
Natural Variant E610G Is a Semi-dominant Suppressor of IAP-Induced RNA Processing Defects
Transposable elements, including endogenous retroviruses, have long been hypothesized as a substrate for creating or modulating gene regulatory networks, particularly through effects on transcription. However, several classes of elements are also known to affect alternative RNA processing events. We previously showed that the major allele of nuclear export factor Nxf1 in Mus musculus castaneus mice acts as a semi-dominant suppressor of de novo mutations caused by intracisternal A particle (IAP) endogenous retroviruses that integrate into introns, disrupting normal RNA processing. Here we show that this suppressor allele of Nxf1 can coordinately modify gene expression phenotypes at several endogenous loci in the C57BL/6 mouse reference genome that contain IAP sequences in their introns. This quadruples the number of known insertional events modified by Nxf1 and extends the effect beyond overt mutations. We previously used transgenic mice and viral vector mediated overexpression to demonstrate Nxf1 as the modifier gene for de novo insertions. Here we use direct genome editing in mouse one cell embryos to create custom germline alleles at the endogenous Nxf1 locus to show that a specific amino acid substitution, E610G, quantitatively accounts for the Nxf1 modifier gene activity.
Published in the journal:
. PLoS Genet 11(4): e32767. doi:10.1371/journal.pgen.1005123
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005123
Summary
Transposable elements, including endogenous retroviruses, have long been hypothesized as a substrate for creating or modulating gene regulatory networks, particularly through effects on transcription. However, several classes of elements are also known to affect alternative RNA processing events. We previously showed that the major allele of nuclear export factor Nxf1 in Mus musculus castaneus mice acts as a semi-dominant suppressor of de novo mutations caused by intracisternal A particle (IAP) endogenous retroviruses that integrate into introns, disrupting normal RNA processing. Here we show that this suppressor allele of Nxf1 can coordinately modify gene expression phenotypes at several endogenous loci in the C57BL/6 mouse reference genome that contain IAP sequences in their introns. This quadruples the number of known insertional events modified by Nxf1 and extends the effect beyond overt mutations. We previously used transgenic mice and viral vector mediated overexpression to demonstrate Nxf1 as the modifier gene for de novo insertions. Here we use direct genome editing in mouse one cell embryos to create custom germline alleles at the endogenous Nxf1 locus to show that a specific amino acid substitution, E610G, quantitatively accounts for the Nxf1 modifier gene activity.
Introduction
Endogenized retroviruses and other molecular parasites frequently influence expression of host genes at sites of insertion. Chromosomal insertions of these mobile elements can alter initiation, splicing, or termination of host gene transcripts, in quality or amount. Remnants of ancient insertion and transposition events that survived selection are thought to have shaped gene expression patterns in modern animals substantially [1–3]. In populations where mobile elements remain highly active, such events can account for a substantial fraction of functional polymorphism and spontaneous mutations. Two families of elements in laboratory mice, Intracisternal A Particle (IAP) and MusD/Early Transposon (ETn) families account for 10–20% of spontaneous mutations [4–6], depending on strain background [6]. While some of these mutations interrupt coding exons [7] or induce novel patterns of transcription [8–11], the majority comprise intronic insertions that introduce alternative splicing or transcript termination (or both), resulting in quantitative loss of normal host gene products.
Alternative processing of nascent transcripts is regulated at several levels [12]. Pre-mRNA splicing typically occurs co-transcriptionally, regulated by a variety of DNA and RNA binding factors that together defines and acts on constitutive exons. Transcriptional initiation complexes may assemble splicing factors on the Pol II complex, resulting in promoter-dependent alternative splicing [13–15]. Elongation rate of the RNA polymerase complex may influence alternative splicing by regulating the appearance of downstream acceptor sites relative to the splicing kinetics for weaker upstream sites [16, 17]. In addition, recent single-molecule imaging data supports post-transcriptional splicing for at least some alternative splice sites [18]. Identification of post-transcriptional alternative splicing events suggests opportunities for regulation by nuclear ribonucleoprotein (RNP)-associated proteins that are not normally found in the nuclear speckles associated with constitutive splicing.
We previously reported a wild-derived variant of the mouse mRNA nuclear export factor gene Nxf1 within the Modifier-of-vibrator-1 (Mvb1) locus as a genetic modifier for each of six mutations caused by insertion of an IAP element into an intron of a host gene [19, 20]. The suppressing allele, Nxf1CAST, defines the major haplotype in wild isolates of Mus musculus castaneus. For each of the six mutations, genetic suppression altered the balance of alternative RNA processing between virus-dependent isoforms and splicing to the 3’ exon. This mechanism appeared highly selective, operating on sense-strand IAPs (six out of seven tested), but not MusD/ETn (0/6), MuLV (0/1), VL30 (0/1), or L1 LINE (0/1) insertions. These studies also identified an exception to the rule: AtrnmgL, which includes a “full-length” IAP, was not suppressed, in contrast to the more frequent IΔ1 class [21] in the six suppressed mutations [19]. We proposed that sequences deleted in IΔ1 elements might in full-length elements mediate additional repressive events that are epistatic to the apparent splicing defects modified by Nxf1CAST, but acknowledged that local genomic context could also play a role. We also proposed that Nxf1CAST effects on alternative splicing are highly specific, but genome-wide analysis of Nxf1CAST effects on either general alternative splicing or alternative splicing at genes whose reference allele includes an IAP element have not been reported.
Here we show that Nxf1CAST has modifier gene activity toward IAP insertion alleles present in the C57BL/6J (B6) reference genome–this includes several full-length IAPs, an IAP in Adamts13 with a novel deletion but otherwise high sequence similarity to the non-suppressed element in AtrnmgL, and two other IAP deletion classes outside of the IΔ1 group. Quantification of well-characterized alternative splicing events, including cassette exon, retained intron, alternative splice donor, and alternative splice acceptor sites detect no Nxf1 modifier effect at non-IAP introns, further supporting the specificity of Nxf1CAST activity toward IAP insertion alleles. We also demonstrate by genome editing that a single nucleotide substitution, encoding a glutamate to glycine amino acid replacement in the carboxyterminal UBA-like domain, is sufficient to explain the semi-dominant modifier phenotype of Nxf1CAST. These results expand the range of IAP family elements modified by Nxf1 variant alleles and identify a single residue in Nxf1 protein whose effect on stability, kinetics, or interactions explain the modifier mechanism.
Results
Nxf1CAST modifies Adamts13S, an atypical deleted-IAP allele in B6 mice
Because congenic Nxf1CAST showed modifier activity against several insertions of the IAP IΔ1 subfamily, but not against a single example of a full-length element [19, 20], we tested its activity on a well-studied IAP allele of a different class (Fig. 1). Adamts13 encodes a large circulating protease responsible for processing multimeric von Willebrand factor (vWF) in vivo [22, 23]; mutations in human ADAMTS13 cause thrombotic thrombocytopenic purpura [24] and variations in ADAMTS13 activity are associated with other thrombotic abnormalities [25, 26]. In the Adamts13S allele present in several inbred strains, an IAP insertion into intron 24 results in an alternatively spliced, stable transcript that terminates in viral sequences, but lacks downstream exons encoding the final two thrombospondin repeats and two CUB (C1r/C1s, Uegf, Bmp1) domains [27]. A very small amount of residual exon 24 to exon 25 splicing can be detected by a quantitative reverse transcription–polymerase chain reaction (qRT-PCR) assays (Ref. 27 and Fig. 1A). The Adamts13S IAP sequence is highly similar to the AtrnmgL IAP (Fig. 1B), but differs in having a 1131-bp deletion in the pol coding region (Fig. 1B), distinct from and smaller than the classical IΔ2 deletion [21]. Of 2329 bp among 107 aligned sites that distinguish the AtrnmgL IAP from the IΔ1 elements, only 33 sites (61 bp) are not shared between AtrnmgL and Adamts13S IAPs.

Congenic Nxf1CAST alleles increased the amount of Adamts13 RNA that is correctly spliced from exon 24 to exon 25. Since Adamts13 is most robustly expressed by hepatic stellate cells [28, 29], we assayed RNA from replicate sets of liver samples. Measurements in previously collected samples from 29 F2 progeny of a BALB/c X C57BL/6J–Nxf1B6/Nxf1CAST intercross [19], in which the Adamts13S allele was fixed, showed a significant increase in full-length Adamts13 relative to standard reference genes (Gapdh or Ppia) in animals with the Nxf1CAST allele, although variance in the measurement was unusually high (Fig. 1C). To clarify the effect of Nxf1CAST, we examined qRT-PCR from liver RNA of C57BL/6J–Nxf1CAST congenic stock, using Desmin as a cell type-specific reference gene to control for proportion of hepatic stellate cells. This confirmed an increase in fully spliced Adamts13 in Nxf1CAST animals, with much lower variance (Fig. 1D). The two experiments together indicate ~2-4-fold elevation in abundance of full-length Adamts13 in Nxf1CAST compared with Nxf1B6.
Nxf1CAST suppresses IAPs that correlate with reduced expression
The C57BL/6J reference genome includes more diverse examples of sense-oriented IAPs in host gene introns. To allow a more comprehensive survey of IAP structural classes that can be modified by Nxf1CAST, we systematically identified IAP elements using a combination of sequences that are conserved across IAP subfamilies and RepBase annotations (S1 Fig). Comparison of IAP and host gene strand orientation confirms the well-known orientation bias [20, 30] against sense-strand insertions (323 sense vs. 969 antisense) and shows a significant difference in size distribution, with sense-oriented insertions having a larger proportion of elements ≤ 1 kb (210/323, compared with 525/969 antisense; p = 7.3x10-4, Fisher’s exact test). The set of sense-oriented insertions was manually curated to remove non-intronic events (mostly short IAP sequences in 3’ ends that were otherwise homologous with 3’ ends in corresponding human or rat genes) and likely annotation errors (mostly tandem gene duplications with the IAP placed between duplicate copies). Of these, 85 were > 3 kb, 58 of which had been previously reported [31] (S2 Fig).
Intronic IAP elements in the reference genome predict decreased expression relative to strains that lack the insertion (Fig. 2). We designed unique qRT-PCR assays for exon-exon splicing across 49 IAP-containing introns, of which 41 passed initial quality control measures (4 solo LTRs and 36 larger elements). Commercial hydrolysis probe (TaqMan) assays were purchased for 5 additional sites (1 solo LTR and 4 larger elements). In draft assemblies of A/J, BALB/c, and 129S1 genomes available from the Wellcome Trust Sanger Institute [32, 33], 39 insertions >3 kb and all 5 solo LTR insertions were absent in at least one strain. While other factors may influence strain-dependent expression of any given locus, the presence of sense-strand insertions >3 kb correlated with lower levels of the assayed splice junction. Among 39 polymorphic sites, 20 had significantly lower expression in strains with the insertion, 2 had nominally significant differences in the opposite direction, and 17 were not significantly different. Effect sizes of significant difference ranged from 1.2 to 1000-fold in relative expression. These results confirmed that intronic IAPs fixed in the B6 genome correlate with reduced expression of the correctly processed RNA.

Congenic Nxf1CAST alleles increased relative expression of processed RNA at these same sites. We assayed correctly spliced expression at 44 sites in paired brain samples homozygous for either Nxf1B6 or Nxf1CAST alleles (Fig. 2). The effect of Nxf1 genotype on level of correctly spliced RNA was independently significant at 14 sites, including full-length, IΔ1, and other deletion classes of IAPs. Class-specific statistics and false discovery rates are listed in Table 1 and S1 Table. Notably, Nxf1CAST also suppressed the effect of an IAP in an AT/AC intron in Gbe1, suggesting that suppression is not dependent on the major spliceosome. The magnitude of statistically supported effects ranged from 1.1 (Cdh19) to 7.8 (Zfp69) in these assays. Furthermore, pooled data from full-length and IΔ1 insertion sites that showed a significant difference between inserted and uninserted strains, but not individually between Nxf1 alleles, nonetheless showed group-wise significance for suppression by Nxf1CAST (p = 0.0012, Wilcoxon signed rank test with continuity correction). These results demonstrate that the Nxf1 congenic alleles have genetic modifier activity toward a diverse set of IAP elements and genomic contexts.

Nxf1 genotype does not influence alternative splicing choice at non-IAP sites
To test further the specificity of Nxf1 variant effect on alternative splicing, we examined 12 alternative splicing choices at well-characterized alternative splice sites [34] and alternative splice sites in genes with known functional connections to Nxf1, reasoning that these might be most likely to be influenced by variation in Nxf1. None of these introns contain sense-oriented IAP sequences. RT-PCR competition assays that amplify both alternative products were used and the relative ratio of alternative products in each sample quantified by fluorescence imaging after gel electrophoresis (Figs. 3A and S3). No significant differences were observed among 12 candidate sites tested.
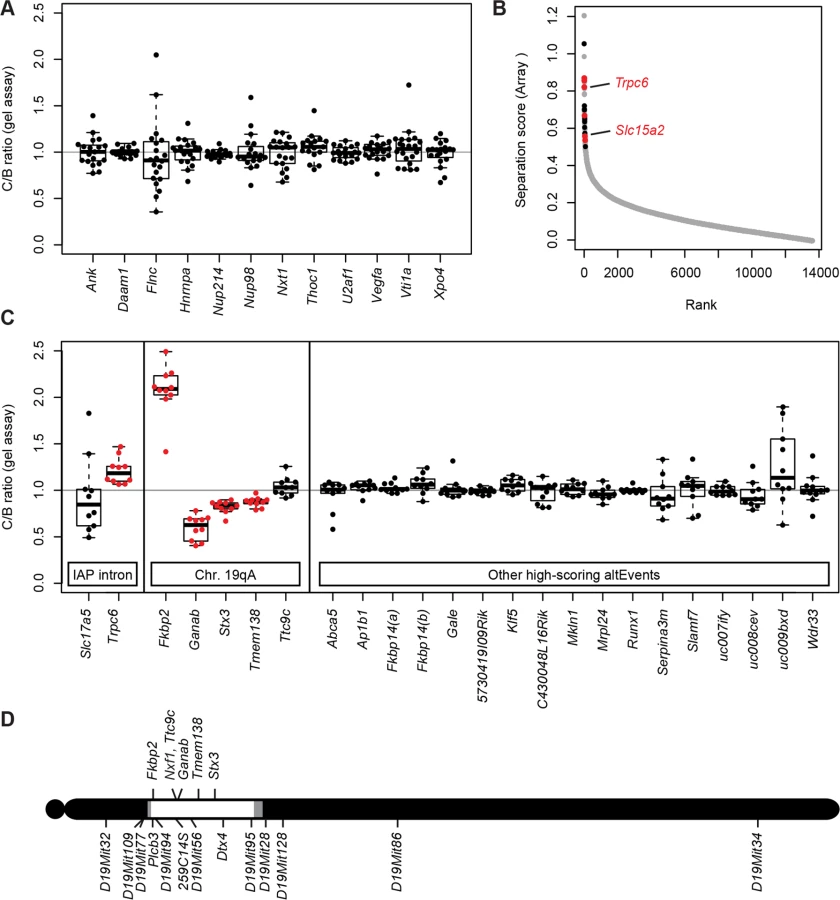
As a more general test of modifier activity on non-IAP alternative splicing choices, we measured brain RNA samples from three same-sex littermate pairs from the Nxf1 congenic stock using genome-wide splicing-sensitive microarrays (Fig. 3B). The Affymetrix MJAY arrays interrogated 13,002 known or imputed alternative junctions in mouse genes, including alternative splice site choices and alternative 5’ and 3’ ends. Approximately 1868 events were called with high confidence (q≤0.05) in this experiment. Using established computational methods [35] to analyze this data, we identified only 43 sites with significant deviations between paired samples (Separation score absolute value >0.5 [~1.4-fold change] and False Discovery Rate q<0.05). Of these, 10 were within the CAST/Ei congenic interval on chromosome 19; due to the high polymorphism rate between B6 and CAST, these are likely due to either mismatch with the probe sequence or to intrinsic allelic differences independent of Nxf1. Interestingly, three of the remaining 33 sites included sense-oriented IAP insertions in the corresponding intron: Trpc6, Slc15a2, and Slc17a5, although most IAP-related alternative RNA junctions were not represented in the array.
To test whether the sites implicated by the array analysis were replicable differences rather than probe sequence polymorphisms or statistical fluctuation across a large number of measurements, we designed gel-based RT-PCR assays across each intron for 12 of the most extreme scores on each end of the distribution (Fig. 3C). Of these, four sites tested from chromosome 19 (Fkbp2, Ganab, Stx3, and Tmem138), showed significant differences, suggesting an intrinsic processing difference between B6 and CAST/Ei alleles in those genes. Of three IAP-containing introns detected by the arrays Slc15a2 and Trpc6 were confirmed by either qRT-PCR (Fig. 2) or both qRT-PCR and the gel assay, while Slc17a5 could not be confirmed. Thus, 6 out of 8 IAP or chromosome 19 sites identified by the MJAY arrays replicated in an independent assay. By contrast, none of 17 assays for unlinked, non-IAP splice sites showed a nominally significant difference between Nxf1 alleles. Lack of replication for these events, compared with very high replication rates for IAP-containing events and sites within the Nxf1-congenic interval on chromosome 19 (Fig. 3D), further suggest that very few if any non-IAP sites are sensitive to this Nxf1 allele, consistent with discovery rates seen in previous experiments with this array platform [36–39]. While these data cannot rule out the possibility of a non-IAP alternative processing target for Nxf1 modifier activity at an unmonitored site or below the limit of detection, together they show that Nxf1CAST modifier activity does not act broadly on alternative splicing and provides further evidence for very strong IAP selectivity.
Construction of Nxf1 E610G mice by genome editing in mouse one cell embryo
The precise genetic variant or variants responsible for the modifier activity of Nxf1 have not been previously determined. As Nxf1 steady-state RNA and protein levels did not appear different between alleles, we reasoned that one or both of two amino acid substitutions that differentiate the alleles was likely causal. Because the E610G variant occurred in a highly-conserved domain and appeared to be the last variant that arose in a 19-marker haplotype before that haplotype rose to high frequency in wild mice [20], we targeted this site for genome editing in mouse one cell embryos, using a synthetic Cas9 mRNA and a single guide RNA (Fig. 4A) co-injected with an oligonucleotide template for homology-dependent repair (Fig. 4B), essentially as described [40]. Two potential founders survived. Each survivor was male and heterozygous for two distinctly edited alleles–(1) a correctly edited allele containing both the E610G polymorphism and a silent polymorphism in the sgRNA homology to deter further cleavage, and (2) either a “pseudo-edited” allele, carrying the induced silent polymorphism, or the E610G edit together with a 3-bp deletion of the adjacent valine codon (Fig. 4C). Both males bred and transmitted each allele. All four transmitted alleles were detected by PCR sequencing tail-clip DNA from the founders and their offspring. Sequencing of 17 predicted [41] off-target cleavage sites from the same samples identified no additional mutations, suggesting that off-target editing was not frequent in either founder.

Nxf1 E610G edited allele recapitulates the Modifier of vibrator
The hypothesis that the E610G polymorphism is sufficient for the original modifier phenotype makes three explicit predictions: that the edited allele should suppress vibrator phenotypes relative to the background Nxf1B6 (and silent, pseudo-edited) allele, that it should act semi-dominantly (and therefore be measurable in heterozygotes), and that it should be indistinguishable from the congenic Nxf1CAST allele in magnitude of effect. We examined 68 sequential vibrator (Pitpnvb/vb) mutant and 47 control G2 animals in backcrosses of our edited alleles to the B6–Pitpnavb/+, Nxf1B6/CAST stock (Fig. 5A). The E610G edited allele, but not the pseudo-edited control allele, altered the tremor phenotype of vibrator mutants–scored by observers blind to genotype–relative to the Nxf1B6 allele (S1_Video and S2_Video). This held for heterozygotes in trans to the Nxf1B6 allele (p = 7.9x10-6, Wilcoxon Rank Sum Test) and in trans to the Nxf1CAST allele (p = 2.0x10-4). The E610G edited allele was indistinguishable from the congenic Nxf1CAST allele in each context (Fig. 5A).

In a subset of animals followed for survival analysis, the edited allele prolonged the viability of vibrator mutants to the same degree as the original congenic allele, while the pseudo-edited allele did not (Fig. 5B). A Cox proportional hazards model showed significantly better survival with the edited allele (p = 0.0013) but not the pseudo-edited allele (p = 0.27) compared with the parental Nxf1B6 allele when heterozygous with respect to Nxf1B6. Similarly, among animals heterozygous to the congenic Nxf1CAST allele the observed deaths all occurred in unedited animals. Survival in unedited animals was different from non-mutant control and Nxf1CAST homozygotes (p = 0.046) under the proportional hazards model, while edited animals were not (p>0.99).
Most of the backcross animals were processed for gene expression analysis. The edited allele increased the expression of correctly spliced Pitpna mRNA from vibrator mutant alleles, again comparable to the congenic allele, as measured by qRT-PCR from brains of the same animals (Fig. 5C). Non-parametric tests supported significantly increased expression in each of the four predicted pair-wise comparisons and in each combination the edited allele was indistinguishable from the congenic allele. These data together show that the single amino acid substitution E610G is sufficient to account quantitatively for the previously observed Modifier of-vibrator (Mvb1) activities of the Nxf1CAST allele in vivo.
Nxf1 E610G edited allele is a general suppressor of IAP insertions
The discovery of B6-endogenous targets of the modifier activity in our original congenic stock (Figs. 1 and 2) predicts that if E610G amino acid substitution allele explains the modifier activity, it should act on expression level of those genes as well. We tested this prediction with 12 qRT-PCR assays on F1 samples that were heterozygous for the edited allele, but without potentially confounding Pitpnvb mutations (Fig. 6). Both Adamts13 and Nsdhl showed suppression by the edited allele equivalent to the congenic allele in liver, where both are well-expressed (Fig. 6A). In brain cDNA, six of seven genes that were suppressed by the congenic stock (Agbl4, Agtr1a, Cdh19, L3mbtl4, Pla2g4e, and Slc15a2) showed effects of the heterozygous edited allele, while the seventh (Fhit) did not approach independent significance in the heterozygote samples (Fig. 6B). In addition, two of three genes for which the congenic experiments showed a trend but did not provide significant support (Sntg1 and Dph5) also showed significant evidence of suppression by the edited allele (Fig. 6B). Each event survived correction for false discovery rate below or near q = 0.05 (S1 Table). The tested sites, including elements closely related to the non-suppressed insertion in AtrnmgL, other full-length elements, IΔ1 elements, and other deletion classes, were significantly increased in a semi-dominant manner by the edited allele. Alternative processing events on chromosome 19 that differed between congenic stocks (Fig. 3C,D) were not significantly different between edited and unedited alleles (S4 Fig), confirming that they represented strain variations in each gene rather than effects of the modifier locus. Together, these measurements confirm that the E610G variant is able to suppress IAP-related gene expression changes at the wide array of sites detected for the congenic allele by our genome-wide screen and explains the modifier effect of previous congenic and transgenic strains [19, 20].

Most of the IAP sequence is not required for mutation or suppression by Nxf1
We next asked whether identifiable sequence features correlated with expression level in inserted strains or response to Nxf1 genotype. The ratio of expression in uninserted vs. inserted strains (U/I ratio) was nominally correlated with IAP length (Spearmen’s ρ = 0.32, p = 0.05) primarily due to lack of effect among LTR-only events and extreme values for Adamts13 and Zfp69 (S5 Fig). Intron size, flanking exon size, distance between the element and each exon, and annotation features of the exons for potential alternative events had no significant correlation.
The expression ratio between Nxf1CAST vs. Nxf1B6 alleles (C/B ratio) was positively correlated with U/I ratio (Spearman’s ρ = 0.44, p = 0.0058). Intron length, distance from 5’ exon to the IAP, IAP length, length of each flanking exon, and whether 5’ or 3’ splice site showed evidence for alternative usage in UCSC annotations showed no significant correlation with the expression ratio between Nxf1 alleles (S6 Fig). However, distance between IAP and 3’ exon approached a nominal significance threshold (ρ = -0.30, p = 0.051) before correction for multiple tests.
A simple alignment among elements that had shown both reduced expression in inserted strains and suppression by either Nxf1CAST or E610G alleles showed that IAP protein coding sequences were not required for either effect, leaving only LTRs, a small sequence 5’ to gag, ~60 bp in four small fragments within pol, and sequences 3’ to pol that include both RNA transport element (RTE) and the polypurine tract (ppt). For classification of elements as suppressed, we used Fisher’s combined probability test as a meta-analysis across the congenic homozygous and genome-edited heterozygous samples (S1 Table). We further characterized each element for presence and sequence divergence in eight segments: LTR, segment 2 (between LTR and gag), gag CDS, prt CDS, pol CDS, segment 6 (after pol), RTE, and polypurine tract. Phylogenetic trees for either full length or discreet segments of the IAP element did not correlate with either differences between inserted and uninserted strains or effect of Nxf1 genotype (S7 Fig). Although suppressed insertions appear somewhat clustered with respect to RTE, identical RTE sequences were associated with divergent effects on host gene RNA (S7 Fig, panel J). Predicted RNA folding also did not show any strict requirement for suppression or lack of suppression by Nxf1 alleles (S8 Fig). These data together show that most of the IAP consensus sequence is not required and suggest that a minimum element for both mutation and Nxf1-mediated suppression might require less than 2 kb (837 bp 5’ in Cdh19 and 1108 bp 3’ in Sntg1), but that efficiency might be influenced by other factors–possibly including elements size, distance to the 3’ exon, and factors not readily predicted from sequence alone.
Discussion
As far as we are aware, Nxf1 remains the most highly-connected node for genetic interactions among modifier genes that act on either natural or chemically-induced mutations in mice [19, 42]. The current results add three times as many allele-specific genetic interactions for Mvb1 alleles of Nxf1 (CAST and E610G) as had previously been reported. We previously showed that the most common Mus musculus castaneus allele of Nxf1, containing many polymorphic sites, suppressed the classical vibrator mutation [20] and that a congenic stock including this allele suppressed several other IΔ1 IAP insertional mutations [19]. Our results extend the diversity of IAP insertions susceptible to Nxf1 substantially beyond the IΔ1 subfamily (Fig. 2) and provide further evidence against activity on alternative processing at non-IAP introns (Fig. 3). Alternative events suppressed by Nxf1 alleles include known bleeding exons with alternative termination, such as Slc15a2 [43] and Trpc6 [30], and IAP-dependent alternative splicing, such as Adamts13 [27, 44] and Cpne8, as well as introns for which public transcriptome data do not indicate a predominant alternative variant. Comparison of IAP elements whose effects can be suppressed by Nxf1 alleles further suggests minimum sequence requirements for both mutation and suppression lie in <1 kb in the 5’ end and ~1 kb in the 3’ end of the IAP (Fig. 7), although construction and experimental measurements of a minimal element will be required to test this idea.

In contrast to most de novo mutations caused by IAP insertions, the magnitude of gene expression changes is relatively modest for most of the B6-endogenous IAP sites we tested (Fig. 2). Indeed, recent evaluation of mouse transposable elements concluded that insertion events that strongly affected gene expression were rapidly purged from populations, hence remaining elements tend toward more modest effect sizes [45]. Adamts13 is a notable exception, with ~1000-fold change in completion of the transcript across the inserted intron (Fig. 1). However, as the truncated Adamts13 encoded by the major B6 transcript retains substantial function [44, 46] and the absolute level of full-length transcript recovered by suppression is modest, little physiological consequence is expected from this increase in full-length Adamts13 protein above the level of function provided by the abundant truncated form. Taken all together the data suggest that using IAP insertional mutations in combination with Nxf1 suppressing alleles should allow relatively clean titrations of intermediate gene doses. This may have application in modeling genetic disorders (and therapeutics) in mice.
Our results using genome-edited mice provide molecular precision to the definition of the modifier allele. Although several polymorphisms distinguish Nxf1B6 and Nxf1CAST alleles in both transgenic [20] and congenic [19] experiments, the E610G single amino acid substitution (Fig. 4) was sufficient for the full range of genetic modifier activity previously ascribed to the congenic locus and published 16 kb transgene. The point mutation modified all of the principle features of the vibrator mutation on which the modifier activity was first described [47] with the characteristic semi-dominant effect (Fig. 5). The ability to assay the edited allele as a heterozygote with respect to congenic Nxf1B6 and Nxf1CAST alleles and the specificity of multiple phenotypes assayed (Figs. 5, 6) also provides an additional safeguard against potential off-target modifications in the editing process.
Our results demonstrate that a single amino acid substitution in the C-terminal, UBA-like domain of Nxf1 simultaneously enhances expression of full-length mRNA at more than a dozen loci in the mouse reference genome at the level of pre-mRNA processing. This illustrates a potential post-transcriptional mechanism for evolution of gene regulatory networks selected from events generated by molecular parasites [3] and provides a post-transcriptional tool for modulating expression level of any mouse gene into which an IAP element can be introduced.
Methods
Ethics statement
All animal procedures were approved by the University of California, San Diego Institutional Animal Care and Use Committee (IACUC). Tissues were obtained after terminal anesthesia with tribromoethanol (avertin)
Mice
Congenic C57BL/6J (B6)–Nxf1CAST mice were developed and maintained in our laboratory by serial backcross and genotyped by PCR as previously described [19]. Mice in this report were at backcross generation N30 or later. A/J, BALB/cJ and 129S1/SvImJ were purchased from The Jackson Laboratory and bred locally in the same vivarium room with our congenic stocks. Adamts13 genotypes were determined by PCR using a three primer combination: ACCTCTCAAGTGTTTGGGATGCTA, TCAGCGCCATCTTGTGACGGCGAA, and TGCCAGATGGCCATGATTAACTCT [27]. Tissues were from paired F2 animals from a B6 x BALB/c intercross previously described [19] (Fig. 1C) or B6 congenic or co-isogenic stocks (all others). Five male and five female pairs were used for measurements from congenic animals. No sex-specific differences were noted. Genome-edited mice were produced in the Moores UCSD Cancer Center Transgenic Mouse Shared Resource by co-injection of mouse C57BL/6 one-cell embryos with sgRNA, Cas9 mRNA, and unpurified oligonucleotide as a homology-dependent repair donor essentially as described [40]. Oligonucleotide sequences for sgRNA, Cas9 amplification, and homology dependent repair are given in S2 Table. Measurements from genome edited mice used unpaired samples collected as a cohort and comprising ~equal numbers of each sex.
Identification of B6 alleles with IAP element insertions
Two independent approaches identified intronic IAP elements. IAP annotations from the Repbase Update definitions were mined from the RepeatMasker track data table in the UCSC Genome Browser (S1 Fig). A custom table was also developed by iterative BLAT searches with a panel of short (35 bp) sequences conserved in subfamilies of IAP elements for which we had previously observed Nxf1-related suppression (S3 Table). Both sets were filtered by strand orientation in Galaxy [48] and compared to UCSC intron or RefSeq gene boundary annotations. After manual curation to remove likely annotation errors, the oligonucleotide-derived set identified 70 and the Repbase Update annotations identified a superset of 85 IAP-family elements within well-defined introns or 3’ ends that were premature relative to human or rat orthologs. Alignments of IAP sequences were performed in MUSCLE (http://www.ebi.ac.uk/Tools/msa/muscle/) using default parameters.
Quantitative RT-PCR assays
PCR assays for transcripts that are correctly spliced around the IAP insertion and for reference genes were designed relative to exon junctions using Primer3 [49]. Assays were selected for use if they produced a single product by melt profile, single band by gel imaging, and low coefficient of variation among replicate samples. Specific primers used are provided in S4 Table. RNA was prepared from tissues homogenized in Trizol, converted to cDNA by reverse transcription, and assayed by quantitative PCR using SYBR green fluorescence in a Bio-Rad CFX instrument as previously described [19]. Normalization was performed by geometric averaging among the indicated reference genes, as described [50], and implemented in software supplied by the manufacturer. Four reference gene assays, Gapdh, Ppia, Sdha, and Pitpna were selected based on high cycle efficiencies, accurate dose titration, and low quantitative variation across tested conditions (only the first three were used for comparisons including vibrator samples, which express ~18% normal level of Pitpna).
Gel-based assays
Relative quantification of alternative splicing products was carried out by gel electrophoresis of products from non-saturating 3-primer competitive PCR amplifications based on pilot experiments for each assay. Samples for comparison were amplified side-by-side and resolved on the same gel. Relative fluorescence intensities of resolved bands were quantified from non-saturated. tiff image files using ImageJ software.
Splicing-sensitive array profiling
Affymetrix MJAY microarrays (GEO platform GPL13185) were hybridized and processed as described [35, 39], for brain RNA from 3 pairs of littermates (two female pairs, one male pair) with opposite Nxf1 genotypes. The separation score was defined as the log2 (ratio of alternative events in Nxf1CAST samples / ratio of alternative events in Nxf1B6 samples) [36]. Events with a separation score >0.5 (~1.4-fold change) and false discovery rate q<0.05 were deemed significant.
Statistics
Statistical tests were conducted in R. Non-parametric rank tests were performed in package exactRankTests or coin; both packages gave identical results for repeated analyses. Non-parametric correlation tests with ties were performed in the package Hmisc. Correlation analyses and standard hypothesis tests were conducted in R 2.8.1 or later running on Mac OS 10.5.8 or later.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Cowley M, Oakey RJ. Transposable elements re-wire and fine-tune the transcriptome. PLoS genetics. 2013;9(1):e1003234. Epub 2013/01/30. doi: 10.1371/journal.pgen.1003234 23358118
2. Gifford WD, Pfaff SL, Macfarlan TS. Transposable elements as genetic regulatory substrates in early development. Trends Cell Biol. 2013;23(5):218–26. Epub 2013/02/16. doi: 10.1016/j.tcb.2013.01.001 23411159
3. Peter IS, Davidson EH. Evolution of gene regulatory networks controlling body plan development. Cell. 2011;144(6):970–85. Epub 2011/03/19. doi: 10.1016/j.cell.2011.02.017 21414487
4. Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420(6915):520–62. 12466850
5. Hamilton BA, Frankel WN. Of mice and genome sequence. Cell. 2001;107(1):13–6. 11595181
6. Maksakova IA, Romanish MT, Gagnier L, Dunn CA, van de Lagemaat LN, Mager DL. Retroviral elements and their hosts: insertional mutagenesis in the mouse germ line. PLoS genetics. 2006;2(1):e2. 16440055
7. Vasicek TJ, Zeng L, Guan XJ, Zhang T, Costantini F, Tilghman SM. Two dominant mutations in the mouse fused gene are the result of transposon insertions. Genetics. 1997;147(2):777–86. 9335612
8. Duhl DM, Vrieling H, Miller KA, Wolff GL, Barsh GS. Neomorphic agouti mutations in obese yellow mice. Nature Genet. 1994;8(1):59–65. 7987393
9. Lugani F, Arora R, Papeta N, Patel A, Zheng Z, Sterken R, et al. A retrotransposon insertion in the 5' regulatory domain of Ptf1a results in ectopic gene expression and multiple congenital defects in Danforth's short tail mouse. PLoS genetics. 2013;9(2):e1003206. Epub 2013/02/26. doi: 10.1371/journal.pgen.1003206 23437001
10. Semba K, Araki K, Matsumoto K, Suda H, Ando T, Sei A, et al. Ectopic expression of Ptf1a induces spinal defects, urogenital defects, and anorectal malformations in Danforth's short tail mice. PLoS genetics. 2013;9(2):e1003204. Epub 2013/02/26. doi: 10.1371/journal.pgen.1003204 23436999
11. Vlangos CN, Siuniak AN, Robinson D, Chinnaiyan AM, Lyons RH Jr., Cavalcoli JD, et al. Next-generation sequencing identifies the Danforth's short tail mouse mutation as a retrotransposon insertion affecting Ptf1a expression. PLoS genetics. 2013;9(2):e1003205. Epub 2013/02/26. doi: 10.1371/journal.pgen.1003205 23437000
12. Bentley DL. Coupling mRNA processing with transcription in time and space. Nat Rev Genet. 2014;15(3):163–75. Epub 2014/02/12. doi: 10.1038/nrg3662 24514444
13. Auboeuf D, Honig A, Berget SM, O'Malley BW. Coordinate regulation of transcription and splicing by steroid receptor coregulators. Science. 2002;298(5592):416–9. 12376702
14. Cramer P, Pesce CG, Baralle FE, Kornblihtt AR. Functional association between promoter structure and transcript alternative splicing. Proc Natl Acad Sci U S A. 1997;94(21):11456–60. Epub 1997/10/23. 9326631
15. Kornblihtt AR. Promoter usage and alternative splicing. Curr Opin Cell Biol. 2005;17(3):262–8. 15901495
16. de la Mata M, Kornblihtt AR. RNA polymerase II C-terminal domain mediates regulation of alternative splicing by SRp20. Nature structural & molecular biology. 2006;13(11):973–80.
17. Kornblihtt AR. Chromatin, transcript elongation and alternative splicing. Nature structural & molecular biology. 2006;13(1):5–7.
18. Vargas DY, Shah K, Batish M, Levandoski M, Sinha S, Marras SA, et al. Single-molecule imaging of transcriptionally coupled and uncoupled splicing. Cell. 2011;147(5):1054–65. Epub 2011/11/29. doi: 10.1016/j.cell.2011.10.024 22118462
19. Concepcion D, Flores-Garcia L, Hamilton BA. Multipotent genetic suppression of retrotransposon-induced mutations by Nxf1 through fine-tuning of alternative splicing. PLoS genetics. 2009;5(5):e1000484. doi: 10.1371/journal.pgen.1000484 19436707
20. Floyd JA, Gold DA, Concepcion D, Poon TH, Wang X, Keithley E, et al. A natural allele of Nxf1 suppresses retrovirus insertional mutations. Nature Genetics. 2003;35 : 221–8. 14517553
21. Kuff EL, Lueders KK. The intracisternal A-particle gene family: structure and functional aspects. Advances in cancer research. 1988;51 : 183–276. 3146900
22. Fujikawa K, Suzuki H, McMullen B, Chung D. Purification of human von Willebrand factor-cleaving protease and its identification as a new member of the metalloproteinase family. Blood. 2001;98(6):1662–6. 11535495
23. Gerritsen HE, Robles R, Lammle B, Furlan M. Partial amino acid sequence of purified von Willebrand factor-cleaving protease. Blood. 2001;98(6):1654–61. 11535494
24. Levy GG, Nichols WC, Lian EC, Foroud T, McClintick JN, McGee BM, et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. 2001;413(6855):488–94. 11586351
25. Nolasco LH, Turner NA, Bernardo A, Tao Z, Cleary TG, Dong JF, et al. Hemolytic uremic syndrome-associated Shiga toxins promote endothelial-cell secretion and impair ADAMTS13 cleavage of unusually large von Willebrand factor multimers. Blood. 2005;106(13):4199–209. 16131569
26. Fujioka M, Hayakawa K, Mishima K, Kunizawa A, Irie K, Higuchi S, et al. ADAMTS13 gene deletion aggravates ischemic brain damage: a possible neuroprotective role of ADAMTS13 by ameliorating postischemic hypoperfusion. Blood. 2010;115(8):1650–3. doi: 10.1182/blood-2009-06-230110 19965676
27. Banno F, Kaminaka K, Soejima K, Kokame K, Miyata T. Identification of strain-specific variants of mouse Adamts13 gene encoding von Willebrand factor-cleaving protease. J Biol Chem. 2004;279(29):30896–903. 15136581
28. Uemura M, Tatsumi K, Matsumoto M, Fujimoto M, Matsuyama T, Ishikawa M, et al. Localization of ADAMTS13 to the stellate cells of human liver. Blood. 2005;106(3):922–4. 15855280
29. Zhou W, Inada M, Lee TP, Benten D, Lyubsky S, Bouhassira EE, et al. ADAMTS13 is expressed in hepatic stellate cells. Laboratory investigation; a journal of technical methods and pathology. 2005;85(6):780–8. 15806136
30. Zhang Y, Romanish MT, Mager DL. Distributions of transposable elements reveal hazardous zones in mammalian introns. PLoS Comput Biol. 2011;7(5):e1002046. Epub 2011/05/17. doi: 10.1371/journal.pcbi.1002046 21573203
31. Zhang Y, Maksakova IA, Gagnier L, van de Lagemaat LN, Mager DL. Genome-Wide Assessments Reveal Extremely High Levels of Polymorphism of Two Active Families of Mouse Endogenous Retroviral Elements. PLoS genetics. 2008;4(2):e1000007 doi: 10.1371/journal.pgen.1000007 18454193
32. Yalcin B, Wong K, Agam A, Goodson M, Keane TM, Gan X, et al. Sequence-based characterization of structural variation in the mouse genome. Nature. 2011;477(7364):326–9. Epub 2011/09/17. doi: 10.1038/nature10432 21921916
33. Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 2011;477(7364):289–94. Epub 2011/09/17. doi: 10.1038/nature10413 21921910
34. Barash Y, Calarco JA, Gao W, Pan Q, Wang X, Shai O, et al. Deciphering the splicing code. Nature. 2010;465(7294):53–9. doi: 10.1038/nature09000 20445623
35. Huelga SC, Vu AQ, Arnold JD, Liang TY, Liu PP, Yan BY, et al. Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell reports. 2012;1(2):167–78. Epub 2012/05/11. doi: 10.1016/j.celrep.2012.02.001 22574288
36. Du H, Cline MS, Osborne RJ, Tuttle DL, Clark TA, Donohue JP, et al. Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nature structural & molecular biology. 2010;17(2):187–93. Epub 2010/01/26. doi: 10.1038/nsmb.1720
37. Gehman LT, Stoilov P, Maguire J, Damianov A, Lin CH, Shiue L, et al. The splicing regulator Rbfox1 (A2BP1) controls neuronal excitation in the mammalian brain. Nat Genet. 2011;43(7):706–11. Epub 2011/05/31. doi: 10.1038/ng.841 21623373
38. Lagier-Tourenne C, Polymenidou M, Hutt KR, Vu AQ, Baughn M, Huelga SC, et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nature neuroscience. 2012;15(11):1488–97. Epub 2012/10/02. doi: 10.1038/nn.3230 23023293
39. Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nature neuroscience. 2011;14(4):459–68. Epub 2011/03/02. doi: 10.1038/nn.2779 21358643
40. Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153(4):910–8. Epub 2013/05/07. doi: 10.1016/j.cell.2013.04.025 23643243
41. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nature protocols. 2013;8(11):2281–308. Epub 2013/10/26. doi: 10.1038/nprot.2013.143 24157548
42. Hamilton BA, Yu BD. Modifier genes and the plasticity of genetic networks in mice. PLoS genetics. 2012;8(4):e1002644. Epub 2012/04/19. doi: 10.1371/journal.pgen.1002644 22511884
43. Li J, Akagi K, Hu Y, Trivett AL, Hlynialuk CJ, Swing DA, et al. Mouse endogenous retroviruses can trigger premature transcriptional termination at a distance. Genome Res. 2012;22(5):870–84. Epub 2012/03/01. doi: 10.1101/gr.130740.111 22367191
44. Zhou W, Bouhassira EE, Tsai HM. An IAP retrotransposon in the mouse ADAMTS13 gene creates ADAMTS13 variant proteins that are less effective in cleaving von Willebrand factor multimers. Blood. 2007;110(3):886–93. 17426255
45. Nellaker C, Keane TM, Yalcin B, Wong K, Agam A, Belgard TG, et al. The genomic landscape shaped by selection on transposable elements across 18 mouse strains. Genome Biol. 2012;13(6):R45. Epub 2012/06/19. doi: 10.1186/gb-2012-13-6-r45 22703977
46. Banno F, Chauhan AK, Kokame K, Yang J, Miyata S, Wagner DD, et al. The distal carboxyl-terminal domains of ADAMTS13 are required for regulation of in vivo thrombus formation. Blood. 2009;113(21):5323–9. doi: 10.1182/blood-2008-07-169359 19109562
47. Hamilton BA, Smith DJ, Mueller KL, Kerrebrock AW, Bronson RT, van Berkel V, et al. The vibrator mutation causes neurodegeneration via reduced expression of PITP alpha: positional complementation cloning and extragenic suppression. Neuron. 1997;18(5):711–22. 9182797
48. Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P, et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 2005;15(10):1451–5. Epub 2005/09/20. doi: 10.1101/gr.4086505 16169926
49. Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods in molecular biology (Clifton, NJ. 2000;132 : 365–86. 10547847
50. Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome biology. 2002;3(7):RESEARCH0034. Epub 2002/08/20. 12184808
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 4
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Lack of GDAP1 Induces Neuronal Calcium and Mitochondrial Defects in a Knockout Mouse Model of Charcot-Marie-Tooth Neuropathy
- Proteolysis of Virulence Regulator ToxR Is Associated with Entry of into a Dormant State
- Frameshift Variant Associated with Novel Hoof Specific Phenotype in Connemara Ponies
- Ataxin-2 Regulates Translation in a New BAC-SCA2 Transgenic Mouse Model
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy