-specific. These screens identified two new genes necessary for filamentous growth as well as ten genes that function in essential processes. We also screened 57 chemically diverse compounds that more potently inhibited growth of C. albicans versus S. cerevisiae. For four of these compounds, we examined the genetic basis of this differential inhibition. Notably, Sec7p was identified as the target of brefeldin A in C. albicans screens, while S. cerevisiae screens with this compound failed to identify this target. We also uncovered a new C. albicans-specific target, Tfp1p, for the synthetic compound 0136-0228. These results highlight the value of haploinsufficiency screens directly in this pathogen for gene annotation and drug target identification.
Published in the journal:
. PLoS Pathog 6(10): e32767. doi:10.1371/journal.ppat.1001140
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1001140
Summary
Candida albicans is the most common human fungal pathogen, causing infections that can be lethal in immunocompromised patients. Although Saccharomyces cerevisiae has been used as a model for C. albicans, it lacks C. albicans' diverse morphogenic forms and is primarily non-pathogenic. Comprehensive genetic analyses that have been instrumental for determining gene function in S. cerevisiae are hampered in C. albicans, due in part to limited resources to systematically assay phenotypes of loss-of-function alleles. Here, we constructed and screened a library of 3633 tagged heterozygous transposon disruption mutants, using them in a competitive growth assay to examine nutrient - and drug-dependent haploinsufficiency. We identified 269 genes that were haploinsufficient in four growth conditions, the majority of which were condition-specific. These screens identified two new genes necessary for filamentous growth as well as ten genes that function in essential processes. We also screened 57 chemically diverse compounds that more potently inhibited growth of C. albicans versus S. cerevisiae. For four of these compounds, we examined the genetic basis of this differential inhibition. Notably, Sec7p was identified as the target of brefeldin A in C. albicans screens, while S. cerevisiae screens with this compound failed to identify this target. We also uncovered a new C. albicans-specific target, Tfp1p, for the synthetic compound 0136-0228. These results highlight the value of haploinsufficiency screens directly in this pathogen for gene annotation and drug target identification.
Introduction
Fungal species of the genus Candida generally live communally on and in the human body, yet Candida infections can become systemic and lethal in up to 60% of immunocompromised patients [1], [2]. C. albicans alone accounts for over 50% of all fungal infections [3]. Furthermore, drug resistance to current therapies is becoming increasingly prevalent [4], motivating research efforts to understand the genetic basis of C. albicans' pathogenesis and uncover novel therapeutic targets.
The etiology of C. albicans is particularly complex, and our understanding of this pathogen lags with respect to the model organism Saccharomyces cerevisiae, which is often used as a proxy for Candida species despite the fact that S. cerevisiae diverged from C. albicans between 150–800 million years ago [5], [6]. Notably, S. cerevisiae is rarely pathogenic [7] and lacks the multiple morphogenic forms that define C. albicans pathogenicity. C. albicans also exists as an obligate diploid and lacks a traditional meiotic cycle [8]; as a result, many of the genetic tools developed in S. cerevisiae are not easily applied. Finally, only 58% of predicted C. albicans proteins share an ortholog with S. cerevisiae [9], [10], underscoring the need for direct study of C. albicans.
An effective approach for such analyses would be to extend high-throughput methodologies developed in S. cerevisiae to C. albicans. For example, experimental multiplexing, in which a genome-wide collection of deletion mutants is pooled and grown competitively to determine the fitness of each mutant in an experimental condition, has been particularly effective in S. cerevisiae (reviewed in [11] and [12]). Strain tracking and quantitation is enabled by the presence of unique DNA sequences, or tags, introduced during the construction of each deletion mutant [13]. To measure strain abundances in pooled growth experiments, these strain-specific tags can be amplified and hybridized to a microarray containing the tag complements, or sequenced directly [14], [15]. The S. cerevisiae deletion collection and pooled phenotypic profiling have been invaluable for examining gene function [14], genetic interactions [16], and for the identification of drug targets and their mechanism of action [17], [18], [19], [20].
Strategies for large-scale mutant screening in C. albicans include two studies using transposon mutagenesis to rapidly generate large numbers of mutants [21], [22], and a third study using targeted deletions combined with a regulatable promoter [23]. While each has been used to uncover novel biological insights, several factors have limited their widespread utility to the C. albicans research community. For instance, mutants in one heterozygous disruption collection [22] are largely unsequenced, making predictions regarding its coverage difficult. The homozygous transposon disruption collection [21], although sequenced and archived as individual mutants, is unavailable as an entire collection and its strains are not tagged; thus experiments must be conducted individually. A proprietary tagged deletion collection has been used to identify novel antifungal targets [23], [24], [25], [26], but the composition of this collection is limited to 2868 strains that share homology to genes essential in S. cerevisiae, other fungi, and higher eukaryotes. Accordingly, a majority of C. albicans-specific genes are not interrogated.
Our goal was to create an unbiased, open-access collection of tagged C. albicans mutants useful for high-throughput experimental multiplexing. We previously reported the creation of a pilot pool of 1290 tagged C. albicans mutants, using universal “TagModules” to label transposons that were subsequently used to simultaneously generate mutants and integrate a pair of DNA tags at the insertion site [27]. Here, we describe the creation and validation of a genome-wide C. albicans tagged transposon mutant collection, using TagModule-based transposon mutagenesis to generate 4252 mutant strains, 3633 of which were detectable by microarray in a pooled growth assay.
To demonstrate the utility of this collection, we investigated nutrient-specific and drug-induced haploinsufficiency. By screening four different media conditions and 57 primarily synthetic inhibitory compounds, we 1) identified genes functioning in core or essential processes, 2) uncovered genes specific to growth in a particular nutrient condition, and 3) demonstrated the utility of this collection in antifungal screening to determine mechanism of action of inhibitory compounds. This collection represents a public, archived resource of tagged C. albicans mutants that can be used to examine gene function either individually or multiplexed in a pool.
Results
Construction and validation of a tagged C. albicans collection
To circumvent the resource-intensive approach of generating gene-specific deletion cassettes to knock out gene function, we used tagged transposon mutagenesis to generate mutants. To incorporate tags into the transposon, we used 4280 Gateway-compatible TagModules developed in a previous study as a source of sequence-verified tags [27]. Each TagModule contains a pair of DNA tags, an uptag and a downtag, flanked by common priming sites for amplification of the tags to determine strain abundance. These TagModules were transferred to a Gateway-compatible transposon modified to contain the UAU1 selectable marker, which allows heterozygous (Arg+) as well as homozygous disruption mutants (Arg+, Ura+) to be generated [28]. The design of the tagged transposons is shown in Figure 1A.
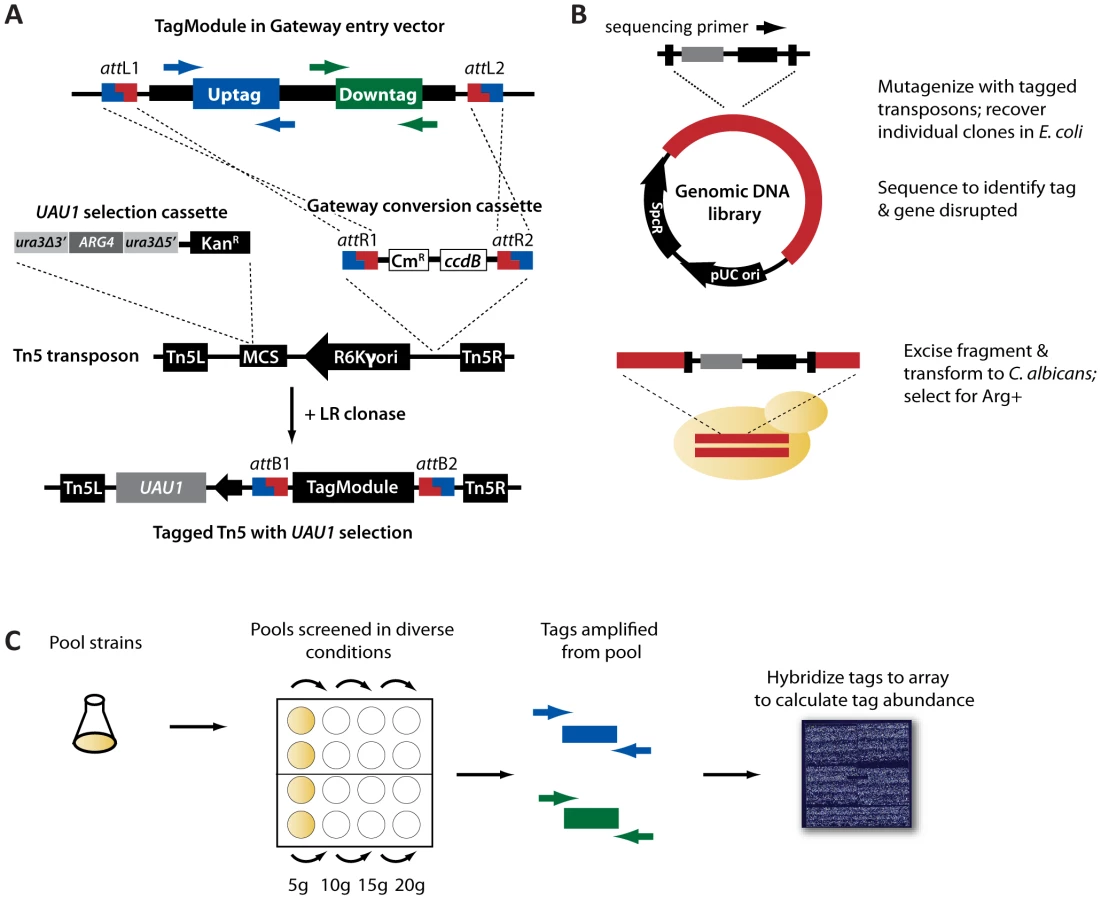
Following the approach of previous transposon mutagenesis studies [21], [22], these tagged transposons were used to mutagenize a C. albicans genomic library in vitro. Following in vitro mutagenesis, unique insertions were recovered in E. coli and sequenced individually to determine the disrupted gene and its linked TagModule (Figure 1B). We recovered a total of 21,468 usable insertion events (see Methods S1 for criteria) representing 4827 unique genes (∼78% of predicted open reading frames). 4239 of these were successfully transformed (for criteria, see Methods) into C. albicans via homologous recombination to create a uniquely tagged, heterozygous disruption mutant (Table S2).
To examine the quality of our collection, we pooled the 4239 strains, amplified their tags, and hybridized them to an Affymetrix TAG4 array (Figure 1C). At a zero timepoint (i.e., with no competitive outgrowth), 3633 strains were detected (tag intensity above 3X median background), with 619 strains falling below background (Figure 2A). Failure to detect strains may result from either a failure of the transformant to re-grow, or low abundance prior to pooling. If the latter, these strains could assayed as a separate subpool, or alternatively, detected using high-throughput sequencing, which provides increased sensitivity [15]. After twenty generations of growth in a pooled assay, biological replicates were highly correlated (Figure 2B; R = 0.98, p<10−16) and strains showed low cross-reactivity with other features on the array; the vast majority (12292/12686, or 97%) of unused tags on the array had signal intensity below our cutoff of 3X background (Figure S3). Thus, our tagged transposon mutants have robust and reproducible performance in a pooled format.

Nutrient-dependent haploinsufficiency profiling of tagged mutants
Genes that have a growth defect when reduced in copy number from two to one (termed haploinsufficiency) are of interest for their potential as drug targets [12]. We therefore sought to use the pooled growth assay to define genome-wide haploinsufficiency in C. albicans, particularly for C. albicans-specific processes and those required for growth in hyphae-inducing conditions, a determinant of virulence. We screened the C. albicans pool in four different nutrient conditions at 30°C: i) rich YPD media, a standard laboratory growth condition, ii) a synthetic media used for the selection of transformants (SC), iii) minimal media, which has been used to induce formation of pseudohyphae and consists of 2% glucose and yeast nitrogen base (YNB), and iv) a low-nitrogen minimal media (synthetic low ammonium dextrose, or SLAD), which can induce pseudohyphal/hyphal growth in C. albicans.
To assess the effect of each gene disruption on growth in these conditions, we tracked tag abundance for each of the detectable 3633 strains, assaying after five, ten, fifteen, and twenty generations of growth with biological replicates. Following calculation of tag abundance by hybridization of amplified tags to a microarray, we fitted a linear regression to each strain's abundance over a timecourse and used the slope of the regression to measure strain sensitivity. Based on Deutschbauer et al. [29], we defined a strain as having a growth defect if its slope was <0, p<0.05 (Table S3).
We found that regardless of media condition, similar proportions of strains were haploinsufficient (Figure 3A). Overall, 145 (4%) strains were haploinsufficient in rich YPD, 105 (2.9%) in SC, 97 (2.7%) in YNB, and 140 (3.9%) in the low-nitrogen SLAD, representing 269 (7.4%) unique genes. Only 9% (25) of these genes were haploinsufficient in all conditions; the majority (55%) of these 269 genes were haploinsufficient in a single condition, suggestive of condition-dependent haploinsufficiency (Figure 3B). This observation suggests that a substantial portion of the C. albicans genome may be amenable to haploinsufficiency profiling under certain conditions.

Finally, comparing our haploinsufficiency profiling data in rich media to that of S. cerevisiae [29], we found that while the overall proportion of haploinsufficient strains was similar, we found a number of biological differences (Figure 3C). 48/145 (33%) of C. albicans haploinsufficient genes in YPD had no S. cerevisiae ortholog. Of those C. albicans genes that did have an ortholog, 26 are orthologous to S. cerevisiae genes that were essential or haploinsufficient in YPD, and an additional 22 orthologs exhibited a growth defect as a homozygous deletion in YPD. 49 (34%) orthologs had no phenotype in YPD. Of the 269 haploinsufficient genes in C. albicans, 97 (36%) had no S. cerevisiae ortholog. A second striking difference between the S. cerevisiae and C. albicans haploinsufficient gene sets is the number of transcription factors (TFs) haploinsufficient in C. albicans. Few transcription factors (excluding general regulatory factors) were haploinsufficient in S. cerevisiae [29]. In contrast, we identified seven TFs as haploinsufficient in YPD (ZCF9, HAC1, LYS142, IRO1, SUA71, BDF1, RBF1), and a total of 13 in the complete haploinsufficient dataset (FGR17, SEF1, SUA72, orf19.6623, orf19.5368, MSN4). Their functions range from iron utilization to regulators of filamentous growth or stress response. These findings highlight the need for direct study of C. albicans, as our assay uncovers novel phenotypes for orthologs as well as a large number of C. albicans-specific genes.
To search for functional enrichment among the 25 genes haploinsufficient in all conditions (Table S4), we used the Gene Ontology Term Finder from the Candida Genome Database (http://www.candidagenome.org/cgi-bin/GO/goTermFinder). These genes were significantly enriched for the GO processes “translation” and GO functions “structural constituent of ribosome” and “structural molecule activity” (Table 1). We also observed genes in this set that function in “core” cellular processes that are likely to be either essential or necessary for growth regardless of condition; for example, POL2 (DNA polymerase epsilon) and orf19.736, whose S. cerevisiae ortholog Sc-SRB8 is a subunit of RNA polymerase II required for transcriptional regulation [30]. We also identified a putative permease (orf19.3293), which may play a role in nutrient sensing, two genes whose products protect against oxidative stress (SOD3 and orf19.5553, based on S. cerevisiae orthology), and a putative 5′-monophosphate 5′-nucleotidase, ISN1, which is a fungal-specific gene whose S. cerevisiae ortholog has been implicated in nucleotide scavenging [31]. Interestingly, the S. cerevisiae orthologs for these four genes are not haploinsufficient, suggesting that S. cerevisiae and C. albicans have diverged with respect to the relative importance of these genes for survival, or that these genes are required at greater than heterozygote gene doses in C. albicans.

Because the 25 genes that were haploinsufficient in all conditions (“4 or more” subset) were enriched for genes involved in fundamental cellular processes, we propose that the unverified genes in this set (those which lack annotation, or those whose annotation has not been confirmed experimentally) are also involved in core or essential processes. As the GO enrichments for genes haploinsufficient in 3 or more conditions (Table S5) were similar to the 4 or more subset (Table 1), we expanded our list of “core” genes to include these genes as well, for a total of 70 genes. Consistent with our observations in the 4 or more subset, this 3 or more group included additional permeases (MUP1 and orf19.5826) and two genes with predicted involvement in oxidative stress response (TRR1 and POS5).
Assigning function to the core haploinsufficient set by complementation
To ask if we could assign functions to this unverified “core” gene set, we selected 17 genes whose S. cerevisiae orthologs (or if an ortholog was not found, the best BLAST hit was used) are essential, so we could assess function by complementation testing in S. cerevisiae. Although an imperfect test for C. albicans function (and susceptible to false negatives), positive results strongly suggest functional similarity.
We used two approaches to test for complementation. First, we cloned these 17 C. albicans ORFs into a CEN/ARS vector with a constitutive promoter and transformed them into the corresponding S. cerevisiae heterozygous mutants (YKO). Sporulation of the heterozygous deletion strain followed by selection for a haploid knockout should yield no growth unless complemented by the plasmid-borne C. albicans gene. To generate homozygous null S. cerevisiae knockouts, we used the Magic Marker strains [32], which use sequential selections to generate haploid deletion mutants. 6/15 C. albicans ORFs with available corresponding Magic Marker strains showed complementation, as did 9/13 S. cerevisiae ORFs tested as a control. Complementation was assessed based on a significant increase in the number of colonies on the overexpression plate versus the vector-only plate (Figure 4A, Figure S5).

Second, we confirmed all negative Magic Marker results by tetrad dissection (Figure 4B, Figure S5) and found that a total of 10/17 C. albicans ORFs (two additional YKOs were available for this test) and 13/13 S. cerevisiae ORFs complemented their corresponding deletion allele. In instances in which we observed complementation, segregation of spore viability improved to 3∶1 or 4∶0 (from 2∶2 in controls), indicating rescue of one or both of the haploid null spores. 3∶1 segregants likely result from incomplete segregation of the CEN/ARS-based plasmid, which generally exists in one or more copies per cell, to all four spores. Interestingly, we also observed two small-size spores in the case orf19.7615/Sc-TRS31, indicating partial complementation. Based on these results, we conclude that 10/17 C. albicans ORFs have the same (or very similar) cellular roles as their S. cerevisiae ortholog and propose the following changes to the description of these ORFs: a change of “Feature Type” from “uncharacterized” to “verified”, and a transfer of function from the S. cerevisiae description (summarized in Table 2).

Confirmation of haploinsufficient phenotypes
Finally, to test whether this group of “core” genes tested via complementation also represents essential C. albicans genes, we obtained GRACE strains [33] for 12/17 of the “core” group genes. Testing the GRACE strains, which are conditional heterozygous knockouts that can be converted (functionally) into homozygous knockout mutants by repression of the second allele with doxycycline, served two purposes: first, to determine whether these strains are viable as homozygotes, and second, to validate that the phenotypes of our mutants represent true positives by validating growth defects in an alternative strain background. This is particularly relevant because it is possible that synthetic effects with BWP17′s uracil or histidine auxotrophies could contribute to the observed growth phenotypes.
We examined the growth of these 12 GRACE strains as well as our 17 transposon-derived mutants in selective media. Overall, after 15–20 generations of growth, the majority of our transposon mutants recapitulated a growth defect as observed in the pooled growth assay (Figure S4A). While the growth phenotypes of the GRACE mutants were generally less severe (Figure S4B), we also observed haploinsufficiency in 11/12 of these strains (defined as <98% of BWP17′s growth rate using the metric AvgG [34]). The difference in the degree of haploinsufficiency could be the result of either a “leakiness” in the tetracycline-regulated promoter of the GRACE strains [33] causing production of additional gene product and thereby alleviating the growth phenotype, or could reflect some contribution of synthetic interactions with the histidine and uridine auxotrophies mentioned above.
When the GRACE strains were grown in the presence of 100 µM doxycycline, 10/12 (FOL3 and TIF34 GRACE mutants excepted) showed a severe to complete growth defect in all media types, suggesting that they are essential in C. albicans (Figure S4C). The fact that two strains were not essential under the GRACE test but were able to complement their essential S. cerevisiae ortholog could be the result of promoter leakiness, or could reflect that these genes, while capable of performing a similar function as their S. cerevisiae orthologs, are not essential in C. albicans.
Analysis of condition-dependent haploinsufficiency
We noted that haploinsufficiency in C. albicans is primarily condition-dependent; 55% of the 269 haploinsufficient genes had a growth defect in a single condition, and 19% of the 269 were identified in only two conditions. Examining these genes, we found two general categories. The first category, identified in rich media, includes genes involved in oxidative metabolism, such as COX2, NAD5, ABC1, KGD2, and (by annotation transfer from S. cerevisiae orthologs) orf19.4204/Sc-PET123. We also observed haploinsufficiency in the C. albicans-specific alternative oxidases AOX1 and AOX2 (GO:0016682, n = 2, 1.4% vs 0% in the genome; hypergeometric p = 0.046), which are thought to function in maintaining turnover of the TCA cycle, relieving saturation in oxidative metabolism [35].
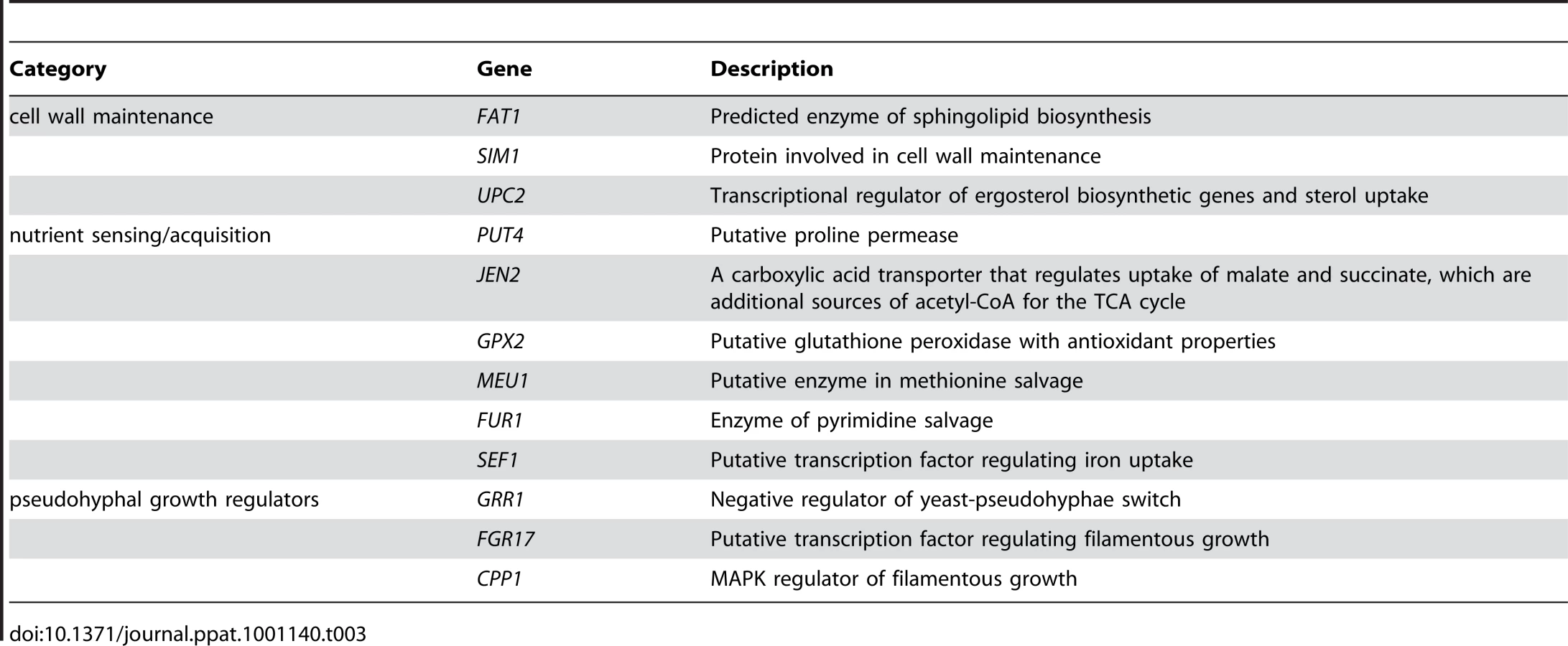
In reduced nutrient conditions, we observed a class of haploinsufficient genes related to growth in low nitrogen. Genes in this category, outlined in Table 3, were involved in 1) cell wall maintenance (FAT1, SIM1, UPC2), 2) nutrient sensing/acquisition (PUT4, JEN2, GPX2, MEU1, FUR1, SEF1), and 3) pseudohyphal growth regulators (GRR1, FGR17, CPP1). orf19.1617, an uncategorized gene in both C. albicans and S. cerevisiae (Sc-YDR282C), has also been shown to have a filamentation defect [22]. Because these categories encompass the roles of nutrient scavenging, initiation of filamentation, and cell wall remodeling necessary to produce hyphae, we asked if filamentation may be one mechanism by which C. albicans thrives in low-nutrient conditions.
We investigated if these defects in growth rate correlate with defects in filamentation by testing if the 77 genes necessary for growth in the nutrient-limiting conditions (Table S6) manifested a filamentous defect. Only 3/77 (BET2, SHE3, and RPS18) mutants had a distinct filamentous defect on Spider agar (Figure 5), one condition that elicits a filamentous phenotype. SHE3 and RPS18 had a completely smooth appearance with no peripheral filaments compared to wild-type. BET2 had a smooth appearance with some peripheral filaments. SHE3 has previously been shown to be necessary for filamentous growth on Spider media [36]. The other two genes (RPS18, a putative ribosomal protein, and BET2) have not previously been implicated in a filamentous phenotype. BET2′s S. cerevisiae ortholog functions in vesicle transport, and in C. albicans BET2 is regulated by Mig1p, a transcriptional repressor that regulates carbon source utilization [37] and is downregulated in hyphal growth [38] but upregulated in biofilms [39]. While we have identified two new genes likely involved in filamentous growth, our results suggest that genes required for growth in low-nutrient conditions, as identified in our SLAD-media screens, are, in general, different from those required for filamentation in Spider agar.

We next performed a pooled screen on solid SLAD media. Briefly, ∼500,000 colonies/pool was plated onto solid SLAD agar plates and grown for 6 days at 37°C. By quantifying cells that had invaded the agarose (pellet) and comparing them to the proportions of cells that had not invaded the agar (supernatant), we were able to determine if particular mutants were defective in invasion. At a cutoff of 2-fold greater abundance in non-invaded fraction (log2(supernatant/pellet) >2), we observed 341 mutants that were invasion-defective (Table S8). As we previously observed, there was little overlap (5/77) between strains that were necessary for full growth in low nitrogen and those that had a defect in agar invasion. However, when we compared the 243 S. cerevisiae orthologs/best hits corresponding to these 341 mutants to invasion-defective mutants of the pseudohyphal S. cerevisiae Sigma 1278B strain (O. Ryan, personal communication), we observed a 26% overlap (62/243), underscoring 1) the ability of our pooled mutants to identify biologically consistent results, and 2) the flexibility of the pooled assay to a solid-media format. Additional experiments on solid media will provide a more detailed genome-wide perspective on this key aspect of C. albicans physiology.
Drug-induced haploinsufficiency profiling
Haploinsufficiency profiling was developed in S. cerevisiae to identify drugs that target gene products essential for growth, based on the premise that lowering copy number of the target gene sensitizes the corresponding heterozygous deletion strain to the drug [40]. To select compounds for screening against the tagged C. albicans mutants, we focused on those that inhibited C. albicans growth more potently than S. cerevisiae. We reasoned that these compounds would be more likely to have a different mechanism of action in the two yeasts, e.g., by having distinct targets, different mechanisms of influx/efflux, or different off-target effects. We screened 1521 compounds (previously titrated to a concentration that inhibits S. cerevisiae by ∼10%, or IC10) against wild-type S. cerevisiae and wild-type C. albicans and measured the ratio of compound-treated growth to that of a control. While we found that the majority of compounds inhibited C. albicans growth at levels similar to S. cerevisiae's, a number of compounds inhibited C. albicans more severely (Figure 6A). For example, the top 40 compounds (highlighted in red) produced 20–90% greater inhibition in C. albicans.

From this screen, we selected for titration and pooled growth screening 67 readily available compounds that inhibited C. albicans more strongly than S. cerevisiae in our initial screen (see Methods for criteria). Comparing the dose response of these compounds for S. cerevisiae and C. albicans, we observed that the compounds that conferred differential growth inhibition fell into two classes of dose response curves (representative curves are shown in Figure 6B, Figure S6). The first class included those with parallel dose response curves, suggestive of a compound that has the same target in both yeasts but different cell permeability or residence time. A second category included those in which the dose-response curves had different slopes, suggestive of a different mechanism of action or distinct secondary effects. Overall, these results from screening a library of 1521 compounds against wild-type C. albicans and S. cerevisiae suggest that certain compounds may have distinct mechanism of action (e.g., a combination of primary and secondary activities) in C. albicans.
To study the genetic basis of the differential action of certain compounds in C. albicans and S. cerevisiae, we screened 57 of the 67 compounds at approximately an IC20 in the pooled growth assay (chemical structures are shown in Figure S7; to demonstrate that these 57 represented chemically diverse compounds, the distribution of their pair-wise similarities, based on Tanimoto scores, is shown in Figure S8). We performed a 20-generation endpoint assay (by analogy to the S. cerevisiae haploinsufficiency profiling) in which the abundance of each strain in a compound-treated pool was compared to its abundance in a set of DMSO-treated controls. To identify drug-induced haploinsufficiency, a normalized z-score was used to examine the response of each strain to a compound, comparing the performance of a strain (proxied by tag intensity) in the compound treatment to its performance in the no-drug controls (Table S7). Positive z-scores indicate increasing sensitivity to the treatment condition; strains with a high z-score were significantly affected by the drug treatment and may be depleted of genes that encode a cellular target. Of the 57 screens, we focused on 25 that had a small number of significantly sensitive outliers, those most likely to be representative of compounds that act through a single or small number of targets. The remaining 32 compounds exerted either widespread or few fitness defects in the pool. An overview of the z-scores for these 25 compounds is shown in Figure 7A.

We found that the GTP exchange factor SEC7, which activates formation of transport vesicles, was the most sensitive strain in screens with two compounds, brefeldin A and nigericin (Figure 7B). In addition, analysis of the 30 most sensitive strains from the nigericin screen in C. albicans showed GO enrichment of a number of vesicle transport-related processes (e.g., Golgi vesicle transport (GO:0048193), 20.7% (n = 6) versus 2.4% in the genome, hypergeometric p<0.008). As nigericin affects ion gradients across lipid membranes (as opposed to having a direct protein target [41]), we speculate that changes in membrane permeability, and by extension intracompartmental pH (see below), interact synthetically with defects in vesicle transport to cause a fitness defect. Brefeldin A, whose protein target is widely considered to be Sec7p [23] had a less pronounced effect on vesicle transport; brefeldin A-sensitive strains were GO-enriched for function “GTPase regulator activity” (GO:0005085, 6.7% (n = 2) versus 0.4% in the genome, hypergeometric p<0.07). Interestingly, neither compound induced SEC7 haploinsufficiency in S. cerevisiae (Figure 7B), even though brefeldin A has been shown to inactivate S. cerevisiae Sec7p complexes in vitro [42]. These results 1) suggest that S. cerevisiae has additional targets that become rate-limiting for growth prior to the effects of inhibition of SEC7, and 2) confirm the utility of our pool for validating C. albicans-specific targets.
Two synthetic, uncharacterized compounds, 1187–1561 and 0136–0228, also inhibited vesicle transport-related functions in C. albicans (Figure 8A). We confirmed sensitivity of these strains by growth in individual culture (Figure 8B). The most sensitive strain in a screen with 1187–1561 was orf19.2411::Tn5/orf19.2411. The S. cerevisiae ortholog Sc-SYN8 (non-essential in S. cerevisiae) is a SNARE protein that functions in vesicle fusion [43]. 0136–0228 induced a strong growth defect in a tfp1 mutant, which has no ortholog in S. cerevisiae, but is computationally predicted to encode a V-type ATPase that regulates intracompartmental pH. Because its best BLAST hit is Sc-TFP1 and because these two genes share a fungal orthogroup [44], we surmise that they also share function. Consistent with an effect on intracompartmental pH, many of the mutants most sensitive to this compound are also sensitive to nigericin (vesicle transport related ORFs: TFP1, BTS1, orf19.2078, orf19.880, orf19.6558, and others: orf19.9, FAA21, SPT5, and orf19.6435). As noted above, it is likely that the genes disrupted in nigericin-sensitive strains are interacting synthetically with defects caused by altered intracompartmental pH.

We then performed a genetic test to see if 0316–0028 may interact with Tfp1p. We overexpressed C. albicans Tfp1p in a wild-type S. cerevisiae BY4743 and an Sc-Δtfp1 homozygous null mutant background. Because we saw no effect with wild-type S. cerevisiae (Figure 8C), we also tested an Δerg6 null mutant to account for the possibility that the compound was not penetrating the cell. Deletion of ERG6 has been shown to increase compound penetration due to defects in the cell membrane and has been used to sensitize S. cerevisiae to brefeldin A [42]. First, we observed that Tfp1p overexpression ameliorated the growth defect of the Δtfp1 null mutant (and interestingly, also the Δerg6 null mutant) even in the absence of drug. This is consistent with results that show that Sc-ERG6 and Sc-TFP1 interact synthetically [45]. Second, when strong growth inhibition was applied with 0136–0228, we observed partial rescue of the growth defect in both the Δtfp1 and Δerg6 backgrounds, most distinctly in the drug-sensitized Δerg6 background (Figure 8C). That we observed rescue in a heterologous system is strong evidence that this protein is a functional target of this compound, and the fact that 0136–0228 also inhibits the Δtfp1 null suggests that it has additional targets in the cell. We thus propose that Tfp1p is a principal protein target of 0136–0228, and that the other sensitive strains in its chemogenomic profile appear as a secondary consequence of altered intracompartmental pH (see Discussion). Haploinsufficiency profiling in S. cerevisiae with these compounds would have failed to reveal these gene-compound interactions (Figure 8A).
Discussion
Experimental multiplexing using DNA tags was one of the essential attributes of the pioneering S. cerevisiae deletion collection, enabling high-throughput genome annotation, genetic analysis, and antifungal discovery [14], [17], [46]. A publically available, archived, tagged mutant collection for C. albicans has the potential to similarly accelerate research and drug discovery in an organism directly relevant to public health. Using tagged transposon mutagenesis, we constructed a tagged C. albicans mutant collection that is fully sequenced identified and archived as individual mutants. We note that our collection has some caveats. First, our mutants were created in the –Arg –Ura –His strain BWP17 and so remain auxotrophic for uracil and histidine. Synthetic effects with these auxotrophies could produce false positives in some screens and follow-up assays. In such cases, complementation of HIS1 and URA3, or validation of the phenotype in an alternative strain background can verify the phenotype. Our results sampling for haploinsufficiency in an alternatively constructed strain both supports that our results in BWP17-derived strains represent true positives and also suggests that there are subtle but detectable synthetic effects contributed by these auxotrophies. A second issue is that because these strains are transposon mutants, they are not likely to represent complete loss of function alleles. This latter case has the advantage that multiple insertion events with different degrees of functional disruption can be interrogated for a particular gene. The other advantage is that this approach is scalable. We report the creation of 4239 uniquely tagged gene disruptions, representing 68% of 6197 predicted ORFs. Additional mutagenesis can be used to create additional mutants, or, given their compatibility, the TagModules can be integrated into deletion cassettes to create the remaining mutants via homologous recombination.
There are many potential applications of a C. albicans collection; here we investigated haploinsufficiency, the phenomenon in which a single gene copy in a diploid organism results in a fitness defect. We applied the tagged mutant collection in a competitive growth assay to identify haploinsufficient genes in four different nutrient conditions, identifying 269 haploinsufficient genes across four media conditions. This dataset represents a resource for further study of their involvement in growth, morphogenesis, and potential druggable targets. We found that C. albicans has a unique profile of haploinsufficient genes, highlighting the importance of niche (or C. albicans)-specific processes for maintaining wild-type fitness. For example, C. albicans relies more heavily on oxidative metabolism, nutrient sensing (e.g., permeases and nutrient scavenging mechanisms), and resistance to oxidative stress for optimal growth. Consistent with this observation, the host immune response via neutrophils and macrophages involve superoxide production to kill C. albicans [47], suggesting that these protective mechanisms may be necessary for full growth. Moreover, dependence on oxidative metabolism is consistent with a requirement for efficient energy production for rapid growth. Because it has evolved within a human host, C. albicans may rely more heavily on oxidative metabolism, because carbon sources in the form of fat or proteins may be more accessible than simple sugars. Metabolites of both fat and proteins are shunted into the tricarboxylic acid (TCA) cycle as acetyl-CoA for subsequent breakdown in oxidative phosphorylation. Consistently, we found a predicted fatty acyl-coA synthetase (FAA21) haploinsufficient in 3 conditions. Targeting C. albicans-specific metabolic processes may be a useful approach for identifying novel antifungals.
We identified several transcription factors as haploinsufficient in C. albicans, a notable distinction from S. cerevisiae, suggesting that transcriptional regulation may be less flexible in C. albicans with heterozygous alleles manifesting haploinsufficiency. One possible explanation for this observation is that C. albicans is less tolerant of changes in gene dosage as it generally exists in a diploid state. In contrast, S. cerevisiae exists as both a haploid and a diploid, and so these changes in dosage can be tolerated without a reduction in fitness. A second possibility is that because C. albicans is an obligate diploid that lacks a traditional meiotic cycle, two alleles of a transcription factor have diverged such that they are no longer functionally equivalent. This is supported by extensive allelic heterozygosity observed during assembly of the C. albicans genomic sequence [48]. Functional allelic variation has also been observed in C. albicans in a number of small-scale studies, for example in drug pumps [49], or for HWP1, in which the two alleles are differentially expressed under biofilm conditions [50].
We next used our dataset to identify which genes function in core cellular processes, and which are haploinsufficient in a specific nutrient condition. Selecting from candidates generated through genome-wide screens, we followed up on a subset of 17 genes from a “core” set of 70 haploinsufficient genes using complementation testing and individual strain growth analysis. Although complementation testing in S. cerevisiae is an imperfect test of function, it is useful for generating hypotheses that can be used to infer function for the remaining uncharacterized genes in the “core” dataset of putative essential genes. We found that 10 of these 17 genes were able to complement their essential S. cerevisiae ortholog, strongly suggestive of functional similarity for these conserved processes. For those genes that did not complement, it is possible that their phenotypes arose from the influence of strain auxotrophies. Alternatively, failure to complement can also result from result of alternative codon usage in C. albicans. Interestingly, we found that both complementing and non-complementing genes contained the alternative CUG codon (Table 2)).
We also identified a set of genes necessary for growth in nutrient-limiting conditions, and found that while necessary for growth in limited nutrients, these genes were generally not necessary for filamentation. As filamentation in fungi is a well-documented response to low-nitrogen conditions (presumably an adaptation to improve nutrient acquisition) [51], we anticipated that filamentation might play a role in growth in nutrient-limited conditions. Contrary to this expectation, our results suggest a disconnect between growth rate (which may require optimal nutrient utilization) and filamentation (which may require specific nutrient sensing) under the conditions that we tested. From a biological perspective, filamentation may be the preferred lifestyle for tissue invasion or macrophage evasion in which the ability to grow rapidly is less important. C. albicans cells that are unable to filament in vivo are avirulent, and null mutants of EFG1, a transcriptional regulator of filamentation, have normal growth [52]. However, whether all mutants that have a filamentous defect display wild-type growth has not been systematically determined. From the standpoint of developing new treatment strategies, identifying both fungicides, which can be identified by growth inhibition screens, and inhibitors of pseudohyphal growth are of value. With the appropriate experimental design, both types of screens can be performed with this collection, as we have exemplified by preliminary results from a SLAD solid media pooled experiment.
We also investigated drug-induced haploinsufficiency in C. albicans, screening the pool with compounds that were most likely to produce a differential drug response by selecting compounds that more potently inhibited C. albicans than S. cerevisiae. Interestingly, we observed that the same chemical inhibitor can have different effects in S. cerevisiae and C. albicans. For instance, wild-type S. cerevisiae is much less sensitive to nigericin or brefeldin A than wild-type C. albicans, and mutants with the highest sensitivity in screens with these compounds did not include Sc-SEC7. This suggests that; 1) other targets in S. cerevisiae have a greater impact on S. cerevisiae's sensitivity to brefeldin A and nigericin, 2) that the drug is detoxified in S. cerevisiae, or 3) that C. albicans has less genetic redundancy for the pathways inhibited by these compounds. We also identified two other synthetic, previously uncharacterized compounds that inhibited vesicle transport in C. albicans, 0136-0228 and 1187–1561. Interestingly, the sensitivity profile of 0136–0228 overlapped that of nigericin, although their chemical structures show no obvious similarity. This result can be explained if 0136–0228 inhibits Tfp1p, a putative V-type ATPase that regulates intracompartmental pH [53]. Support for this scenario came from our complementation experiments, in which the drug-induced growth defect was rescued by overexpression of Tfp1p in S. cerevisiae. Abrogation of intracompartmental pH regulation via disruption of ion flow across vesicle membranes likely results in growth defects similar to those produced by nigericin, which produces a similar disregulation of intracompartmental pH. Interestingly, no orthologs or proteins of similar function were found in S. cerevisiae screens with this compound, again underscoring the need for direct study of C. albicans to identify novel treatment strategies.
The approach of using tagged transposon mutagenesis to generate mutant collections can be applied to a wide range of fungi of medical interest. The in vitro mutagenesis method allows flexibility because organism-specific transposons are not required, although a means for homologous recombination is needed. However, this approach could also be adapted to an in vivo format if the transposon could be electroporated directly into the cell (e.g., using commercially available transposome technology [54]), or if it could be expressed endogenously. Both of these approaches could bypass the transformation step, although they may be subject to insertion bias. Additionally, the TagModules can be readily adapted to a targeted deletion system for fungi with a compatible recombination system. While model organisms such as S. cerevisiae have been invaluable in initiating research in a range of microorganisms of medical interest, ultimately, it will be most fruitful to identify novel treatment strategies using the pathogen itself, owing to pathogen-specific differences. In summary, we have generated a uniquely tagged, publically available and archived disruption collection in C. albicans that can be used in multiplexed phenotypic assays or in individual experiments to identify potential new biology, therapeutic targets and mechanisms of pathogenesis.
Methods
All primers, plasmids, and strains are detailed in Tables S9, S10, and S11. Additional details are available on the supplementary website (http://chemogenomics.med.utoronto.ca/supplemental/transposon/) and as a text file (Methods S1). GO analysis (http://www.candidagenome.org/cgi-bin/GO/goTermFinder) was performed between 01/2010-02/2010.
Library construction
C. albicans genomic DNA was isolated from strain BWP17 (ura3Δ:: λ imm434/ura3Δ::λimm434 his1::hisG/his1::hisG arg4::hisG/arg4::hisG) [55] and partially digested with one or more of the following enzymes: XbaI, EcoRV, SpeI, XbaI/SpeI, XbaI/EcoRV, or BsrBI. The partially digested DNA fragments were gel purified to approximately 2–8 kb in size and ligated into a library backbone cut with the corresponding enzyme and phosphatase-treated. Backbones used were pCR 8/GW/TOPO + linker for XbaI, EcoRV, and BsrBI-cut genomic DNA, and pUC19 + linker for SpeI, XbaI/EcoRV, and SpeI/XbaI-cut genomic DNA. Construction of these vectors was previously described [27]. Each library contained on average 20,000 clones. We also used a commercially available C. albicans genomic library (Open Biosystems; [56]).
Transposon mutagenesis
The transposon destination vectors Tn7-UAU1-A and Tn5-UAU1-C.1, modified from Tn7-UAU1 [21] and EZ-Tn5 pMOD-3 (Epicentre Biotechnologies) to contain the Gateway recombination sites, were obtained from our previous study [27]. The Gateway-compatible TagModules [27] were pooled and transferred to the Tn7-UAU1-A or the Tn5-UAU1-C.1 vectors as previously described [27], and the resulting tagged transposons were used in the mutagenesis of C. albicans genomic libraries as described in the same study. The majority of insertions were generated using the Tn5-UAU1 due to technical difficulties with the Tn7. Each insertion was sequenced using D2_revcomp (Tn7-based) or U1 (Tn5-based) to identify the gene disrupted and the tag associated with each disruption. Gene-tag pairs were sorted to maximize i) the number of unique TagModules, ii) unique gene mutagenesis events, and iii) highest % gene disrupted (# base pairs from transposon junction to gene start)/(total gene length). Overall genome coverage is represented in Figure S1. Select insertion plasmids were then plasmid prepped with Seqprep 96 (Edge Biosystems), and the genomic fragment containing the tagged insertion was excised with the appropriate enzyme and chemically transformed into BWP17 in 96-well format.
At this step, two pools were created, one with unique uptags and one with unique downtags (for details, see Figure S2 and Methods S1; for pool characteristics, see Table S1). We separately arrayed transformants for each pool to agar plates and scraped the colonies into SC - Arg + uridine +15% glycerol to create the C. albicans pools, which we then stored as 50 µL aliquots at −80°C. For validation, we grew the pools independently in YPD for 20 generations as described in [34] minus the 10-generation recovery time, and extracted genomic DNA using the YeaStar Genomic DNA Kit (Zymo Research). PCR amplification of the uptags and downtags was performed separately with the common primers U1′ & BTEG-U2′, and D1′ & BTEG-D2′, and 30 µL each of uptag and downtag product was hybridized to an Affymetrix TAG4 microarray as described [57].
Pooled growth assays
For haploinsufficiency experiments, YPD, SC – Arg + uridine, YNB + uridine, and SLAD + uridine were made as defined in [58]. Growth assays were performed in duplicate in each of the media conditions, and samples were recovered at 5, 10, 15, and 20 generations of growth. Genomic DNA extraction, tag amplification, and hybridization were performed as described above. In total, we performed a series of eight hybridizations per media condition plus two hybridizations for a common zero timepoint. For the zero timepoint, ∼2 OD600 of frozen cells were used as the cell template for the genomic extraction.
For array data pre-processing, we followed the protocol outlined in [57], [59]. Briefly, outliers were masked and removed, and the average of the unmasked replicates were calculated for each tag. Uptags and downtags were mean-normalized, and low-quality tags (those below 3X background intensity units) were removed. To identify strains with reduced fitness in each of these growth conditions, we used a linear regression model to track decreases in tag hybridization intensity as a function of time. Linear regression of the log2(tag intensity) as a function of generations of growth (0, 5, 10, 15, and 20) was implemented using the lm() function in the statistical program R. A negative regression slope indicates that the tag signal decreases over time, inferring that the strain has a fitness defect in this condition with respect to the pool as a whole. To correct for multiple testing, the p-value for each regression was adjusted using the false discovery rate (“fdr”) option of the R function p.adjust(). For comparisons of regression slope between media types, an F-statistic was calculated and a p-value derived from the distribution. Our criteria for strain sensitivity was slope <0, p<0.05. This is a slightly less stringent cutoff than reported in Deutschbauer et al. (2005) as we have observed that the competitive growth assay is capable of detecting even slight defects in growth.
Complementation testing
For complementation testing, we used several pre-existing S. cerevisiae resources for the strain background. We picked individual strains from the Magic Marker collection (Open Biosystems, [32]) of yeast heterozygous deletions containing the selectable matA haploid marker. As the Magic Marker strains have some reversion rate, as evidenced by some colonies observed on the no-vector plates, we confirmed a few of these results with traditional tetrad dissection of the sporulated yeast knockout (YKO) strain, which has no Magic Marker cassette. For overexpression, we used as our destination vector the Gateway compatible destination vector pAG416GPD-ccdB (Addgene, [60]), which is a Ura+ CEN plasmid under control of a constitutive promoter.
For cloning C. albicans ORFs, we PCR amplified approximately 500 bp up and downstream of the start codon using primers specific to each gene and Platinum PCR SuperMix High Fidelity Primer Solution (Invitrogen). PCR products were TOPO cloned into the Gateway entry vector pCR 8/GW/TOPO (Invitrogen). Individual clones were sequenced using primers GW1 and GW2. Correct clones were then transferred to the pAG416GPD-ccdB destination vector using the LR clonase reaction (Invitrogen), and resequenced as described. For S. cerevisiae overexpression plasmids, we picked clones from the Molecular Barcoded Yeast (MoBY) ORFs (Open Biosystems, [61]). As these were already Gateway compatible, we transferred them to the overexpression plasmids using the LR clonase reaction and sequence verified them using primers GPD_ProF and pBluescriptSK.
Overexpression plasmids were chemically transformed into the corresponding Magic Marker or YKO strain, selecting for Ura+ transformants. Individual colonies were then inoculated to SC –Ura and grown overnight. Cultures were then harvested, washed twice with water, and then resuspended in sporulation media (2% potassium acetate) at a density of 1–1.5 OD600/mL. After sporulation for 5 days, 4.5×10−4 OD600 of sporulated Magic Marker culture was plated to Magic Marker selection media –Ura and grown for 2–3 days at 30°C. YKO-based strains were tetrad dissected and plated onto SC –Ura + G418. Tetrad dissected plates were then replica plated onto 5-FOA and YPD + G418 agar plates to confirm complementation.
Individual growth experiments
For individual growth experiments, we picked mutants from the frozen stock and grew them to saturation in SC –Arg + uridine. Each strain was then diluted to a working OD600 of ∼0.6 in water. For 5 generation experiments, each strain was diluted to a final OD600 of 0.06 in YPD, SC –Arg + uridine, YNB, or SLAD in 96-well plates. For 20 generation experiments, each strain was diluted to a final OD of 0.03 in the media of interest in 48-well plates, and growth was measured every 15 minutes robotically diluting every 5 generations as in the pooled growth assay. To measure growth of the GRACE strains, 12 of 17 of the core haploinsufficient strains identified in our screens were obtained as GRACE alleles (5 were not constructed), in which heterozygotes were constructed such that the remaining allele is under the control of a tetracycline-repressible promoter [33]. 400 cells of each strain (as determined by hemocytometry) were inoculated into 650 µL of media in 48-well plates (Greiner) and grown with constant shaking at 30°C as described [57]. In these conditions, individual cultures underwent ∼14 generations of growth without necessitating re-inoculation during the course of the experiment. Each strain was assayed in triplicate in selective SC –Arg + uridine media in the presence or absence of the tetracycline analog doxycycline (100 µM). AvgG (a metric of growth) was calculated as previously described [34].
Test for filamentation on solid media
Individual strains were picked from the frozen stock and grown overnight in SC –Arg + uridine. Strains were then stamped in triplicate to Spider agar media and grown for 5 days at 30°C, and colonies were then examined microscopically for filamentation. Methods for the pooled assay on solid SLAD media are described in Methods S1.
Drug screening
A diversity library was obtained from ChemDiv, Inc. and dissolved in DMSO. Other compounds were obtained from Sigma with the exception of itavastatin ca (Sequoia Research Products) and enantio-paf C-16 (Enzo Life Sciences). Each compound had previously been titrated to an inhibitory level of ∼10% in S. cerevisiae in YPD buffered to a pH of 6.8 with HEPES [62]. We grew C. albicans strain BWP17 and S. cerevisiae strain HO-1 in the presence of these 1521 distinct compounds under the same conditions, measuring AvgG as previously described [34]. Titration experiments were performed in microtitre plates, growing BWP17 and HO-1 in 100 µL buffered YPD plus either 1% DMSO (control) or 2 µL of compound at stock concentration. Growth rate in compound dilutions of up to 1/64th original concentration (or further, if necessary) were measured as previously described. AvgG from each microtitre plate was normalized to the 12 controls on each plate.
Selected compounds were then chosen for follow-up in dose response and pooled growth assays. First, compounds were ranked in order of greatest difference in inhibition between C. albicans and S. cerevisiae (Δ(normalized AvgG)). As a number of the compounds which produced the greatest difference in inhibition were not readily available or obtainable, and some hybridizations in the pooled growth assay failed, we focused on ∼67 available compounds for follow-up. The majority of these produced greater inhibition of C. albicans in the dose response assays (Figure S6). For pooled growth assays with 57/67 of these compounds, 20-generation pooled growth assays were performed as described [34], [62]. To determine sensitive strains, the experimental array was compared to a matched control set comprised of 11 no-drug arrays [18]. We then calculated z-scores and associated p-values as a metric of sensitivity as described [18].
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Nolla-SalasJ
Sitges-SerraA
Leon-GilC
Martinez-GonzalezJ
Leon-RegidorMA
1997 Candidemia in non-neutropenic critically ill patients: analysis of prognostic factors and assessment of systemic antifungal therapy. Study Group of Fungal Infection in the ICU. Intensive Care Med 23 23 30
2. PfallerMA
DiekemaDJ
2007 Epidemiology of invasive candidiasis: a persistent public health problem. Clin Microbiol Rev 20 133 163
3. WisplinghoffH
BischoffT
TallentSM
SeifertH
WenzelRP
2004 Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis 39 309 317
4. AkinsRA
2005 An update on antifungal targets and mechanisms of resistance in Candida albicans. Med Mycol 43 285 318
5. HeckmanDS
GeiserDM
EidellBR
StaufferRL
KardosNL
2001 Molecular Evidence for the Early Colonization of Land by Fungi and Plants. Science 293 1129 1133
6. LottTJ
FundygaRE
KuykendallRJ
ArnoldJ
2005 The human commensal yeast, Candida albicans, has an ancient origin. Fungal Genetics and Biology 42 444 451
7. McCuskerJH
ClemonsKV
StevensDA
DavisRW
1994 Genetic characterization of pathogenic Saccharomyces cerevisiae isolates. Genetics 136 1261 1269
8. BennettRJ
JohnsonAD
2003 Completion of a parasexual cycle in Candida albicans by induced chromosome loss in tetraploid strains. EMBO J 22 2505 2515
9. O'BrienKP
RemmM
SonnhammerEL
2005 Inparanoid: a comprehensive database of eukaryotic orthologs. Nucleic Acids Res 33 D476 480
10. SkrzypekMS
ArnaudMB
CostanzoMC
InglisDO
ShahP
2010 New tools at the Candida Genome Database: biochemical pathways and full-text literature search. Nucl Acids Res 38 D428 432
11. CostanzoM
GiaeverG
NislowC
AndrewsB
2006 Experimental approaches to identify genetic networks. Curr Opin Biotechnol 17 472 480
12. HoonS
St OngeRP
GiaeverG
NislowC
2008 Yeast chemical genomics and drug discovery: an update. Trends Pharmacol Sci 29 499 504
13. ShoemakerDD
LashkariDA
MorrisD
MittmannM
DavisRW
1996 Quantitative phenotypic analysis of yeast deletion mutants using a highly parallel molecular bar-coding strategy. Nat Genet 14 450 456
14. GiaeverG
ChuAM
NiL
ConnellyC
RilesL
2002 Functional profiling of the Saccharomyces cerevisiae genome. Nature 418 387 391
15. SmithAM
HeislerLE
MellorJ
KaperF
ThompsonMJ
2009 Quantitative phenotyping via deep barcode sequencing. Genome Res
16. OoiSL
ShoemakerDD
BoekeJD
2003 DNA helicase gene interaction network defined using synthetic lethality analyzed by microarray. Nat Genet 35 277 286
17. GiaeverG
FlahertyP
KummJ
ProctorM
NislowC
2004 Chemogenomic profiling: identifying the functional interactions of small molecules in yeast. Proc Natl Acad Sci U S A 101 793 798
18. HillenmeyerME
FungE
WildenhainJ
PierceSE
HoonS
2008 The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science 320 362 365
19. LumPY
ArmourCD
StepaniantsSB
CavetG
WolfMK
2004 Discovering modes of action for therapeutic compounds using a genome-wide screen of yeast heterozygotes. Cell 116 121 137
20. ParsonsAB
LopezA
GivoniIE
WilliamsDE
GrayCA
2006 Exploring the mode-of-action of bioactive compounds by chemical-genetic profiling in yeast. Cell 126 611 625
21. DavisDA
BrunoVM
LozaL
FillerSG
MitchellAP
2002 Candida albicans Mds3p, a conserved regulator of pH responses and virulence identified through insertional mutagenesis. Genetics 162 1573 1581
22. UhlMA
BieryM
CraigN
JohnsonAD
2003 Haploinsufficiency-based large-scale forward genetic analysis of filamentous growth in the diploid human fungal pathogen C.albicans. EMBO J 22 2668 2678
23. XuD
JiangB
KetelaT
LemieuxS
VeilletteK
2007 Genome-wide fitness test and mechanism-of-action studies of inhibitory compounds in Candida albicans. PLoS Pathog 3 e92
24. JiangB
XuD
AlloccoJ
ParishC
DavisonJ
2008 PAP inhibitor with in vivo efficacy identified by Candida albicans genetic profiling of natural products. Chem Biol 15 363 374
25. Rodriguez-SuarezR
XuD
VeilletteK
DavisonJ
SillaotsS
2007 Mechanism-of-action determination of GMP synthase inhibitors and target validation in Candida albicans and Aspergillus fumigatus. Chem Biol 14 1163 1175
26. XuD
SillaotsS
DavisonJ
HuW
JiangB
2009 Chemical genetic profiling and characterization of small-molecule compounds that affect the biosynthesis of unsaturated fatty acids in Candida albicans. J Biol Chem 284 19754 19764
27. OhJ
FungE
PriceMN
DehalPS
DavisRW
2010 A universal TagModule collection for parallel genetic analysis of microorganisms. Nucleic Acids Res 38 e146
28. EnloeB
DiamondA
MitchellAP
2000 A single-transformation gene function test in diploid Candida albicans. J Bacteriol 182 5730 5736
29. DeutschbauerAM
JaramilloDF
ProctorM
KummJ
HillenmeyerME
2005 Mechanisms of haploinsufficiency revealed by genome-wide profiling in yeast. Genetics 169 1915 1925
30. HengartnerCJ
ThompsonCM
ZhangJ
ChaoDM
LiaoSM
1995 Association of an activator with an RNA polymerase II holoenzyme. Genes Dev 9 897 910
31. ItohR
Saint-MarcC
ChaignepainS
KatahiraR
SchmitterJM
2003 The yeast ISN1 (YOR155c) gene encodes a new type of IMP-specific 5′-nucleotidase. BMC Biochem 4 4
32. PanX
YuanDS
XiangD
WangX
Sookhai-MahadeoS
2004 A robust toolkit for functional profiling of the yeast genome. Mol Cell 16 487 496
33. RoemerT
JiangB
DavisonJ
KetelaT
VeilletteK
2003 Large-scale essential gene identification in Candida albicans and applications to antifungal drug discovery. Mol Microbiol 50 167 181
34. LeeW
St OngeRP
ProctorM
FlahertyP
JordanMI
2005 Genome-wide requirements for resistance to functionally distinct DNA-damaging agents. PLoS Genet 1 e24
35. HuhWK
KangSO
2001 Characterization of the gene family encoding alternative oxidase from Candida albicans. Biochem J 356 595 604
36. ElsonSL
NobleSM
SolisNV
FillerSG
JohnsonAD
2009 An RNA Transport System in Candida albicans Regulates Hyphal Morphology and Invasive Growth. PLoS Genet 5 e1000664
37. ZaragozaO
RodriguezC
GancedoC
2000 Isolation of the MIG1 gene from Candida albicans and effects of its disruption on catabolite repression. J Bacteriol 182 320 326
38. MenonV
BernardisFD
CalderoneR
ChauhanN
2008 Transcriptional profiling of the Candida albicans Ssk1p receiver domain point mutants and their virulence. FEMS Yeast Research 8 756 763
39. Garcia-SanchezS
AubertS
IraquiI
JanbonG
GhigoJM
2004 Candida albicans biofilms: a developmental state associated with specific and stable gene expression patterns. Eukaryot Cell 3 536 545
40. GiaeverG
ShoemakerDD
JonesTW
LiangH
WinzelerEA
1999 Genomic profiling of drug sensitivities via induced haploinsufficiency. Nat Genet 21 278 283
41. NichollsDG
FergusonSJ
2001 A standard and up-to-date textbook with a comprehensive introduction to membrane bioenergetics and the chemiosmotic theory. Bioenergetics 3 17 26
42. PeyrocheA
AntonnyB
RobineauS
AckerJ
CherfilsJ
1999 Brefeldin A acts to stabilize an abortive ARF-GDP-Sec7 domain protein complex: involvement of specific residues of the Sec7 domain. Mol Cell 3 275 285
43. LewisMJ
PelhamHRB
2002 A New Yeast Endosomal SNARE Related to Mammalian Syntaxin 8. Traffic 3 922 929
44. WapinskiI
PfefferA
FriedmanN
RegevA
2007 Natural history and evolutionary principles of gene duplication in fungi. Nature 449 54 61
45. CostanzoM
BaryshnikovaA
BellayJ
KimY
SpearED
2010 The genetic landscape of a cell. Science 327 425 431
46. WinzelerEA
ShoemakerDD
AstromoffA
LiangH
AndersonK
1999 Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285 901 906
47. VazquezN
WalshTJ
FriedmanD
ChanockSJ
LymanCA
1998 Interleukin-15 augments superoxide production and microbicidal activity of human monocytes against Candida albicans. Infect Immun 66 145 150
48. JonesT
FederspielNA
ChibanaH
DunganJ
KalmanS
2004 The diploid genome sequence of Candida albicans. Proc Natl Acad Sci U S A 101 7329 7334
49. HolmesAR
TsaoS
OngSW
LampingE
NiimiK
2006 Heterozygosity and functional allelic variation in the Candida albicans efflux pump genes CDR1 and CDR2. Mol Microbiol 62 170 186
50. PadovanACB
ChavesGM
ColomboAL
BrionesMRS
2009 A novel allele of HWPI, isolated from a clinical strain of Candida albicans with defective hyphal growth and biofilm formation, has deletions of Gln/Pro and Ser/Thr repeats involved in cellular adhesion. Medical Mycology 47 824 835
51. GimenoCJ
LjungdahlPO
StylesCA
FinkGR
1992 Unipolar cell divisions in the yeast S. cerevisiae lead to filamentous growth: Regulation by starvation and RAS. Cell 68 1077 1090
52. LoHJ
KohlerJR
DiDomenicoB
LoebenbergD
CacciapuotiA
1997 Nonfilamentous C. albicans mutants are avirulent. Cell 90 939 949
53. ForgacM
1999 Structure and properties of the vacuolar (H+)-ATPases. J Biol Chem 274 12951 12954
54. GoryshinIY
JendrisakJ
HoffmanLM
MeisR
ReznikoffWS
2000 Insertional transposon mutagenesis by electroporation of released Tn5 transposition complexes. Nat Biotechnol 18 97 100
55. WilsonRB
DavisD
MitchellAP
1999 Rapid Hypothesis Testing with Candida albicans through Gene Disruption with Short Homology Regions. J Bacteriol 181 1868 1874
56. KadoshD
JohnsonAD
2001 Rfg1, a protein related to the Saccharomyces cerevisiae hypoxic regulator Rox1, controls filamentous growth and virulence in Candida albicans. Mol Cell Biol 21 2496 2505
57. PierceSE
DavisRW
NislowC
GiaeverG
2007 Genome-wide analysis of barcoded Saccharomyces cerevisiae gene-deletion mutants in pooled cultures. Nat Protoc 2 2958 2974
58. ChauhanN
KruppaMD
2009 Standard growth media and common techniques for use with Candida albicans. Methods Mol Biol 499 197 201
59. PierceSE
FungEL
JaramilloDF
ChuAM
DavisRW
2006 A unique and universal molecular barcode array. Nat Methods 3 601 603
60. AlbertiS
GitlerAD
LindquistS
2007 A suite of Gateway cloning vectors for high-throughput genetic analysis in Saccharomyces cerevisiae. Yeast 24 913 919
61. HoCH
MagtanongL
BarkerSL
GreshamD
NishimuraS
2009 A molecular barcoded yeast ORF library enables mode-of-action analysis of bioactive compounds. Nat Biotechnol 27 369 377
62. HoonS
SmithAM
WallaceIM
SureshS
MirandaM
2008 An integrated platform of genomic assays reveals small-molecule bioactivities. Nat Chem Biol 4 498 506
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 10
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Retroviral RNA Dimerization and Packaging: The What, How, When, Where, and Why
- Viral Replication Rate Regulates Clinical Outcome and CD8 T Cell Responses during Highly Pathogenic H5N1 Influenza Virus Infection in Mice
- Antimicrobial Peptides: Primeval Molecules or Future Drugs?
- Crystal Structure of DotD: Insights into the Relationship between Type IVB and Type II/III Secretion Systems
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy