
The Evolutionary Origination and Diversification of a Dimorphic Gene Regulatory Network through Parallel Innovations in and
The genomic content of regulatory genes such as transcription factors is surprisingly conserved between diverse animal species, raising the paradox of how new traits emerge, and are subsequently modified and lost. In this study we make a connection between the developmental basis for the formation of a fruit fly trait and the evolutionary basis for that trait’s origin, diversification, and loss. We show how the origin of a novel pigmentation trait is associated with the evolution of two regulatory sequences that control the co-expression of two key pigmentation genes. These sequences interact in unique ways with evolutionarily conserved Hox transcription factors to drive gene co-expression. Once these unique connections evolved, the alteration of this trait appears to have proceeded through changes to regulatory genes rather than regulatory sequences of the pigmentation genes. Thus, our findings support a scenario where regulatory sequence evolution provided new functions to old transcription factors, how co-expression can emerge from different utilizations of the same transcription factors, and that trait diversity was surprisingly shaped by changes in some manner to the deeply conserved regulatory genes.
Published in the journal:
. PLoS Genet 11(4): e32767. doi:10.1371/journal.pgen.1005136
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005136
Summary
The genomic content of regulatory genes such as transcription factors is surprisingly conserved between diverse animal species, raising the paradox of how new traits emerge, and are subsequently modified and lost. In this study we make a connection between the developmental basis for the formation of a fruit fly trait and the evolutionary basis for that trait’s origin, diversification, and loss. We show how the origin of a novel pigmentation trait is associated with the evolution of two regulatory sequences that control the co-expression of two key pigmentation genes. These sequences interact in unique ways with evolutionarily conserved Hox transcription factors to drive gene co-expression. Once these unique connections evolved, the alteration of this trait appears to have proceeded through changes to regulatory genes rather than regulatory sequences of the pigmentation genes. Thus, our findings support a scenario where regulatory sequence evolution provided new functions to old transcription factors, how co-expression can emerge from different utilizations of the same transcription factors, and that trait diversity was surprisingly shaped by changes in some manner to the deeply conserved regulatory genes.
Introduction
The complexity of developmental processes often hinders our ability to trace their evolutionary history. Genetic programs of development are structured into convoluted networks of genes, interconnected at the level of transcriptional regulation [1]. Each network connection, or regulatory linkage, is formed through interactions between a transcription factor protein and binding site sequences within a cis-regulatory element (CRE). The collection of regulatory linkages possessed by a CRE encodes the pattern of gene expression driven by the CRE. Networks culminate in the regulation of differentiation genes whose encoded products generate cell type-specific phenotypes. Hence, to understand how a developmental program originated or was diversified, one must trace how individual connections were formed between transcription factors and the CREs of the network.
CRE evolution is suspected to be a prime mode of trait evolution [2–4], and in recent years several case studies have described CREs that have been modified to generate phenotypic consequences [5–19]. However, our current understanding of network evolution is hampered by the general difficulty of resolving the direct regulatory linkages within CREs and how CRE mutations alter specific linkages [20]. For example, when genes are coordinately expressed, how similar are the encoded regulatory linkages within their CREs? What factors preside over the tendency of a network to evolve at upper level regulators or the terminal differentiation genes? The answers to these questions require studies of well-defined networks that govern morphologies that have diversified during recent evolutionary history.
The diverse abdominal pigmentation patterns of fruit fly species represent an optimal model to study the evolution of morphological characteristics [21]. The model organism Drosophila (D.) melanogaster belongs to the fruit fly subgenus Sophophora [22], which contains species with a wide diversity of abdominal pigmentation patterns (Fig 1). A central theme that typifies D. melanogaster and its close relatives (the melanogaster species group) is dimorphism, in which darkly pigmented males differ substantially from the generally unpigmented females. An ancestral character reconstruction analysis supports (84% posterior probability) an evolutionary scenario in which the most recent common ancestor of the melanogaster species group possessed a male-specific pattern of abdomen pigmentation (Fig 1, node 3)[23]. Unlike the melanogaster species group, monomorphic pigmentation is predominant among extant species in the more distantly-related obscura (e.g. D. pseudoobscura) and willistoni (e.g. D. willistoni) species groups, supporting the scenario that the most recent common ancestor of the melanogaster, obscura, and willistoni species groups had a monomorphic pattern of pigmentation (Fig 1, node 1) [23]. However, it remains uncertain whether the most recent common ancestor of the melanogaster and obscura group species (Fig 1, node 2) had dimorphic or monomorphic abdomen pigmentation. Once dimorphic pigmentation arose, the patterns diversified, expanding and contracting along the body axis in Oriental (e.g. D. melanogaster), montium (e.g. D. auraria), and ananassae (e.g. D. malerkotliana) clades (Fig 1) [23–25]. Within the melanogaster species group, several instances of reversion to the monomorphic state occurred, as exemplified by D. kikkawai (montium subgroup) and D. ananassae (ananassae subgroup) (Fig 1, nodes 4 and 5). Collectively, Sophophora tergite pigmentation provides an optimal model to investigate trait evolution, especially considering the extensively characterized network and CREs governing the development of pigmentation in D. melanogaster.

Within the abdomen of D. melanogaster, coloring of the dorsal cuticular plates (tergites) requires the co-expression of the genes tan and yellow in the underlying epidermal cells (Fig 1B’ and 1B”) [26]. Robust expression of tan and yellow in the male A5 and A6 segments are respectively regulated by CREs known as the tan male specific element (t_MSE) [26] and the yellow body element (yBE) [23,27]. The Hox protein Abd-B, expressed in the pigmented A5 and A6 segments, is a direct activator of the yBE [23], and represents a likely regulator of tan. However, little else is known about the regulatory linkages encoding the spatial and temporal activities for these CREs.
In this study we investigated the evolutionary histories and regulatory encodings of the yBE and t_MSE that coordinate expression of the Tan and Yellow pigmentation enzymes. Our results indicate that these CREs originated at different time points in the lineage leading to the common ancestor of the melanogaster species group. Our data supports a scenario where expansions, contractions, and losses of male-specific pigmentation evolved through a preponderance of trans-regulatory changes to the abdominal pigmentation gene network. In dissecting trans-regulatory inputs to the yBE and t_MSE, we discovered that these two CREs respond differently to alterations of the trans-landscape, notwithstanding their superficially similar patterns of expression. Lastly, our results indicate that these differences in responsiveness may be due to unique binding site architectures at these CREs, including a novel mechanism for the regulation of the t_MSE by the Hox protein Abd-A.
Results
The evolution of tan and yellow expression patterns
In D. melanogaster, tan and yellow are required for the pigmentation that develops on the male A5 and A6 segment tergites [26,28,29], and these genes’ expression patterns in the dorsal epidermis underlying the tergites closely matches the pattern of pigmentation (compare Fig 1B’ and 1B” to Fig 1B). For D. melanogaster, this is both the first report of tan expression and the first report of the RNA expression pattern for yellow. It has been shown that a similar expansion in tan and yellow expression occurs in D. prostipennis, a species that displays pigmentation extending into the male A4 tergite [30]. Additionally, the loss of pigmentation in D. santomea was accompanied by the joint loss of yellow and tan expression [23,26]. Thus, we anticipated that the origin, diversification, and loss of male pigmentation among the Sophophora subgenus will have been driven in part by physically corresponding changes in tan and yellow expression.
For D. auraria, we found that tan and yellow expression is limited to the dorsal epidermis of the male A6 segment (Fig 1C’ and 1C”), the only segment in this species that manifests male-specific pigmentation. While yellow expression occurs throughout the A6 segment, the hemispherical pattern of tan expression more-closely matches that of the pigmentation pattern, suggesting that tan plays an important role in the spatial-limitation of this male pattern element. For D. malerkotliana, we found that the expression of yellow (Fig 1E”) extends into the A4 segment that exhibits expanded pigmentation (Fig 1E). However, tan expression remained restricted to the A5 and A6 segments (Fig 1E’), suggesting that the evolved pattern of yellow expression plays a role in this derived phenotype, and the absence of tan expression may explain why the A4 tergite has less intense pigmentation than that present on the A5 and A6 tergites.
Pigmentation has been secondarily lost from D. kikkawai and D. ananassae males, which may have resulted from the loss of expression of tan and yellow. While yellow expression was found to be absent from the abdomen of D. kikkawai (Fig 1D”), tan expression was observed in the A6 segment in a pattern similar to that for D. auraria (compare Fig 1C’ and 1D’). This implies that the loss of male-specific pigmentation for this species included the loss of yellow expression, but did not require the inactivation of tan expression. For D. ananassae, neither tan nor yellow were found to be expressed in the male abdominal epidermis (Fig 1F’ and 1F”). It remains possible that these cases of pigmentation loss were initially due to a mechanism that had no bearing on yellow and tan expression. At a minimum, our results suggest that pigmentation loss at some point was accompanied by changes that eliminated these genes expression from the abdominal epidermis. This outcome was previously shown to have occurred in D. santomea [23,26], a third Sophophora species for which male tergite pigmentation was independently lost [31].
The aforementioned expression patterns support a role for changes in tan and yellow expression in the diversification and secondary loses of male pigmentation within the melanogaster species group. To date though, the expression of these pigmentation genes had not been investigated in species from the obscura and willistoni species groups in which most known extant species possess sexually monomorphic patterns of tergite pigmentation. For D. pseudoobscura of the obscura species group, we found that tan and yellow are both expressed throughout the male abdominal epidermis (Fig 1G’ and 1G”), a pattern that corresponds with this species’ dark coloration (Fig 1G). For D. willistoni, of the willistoni species group, we found that neither yellow nor tan are expressed at appreciable levels in the abdominal epidermis during the stages when tergite pigmentation is being specified (Fig 1H’ and 1H”). This suggests that D. willistoni lacks pigmentation in part due to the absence of these genes’ expression, and this monomorphic absence may reflect the ancestral state from which male-specific pigmentation evolved, as suggested elsewhere [6,16,23,32,33].
Collectively, our expression analyses support the hypothesis that the origin, diversification, and loss of male-specific pigmentation involved numerous alterations to the expression of tan and yellow. The mutational and mechanistic basis for CRE evolution remains poorly understood, especially in cases for genes whose expression patterns physically correspond. Thus, we next sought to determine the CRE basis for these evolved gene expression patterns and phenotypic differences.
The evolutionary origin of CREs controlling tan and yellow expression
The recent evolution of sexually dimorphic pigmentation within the subgenus Sophophora provides a model trait whose origination can be resolved to the level of CREs activating pertinent genes within a network. We sought to elucidate the evolutionary histories of the yBE and t_MSE that respectively control the coordinated expression of the yellow and tan genes. The sequences orthologous to these tan and yellow abdominal CREs were isolated from species with the derived male-specific tergite pigmentation, as well as species from more distantly-related lineages and that possess monomorphic patterns of tergite pigmentation. For these sequences, we directly compared their regulatory activities in in vivo reporter transgene assays in D. melanogaster (Fig 2).
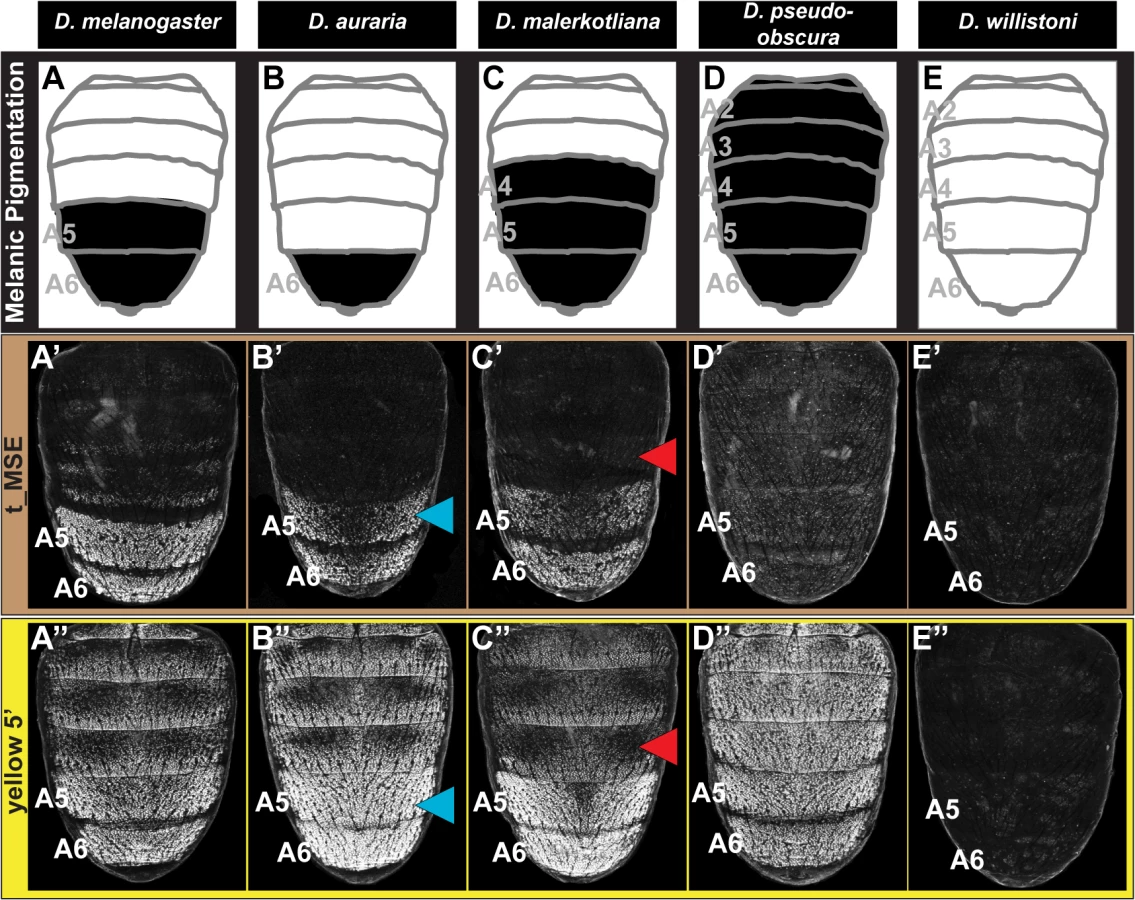
The region orthologous to the t_MSE (S1A Fig) was isolated from the dimorphically pigmented species D. auraria (montium subgroup), and D. malerkotliana (ananassae subgroup), and tested for abdominal activity in transgenic D. melanogaster pupae. In each case, robust reporter expression was observed in the male A5 and A6 abdomen segments (Fig 2B’ and 2C’). Likewise, orthologous sequences containing the yBE CREs derived from these same species each drove a male-specific pattern of reporter gene expression (Fig 2B” and 2C”). These similarities in regulatory activities to the D. melanogaster CREs support a scenario in which the common ancestor of the melanogaster species group (Fig 1, node 3) possessed male-specific tan and yellow gene expression driven respectively by an ancestral t_MSE and yBE.
To determine the timing and mechanism by which the novel trait of sexually dimorphic pigmentation arose, we isolated and tested orthologous sequences from the genomes of D. pseudoobscura and D. willistoni, two species from inferred ancestrally monomorphic lineages. As the t_MSE lies in between two upstream genes (CG1537 and Gr8a, S1A Fig), we confirmed their syntenic organization in these species (S2 Fig), suggesting a conserved gene order in the common ancestor of these disparate Sophophora lineages. However, the D. pseudoobscura and D. willistoni intergenic sequences between CG1537 and Gr8a had little-to-no t_MSE-like CRE activity (Fig 2D’ and 2E’). The absence of CRE activity parallels our observations of tan expression for D. willistoni (Fig 1H’), but contrasts with the monomorphic pattern of expression observed for the monomorphically pigmented D. pseudoobscura (Fig 1G’). These results are consistent with a scenario where the t_MSE originated to generate dimorphic expression in the lineage leading to the melanogaster species group after it diverged from the obscura group lineage (Fig 1, node 3). Moreover, the monomorphic expression of tan for D. pseudoobscura seemingly would be driven by another regulatory sequence or sequences.
The sequence 5’ of the D. pseudoobscura yellow gene possessed abdominal CRE activity that was enhanced in males compared to females (S3 Fig), though the domain of activity spanned all abdominal segments (Fig 2D”). Previously, a pan-abdomen CRE activity was observed for D. pseudoobscura [34] and for the orthologous gene region from D. subobscura, another obscura group species [23]. The results here show that the D. pseudoobscura sequence has abdominal regulatory activity that is enhanced in males when assayed in the D. melanogaster trans-regulatory environment. This suggests that a CRE with spatial and sex-specific inputs was present in the yellow gene of the most recent common ancestor of the obscura and melanogaster species groups (Fig 1, node 2).
To determine whether the yBE has an even deeper Sophophora ancestry, we inspected the regulatory capability of sequences 5’ of the D. willistoni yellow gene (S4 Fig). Since little-to-no sequence conservation is detectable in comparisons of D. willistoni yellow 5’ sequence to that of D. pseudoobscura and D. melanogaster, we evaluated the regulatory activity of two partially overlapping (1.2 kilobase, or kb, overlap) sequences that collectively span the first 5.1 kb of sequence 5’ of yellow exon 1 (S4A Fig). The proximal 3 kb to yellow exon 1 (called y wil 5’ 2) lacked abdominal regulatory activity (S4C Fig), whereas the more distal (y wil 5’ 1) sequence had CRE activity limited to monomorphic stripes at the posterior edges of each abdominal segment (S4B Fig) and throughout the pupal wing (S4B’ Fig). These stripe and wing activities are characteristic of the D. melanogaster wing element CRE that is similarly positioned more distal to the 1st exon of yellow than the yBE [27] and corresponds with the location of this species tergite pigmentation (Fig 1H). Thus, the D. willistoni yellow locus possesses an orthologous wing element, but lacks a CRE with activity characteristic of the yBE. This D. willistoni CRE architecture can also be inferred from a previous study that looked at 5.9 kb of 5’ sequence in a single reporter transgene [34]. These results support an evolutionary scenario where the most recent common ancestor of monomorphic and dimorphic Sophophora lineages (Fig 1, node 1) lacked an orthologous body element. Moreover, the evolution of sexually dimorphic pigmentation was accompanied by the origination of novel CRE activities of yellow and tan that integrate spatial and sex-specific regulatory inputs, and the t_MSE appears to be of more recent origin than the yBE.
Expansion and contraction of pigment patterns through trans evolution
Following the origin of male-specific tergite pigmentation, the number of pigmented tergites expanded and contracted to range from the single A6 segment of D. auraria, to the A5 and A6 segments of D. melanogaster, and the A4-A6 segments seen for D. malerkotliana (Fig 1). These phenotypic changes correspond with expansions and retractions in the expression of tan (Fig 1B’, 1C’, and 1E’) and yellow (Fig 1B”, 1C”, and 1E”) along the anterior-posterior axis. A priori, such changes in spatial expression could originate from sequence changes in the t_MSE and yBE (hereafter referred to as “cis-evolution”) or through changes in an upstream regulatory gene or genes (referred to hereafter as “trans-evolution”). We found that the t_MSE and yellow 5’ regulatory sequences from D. auraria each drove reporter gene expression in the male A5 and A6 segments of transgenic D. melanogaster (Fig 2B’ and 2B”). This domain of activity matches the output driven by the D. melanogaster CREs, yet extends one segment anterior relative to their endogenous expression in D. auraria (compare Fig 2B’ and 2B” to Fig 1C’ and 1C”). Similarly, the t_MSE and yellow 5’ sequences for D. malerkotliana each drove reporter expression in the A5 and A6 segments of transgenic D. melanogaster (Fig 2C’ and 2C”), a domain that is shifted posterior by one segment compared to the pigmentation phenotype and that for the endogenous pattern of yellow expression in D. malerkotliana (Fig 1E”). The up-regulation of yellow in the D. malerkotliana A4 segment is modest relative to the A5 and A6 segments, matching the lighter phenotype of this segment (Fig 1E). However, the D. malerkotliana yellow 5’ sequence is functionally-indistinguishable from the D. melanogaster CRE in the A4 segment. This similarity could be explained by “cis” evolution elsewhere in the yellow locus, such as the wing element, promoter, or intron. However, the yellow 5’ sequences evaluated here included the wing element and the putative D. malerkotliana promoter region. Thus, such a “cis-elsewhere” scenario would have to be due to an evolved intronic or more distally-located CRE. We favor the interpretation that the D. malerkotliana expression phenotype arose by evolution in trans, and more broadly that diversification in male tergite pigmentation evolved in large part from changes in trans to tan and yellow. In the future, the reciprocal test of the orthologous sequences as reporter transgenes in D. malerkotliana could provide more definite evidence for either the cis-elsewhere in yellow or trans-evolution scenarios.
Pigmentation loss through a mosaic of cis - and trans-evolution
Male-specific pigmentation has been lost several times within the melanogaster species group [23]. We sought to trace the paths by which this has occurred independently at the network level in two monomorphic species, D. kikkawai and D. ananassae (Fig 1). Previously, the D. kikkawai yBE was found to lack regulatory activity due to cis-evolution, in which a key binding site for Abd-B was lost [23]. Our in situ hybridization results confirm that yellow expression is indeed absent in the abdomen of D. kikkawai (Fig 1D”). We were curious whether the expression of tan had similarly been lost through cis-regulatory changes to its CRE. Surprisingly, tan expression was detected in the A6 body segment of D. kikkawai males, in a pattern reminiscent of D. auraria, a second montium species (Compare Fig 1C’ and 1D’), and male-limited like that seen for the expanded expression in males of species from the outgroup Oriental clade (D. melanogaster) and ananassae subgroup (D. malerkotliana) (Fig 1B’ and 1E’). Consistent with the endogenous expression pattern, the D. kikkawai t_MSE drove robust expression in the A5 and A6 segments of transgenic D. melanogaster pupae (Fig 3A’). These results indicate that the loss of pigmentation in D. kikkawai has proceeded without altering the ancestral expression of tan, suggesting that evolution of this trait occurred through other genes.

For D. ananassae, we found that the loss of male pigmentation was accompanied by the loss of yellow and tan expression in males (Fig 1F’ and 1F”). Interestingly, the CRE targets for cis - and trans-evolution were distinct from the case of D. kikkawai. Specifically, the orthologous yellow 5’ regulatory region retained regulatory activity in the male A5 and A6 segments in transgenic D. melanogaster (Fig 3B”), whereas the t_MSE lacked activity (Fig 3B’). As the most recent common ancestor of the melanogaster species group likely possessed a male pattern of abdomen pigmentation (Fig 1, node 3) [23], our results indicate that dissimilar modifications to this ancestrally dimorphic abdominal pigmentation network were responsible for these similar morphological outcomes. These divergent evolutionary paths may reflect the use of trans-regulatory inputs that differ between the yBE and t_MSE CREs. In order to understand how the evolution of tan and yellow expression has been individualized, we sought to characterize the regulatory linkages that control these CREs in D. melanogaster.
Distinct combinations of Hox factors and co-factors control the coordinated activities of the t_MSE and the yBE
Previously, Abd-B was shown to be a direct activator of yellow expression in the A5 and A6 segments through its interaction with two binding sites in the ~1.6 kb yBE reporter (S1G Fig, vertical blue lines) [23]. With our ultimate goal being to functionally characterize the regulatory inputs responsible for yellow expression in the abdominal epidermis; we sought to define a more minimal CRE sequence capable of directing robust expression in the male A5 and A6 abdominal segments (S1H Fig). Thus, we created three progressively truncated forms of the yBE centered on the two Abd-B binding sites (S1G Fig). The 1.1 and 0.9 kb sequences each drove robust but ectopic EGFP reporter gene expression (S1I and S1J Fig). The third truncated version of 0.6 kb drove reporter expression in a pattern limited to the A5 and A6 segments (S1K Fig). We refer to this sequence as yBE0.6, and this was the sequence that we chose to further characterize.
The coordinated patterns of tan and yellow expression in the A5 and A6 segments may be explained through the yBE0.6 and t_MSE possessing the same regulatory inputs and equivalent regulatory activities, or alternatively by these two CREs possessing unique regulatory inputs. The only known direct regulator of a pigmentation gene CRE is Abd-B’s interaction with the yBE. Genetic evidence for this regulatory input can be seen in the Transabdominal (Tab) genetic background, where ectopic Abd-B expression occurs in the A4 and A3 segments [35]. Consistent with Abd-B functioning as an upstream trans-activator of the yBE0.6, EGFP expression occurred ectopically in the male A4 and A3 segments of Tab mutants (Fig 4B’, blue arrowheads). When the t_MSE reporter gene was evaluated in the Tab background, a similar expansion of regulatory activity was seen (Fig 4B, blue arrowheads), suggesting that like yellow, Abd-B is an upstream activator of tan expression.

During development the Hox genes abd-A and Abd-B are both required to specify the identities of the A5 and A6 segments [36,37]. While Abd-B expression occurs in both the A5 and A6 segments [38], the range of Abd-A expression includes the A2-A6 segments [39]. Previously we found that Abd-A is expressed in and required for the male-specific pattern of pigmentation and t_MSE activity in the A5 and A6 segments [39]. It seemed plausible that Abd-A and Abd-B are part of a shared Hox-regulatory circuit that directs the coordinated expression of tan and yellow. To test this possibility, we used the pnr-GAL4 chromosome to drive dorsal midline expression of an RNA interference (RNAi) transgene that specifically silences abd-A expression. In this genetic background, EGFP expression driven by the t_MSE was markedly reduced compared to a control genetic background in which a luciferase transgene was ectopically expressed (compare Fig 4C and 4D). To our surprise, EGFP expression driven by the yBE0.6 was not noticeably altered in the abd-A silenced genetic background compared to the control genetic background (compare Fig 4C’ and 4D’). These outcomes indicate that abd-A functions in the abdomen as an upstream regulator of tan, but has little-to-no effect on yellow expression as directed by the yBE0.6 CRE. Hence, the regulatory wiring responsible for coordinated expression of tan and yellow seem to substantially differ.
The in vivo selectivity of Hox proteins for their target gene CREs has been found in several cases to be enhanced through cooperative binding with Hox cofactor proteins [40]. For Drosophila, the best studied Hox cofactors are the transcription factors Hth and Exd. RNAi-mediated suppression of hth and exd results in ectopic pigmentation of the male A3 and A4 abdominal segments, suggesting that these Hox cofactors operate as upstream repressors of male tergite pigmentation in the A3 and A4 abdomen segments [39]. Moreover, RNAi-mediated suppression of hth and exd in the dorsal abdomen midline results in ectopic regulatory activity for the t_MSE in the male A3 and A4 segments (Fig 4E and 4F, blue arrowheads). In contrast, we found that the yBE0.6 failed to drive a comparable expanded reporter gene expression upon RNAi knockdown of hth or exd expression (Fig 4E’ and 4F’), highlighting that the coordinated activation of the yBE0.6 and t_MSE occur through distinct mechanisms that differ in Hox co-factor dependence.
We were concerned that the different utilizations of Hox and Hox cofactor inputs for the t_MSE and yBE0.6 might be an artifact of truncating down the full yellow regulatory sequence to a minimal element lacking key regulatory inputs. Thus, we evaluated a larger yellow gene 5’ sequence which contains both the wing element and body element in the same genetic backgrounds where Hox and Hox cofactor expression were modified. This larger sequence had ectopic expression in the Abd-B misexpression background as seen for the yBE0.6 element (compare Fig 4B” to 4B’). Like the yBE0.6 element, little-to-no change in reporter expression was observed in the abd-A and exd RNAi background (Compare Fig 4D” and 4F” to 4D’ and 4F’), results that contrast with the prominent alteration that occurs for t_MSE-directed reporter expression (Fig 4D and 4F). The only difference we observed was a modest up-regulation of yellow 5’-driven reporter expression in the A3 and A4 segments of the hth RNAi background. This suggests that yellow expression is responsive to hth through some regulatory sequence outside of the yBE0.6 element, perhaps through the wing element which drives a low level of expression in the epidermis [27].
Spatial-mapping of yBE0.6 and t_MSE regulatory inputs
In order to more comprehensively characterize the regulatory linkages within the yBE0.6, we created 10 scanning mutant (SM1—SM10) versions of the yBE0.6 (S5 and S6 Fig). For each scanning mutant, a single contiguous block of ~70 bp was altered at every other base pair to its non-complementary transversion (S5 Fig). The regulatory activity characteristic of the yBE0.6 was not notably altered by the SM8 and SM9 mutations. For several scan mutants, activity was either reduced (Fig 5C and 5D; SM2 and SM3) or lost (Fig 5F and 5G; SM5 and SM6), indicating that the mutated blocks likely encode binding sites for activating transcription factor inputs. The SM5 and SM6 mutations spanned CRE sequences that include the bona fide binding sites for Abd-B [23], though we left these binding sites unaltered in these two scanning mutants (S5 Fig). The diminished regulatory activity caused by both SM5 and SM6 indicates that Abd-B collaborates with other adjacent binding transcription factors to activate gene expression in the pupal abdomen. The reduced regulatory activity of the SM2 and SM3 mutants demonstrates that additional activating inputs reside outside of the known Abd-B sites.

In addition to scanning mutations resulting in reductions in yBE0.6 regulatory activity, three resulted in gains in regulatory activity suggesting that the mutated sequences disrupted binding sites for repressive transcription factor inputs. yBE0.6 regulatory activity in the male A2-A6 abdomen segments was notably increased by the SM4 alteration (Fig 5E). The yBE0.6 SM8 and SM10 CREs each exhibited a modest ectopic regulatory activity in the male A4 and A3 segments (S6I and S6K Fig). Collectively, these results suggest that repressing and activating inputs are distributed throughout the 660 base pairs of yBE0.6.
A similar scanning mutagenesis strategy was carried out for the 860 bp t_MSE (Fig 5A, and S7 and S8 Fig), in which each scanning mutation spanned ~80 bp. While the t_MSE regulatory activity was not noticeably altered in 8 of 10 scanning mutants (S8 Fig), the SM5 and SM6 alterations each resulted in a dramatic reduction of EGFP reporter expression in the male A5 and A6 segments (Fig 5I and 5J). In order to more precisely localize activation inputs, we generated a series of fine-scale scanning mutations (S9 and S10 Fig) within a minimal 351 bp subfragment of the t_MSE that reproduces its activity (“t_MSE2”, S1 Fig). For the 351 bp t_MSE, we generated 10 scanning mutants in which each mutation spanned ~20 bp and collectively covered the entire SM5 and SM6 region (S9 Fig). While the 5i1, 5i3, 5i4, and 6i1–6i3 scanning mutants had no noticeable effect on t_MSE2 regulatory activity, scanning mutants 5i2, 6i4, 6i5, and 6i6 consistently drove reduced reporter expression in the male A5 and A6 segments (S10 Fig). Thus, this CRE has activating inputs located within the SM5 and SM6 regions. We sought to determine if these activating inputs include binding sites for Abd-A and Abd-B.
The t_MSE is an indirect and direct target for posterior Hox proteins
Abd-B is a key direct regulatory input for the yBE that is necessary to drive yellow expression throughout the male A5 and A6 segments [23], and yet, the yBE has little-to-no response to alterations of Abd-A. To disentangle how tan generates a correlated pattern with yellow, but is genetically downstream of both Abd-B and Abd-A, we sought to determine whether these Hox factors directly bind the t_MSE. An in vitro study has demonstrated a preference of TTAT sites for Abd-B binding and TAAT sites for Abd-A [41]. Within the SM5i2 region required for A5 and A6 regulatory activity, resides only a single TTAT site (S9 Fig). However, this site also occurs in the overlapping region for the SM5i1 region which is dispensable for abdominal activity as this mutant CRE has 114±3% of the wild type CRE’s activity in the male A5 segment (S10D Fig). Thus, we did not further consider the SM5i2 region as a location for direct Hox-regulation.
We next considered the SM6 region, for which the scanning mutation (SM6) resulted in a drastic reduction of male activity to 31±1% of the wild type CRE (compare Fig 6B and 6F). Within this 85 bp SM6 region resides 5 sequences matching either TTAT or TAAT sites, or both (Fig 6A, sites 1–5 or S1-S5). We generated and tested two mutant t_MSE2 reporter constructs, one in which the three TTAT sequences were mutated and the other for which the TTAT and TAAT sites were mutated (S11 Fig). To our surprise, the activity of the TTAT site mutant was only modestly reduced to 89±6% of the wild type CRE in the A5 segment (compare Fig 6B and 6C). Moreover, when the two TAAT sites were additionally mutated, regulatory activity was measured at 88±3% of wild type (Fig 6D). This lack of a pronounced regulatory effect contrasts with that caused by the 85 bp scanning mutation 6 (Fig 6F). Thus, with respect to the posterior male segment activity of the t_MSE2, it appears that Abd-B and Abd-A play little-to-no role as direct activators.

While the TTAT and TAAT sites had little-to-no importance regarding the activity of the t_MSE2 in the A5 and A6 segments, these sites were important for limiting reporter expression to these more posterior abdomen segments. When the TTAT sites were mutated, the regulatory activity in the A4 and A3 segments respectively increased to 169±4% and 261±6% of the wild type sequence (Fig 6C, blue arrowheads). When all TTAT and TAAT sites were mutated, more pronounced increases in regulatory activity were observed in the A4 (207±2%) and A3 (281±2%) segments (Fig 6D, blue arrowheads). These effects contrast with the modest reductions in activity that occurred for the scanning mutation 6 (Fig 6F; 64±1% for A4 and 73±1% for A3). Our results suggest that these sequences function as Hox binding sites to repress tan expression in abdominal segments anterior to that of A5. Of note, these anterior segments express Abd-A, but not Abd-B. Hence, unlike the yBE, the t_MSE appears to have co-opted direct Abd-A regulation in a unique way.
To further address whether Abd-A directly binds to and regulates the t_MSE, we focused our attention on sites S3, S4, and S5, as these sites were closely associated with a sequence resembling a site for Exd and one resembling a site for Hth (Fig 6A). We found that Abd-A bound to probes with the wild type sites but little-to-no binding occurred with the mutant versions (Fig 6G–6I), indicating that Abd-A can specifically bind to these sequences. A similar specific binding was shown for Abd-B, suggesting that these sequences can also be bound by this Hox protein.
It seemed plausible that the adjacent cofactor sites might be necessary to repress t_MSE activity in the anterior abdominal segments in conjunction with Abd-A. As a cursory test of this possibility, we mutated the putative Hth-site within the context of the t_MSE2 sequence (S11 Fig) and tested this sequence’s capability to regulate EGFP expression in transgenic pupa (Fig 6E). Consistent with this site being necessary for repression, the regulatory activity in the A3 and A4 segments respectively increased to 236±6% and 276±4% of the wild type sequence (Compare Fig 6B to 6E). Collectively, the t_MSE’s abdominal regulatory activity occurs through an encoding distinct from that responsible for the similar pattern of yellow expression directed by the yBE0.6 CRE.
Discussion
Here, we have traced the evolutionary history of two CREs required for a novel trait, and show that they have recently evolved similar expression patterns through remarkably different architectures in a common trans-regulatory landscape. Our data indicates that the tergite-wide activities of the yBE and t_MSE did not exist in the monomorphic ancestor for Sophophora, but evolved in the lineage leading to the common ancestor of the melanogaster species group. Our results support a scenario where the subsequent expansion and contraction of male pigmentation pattern was driven primarily by alteration of the trans-regulators, whereas repeated losses involved both cis - and trans-evolution with respect to these CREs. Though the t_MSE and yBE drive coordinated patterns of gene expression, we found striking differences in their upstream regulators and direct regulatory linkages (Fig 7). These results bear on our understanding of how new gene regulatory networks form, diversify, and how coordinated regulatory activities can arise through the independent evolution of unique regulatory codes.

Inferring a mechanism for a nascent Hox-regulated genetic switch
Hox transcription factors play a prominent role in generating the differences in serially homologous animal body parts, and the origin of novelties [42]. The diversification of homologous parts can be driven by changes in the spatial domains of Hox protein expression, as has been shown for crustacean appendage morphology [43], snake limblessness [44], and for the water strider appendage ground plan [45]. Changes in the downstream Hox targets are evident in cases such as the hindwings of insects [46], and for fruit fly tergite pigmentation [23]. The origin of novel structures can also be traced to the co-option of Hox proteins, as exemplified by cases such as the Photuris firefly lantern [47] and the sex combs residing on the forelegs of certain Drosophila species [48,49]. For many of these evolved traits, the molecular mechanisms by which Hox expression patterns and target genes evolve remain unknown.
While mechanistic studies on the evolution of Hox-regulated CREs remain limited, several target gene CREs have been thoroughly characterized and serve as exemplars of Hox-regulation during development [40]. Hox proteins can interact with CRE binding sites as monomers [50] or through cooperative interactions with Hox-cofactors [51–53]. The activity of these bound complexes can be further modulated through interactions with collaborating transcription factors. However, to date, few direct Hox target linkages have been traced to their evolutionary beginnings. Expression of yellow in the male A5 and A6 segments required the gain of two binding sites for Abd-B [23], but it remains uncertain whether these binding events require cooperative interactions with Hox cofactors and which transcription factors are acting as collaborators.
The t_MSE presented an opportunity to study how a second Hox-responsive CRE evolved in parallel to the activity at yellow. In this study, we show that Abd-A and Abd-B respectively are necessary and sufficient for t_MSE regulatory activity. However, we show that the ablation of the resident Hox sites had little effect on this CRE’s activity in the A5 and A6 segments, though mutations to nearby CRE sequences resulted in dramatically reduced activity. This result strongly implies that both Abd-A and Abd-B indirectly activate the t_MSE through a downstream factor or factors. While it can’t be entirely ruled out that these factors are operating directly through other non-canonical Hox sites, our gel shift assays did not provide convincing evidence that such sites exist. While the Hox sites were not necessary for activation in the A5 and A6 segments, their ablation resulted in a drastic gain of regulatory activity in the A4 and A3 segments, a setting in which Abd-A is the only Hox protein present. This indicates that Abd-A is a direct repressor of t_MSE function in these anterior abdomen segments. The observed dichotomy in Abd-A function can be explained by at least two—not necessarily mutually exclusive—scenarios. First, in the A5 and A6 segments Abd-B may not act as a direct activator of the t_MSE but its occupancy of Hox sites might preclude the direct repressive effects of Abd-A. Secondly, Abd-A may interact cooperatively or collaboratively with other transcription factors in the more anterior segments to impart repression. Our results with Hth support this second scenario.
The Hox co-factors Hth and Exd were prime candidates to mediate the context-dependent modulation of Abd-A activity. First, RNAi suppression of hth and exd expression each resulted in ectopic pigmentation [39] and t_MSE activity in the male A4 and A3 segments (Fig 4). Furthermore, inspection of the t_MSE sequence revealed sites characteristic of Hth (AGACAG) and Exd (GATCAT) binding that reside in close proximity to Hox sites (Fig 6A). This site content and arrangement is strikingly similar to that found in an abdominal-repressive module for the CRE controlling thoracic Distalless expression [51,54]. Along a similar vein, we show that the ablation of the Hth-like site led to an anterior expansion in t_MSE activity similar to that induced by the Hox site mutations (Fig 6). This outcome supports the interpretation that the more recent origin of the t_MSE involved the formation of novel regulatory linkages with Hox proteins and Hox cofactors.
The origins of a network controlling a sexually dimorphic trait
Morphological traits result from the activities of gene regulatory networks, in which each network is governed by a trans-regulatory tier of transcription factors and cell signaling components that ultimately regulate the expression of a set of differentiation genes [1,55,56]. For animals, the trans-regulatory genes are remarkably conserved [2,57]. It is plausible that the origin of new morphologies occurs through the formulation of new gene regulatory networks, while diversification and losses in traits would likely occur through the modification and dismantling of extant networks. The empirical evaluation of such trends of network evolution necessitates the study of trait evolution at the level of networks, CREs, and their encoded binding sites for multiple animal lineages, traits, and evolutionary time frames. The Drosophila pigmentation system is particularly well poised to make pioneering contributions to this growing body of knowledge.
The most recent common ancestor of monomorphic and dimorphic Sophophora lineages was inferred to have possessed monomorphic tergite pigmentation (Fig 1, node 1) [23], in the context of an otherwise invariant morphological landscape, in which segment number and form has remained conserved at the genus level. Hence, the origin of this novel pigmentation trait may be expected to have co-opted spatial and sex-specific patterning mechanisms that shape the conserved abdomen features. Our comparative analysis of orthologous yellow and tan non-coding sequences indicate that these co-option events involved the origination of novel CRE activities that connected a trans-regulatory tier of Hox, Hox-cofactors, and the Bab proteins to these key differentiation genes that encoded pigmentation enzymes (Fig 7).
The patterns of regulatory activity for the orthologous tan and yellow sequences support some additional inferences about the early events in this dimorphic trait’s origin. While the t_MSE abdominal activity was strikingly lower in D. pseudoobscura and D. willistoni, the D. pseudoobscura yellow body element was active (albeit with expanded activity). These outcomes support at least two evolutionary scenarios. One scenario is a sequence of events where the origination of the t_MSE and y_BE in the lineage of D. pseudoobscura (Fig 1, node 2) was followed by a secondary loss of the t_MSE. This scenario is supported by our previous observation of dimorphic Bab expression in the D. pseudoobscura abdomen [33], backing the notion that this species’ broad pattern of monomorphic abdominal pigmentation evolved from a dimorphic ancestral state. For the other scenario, the body element-like regulatory activity of D. pseudoobscura could be due to this CRE’s origin preceding (Fig 1, node 2) that of the t_MSE (Fig 1, node 3). Distinguishing between these two scenarios will require a more rigorous comparison of the pigmentation phenotypes and networks within the melanogaster and obscura species groups. The outcomes would provide a more nuanced understanding of the early evolutionary history for the derived sexually dimorphic pigmentation network.
Diversification and deconstruction of sexually dimorphic pigmentation
Tergite pigmentation evolution in the Sophophora subgenus has been relatively well-studied, and the accumulated results frame an extended perspective of trait evolution within a common network (Table 1). Trans-evolution at the bric-à-brac (bab) locus has been found to be a major driver for the diversification of female tergite pigmentation [16,58,59]. This study, in addition to previous studies, indicates that trans-evolution at as of yet unidentified loci may have played prominent roles in the diversification of male-limited tergite pigmentation [26,30]. Regarding the repeated losses in male pigmentation, our results are consistent with a scenario where both trans - and cis-evolution occurred, though the targets of cis-evolution have alternated between tan and yellow [23,26]. While cis-evolution has been identified for a case of monomorphic gain (ebony) in tergite pigmentation [8], and for a case of monomorphic loss (ebony and tan) [60], the full wealth of case studies portend to a more prominent role for evolutionary changes in the trans-regulatory tier of the pigmentation gene network. However, it is important to note that many of these case studies only assessed the activities of transgenes in D. melanogaster. While similarities in CRE activity might be indicative that expression divergence occurred through trans-evolution, it does not rule out the possibility that cis-changes occurred at other regions in the pigmentation enzyme gene loci, or that expression divergence results from combined cis - and trans-changes. In the future, it will be important to validate or reject the prominent role for trans-regulatory evolution by the reciprocal tests of CREs in species with the contrasting patterns of pigmentation. Two studies where CREs were tested in species with contrasting pigmentation phenotypes, showed that trans-regulatory evolution was a major driver for diversification of fruit fly wing spot patterns by modifying Distalless and wingless expression [10,61]. Thus it appears the notion of a “conserved trans-landscape” requires more scrutiny.

In this study, and elsewhere, experiments indicate that pigmentation losses are associated with and perhaps result from both changes in the trans-regulatory tier and in the cis-regulatory regions of the yellow and tan genes (Table 1). Interestingly, some instances of trans-regulatory modifications that cause loss of gene expression appear to leave perfectly good CREs intact. Our data provides a second instance in which loss of expression occurred without the loss of the encoded CRE. The yBE was found to be conserved in D. santomea, which diverged from D. yakuba ~400,000 years ago [23]. The activity for this CRE has also remained for D. ananassae since its divergence from a pigmented ancestor. In contrast, D. kikkawai has lost pigmentation while still expressing tan in the abdomen through a perfectly active t_MSE. These results suggest that these CREs were maintained within the population for long periods of time, perhaps indicating additional functions that promote the preservation of these CREs’ ancestral potential [62–64]. Furthermore, the observed heterogeneity of changes in cis and trans to yellow and tan were at first surprising. However, our study of the binding site architecture at the yBE and t_MSE provided key clues as to why their evolution may often be uncoupled.
Coordinate expression patterns through discordant CREs
The coordinated expression of genes is a ubiquitous theme in developmental biology. Gene expression is finely regulated during development through the activities of CREs that are individually encoded as evolved combinations of transcription factor binding sites (regulatory logic). A compelling question is whether such synchronized expression results from the independent evolution of CREs with similar logics. This question was previously pursued for CREs of regulatory genes coordinately expressed in the developing fruit fly neurogenic ectoderm [65]. In this case, the coordinately activated CREs are encoded by a common regulatory logic, or a so called “cis-regulatory module equivalence class” [66]. However, the neurogenic ectoderm CREs are deeply conserved, and arose in the distant past (over 230 million years ago).
The recently evolved male-specific expression patterns for tan and yellow present a case in which the evolutionary formation of coordinated regulation can be observed over shorter time-scales. Though both the t_MSE and yBE0.6 drive reporter expression in the dorsal A5 and A6 segment epidermis of males during late pupal development, we found their regulatory logic to be surprisingly dissimilar. Whereas the yBE0.6 is directly activated by Abd-B, our results indicate that the t_MSE is indirectly activated by Abd-B and Abd-A, and is directly repressed in more anterior body segments by Abd-A and seemingly Hth. Thus, this study provides an example that illustrates how coordinated expression evolved through the evolution of very different binding site architectures and logic.
The disparity of regulatory logic governing the yBE0.6 and t_MSE sheds light on the evolutionary tendencies of gene regulatory networks. The incipient stages of the dimorphic pigmentation network’s origin involved the derivation of CREs that generate similar patterns through distinct combinations of binding sites. This evolutionary history establishes a “branched” network in which several of the possible trans-regulatory alterations are incapable of generating coordinated shifts in the expression patterns for co-expressed genes. Hence, an emerging theme from the work in this system is that the differences in regulatory logic of yBE and t_MSE may necessitate changes in one CRE or the other, but is unable to be altered through a common trans regulator that influences both CRE’s patterning. Future studies are needed to substantiate the occurrence and identity of the trans changes altering this network’s structure. As other recently derived morphological traits are resolved to the level of binding sites within their networks, it will be instructive to see whether similar branched networks and paths of cis and trans evolution permeate their origin and diversification. The net results may reveal general principles of gene regulatory network evolution.
Materials and Methods
Fly stocks and genetic crosses
Fly stocks were maintained at 25°C on a sugar food medium that was previously described [33]. CRE sequences were obtained from the D. melanogaster (14021–0231.04), D. kikkawai (14028–0561.14), D. malerkotliana (14024–0391.00), D. ananassae (14024–0371.33), and D. willistoni (14030–0811.24) species stocks from the San Diego Drosophila Stock Center, and D. biarmipes, D. auraria, and D. pseudoobscura species stocks that were obtained from Dr. Sean B. Carroll. Tests for genetic interactions with Abd-B, abd-A, exd, and hth were done using the yBE0.6-EGFP transgene that was inserted into the attP40 site on chromosome 2 [67] and the yellow 5’-EGFP and t_MSE-EGFP transgenes were inserted into the 51D attP site on chromosome 2 [68]. All other reporter transgenes used in this study were inserted into the attP2 site on chromosome 3 [69].
D. melanogaster stocks possessing the Abd-Biab9Tab (BDSC ID#8620) allele, and UAS-abd-A RNAi (BDSC ID#28739), UAS-exd RNAi (BDSC ID#34897), UAS-hth RNAi (BDSC ID#27655), UAS-luciferase control (BDSC#35788), and pnr-GAL4 (BDSC ID#3039) transgenes were obtained from the Bloomington Drosophila Stock Center. The effects of ectopic Abd-B on tan and yellow loci CRE activities was observed for flies of genotype CRE-EGFP/+;Abd-Biab9Tab/+. The effects of reduced abd-A/exd/hth expression on CRE activity were observed for flies of genotype CRE-EGFP/+;UAS-gene specific RNAi/pnr-GAL4. The pnr-GAL4 stock has a chromosome where the GAL4 gene is inserted in the pannier (pnr) locus resulting in GAL4 expression in the dorsal-medial abdomen [70].
in situ hybridizations
in situ hybridizations were carried out as previously described [26]. Briefly, digoxigenin labeled riboprobes for yellow and tan were prepared through in vitro transcription of PCR templates amplified from each species (S1 Table for primers). Pupal abdomens were dissected at optimal time points for the visualization of yellow (70–80 h APF) and tan (85–95 h APF). Probe hybridization was visualized through an anti-digoxigenin antibody (Roche Diagnostics), detected by an alkaline phosphatase reaction using BCIP/NBT (Promega).
DNA sequence alignments
The contiguous genomic DNA sequence spanning from the first exon of Gr8a through the last exon (exon 8) of tan was obtained from the D. melanogaster genome version FB2013_05. Orthologous contigs were retrieved by BLAST searches of fruit fly genomes using the tan exon 8 as the query sequence [71,72]. The following GenBank accessions were identified as possessing the genome sequences orthologous to tan: D. biarmipes (AFPP01032826), D. yakuba (CM000162), D. kikkawai (AFFH02000000), D. ananassae (AAPP01016557), D. pseudoobscura (AADE01002480), and D. willistoni (AAQB01008786). Sequences were aligned to the annotated tan locus of D. melanogaster using mVISTA comparative genomics tool (S2 Fig) [73]. Sequence visualizations for the tan and yellow loci of D. melanogaster (S1 Fig) were made using the GenePalette tool [74].
EGFP reporter transgenes
EGFP reporter transgenes were used as a surrogate for the endogenous expression of the yellow and tan genes. Reporter transgene were assembled by cloning CREs into the AscI and SbfI restriction enzyme sites of the S3aG vector [75]. Each reporter transgene includes a CRE sequence cloned 5’ of a minimal hsp70 promoter and the coding sequence of the EGFP-NLS reporter protein [76]. All reporter transgenes analyzed were integrated into an attP landing site using ϕC integrase methods (Best Gene Inc.) [69]. The primer pairs used to clone D. melanogaster yellow and tan CREs are presented in the S2 Table. The primer pairs used to clone orthologous CREs are presented in the S3 Table. Scanning mutant sequences for the yBE0.6 (S5..), t_MSE (S7 Fig), and t_MSE2 (S9 Fig), the t_MSE2 with putative Hox sites mutated (S11 Fig), and the D. ananassae t_MSE were synthesized by GenScript USA Inc. These synthesized sequences were flanked by an AscI and SbfI restriction enzyme sites for cloning CREs into the S3aG vector.
Quantitative comparisons of the levels of EGFP reporter gene expression driven by the t_MSE2, t_MSE2 5i1, t_MSE2 SM6, t_MSE2 TTAT sites knockouts (KO), t_MSE2 TTAT +TAAT sites KO, and t_MSE2 Hth site KO CRE sequences were performed similar to that previously described for another CRE [16,75]. For each transgene, EGFP expression was imaged from five independent replicate specimens using a confocal microscope with settings for which few pixels were saturated when EGFP expression was driven by the wild type t_MSE2. For each confocal image, a separate pixel value statistic was determined for the dorsal epidermis of the A3, A4, and A5 segments using the Image J program [77]. For each reporter transgene, the regulatory activity was calculated as the mean pixel value and standard error of the mean (SEM). Activities reported in Fig 6 were normalized to the activity for the wild type t_MSE2.
Imaging of fly abdomens
Images of fruit fly abdomen pigmentation patterns were taken using an Olympus SZX16 Zoom Stereoscope and Olympus DP72 digital camera. Specimens were prepared for 5–10 day old adults. Projection images for EGFP-NLS reporter transgene expression were generated with an Olympus Fluoview FV 1000 confocal microscope and software. The regulatory activities of the t_MSE and t_MSE2 sequences were evaluated at ~90 hours after puparium formation (hAPF), a time point during pupal development when dimorphic expression of tan is first observed in the abdomen [26]. The regulatory activities for the yellow gene CREs were evaluated at ~85 hAPF, a time point when endogenous sexually dimorphic yellow expression is observed [23]. In each figure comparing CRE activities, a representative image was selected from replicate specimens (n≥6) and that were processed through the same modifications using Photoshop CS3 (Adobe). In situ hybridization images were taken on the same day using the same microscope and camera, and representative images were selected for Fig 1 and processed through the same modifications.
Gel shift assays
Reverse complementary oligonucleotides were synthesized (Integrated DNA Technologies) that contain t_MSE2 sequence with wild type or mutant Hox sites (S4 Table). Each oligonucleotide was biotin-labeled on their 3’ end using the DNA 3’ End Biotinylation Kit (Thermo Scientific) and complementary oligonucleotides were annealed by standard protocol. Labeling efficiency for each binding site was determined using a quantitative Dot Blot assay (DNA 3’ End Biotinylation Kit, Thermo Scientific). Each probe was separately tested for binding with a GST-Abd-B DNA Binding Domain (DBD) [6,23] and GST-Abd-A DBD fusion proteins. The coding sequence for amino acids 136–209 of D. melanogaster abd-A was cloned 3’ to that for GST in the EcoRI and NotI sites of the pGEX4T1 vector (Amersham). This abd-A sequence was amplified using the primers: ACCGgaattcTGTCCACGAAGGCGCGGTCGC and AGCCgcggccgcTCATTAGCGTCGCGCCTGTTCATTTATTTCC. All gel shift reactions included 20 fmol of one labeled binding site and GST-fusion protein in General Footprint Buffer (50 mM HEPES pH 7.9, 100 mM KCl, 1 mM DTT, 12.5 mM MgCl2, 0.05 mM EDTA, 17% glycerol) with 400 ng/μl of poly (dI-dC) (Thermo Scientific). For each binding site, a reaction was done that included an amount of GST-fusion protein ranging from 3,000 ng down to 111 ng. For each binding site, a control reaction was done that lacked GST-fusion protein. Binding reactions were carried out for 30 minutes on ice and then separated by a 5% non-denaturing polyacrylamide gel for 2 hours at 200 V. Reactions were then transferred and cross linked to a Hybond-N+ membrane (GE Healthcare Amersham) for chemiluminescent detection using the Chemiluminescent Nucleic Acid Detection Module and manufacture’s protocol (Thermo Scientific). Chemiluminescent images were taken using a BioChemi gel documentation system (UVP).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Davidson EH, Erwin DH (2006) Gene regulatory networks and the evolution of animal body plans. Science 311 : 796–800. 16469913
2. Carroll SB (2008) Evo-devo and an expanding evolutionary synthesis: a genetic theory of morphological evolution. Cell 134 : 25–36. http://www.ncbi.nlm.nih.gov/pubmed/18614008. Accessed 24 July 2011. doi: 10.1016/j.cell.2008.06.030 18614008
3. Wray GA (2007) The evolutionary significance of cis-regulatory mutations. Nat Rev Genet 8 : 206–216. http://www.ncbi.nlm.nih.gov/pubmed/17304246. Accessed 17 July 2011. 17304246
4. Stern DL, Orgogozo V (2008) The loci of evolution: how predictable is genetic evolution? Evolution 62 : 2155–2177. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2613234&tool=pmcentrez&rendertype=abstract. Accessed 10 June 2011. doi: 10.1111/j.1558-5646.2008.00450.x 18616572
5. Martin A, Orgogozo V (2013) The Loci of repeated evolution: a catalog of genetic hotspots of phenotypic variation. Evolution 67 : 1235–1250. http://www.ncbi.nlm.nih.gov/pubmed/23617905. Accessed 6 August 2013. doi: 10.1111/evo.12081 23617905
6. Williams TM, Selegue JE, Werner T, Gompel N, Kopp A, et al. (2008) The regulation and evolution of a genetic switch controlling sexually dimorphic traits in Drosophila. Cell 134 : 610–623.http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2597198&tool=pmcentrez&rendertype=abstract. Accessed 4 August 2011. doi: 10.1016/j.cell.2008.06.052 18724934
7. Rebeiz M, Jikomes N, Kassner VA, Carroll SB (2011) Evolutionary origin of a novel gene expression pattern through co-option of the latent activities of existing regulatory sequences. Proc Natl Acad Sci U S A 108 : 10036–10043. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3121811&tool=pmcentrez&rendertype=abstract. Accessed 7 July 2011. doi: 10.1073/pnas.1105937108 21593416
8. Rebeiz M, Pool JE, Kassner VA, Aquadro CF, Carroll SB (2009) Stepwise modification of a modular enhancer underlies adaptation in a Drosophila population. Science 326 : 1663–1667. http://www.ncbi.nlm.nih.gov/pubmed/20019281. Accessed 2 August 2011. doi: 10.1126/science.1178357 20019281
9. Shirangi TR, Dufour HD, Williams TM, Carroll SB (2009) Rapid evolution of sex pheromone-producing enzyme expression in Drosophila. PLoS Biol 7: e1000168. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2711336&tool=pmcentrez&rendertype=abstract. Accessed 25 July 2011. doi: 10.1371/journal.pbio.1000168 19652700
10. Arnoult L, Su K, Manoel D, Minervino C, Magrina J, et al. (2013) Emergence and Diversification of Fly Pigmentation Through Evolution of a Gene Regulatory Module. Science 339 : 1423–1426. http://www.sciencemag.org/cgi/doi/10.1126/science.1233749. Accessed 22 March 2013. doi: 10.1126/science.1233749 23520110
11. Prud’homme B, Gompel N, Rokas A, Kassner VA, Williams TM, et al. (2006) Repeated morphological evolution through cis-regulatory changes in a pleiotropic gene. Nature 440 : 1050–1053. http://www.ncbi.nlm.nih.gov/pubmed/16625197. Accessed 10 July 2011. 16625197
12. Gompel N, Prud’homme B, Wittkopp PJ, Kassner VA, Carroll SB (2005) Chance caught on the wing: cis-regulatory evolution and the origin of pigment patterns in Drosophila. Nature 433 : 481–487. http://www.ncbi.nlm.nih.gov/pubmed/15690032. 15690032
13. Cretekos CJ, Wang Y, Green ED, Martin JF, Rasweiler JJ, et al. (2008) Regulatory divergence modifies limb length between mammals. Genes Dev 22 : 141–151. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2192750&tool=pmcentrez&rendertype=abstract. Accessed 21 July 2011. doi: 10.1101/gad.1620408 18198333
14. Prabhakar S, Visel A, Akiyama JA, Shoukry M, Lewis KD, et al. (2008) Human-specific gain of function in a developmental enhancer. Science 321 : 1346–1350. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2658639&tool=pmcentrez&rendertype=abstract. Accessed 30 July 2011. doi: 10.1126/science.1159974 18772437
15. Chan YF, Marks ME, Jones FC, Villarreal G, Shapiro MD, et al. (2010) Adaptive evolution of pelvic reduction in sticklebacks by recurrent deletion of a Pitx1 enhancer. Science 327 : 302–305. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3109066&tool=pmcentrez&rendertype=abstract. Accessed 30 July 2011. doi: 10.1126/science.1182213 20007865
16. Rogers WA, Salomone JR, Tacy DJ, Camino EM, Davis KA, et al. (2013) Recurrent Modification of a Conserved Cis-Regulatory Element Underlies Fruit Fly Pigmentation Diversity. PLoS Genet 9: e1003740. http://dx.plos.org/10.1371/journal.pgen.1003740. Accessed 1 September 2013. doi: 10.1371/journal.pgen.1003740 24009528
17. Spitz F, Gonzalez F, Duboule D (2003) A Global Control Region Defines a Chromosomal Regulatory Landscape Containing the HoxD Cluster. Cell 113 : 405–417. 12732147
18. Guerreiro I, Nunes A, Woltering JM, Casaca A, Nóvoa A, et al. (2013) Role of a polymorphism in a Hox/Pax-responsive enhancer in the evolution of the vertebrate spine. Proc Natl Acad Sci U S A 110 : 10682–10686. http://www.ncbi.nlm.nih.gov/pubmed/23674686. Accessed 14 November 2013. doi: 10.1073/pnas.1300592110 23674686
19. Frankel N, Erezyilmaz DF, McGregor AP, Wang S, Payre F, et al. (2011) Morphological evolution caused by many subtle-effect substitutions in regulatory DNA. Nature 474 : 598–603. http://www.nature.com/doifinder/10.1038/nature10200. Accessed 29 June 2011. doi: 10.1038/nature10200 21720363
20. Rebeiz M, Williams TM (2011) Experimental Approaches to Evaluate the Contributions of Candidate Cis - regulatory Mutations to Phenotypic Evolution. In: Orgogozo V, Rockman M V., editors. Methods in Molecular Biology. Methods in Molecular Biology. Totowa, NJ: Humana Press, Vol. 772. pp. 351–375. http://www.springerlink.com/index/10.1007/978-1-61779-228-1. Accessed 9 November 2011. doi: 10.1007/978-1-61779-228-1_21 22065449
21. Wittkopp PJ, Carroll SB, Kopp A (2003) Evolution in black and white: genetic control of pigment patterns in Drosophila. Trends Genet 19 : 495–504. http://linkinghub.elsevier.com/retrieve/pii/S016895250300194X. Accessed 17 July 2011. 12957543
22. Markow TA, O’Grady PM (2006) Drosophila: A guide to species identification and use. Academic Press.
23. Jeong S, Rokas A, Carroll SB (2006) Regulation of body pigmentation by the Abdominal-B Hox protein and its gain and loss in Drosophila evolution. Cell 125 : 1387–1399. http://www.ncbi.nlm.nih.gov/pubmed/16814723. Accessed 27 August 2011. 16814723
24. Kopp A, True JR (2002) Phylogeny of the Oriental Drosophila melanogaster species group: a multilocus reconstruction. Syst Biol 51 : 786–805. http://www.ncbi.nlm.nih.gov/pubmed/12396591. Accessed 19 November 2012. 12396591
25. Kopp A (2006) Basal relationships in the Drosophila melanogaster species group. Mol Phylogenet Evol 39 : 787–798. http://www.ncbi.nlm.nih.gov/pubmed/16527496. Accessed 21 April 2014. 16527496
26. Jeong S, Rebeiz M, Andolfatto P, Werner T, True J, et al. (2008) The evolution of gene regulation underlies a morphological difference between two Drosophila sister species. Cell 132 : 783–793. http://www.ncbi.nlm.nih.gov/pubmed/18329365. Accessed 10 June 2011. doi: 10.1016/j.cell.2008.01.014 18329365
27. Wittkopp PJ, Vaccaro K, Carroll SB (2002) Evolution of yellow gene regulation and pigmentation in Drosophila. Curr Biol 12 : 1547–1556. http://www.ncbi.nlm.nih.gov/pubmed/12372246. 12372246
28. True JR, Yeh S-D, Hovemann BT, Kemme T, Meinertzhagen IA, et al. (2005) Drosophila tan encodes a novel hydrolase required in pigmentation and vision. PLoS Genet 1: e63. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1285064&tool=pmcentrez&rendertype=abstract. Accessed 7 January 2012. 16299587
29. Wittkopp PJ, True JR, Carroll SB (2002) Reciprocal functions of the Drosophila yellow and ebony proteins in the development and evolution of pigment patterns. Development 129 : 1849–1858. http://www.ncbi.nlm.nih.gov/pubmed/11934851. 11934851
30. Ordway A, Hancuch KN, Johnson W, Wiliams TM, Rebeiz M (2014) The expansion of body coloration involves coordinated evolution in cis and trans within the pigmentation regulatory network of drosophila prostipennis. Dev Biol: 1–10. http://www.ncbi.nlm.nih.gov/pubmed/24907418. Accessed 10 June 2014.
31. Lachaise D, Harry M, Solignac M, Lemeunier F, Bénassi V, et al. (2000) Evolutionary novelties in islands: Drosophila santomea, a new melanogaster sister species from São Tomé. Proc Biol Sci 267 : 1487–1495. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1690712&tool=pmcentrez&rendertype=abstract. Accessed 6 March 2013. 11007323
32. Kopp A, Duncan I, Godt D, Carroll SB (2000) Genetic control and evolution of sexually dimorphic characters in Drosophila. Nature 408 : 553–559. doi: 10.1038/35046017 11117736
33. Salomone JR, Rogers WA, Rebeiz M, Williams TM (2013) The evolution of Bab paralog expression and abdominal pigmentation among Sophophora fruit fly species. Evol Dev 15 : 442–457. http://www.ncbi.nlm.nih.gov/pubmed/24261445. Accessed 24 November 2013. doi: 10.1111/ede.12053 24261445
34. Kalay G, Wittkopp PJ (2010) Nomadic enhancers: tissue-specific cis-regulatory elements of yellow have divergent genomic positions among Drosophila species. PLoS Genet 6: e1001222. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2996884&tool=pmcentrez&rendertype=abstract. Accessed 26 July 2012. doi: 10.1371/journal.pgen.1001222 21151964
35. Celniker SE, Lewis EB (1993) Molecular basis of transabdominal—a sexually dimorphic mutant of the bithorax complex of Drosophila. Proc Natl Acad Sci U S A 90 : 1566–1570. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=45915&tool=pmcentrez&rendertype=abstract. 8094560
36. Sánchez-Herrero E (1991) Control of the expression of the bithorax complex genes abdominal-A and abdominal-B by cis-regulatory regions in Drosophila embryos. Development 111 : 437–449. http://www.ncbi.nlm.nih.gov/pubmed/1680047. 1680047
37. Sanchez-Herrero E, Vernos I, Marco R, Morata G (1985) Genetic organization of Drosophila bithorax complex. Nature 313 : 108–113. 3917555
38. Kopp A, Duncan I (2002) Anteroposterior patterning in adult abdominal segments of Drosophila. Dev Biol 242 : 15–30. http://www.ncbi.nlm.nih.gov/pubmed/11795937. Accessed 19 February 2012. 11795937
39. Rogers WA, Grover S, Stringer SJ, Parks J, Rebeiz M, et al. (2014) A survey of the trans-regulatory landscape for Drosophila melanogaster abdominal pigmentation. Dev Biol 385 : 417–432. http://www.ncbi.nlm.nih.gov/pubmed/24269556. Accessed 18 December 2013. doi: 10.1016/j.ydbio.2013.11.013 24269556
40. Mann RS, Lelli KM, Joshi R (2009) Hox Specificity: Unique Roles for Cofactors and Collaborators. Curr Top Dev Biol 88 : 63–101. doi: 10.1016/S0070-2153(09)88003-4 19651302
41. Noyes MB, Christensen RG, Wakabayashi A, Stormo GD, Brodsky MH, et al. (2008) Analysis of homeodomain specificities allows the family-wide prediction of preferred recognition sites. Cell 133 : 1277–1289. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2478728&tool=pmcentrez&rendertype=abstract. Accessed 17 July 2012. doi: 10.1016/j.cell.2008.05.023 18585360
42. Carroll SB (1995) Homeotic genes and the evolution of arthropods and cordates. Nature 376 : 479–485. 7637779
43. Averof M, Patel NH (1997) Crustacean appendage evolution associated with changes in Hox gene expression. Nature 388 : 682–686. http://www.ncbi.nlm.nih.gov/pubmed/9262403. 9262403
44. Cohn MJ, Tickle C (1999) Developmental basis of limblessness and axial patterning in snakes. Nature 399 : 474–479. 10365960
45. Khila A, Abouheif E, Rowe L (2009) Evolution of a novel appendage ground plan in water striders is driven by changes in the Hox gene Ultrabithorax. PLoS Genet 5: e1000583. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2709915&tool=pmcentrez&rendertype=abstract. Accessed 26 July 2012. doi: 10.1371/journal.pgen.1000583 19649305
46. Weatherbee SD, Nijhout HF, Grunert LW, Halder G, Galant R, et al. (1999) Ultrabithorax function in butterfly wings and the evolution of insect wing patterns. Curr Biol 9 : 109–115. http://www.ncbi.nlm.nih.gov/pubmed/10021383. 10021383
47. Stansbury MS, Moczek AP (2014) The function of Hox and appendage-patterning genes in the development of an evolutionary novelty, the Photuris firefly lantern. Proc Biol Sci 281.
48. Tanaka K, Barmina O, Sanders LE, Arbeitman MN, Kopp A (2011) Evolution of sex-specific traits through changes in HOX-dependent doublesex expression. PLoS Biol 9: e1001131. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3160335&tool=pmcentrez&rendertype=abstract. Accessed 18 April 2014. doi: 10.1371/journal.pbio.1001131 21886483
49. Barmina O, Kopp A (2007) Sex-specific expression of a HOX gene associated with rapid morphological evolution. Dev Biol 311 : 277–286. http://www.ncbi.nlm.nih.gov/pubmed/17868668. Accessed 26 March 2014. 17868668
50. Walsh CM, Carroll SB (2007) Collaboration between Smads and a Hox protein in target gene repression. Development 134 : 3585–3592. http://www.ncbi.nlm.nih.gov/pubmed/17855427. Accessed 18 April 2014. 17855427
51. Gebelein B, McKay DJ, Mann RS (2004) Direct integration of Hox and segmentation gene inputs during Drosophila development. Nature 431 : 653–659. http://www.ncbi.nlm.nih.gov/pubmed/15470419. 15470419
52. Li-Kroeger D, Witt L, Grimes HL, Cook TA, Gebelein B (2008) Hox and senseless antagonism functions as a molecular switch to regulate EGF secretion in the Drosophila PNS. Dev Cell 15 : 298–308. http://www.ncbi.nlm.nih.gov/pubmed/20798606. Accessed 10 April 2012. doi: 10.1016/j.devcel.2008.06.001 18694568
53. Ryoo HD, Marty T, Casares F, Affolter M, Mann RS (1999) Regulation of Hox target genes by a DNA bound Homothorax/Hox/Extradenticle complex. Development 126 : 5137–5148. http://www.ncbi.nlm.nih.gov/pubmed/10529430. 10529430
54. Gebelein B, Culi J, Ryoo HD, Zhang W, Mann RS (2002) Specificity of Distalless repression and limb primordia development by abdominal Hox proteins. Dev Cell 3 : 487–498. http://www.ncbi.nlm.nih.gov/pubmed/12408801. 12408801
55. Stathopoulos A, Levine M (2005) Genomic regulatory networks and animal development. Dev Cell 9 : 449–462. http://www.ncbi.nlm.nih.gov/pubmed/16198288. Accessed 27 July 2011. 16198288
56. Bonn S, Furlong EEM (2008) cis-Regulatory networks during development: a view of Drosophila. Curr Opin Genet Dev 18 : 513–520. http://www.ncbi.nlm.nih.gov/pubmed/18929653. Accessed 14 March 2012. doi: 10.1016/j.gde.2008.09.005 18929653
57. Carroll SB, Grenier J, Weatherbee SD (2005) From DNA to Diversity: Molecular Genetics and the Evolution of Animal Design. 2nd ed. Malden, MA: Blackwell Publishing.
58. Kopp A, Graze RM, Xu S, Carroll SB, Nuzhdin S V (2003) Quantitative Trait Loci Responsible for Variation in Sexually Dimorphic Traits in Drosophila melanogaster. Genetics 787 : 771–787.
59. Bickel RD, Kopp A, Nuzhdin S V (2011) Composite effects of polymorphisms near multiple regulatory elements create a major-effect QTL. PLoS Genet 7: e1001275. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3020931&tool=pmcentrez&rendertype=abstract. Accessed 26 October 2012. doi: 10.1371/journal.pgen.1001275 21249179
60. Wittkopp PJ, Stewart EE, Arnold LL, Neidert AH, Haerum BK, et al. (2009) Intraspecific polymorphism to interspecific divergence: genetics of pigmentation in Drosophila. Science 326 : 540–544. http://www.ncbi.nlm.nih.gov/pubmed/19900891. Accessed 5 July 2011. doi: 10.1126/science.1176980 19900891
61. Werner T, Koshikawa S, Williams TM, Carroll SB (2010) Generation of a novel wing colour pattern by the Wingless morphogen. Nature 464 : 1143–1148. http://www.ncbi.nlm.nih.gov/pubmed/20376004. Accessed 15 July 2011. doi: 10.1038/nature08896 20376004
62. Rajakumar R, San Mauro D, Dijkstra MB, Huang MH, Wheeler DE, et al. (2012) Ancestral developmental potential facilitates parallel evolution in ants. Science 335 : 79–82. http://www.ncbi.nlm.nih.gov/pubmed/22223805. Accessed 13 August 2014. doi: 10.1126/science.1211451 22223805
63. Abouheif E (2008) Parallelism as the pattern and process of mesoevolution. Evol Dev 10 : 3–5. http://www.ncbi.nlm.nih.gov/pubmed/18184352. doi: 10.1111/j.1525-142X.2007.00208.x 18184352
64. Abouheif E, Fave M-J, Ibarraran-Viniegra AS, Lesoway MP, Rafiqi AM, et al. (2014) Eco-Evo-Devo: The Time Has Come. In: Landry CR, Aubin-Horth N, editors. Advances in Experimental Medicine and Biology. Springer. pp. 107–126. doi: 10.1007/978-3-319-06068-2_6 24952181
65. Erives A, Levine M (2004) Coordinate enhancers share common organizational features in the Drosophila genome. Proc Natl Acad Sci U S A 101 : 3851–3856. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=374333&tool=pmcentrez&rendertype=abstract. Accessed 30 November 2012. 15026577
66. Crocker J, Tamori Y, Erives A (2008) Evolution acts on enhancer organization to fine-tune gradient threshold readouts. PLoS Biol 6: e263. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2577699&tool=pmcentrez&rendertype=abstract. Accessed 1 March 2012. doi: 10.1371/journal.pbio.0060263 18986212
67. Markstein M, Pitsouli C, Villalta C, Celniker SE, Perrimon N (2008) Exploiting position effects and the gypsy retrovirus insulator to engineer precisely expressed transgenes. Nat Genet 40 : 476–483. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2330261&tool=pmcentrez&rendertype=abstract. Accessed 17 July 2011. doi: 10.1038/ng.101 18311141
68. Bischof J, Maeda RK, Hediger M, Karch F, Basler K (2007) An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc Natl Acad Sci U S A 104 : 3312–3317. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1805588&tool=pmcentrez&rendertype=abstract. Accessed 21 June 2011. 17360644
69. Groth AC, Fish M, Nusse R, Calos MP (2004) Construction of Transgenic Drosophila by Using the Site-Specific Integrase From Phage phiC31. Genetics 166 : 1775–1782. 15126397
70. Calleja M, Herranz H, Estella C, Casal J, Lawrence P, et al. (2000) Generation of medial and lateral dorsal body domains by the pannier gene of Drosophila. Development 127 : 3971–3980. http://www.ncbi.nlm.nih.gov/pubmed/10952895. 10952895
71. Clark AG, Eisen MB, Smith DR, Bergman CM, Oliver B, et al. (2007) Evolution of genes and genomes on the Drosophila phylogeny. Nature 450 : 203–218. Available: http://www.ncbi.nlm.nih.gov/pubmed/17994087. Accessed 1 March 2012. 17994087
72. Richards S, Liu Y, Bettencourt BR, Hradecky P, Letovsky S, et al. (2005) Comparative genome sequencing of Drosophila pseudoobscura: chromosomal, gene, and cis-element evolution. Genome Res 15 : 1–18. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=540289&tool=pmcentrez&rendertype=abstract. Accessed 12 March 2012. 15632085
73. Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I (2004) VISTA: computational tools for comparative genomics. Nucleic Acids Res 32: W273–W279. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=441596&tool=pmcentrez&rendertype=abstract. Accessed 26 September 2013. 15215394
74. Rebeiz M, Posakony JW (2004) GenePalette: a universal software tool for genome sequence visualization and analysis. Dev Biol 271 : 431–438. 15223345
75. Rogers WA, Williams TM (2011) Quantitative Comparison of cis-Regulatory Element (CRE) Activities in Transgenic Drosophila melanogaster. J Vis Exp: 2–7. http://www.ncbi.nlm.nih.gov/pubmed/22215325. Accessed 22 January 2012.
76. Barolo S, Castro B, Posakony JW (2004) New Drosophila transgenic reporters: insulated P-element vectors expressing fast-maturing RFP. Biotechniques 36 : 436–440, 442. http://www.ncbi.nlm.nih.gov/pubmed/15038159. 15038159
77. Abràmoff MD, Hospitals I, Magalhães PJ, Abràmoff M (2004) Image Processing with ImageJ. Biophotonics Int 11 : 36–42.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 4
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Lack of GDAP1 Induces Neuronal Calcium and Mitochondrial Defects in a Knockout Mouse Model of Charcot-Marie-Tooth Neuropathy
- Proteolysis of Virulence Regulator ToxR Is Associated with Entry of into a Dormant State
- Frameshift Variant Associated with Novel Hoof Specific Phenotype in Connemara Ponies
- Ataxin-2 Regulates Translation in a New BAC-SCA2 Transgenic Mouse Model
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy