
Postnatal Loss of Hap1 Reduces Hippocampal Neurogenesis and Causes Adult Depressive-Like Behavior in Mice
Although the majority of the neurons in the brain are generated during embryonic stage, new neurons are continuously being produced postnatally, and at a much lower rate in adulthood. As postnatal neurogenesis is a key component of the brain maturation process that creates dynamic ‘wirings’ in the brain necessary for an individual to grow, learn, and cope with the external world, attenuated postnatal neurogenesis may affect an individual’s mental stability, rendering a higher susceptibility to depression later in life. In the current study, we genetically ablated the expression of huntingtin-associated protein 1 (Hap1) in mice at various ages or in selective brain regions, and found that early loss of Hap1 significantly reduces postnatal hippocampal neurogenesis, and leads to adult depressive-like behavior. We also found c-kit as an effector to mediate the neurogenesis defect and adult depressive-like phenotype in mice lacking Hap1. The results provide the first genetic evidence to demonstrate the importance of postnatal neurogenesis in adult depression, and may offer new avenues in the prevention and treatment of depression. Our study also has potential implications to other adult-onset mental disorders.
Published in the journal:
. PLoS Genet 11(4): e32767. doi:10.1371/journal.pgen.1005175
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005175
Summary
Although the majority of the neurons in the brain are generated during embryonic stage, new neurons are continuously being produced postnatally, and at a much lower rate in adulthood. As postnatal neurogenesis is a key component of the brain maturation process that creates dynamic ‘wirings’ in the brain necessary for an individual to grow, learn, and cope with the external world, attenuated postnatal neurogenesis may affect an individual’s mental stability, rendering a higher susceptibility to depression later in life. In the current study, we genetically ablated the expression of huntingtin-associated protein 1 (Hap1) in mice at various ages or in selective brain regions, and found that early loss of Hap1 significantly reduces postnatal hippocampal neurogenesis, and leads to adult depressive-like behavior. We also found c-kit as an effector to mediate the neurogenesis defect and adult depressive-like phenotype in mice lacking Hap1. The results provide the first genetic evidence to demonstrate the importance of postnatal neurogenesis in adult depression, and may offer new avenues in the prevention and treatment of depression. Our study also has potential implications to other adult-onset mental disorders.
Introduction
Depression is the most common mental disorder and a leading cause of disability around the world [1, 2]. In the US, the lifetime prevalence for major depression is estimated to be as high as 16.2% [3]. There are a variety of symptoms associated with depression, including anhedonia, depressed mood, fatigue, helplessness, and other cognitive and metabolic abnormalities [4, 5]. Despite its wide influence, the causes of depression have not been made clear, nor have we established effective and long-lasting treatments for it. To gain insight into its etiology, twin studies were conducted to determine whether genetics could play a role in depression. The results revealed that genetic factors account for about 40% of the risk of developing depression, with the remaining 60% being due to environmental factors [6].
Despite various genetic and environmental causes of depression, there must be some common pathways that lead to depressive symptoms. It has been long proposed that deficiency in serotonin (5-HT) level may underlie depression as selective serotonin reuptake inhibitors (SSRIs), the most frequently used antidepressant drugs, work through enhancing extracellular levels of 5-HT [7, 8]. Furthermore, synaptic dysfunction [9, 10], hyperactivity of the hypothalamic-pituitary-adrenal (HPA) axis [11], and expression changes or polymorphisms of brain-derived neurotrophic factor (BDNF) may also be associated with depression [12]. Recently, hippocampal neurogenesis has emerged as an attractive theory of depression [13, 14], largely because many antidepressants are known to enhance hippocampal neurogenesis [15–18], and ablating adult neurogenesis reduces some of the behavioral effects of antidepressants [19]. Of note, many of the common theories of depression such as those aforementioned have also been closely linked to adult neurogenesis [20–23], further accentuating its importance in depressive behavior. Although much of the focus in depression research has been on adult neurogenesis, postnatal neurogenesis, which occurs early in life, is also important as an increasing number of studies found that it is capable of influencing adult depressive behavior [24–27]. Nevertheless, most if not all of these studies used either chemicals or maternal separation to induce a decline in postnatal neurogenesis. To our knowledge, a genetic model for investigating the relationship between postnatal neurogenesis and adult depression is still lacking.
Huntingtin-associated protein a (Hap1) is an intriguing neuronal-enriched protein that interacts with several disease-related proteins, including huntingtin and Ahi1, whose mutations cause Huntington's disease (HD) and Joubert syndrome, respectively [28, 29]. Mounting evidence shows that Hap1 mediates the intracellular transport of several neurotrophic factors and their receptors to support neuronal function and survival, which may require a concerted effort with huntingtin [30–37]. The involvement of Hap1 in BDNF/TrkB trafficking [33–35] may be especially important as BDNF is the most abundant neurotrophic factor in the nervous system and is essential for neuronal activity and the survival of animals. Our recent work revealed that Hap1 regulates early postnatal hypothalamic neurogenesis by stabilizing BDNF/TrkB signaling and that this hypothalamic neurogenesis is critical for the postnatal growth and survival of mice [32]. Earlier work found that neuronal deficiency of the Hap1-binding protein Ahi1 leads to a depressive-like phenotype in adult mice [38]. In the current study, we found that selective depletion of Hap1 expression at postnatal ages also led to adult depressive-like behavior, which was associated with reduced postnatal neurogenesis in the hippocampus. Moreover, we discovered that this decreased neurogenesis is mediated by a different mechanism involving c-kit, a receptor for stem cell factor (SCF) that is also expressed in neural progenitor or stem cells in the rodent brain [39–41]. c-kit is downregulated in Hap1 KO mouse hippocampus because its stability requires Hap1. Our results also demonstrate that overexpression of c-kit in the postnatal hippocampus can augment hippocampal neurogenesis and alleviate adult depression, suggesting a new mechanism by which Hap1 regulates postnatal neurogenesis and consequent adult depressive behavior.
Results
Early Postnatal Depletion of Hap1 Leads to Depressive-Like Behavior in Adult Mice
Since mice with neuronal deficiency of the Hap1-interacting protein Ahi1 show depressive phenotypes [38], we were interested to see whether Hap1 KO mice would display similar phenotypes and whether Hap1 deletion at different ages would affect behavioral outcomes. To test these questions, we used the Cre-LoxP system via tamoxifen (TM) induction to induce Hap1 deletion at different postnatal stages (Fig 1A). Substantial depletion of Hap1 protein expression was achieved in all KO groups as analyzed by western blot (S1 Fig). Two to three months after the TM induction, we subjected the Hap1-depleted mice to the forced swim test (FST) and tail suspension test (TST). These tests showed that early postnatal (Fig 1B and 1C), but not late postnatal (Fig 1D and 1E) or adult (Fig 1F), depletion of Hap1 resulted in significant increases in immobility, which is considered depressive-like behavior. Since the relationship between decreased or altered locomotion and depression has been suggested by previous clinical studies [42–45], we also tested locomotor activity in these mice and found that depletion of Hap1 at an earlier age also resulted in lower locomotor activity in adult mice compared to controls (Fig 1G).

To further strengthen the idea that early postnatal Hap1 depletion leads to an adult depressive phenotype, we crossed floxed-Hap1 mice with camk2a-Cre transgenic mice and obtained Hap1 conditional KO mice in which Hap1 is deleted in camk2a-expressing neurons (Fig 2A). Based on camk2a-promoter activities, Hap1 expression was diminished early postnatally to varying extents in the forebrain areas, with the greatest reduction seen in the cortex and hippocampus (Fig 2B). As camk2a-Hap1 KO mice still maintained a fairly high level of hypothalamic Hap1 expression, which controls feeding and growth [46], we only saw a mild decrease in the body weight gain of these mice at the postnatal stage (Fig 2C). The KO mice displayed a mild decrease in body weight during postnatal life, but the body weight difference between KO and control mice at 4 months old was indiscernible. To see whether camk2a-Hap1 KO mice would behave similarly in the depression tests as the induced Hap1 KO mice, we performed the FST and TST, which demonstrated that camk2a-Hap1 KO mice at 2-month old indeed displayed depressive-like behavior (Fig 2D).

To rule out the possibility that our Hap1 KO mice had any physical impairment that might result in poor performances in depression tests, as well as a locomotor activity assay, we did a rotarod test on both P1 KO and camk2a-Hap1 KO mice; we found no such impairment in either of the KO groups (S2A Fig). Instead, P1 KO mice displayed enhanced performance, which very likely could be the result of their smaller body size allowing them to more readily balance and cling on to the rod. Imipramine, a tricyclic antidepressant, is used in the treatment of major depression and can rapidly reduce depressive phenotypes in mice [38, 47]. To examine whether this drug could rescue the depressive phenotype in Hap1-depleted mice, we delivered imipramine via intraperitoneal injection (i.p.) and found that it substantially increased the mobility of camk2a-Hap1 KO mice in both the FST and TST, more so than it did to the control mice (S2B Fig). As a result, the mobility of the mice after treatment was not significantly different between the genotypes (S2B Fig).
To further validate the depressive phenotype, we assessed anhedonia in camk2a-Hap1 KO mice using the sucrose preference test. Anhedonia, or an inability to experience pleasure, is considered the core feature of major depression [48]. In rodents, the sucrose preference test is used widely to assess anhedonia based on the finding that depressed human patients have higher hedonic responses to sucrose solutions than controls [49]. We found that camk2a-Hap1 KO mice displayed lower preference to 1% sucrose solution than controls, as measurements of fluid intake indicated that, although water intake was not different between the genotypes, camk2a-Hap1 KO mice consumed significantly less sucrose solution and total fluid (Fig 2E). Collectively, these results support a depressive-like phenotype caused by Hap1 depletion.
Early Postnatal Hap1 Depletion Affects Hippocampal Neurogenesis
Hippocampal neurogenesis occurs abundantly after birth and remains active in adulthood. As the involvement of hippocampal neurogenesis in adult depression has been widely demonstrated [13], we next examined whether Hap1 also regulates hippocampal neurogenesis. Thus, we first injected BrdU into P6 Hap1 P1 KO mice and controls, and analyzed BrdU incorporation 24 hours later. BrdU immunostaining and stereological quantification clearly showed a decrease in proliferating cells in the hippocampal DG (Fig 3A and 3B), indicating that early postnatal Hap1 expression may positively regulate hippocampal neurogenesis. We then performed the same assay on Hap1 P1 KO and P21 KO mice at P34 and found that, while Hap1 depletion at P1 still had an effect on DG neurogenesis at P34, late postnatal Hap1 KO (P21 KO) could no longer influence hippocampal neurogenesis (Fig 3C and 3D). Since camk2a-Hap1 KO mice started to deplete Hap1 weakly at the late embryonic stage and much more potently after birth [50, 51], we went on to assess BrdU incorporation in camk2a-Hap1 KO mice to see whether the observed adult depressive phenotype in these mice was associated with a decrease in postnatal neurogenesis. We found that camk2a-Hap1 KO mice also had a reduced number of BrdU+ cells in the DG (Fig 3E), further supporting an important role for early postnatal Hap1 expression in hippocampal neurogenesis.

Neural progenitor cells (NPCs), when undergoing differentiation, can give rise to both neuronal and glial cells. Because of the decreased number of proliferating NPCs as indicated by the number of BrdU+ cells, we also wanted to know whether the differentiation of NPCs was perturbed by the early postnatal loss of Hap1. To explore this, we injected BrdU into Hap1 P1 KO and control mice at P6 and sacrificed them 4 weeks later so that NPCs, which incorporated BrdU at P6, would differentiate into either mature neurons or glia. Co-immunostaining of BrdU with NeuN, a marker for mature neurons, or GFAP, a marker for mature astrocytes, revealed that P1 deletion of Hap1 significantly affected neuronal differentiation, as the ratio of NeuN+/BrdU+ cells in P1 KO mice (53%) is significantly lower than in controls (70%) (Fig 3F, left panel). In contrast, the ratio of GFAP+/BrdU+ cells among BrdU+ cells was increased in P1 KO mice (Fig 3F, right panel). Because Hap1 expression is not seen in mature glial cells [52, 53], we believe that this increase is unlikely to be due to the direct effect of Hap1 on glial cell differentiation, but rather is a reflection of decreased neuronal differentiation in P1 KO mice.
Adult Hap1 Expression Protects Animals against Stress-Induced Depression by Maintaining Hippocampal Neurogenesis
Since neural activities and signal transduction are very different between developing and mature brains, many genes are expected to play differential roles at these two stages. Whether Hap1 plays differential roles in neurogenesis in postnatal and adult brains has gone unexplored. As the hippocampus is a brain region known to be highly responsive to stress [21], we wanted to test the idea that adult-expressed Hap1 may respond to stress in the regulation of hippocampal neurogenesis and animal behavior. To this end, we used Hap1 P21 KO mice, which did not present gross phenotypes, including deficits in neurogenesis, and subjected them to 7-day repeated restraint stress. P21 KO mice did not exhibit impaired hippocampal neurogenesis under normal conditions; however, after repeated stress, they showed a significant reduction in hippocampal neurogenesis, much greater than in control mice (Fig 4A and 4B). Repeated restraint stress is well known to suppress hippocampal neurogenesis, but does not lead to apparent depressive behavior in WT rodents [54–57]. We thought that a certain level of neurogenesis must be maintained in order for animals to battle against the potential behavioral changes caused by stress. Because the lack of Hap1 caused hippocampal neurogenesis to drop to an abnormally low level following stress, it is possible that this reduction might undermine the ability of animals to cope with stress. Using the forced swim test (FST), we found that after repeated restraint stress, Hap1 P21 KO mice displayed a marked increase in immobility compared to non-stressed Hap1 KO and stressed control mice (Fig 4C), suggesting that adult expression of Hap1 is involved in the maintenance of hippocampal neurogenesis in response to certain types of stress, including restraint stress, thereby protecting animals against stress-induced depressive-like behavior.

c-kit Levels Are Decreased in Hap1 KO Mouse Hippocampus
Our results indicate that early postnatal Hap1 deletion reduces hippocampal neurogenesis and leads to an adult depressive-like phenotype. To find a molecular target for Hap1-regulated hippocampal neurogenesis, we prepared P1 WT and Hap1 KO mouse hippocampal lysates and performed mass spectrometry analysis to look for potential targets of Hap1 whose levels were significantly altered between WT and KO samples. Among all the proteins identified, we found that c-kit, or mast/stem cell growth factor receptor, showed nearly 50% down-regulation in Hap1 KO hippocampus (Fig 5A). A previous study indicated that c-kit is expressed in neuroproliferative zones of the rat brain, and in vivo administration of its ligand, stem cell factor (SCF), increases neurogenesis in these regions [40]. We first confirmed the mass spectrometry result by immunofluorescent staining (Fig 5B) and western blot analysis (Fig 5C and 5D) of c-kit, which revealed that c-kit expression in Hap1 KO hippocampus is indeed decreased. Then, we looked at c-kit levels in Hap1 P1 KO and adult KO hippocampal tissues. As compared with controls, c-kit expression was significantly lower in P1 KO, but not adult KO, hippocampus, suggesting that c-kit down-regulation could be associated with the decreased hippocampal neurogenesis and adult depressive-like phenotype displayed in P1 KO mice (S3A and S3B Fig). Our previous results indicate that Hap1 controls postnatal hypothalamic neurogenesis by stabilizing TrkB protein level [32]. To find out whether the same mechanism also underlies Hap1-mediated regulation of postnatal hippocampal neurogenesis, we assessed TrkB levels in germline Hap1 KO as well as P1 and adult KO hippocampal tissues, and found that neither of these KO tissues showed a reduction in TrkB level (Fig 5C, 5D, S3A and S3B Fig), suggesting that Hap1 may regulate hippocampal neurogenesis through different signaling molecules, such as c-kit. We next examined c-kit expression levels by western blot in various tissues from camk2a-Hap1 KO and control mice. Besides the hippocampus, which showed a significant reduction in c-kit level, we also found a trend of decreased c-kit expression in the striatum and hypothalamus of camk2a-Hap1 KO mice (Fig 5E). Notably, c-kit is highly expressed in the hippocampus compared to other tissues examined (Fig 5E), which lends credence to the finding that SCF/c-kit signaling regulates neurogenesis. To look for more evidence that c-kit activation and neurogenesis are perturbed in camk2a-Hap1 KO mouse hippocampus, we assessed the levels of phosphorylated c-kit (pc-kit), a proliferating cell marker, ki67, a neuroblast or immature neuron marker, DCX, and a mature neuron marker, NeuN, via western blot analysis (Fig 5F and 5G). The results showed that all these proteins were significantly reduced, indicating that loss of Hap1 affects c-kit activation by down-regulating its expression level, compromising postnatal hippocampal neurogenesis in camk2a-Hap1 KO mice.

Hap1 Is Partially Coexpressed with c-kit in the DG and Stabilizes Its Level In Vitro
We next looked at the expression level of c-kit in the hippocampus at different ages and found that, very similar to Hap1, the c-kit expression level peaks at the postnatal stage (Fig 6A), which supports a role for both proteins in the regulation of postnatal neurogenesis. To see whether Hap1 directly promotes the c-kit level in hippocampal neurons, we first examined whether these two proteins are expressed in the same neurons. Therefore, we cultured primary hippocampal neurons from P1–P2 WT pups and co-stained these neurons at DIV5 with antibodies for Hap1 and c-kit. We saw that Hap1 and c-kit were coexpressed in most neurons in this in vitro system (Fig 6B). To examine their expressions in vivo, we also performed co-immunofluorescent staining of Hap1 and c-kit in P12 WT mouse brain sections and found that Hap1 and c-kit were partially coexpressed in the subgranular zone of the DG, a region where new neurons are born (Fig 6C). To examine which type of cells in the DG express c-kit, we stained WT mouse brain sections from P7–P15 with antibodies for c-kit and an array of cell type-specific markers. We found that during the early postnatal stage, c-kit was expressed in NPCs, immature neurons, and mature neurons, as there was co-immunostaining of c-kit with nestin, DCX, or NeuN in the same cells (S4A Fig). In mature neurons, c-kit expression was found mainly in GAD67-expressing GABAergic interneurons, but not in prox1-expressing granule cells (S4A Fig). We also saw a similar pattern of expression for Hap1 at this age (S4B Fig). Therefore, it is possible that Hap1 and c-kit may directly regulate the proliferation and/or differentiation of hippocampal NPCs at an early postnatal stage, or that they might be involved in GABAergic control of hippocampal neurogenesis, a phenomenon reported previously [58–61]. In 1-month old mouse hippocampus, however, c-kit expression was largely restricted in GAD67-expressing GABAergic interneurons (S4A Fig). Since Hap1 could not be detected in NPCs in adult rat hippocampus, and more than 60% of Hap1-immunoreactive cells express GABA [62], it is likely that Hap1 and c-kit may mediate GABAergic control of adult hippocampal neurogenesis, which could serve as a buffer mechanism against stress-induced behavioral changes. Hap1 is known to stabilize internalized membrane receptors via its trafficking function between endosomes and lysosomes [29, 31, 32, 63–65]. To verify whether Hap1 directly regulates c-kit levels intracellularly, we measured the stability of transfected c-kit in Neuro2A cells. We found that knocking down endogenous Hap1 expression via siRNA substantially diminished the stability of c-kit (Fig 6D), indicating that direct intracellular regulation of c-kit levels by Hap1 is at least part of the mechanism by which Hap1 stabilizes c-kit.

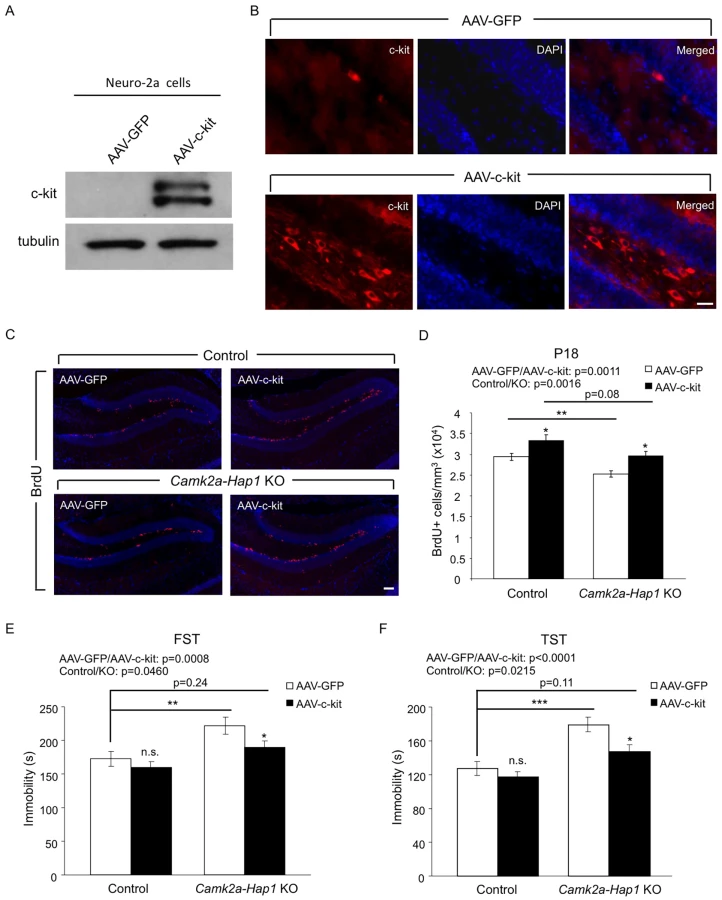
Overexpression of c-kit in the Hippocampus Rescues the Neurogenesis Defect and Adult Depressive-Like Behavior in camk2a-Hap1 KO Mice
We next asked whether increasing the expression of c-kit in the hippocampus of camk2a-Hap1 KO mice could rescue the neurogenesis defect and adult depressive-like behavior displayed by these mice. To this end, we designed adeno-associated virus (AAV) expressing c-kit under the synapsin-1 promoter and validated the successful infection of the virus and expression of c-kit both in Neuro-2a cells (Fig 7A) and mouse hippocampus, which was stereotaxically injected with AAV-c-kit at P3 (Fig 7B). The overexpression of c-kit in the hippocampus resulted in increased BrdU incorporation at P18 for both camk2a-Hap1 KO and control mice (Fig 7C and 7D), suggesting that postnatal hippocampal neurogenesis can be modulated by altering the c-kit level. Furthermore, behavioral examination using the FST (Fig 7E) and TST (Fig 7F) showed that P3 AAV-c-kit injection in the hippocampus had a significant effect on mouse depressive-like behavior compared with control virus. Although c-kit overexpression slightly reduced the immobility of control mice in both tests, this overexpression in the hippocampus was able to significantly improve the performance of camk2a-Hap1 KO mice to a greater extent, thus partially ameliorating the adult depressive-like phenotype in these mice. As the virus directed the expression of c-kit in the hippocampus, the improved behavioral phenotype is very likely a specific result of the enhanced hippocampal neurogenesis by c-kit overexpression. Taken together, these results indicate that suppressed c-kit expression indeed accounts for both the neurogenesis defect and adult depressive behavior in mice lacking Hap1, and upregulation of c-kit or Hap1 levels could be considered as therapeutic options.
Discussion
Since its identification as the first known interacting protein of huntingtin, Hap1 has been investigated in a number of studies for its role in both physiological and disease contexts. It has become evident that the neuronal expression of Hap1 is essential for early postnatal survival of mice, as germline Hap1 KO results in an early postnatal lethal phenotype [30, 66]; however, whether Hap1 depletion induced at different ages has differential effects on adult animal behaviors has gone unexplored. Using an inducible Hap1 KO mouse model, we were able to demonstrate that early postnatal Hap1 depletion leads to reduced neurogenesis in the hippocampus and depressive-like behavior in adult mice. Our studies thus demonstrate for the first time that genetic induction of defective postnatal neurogenesis can lead to adult depressive-like behavior.
Depression is a major mental disorder that affects hundreds of millions of people worldwide [67]. Due to its wide spectrum of symptoms, many genetic and environmental factors may trigger the onset of a depressive episode. These episodes are normally observed in adults, but are also seen in childhood and adolescence, although the latter cases are more difficult to diagnose and often go overlooked [68]. However, the importance of early life experience to adult depression is accentuated by the fact that, in the most severe cases of adult depression, some form of abuse was experienced in childhood [69]. It is therefore apparent that environmental factors in early life can contribute significantly to adult depression; whether genetic mutations also play a role in predisposing people with unpleasant early life experiences or even without such experiences to depression remains largely unknown. Our results using induced Hap1 KO mice show that early loss of a gene involved in neurogenesis can indeed contribute to the etiology of depression, suggesting that early genetic diagnosis could possibly help with prediction of and early intervention for depression, or likely other forms of adult-onset mental disorders.
Hippocampal neurogenesis has emerged as an appealing theory to explain depression; however, a causal relationship between hippocampal neurogenesis and depression has not been established [70, 71]. Because neurogenesis happens more dynamically in the early postnatal brain than adult brain and since Hap1 regulates postnatal hypothalamic neurogenesis [32], we hypothesized that Hap1 may also regulate postnatal hippocampal neurogenesis, and the reduction of which could contribute to adult depressive behavior. We found that Hap1 deletion at P1 significantly reduced hippocampal neurogenesis at the early postnatal stage and caused depressive-like behavior in adults. Since these P1 KO mice also showed severe growth retardation and reduced survival, we used camk2a-Hap1 KO mice as another model for the study, which deletes Hap1 mainly in the cortex and hippocampus from early postnatal life. These mice could survive and grow normally, yet exhibited a postnatal neurogenesis defect as well as adult depressive behavior, suggesting that reduced postnatal neurogenesis is more likely to be a cause of the depressive phenotype. In addition, we also found that loss of Hap1 reduces the expression of c-kit, which is present in progenitor cells for hippocampal neurogenesis. It should be pointed out that in conditional Hap1 KO mice, Hap1 depletion, which is mediated by Cre under the control of camk2a promoter, is not restricted to the hippocampus. To explore whether Hap1 depletion in the postnatal hippocampus plays a pivotal role in adulthood depression, we used stereotaxic injection to overexpress c-kit in P3 mouse hippocampus and found that this overexpression can augment hippocampal neurogenesis and mitigate the depressive phenotype caused by the loss of Hap1, further supporting a causal role of reduced postnatal hippocampal neurogenesis in adult depression in our Hap1-deficient mice. All these findings provide evidence of a new role for Hap1 in adult depressive behavior.
Our earlier study suggested that Hap1 is an essential gene for postnatal survival, but might be dispensable for adults [32]. However, mice used for those experiments were raised in standard housing conditions, which are far different from all the stress and threats animals would experience in the wild. In our case, although late postnatal or adult Hap1 depletion did not lead to overt phenotypes, its expression could be needed under certain stress conditions. Despite a lack of depressive phenotype caused by ablation of adult hippocampal neurogenesis, stress is found to down-regulate adult hippocampal neurogenesis, and adult-born hippocampal neurons are required for the regulation of the hypothalamic–pituitary–adrenal (HPA) axis in buffering stress-induced depressive behaviors [14, 21, 72]. Thus, an investigation of whether adult Hap1 expression influences hippocampal neurogenesis and depressive behavior under stressful conditions was of great interest to us. We found that 7-day repeated restraint stress was able to diminish adult hippocampal neurogenesis in P21 KO mice in which Hap1 depletion occurred after the postnatal period. This finding suggests that Hap1 expression may be required for adult hippocampal neurogenesis under stress and that its loss at least increases susceptibility to stressors in adulthood.
Since postnatal neurogenesis is necessary for the maturation of the central nervous system after birth, its reduction might affect the connectivity of certain critical neural circuitries, rendering animals susceptible to depression later in their lives. It is also possible that adult-expressed Hap1 plays an important role in maintaining a proper level of hippocampal neurogenesis and thus the hippocampal neural circuitry, which is needed for animals to buffer against stress-induced behavioral changes. Hap1 is known to interact with a number of endocytic receptors in a way that their degradation is inhibited and levels are stabilized [29–32, 63–65]. These receptors could be important for neurogenesis or other neuronal functions that are involved in maintaining and regulating hippocampal connectivity and circuitry in response to environmental stress. We conducted mass spectrometry analysis and found that c-kit, a protein that has been suggested to play a role in hippocampal neurogenesis and synaptic potentiation, as well as hippocampal-dependent learning and memory [40, 73, 74], was downregulated in Hap1 KO hippocampus. Since c-kit also undergoes ligand-induced internalization [75, 76], it is likely that Hap1 functions to stabilize endocytic c-kit as it does to those other receptors. Moreover, Hap1 and c-kit both peak their expression levels in the hippocampus at early postnatal stage, and show a continued decrease in expression with age. Although this alteration is in line with the age-dependent decline in neurogenesis, it remains to be investigated whether increasing Hap1 expression or its mediated signaling, e.g., SCF/c-kit pathway in the adult brain can be beneficial to adult neurogenesis and reducing aging-related phenotypes [77, 78]. We found that Hap1 and c-kit are coexpressed in NPCs, immature neurons, and GABAergic interneurons in the DG during early postnatal life, though they become more restricted in interneurons later in life. Such different distributions could account for the differential roles of Hap1 in postnatal and adult neurogenesis and associated behavioral phenotypes. It is also possible that during the postnatal stage, Hap1 and c-kit regulate the proliferation and differentiation of a subpopulation of DG NPCs into interneurons, and once differentiated, they remain expressed in these interneurons, which can also regulate neurogenesis as reported previously [58–60]. As their expressions decrease significantly with age, Hap1 and c-kit might become part of the cellular machinery in response to stress or injuries as supported by the previous finding on c-kit [41]. The differential roles of Hap1 in postnatal and adult life are not unexpected as Hap1 is a multifaceted protein that interacts with different partners. Its association with other proteins and the resulting functions may be cell-type dependent and also depend on posttranslational modulations that can be cell-type and age-dependent. Whether Hap1 regulates c-kit function differently during postnatal and adult life requires further investigations. The neurogenesis and behavioral rescue in camk2a-Hap1 KO mice via c-kit overexpression in the hippocampus suggests that c-kit-mediated signaling pathways are important for postnatal hippocampal neurogenesis and adult depressive behavior.
In conclusion, we have demonstrated a novel role for Hap1 in the regulation of postnatal neurogenesis and adult depressive-like behavior and provided the first genetic model that relates postnatal neurogenesis to adult depression. Our findings may help us better understand the mechanisms of depression, as well as identify potential therapeutic interventions.
Materials and Methods
Ethics Statement
All animal studies were performed in compliance with IACUC (Institutional Animal Care and Use Committee) at Emory University.
Reagents and Antibodies
Dulbecco’s modified Eagle’s medium (DMEM), Neurobasal-A, B27, GlutaMAX-1, D-Hank’s, and fetal calf serum (FCS) were obtained from Life Technologies. Trypsin, poly-D-lysine, BSA, BrdU, imipramine, cycloheximide were all from Sigma. Cell culture dishes, coverslips, plates, and flasks were purchased from Corning and Nunc, Inc. Guinea pig antibody to Hap1 was generated in our laboratory [28, 30]. Rabbit anti-c-kit and phosphorylated-c-kit (Cell Signaling), rabbit anti-NeuN, mouse anti-NeuN, GFAP, nestin, GAD67, prox1 (all from Millipore), goat anti-DCX and guinea pig anti-DCX (Santa Cruz and Millipore), rat anti-BrdU (Accurate Chemical), rabbit anti-Ki67 (Thermal), mouse anti-calretinin (BD Transduction Laboratories), mouse anti-tubulin (Sigma) were used for western blot and immunofluorescent staining. Dilutions of the primary antibodies used can be found in S1 Table. HRP-tagged or fluorescent secondary antibodies were obtained from Jackson ImmunoResearch Laboratories.
Animals
Mice were housed in the Division of Animal Resources at Emory University on a 12-h light (7 am-7 pm)/dark (7 pm-7 am) cycle. All procedures and husbandry were in accordance with the NIH Guide for the Care and Use of Laboratory Animals. Generation of germline Hap1 KO mice (C57BL/6/black Swiss), floxed-Hap1 mice (C57BL/6/SV129), and TM-inducible Cre-ER (C57BL/6/CBA)/floxed-Hap1 mice was described in our previous studies [30, 32, 79]. Transgenic mice expressing Cre under the control of the mouse calcium/calmodulin-dependent protein kinase II alpha (camk2a) promoter (C57BL/6/BALB/C) were kindly provided by Dr. Stephen Warren (Emory University). Camk2a-Hap1 KO mice were generated by crossing the floxed-Hap1 mice with camk2a-Cre transgenic mice. Control mice were floxed-Hap1 mice without transgenic Cre, and were all littermates of the KO mice.
Western Blot Analysis
Mouse brain tissues or harvested cells were lysed in ice-cold RIPA buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1 mM EDTA pH 8.0, 1 mM EGTA pH 8.0, 0.1% SDS, 0.5% DOC, and 1% Triton X-100) containing Halt protease inhibitor cocktail (Thermol Scientific) and phosphatase inhibitors. The lysates were incubated on ice for 30 min, sonicated, and centrifuged at top speed for 10 min. The supernatants were subjected to SDS-PAGE. The proteins on the gel were transferred to a nitrocellulose membrane, which was then blocked with 5% milk/PBS for 1 h at room temperature. The blot was incubated with primary antibodies in 3% BSA/PBS overnight at 4°C. After 3 washes in PBS, the blot was incubated with HRP-conjugated secondary antibodies in 5% milk/PBS for 1 h at room temperature. After 3 washes in PBS, ECL Prime (GE Healthcare) was then used to detect immunoreactive bands on the blot. A detailed list of the primary antibodies used can be found in S1 Table.
Immunofluorescence Microscopy
For immunofluorescent staining of cultured neurons, neurons were washed once with PBS, and fixed with 4% paraformaldehyde (PFA) for 10 min. Fixed cells were washed 3 times with PBS, and then blocked with 3% BSA + 5% normal donkey serum/PBST (0.2% Triton X-100 in PBS) for 1 h at room temperature. Primary antibodies were diluted in blocking buffer and incubated with the cells at 4°C overnight followed by 3 washes with PBS and incubation with fluorescent secondary antibodies and nuclear dye. After 3 washes, the cells were ready for examination using a Zeiss (Axiovert 200M, Germany) microscope with a digital camera (Orca-100; Hamamatsu Photonics, Bridgewater, NJ) and the Openlab software (Improvision, Lexington, MA). Immunofluorescent staining of brain sections was performed as described previously [29, 38]. Briefly, mice were deeply anesthetized, perfused with saline followed by 4% PFA fixation. Brains were postfixed overnight in the same fixative, and switched to 30% sucrose at 4°C. After completely sunk, brains were sectioned at 15 μm (40 μm for BrdU staining) with a cryostat at −19°C and mounted onto gelatin-coated slides. The tissues on the slides were washed, blocked, and immunostained with antibodies using the same method described above for cultured cells. For BrdU immunostaining, sections were first treated with 2 N HCl for 30 min at 37°C and then neutralized with 0.1 M sodium borate (pH 8.5) for 15 min at room temperature. A detailed list of the primary antibodies used can be found in S1 Table.
TM Induction in Mice
TM induction in mice was performed as described previously [32]. Briefly, TM (Sigma T5648) was dissolved in ethanol at 20 mg/ml and stored at -20°C before use. To induce Hap1 depletion, a calculated amount of TM was mixed with corn oil, and ethanol was removed by a vacuum centrifuge. 1.1 mg or 2.2 mg TM per 40 g body weight was used to inject P1 (subcutaneous) or P10 (i.p.) mouse pups for 3 consecutive days. For mice at P15 or older, 4 mg TM per 40 g body weight was used for i.p. injection for 5 consecutive days. Both TM-inducible Cre-ER/floxed-Hap1 mice or camk2a-Hap1 KO mice and their control littermates were given TM injections at the same time.
BrdU Incorporation Assay
For BrdU injection into Hap1 P1 KO mice and controls, mice at P6 were i.p. injected with 50 mg/kg body weight BrdU. The animals were perfused and fixed 24 hours later for the analysis of NPC proliferation, or 4 weeks later for the analysis of neural differentiation. For BrdU injection into camk2a-Hap1 KO or P21 KO mice and controls, mice at P33 (or P17 for AAV-c-kit rescue experiment) were i.p. injected with 50mg/kg body weight BrdU. Twenty four hours later, the mice were perfused and fixed for analyzing the number of proliferating cells.
Stereology and Quantification
Stereological cell counting and quantification were performed as described in our previous study [32]. Briefly, to quantify BrdU+ cells, the optical-fractionator method implemented in Stereo Investigator 9.03.2 (MicroBrightField, Magdeburg, Germany) was used. One-in-six 40-μm serial sections covering the entire hippocampal region were stained to visualize and quantify BrdU+ cells in the DG. A minimum of 3 mice from each genotype and 8 sections from each mouse brain were examined for comparisons. The volume of the DG and the total number of BrdU+ cells in the DG were calculated by Stereo Investigator software. The total number of BrdU+ cells was then divided by the volume to yield cell density presented as the number of BrdU+ cells per mm3. For estimation of the ratios of NeuN+/BrdU+ and GFAP+/BrdU+ cells among BrdU+ cells, sections were co-stained for BrdU and either NeuN or GFAP. Each BrdU+ cell counted was also examined for the presence of NeuN or GFAP labeling, and the double-positive cells were marked and quantified separately. The densities of the double-positive cells were divided by those of the total BrdU+ cells to yield the ratios. Counting of the cells was performed under the 40× lens in a Zeiss AX10 microscope.
Forced Swim Test (FST)
In multiple experiments, conditional Hap1 KO mice and their control littermates (age indicated in figure legends) were placed individually into a round opaque plastic cylinder (18 cm in height, 15 cm in diameter) filled with water (25°C) at a depth of 12 cm. Immobility time, defined as floating or the absence of active behaviors, such as swimming or struggling to escape, was measured. Slight movements of the feet and tail necessary to keep the head above water were excluded as mobility. Each mouse was measured for 6 min by a trained observer who was kept blind to the genotypes of the mice and drug treatment. No pretest training of mice was performed.
Tail Suspension Test (TST)
As for FST, conditional Hap1 KO mice and their control littermates (age indicated in figure legends) were used for TST in multiple experiments. The mice were suspended by taping the tail (~1 cm from tip of tail) to a horizontal bar at a height of 40 cm from the table surface for 6 min. The trial was conducted for a duration of 6 min, and the immobility time was recorded manually via stopwatch by a trained observer who was blind to the genotypes of the mice examined. Mice were considered immobile when they hung passively and motionlessly without escape-oriented behaviors.
Rotarod Test
Motor activity was evaluated using Rotamex (Columbus Instruments). Two-month old Hap1 P1 KO or camk2a-Hap1 KO mice and their control littermates were trained on a rotating rod at a speed of 5 rpm for three 5-min trials on 3 consecutive days. Testing was performed on the fourth day. During the test, the rotating rod was gradually accelerated to 40 rpm over 5 min. Latency to fall from the rotarod was recorded in 3 trials, and the average of the 3 trials was used for each mouse.
Imipramine Treatment
Tricyclic antidepressant imipramine (30 mg/kg, sigma) was freshly made in saline and i.p. injected into 4-month old camk2a-Hap1 KO or control mice 30 min before FST or TST. Saline injection was used as vehicle treatment.
Restraint Stress
One-year old Hap1 P21 KO and control mice were subjected to repeated restraint stress by placement for 4 h per day for 7 consecutive days in ventilated 50 ml conical tubes. After each stress session, mice were immediately returned to their home cages. Control mice were housed in separate cages from the stressed mice, and were deprived of food and water but otherwise untouched during each session. Twenty four hours after the final session, mice were evaluated by FST. For neurogenesis analysis, BrdU (100mg/kg body weight) was i.p. injected before the stress session on each of the last 3 days. Twenty four hours after the final session, mice were perfused and fixed, their brains were then sectioned for BrdU immunostaining.
Locomotor Activity Assay
Locomotor activity was measured using an automated system (San Diego Instruments, La Jolla, CA) with photobeams that record ambulations (consecutive beam breaks). Two-month old Hap1 P1 KO and control mice were individually placed in the chambers under 12-h light-dark cycle with free access to food and water. Mice were allowed 4 h to acclimate to the new environment before recording. Activities were recorded every 30 min for 24 h.
Sucrose Preference Test
The sucrose preference test was conducted as previously described with modification [21]. Briefly, 2-month old camk2a-Hap1 KO and control mice were individually housed with free access to food and two weighed bottles of liquid: one filled with water, the other with 1% sucrose solution. The positions of the two bottles were balanced across animals. After 3 days of acclimation, both bottles were removed and weighed at 12 pm, and then put back in reversed positions at 7 pm. The bottles were weighed again in 1 h for an acute test, and again on the next morning for an overnight test. Sucrose preference was calculated as (Δweightsucrose)/(Δweightsucrose + Δweightwater) × 100.
Hippocampal Neuronal Cell Culture
Hippocampal dissection and neuronal cell culture were performed as previously described [80]. Briefly, hippocampi were dissected from P1 WT mice and placed in a sterile 35-mm petri dish containing ice-cold Hanks’ balanced salt solution (HBSS, Ca2+-and Mg2+-free), chopped into 1 mm3 pieces by microscissors, and digested with 0.125% (w/v) trypsin at 37°C for 25 min. The enzymatic activity was terminated by adding DNaseI (200 U/ml final concentration) and heat-inactivated FCS (20% final concentration) into the solution. The tissue was then dissociated by triturating through a fire-polished Pasteur pipette, spun down and washed twice with culture medium (Neurobasal-A supplemented with B27 and GlutaMAX-1). After resuspension in culture medium, 2 × 105 cells per well were plated onto 6-well plates. Neurons that were cultured for 5 days in vitro (DIV) were fixed by 4% PFA and used for immunofluorescent staining.
Preparation of Adenoviral Hap1 siRNA and AAV-c-kit
Adenoviral Hap1-specific and scramble siRNAs were prepared in our previous study [81]. Viral stocks were adjusted to 1X108 viral particles/μl before use. Neuro2A cells were incubated with adenoviral Hap1 siRNA at a multiplicity of infection of 50. Twenty-four h after infection, the virus-containing medium was removed, and the c-kit plasmid was transfected for another 48 h before performing the protein stability assay. Mouse c-kit cDNA was subcloned into a pAAV-MCS vector (Cell Biolabs). The human synapsin-1 promoter sequence was inserted into the construct to replace the original promoter in the vector. AAV-c-kit (Serotype 9) was packaged and amplified by the viral vector core at Emory University. AAV-GFP (Serotype 9, SignaGen Laboratories) under the same promoter was used as a control.
Stereotaxic Viral Injection
A camk2a-Hap1 KO or control mouse pup at P3 was placed in a latex sleeve and immersed up to the neck in crushed ice and water (2–3°C) for 7–10 min. The pup was then placed on an ice pack (3–4°C) and stabilized on a platform while being injected with 0.5 μl of AAV-c-kit or AAV-GFP viral particles (1X1012 particles/ml) into each side of the hippocampus (1.5 mm lateral from the sagittal suture, 2 mm rostral to the lambda, and 2 mm below the skull) over 2 min from a 5-μl Hamilton syringe and 33-gauge needle. The needle was kept still for another 2 min before withdrawal. The surgical field was illuminated with fiber optic to minimize inadvertent and uncontrollable warming. The pup was then transferred into a clean cage placed on a heat pad (33°C) with nest for 30 min to recover from hypothermia before returning to the home cage. We used 4–6 pups per experimental group.
Statistical Analysis
All data are expressed as mean ±SEM. The statistical significance was determined by two-tailed Student’s t-tests or two-way ANOVA followed when appropriate by post hoc t-tests using GraphPad Prism 5.0 software. A value of p<0.05 was considered statistically significant.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Ferrari AJ, Charlson FJ, Norman RE, Patten SB, Freedman G, Murray CJ, et al. Burden of depressive disorders by country, sex, age, and year: findings from the global burden of disease study 2010. PLoS Med. 2013;10(11):e1001547. doi: 10.1371/journal.pmed.1001547 24223526
2. Kupfer DJ, Frank E, Phillips ML. Major depressive disorder: new clinical, neurobiological, and treatment perspectives. Lancet. 2012;379(9820):1045–55. doi: 10.1016/S0140-6736(11)60602-8 22189047
3. Kessler RC, Berglund P, Demler O, Jin R, Koretz D, Merikangas KR, et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA. 2003;289(23):3095–105. 12813115
4. Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM. Neurobiology of depression. Neuron. 2002;34(1):13–25. 11931738.
5. Belmaker RH, Agam G. Major depressive disorder. N Engl J Med. 2008;358(1):55–68. doi: 10.1056/NEJMra073096 18172175
6. Flint J, Kendler KS. The genetics of major depression. Neuron. 2014;81(3):484–503. doi: 10.1016/j.neuron.2014.01.027 24507187
7. Jacobsen JPR, Medvedev IO, Caron MG. The 5-HT deficiency theory of depression: perspectives from a naturalistic 5-HT deficiency model, the tryptophan hydroxylase 2(Arg)439(His) knockin mouse. Philos T R Soc B. 2012;367(1601):2444–59. doi: 10.1098/rstb.2012.0109 22826344
8. Graeff FG, Guimaraes FS, DeAndrade TGCS, Deakin JFW. Role of 5-HT in stress, anxiety, and depression. Pharmacol Biochem Be. 1996;54(1):129–41. 8728550
9. Duman RS, Aghajanian GK. Synaptic Dysfunction in Depression: Potential Therapeutic Targets. Science. 2012;338(6103):68–72. doi: 10.1126/science.1222939 23042884
10. Kang HJ, Voleti B, Hajszan T, Rajkowska G, Stockmeier CA, Licznerski P, et al. Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat Med. 2012;18(9):1413-+. 22885997
11. Pariante CM, Lightman SL. The HPA axis in major depression: classical theories and new developments. Trends Neurosci. 2008;31(9):464–8. doi: 10.1016/j.tins.2008.06.006 18675469
12. Groves JO. Is it time to reassess the BDNF hypothesis of depression? Mol Psychiatry. 2007;12(12):1079–88. 17700574
13. Sahay A, Hen R. Adult hippocampal neurogenesis in depression. Nat Neurosci. 2007;10(9):1110–5. 17726477
14. Eisch AJ, Petrik D. Depression and hippocampal neurogenesis: a road to remission? Science. 2012;338(6103):72–5. doi: 10.1126/science.1222941 23042885
15. Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci. 2000;20(24):9104–10. 11124987.
16. Malberg JE. Implications of adult hippocampal neurogenesis in antidepressant action. J Psychiatry Neurosci. 2004;29(3):196–205. 15173896; PubMed Central PMCID: PMC400689.
17. Wang JW, David DJ, Monckton JE, Battaglia F, Hen R. Chronic fluoxetine stimulates maturation and synaptic plasticity of adult-born hippocampal granule cells. J Neurosci. 2008;28(6):1374–84. doi: 10.1523/JNEUROSCI.3632-07.2008 18256257
18. Boldrini M, Underwood MD, Hen R, Rosoklija GB, Dwork AJ, John Mann J, et al. Antidepressants increase neural progenitor cells in the human hippocampus. Neuropsychopharmacology. 2009;34(11):2376–89. doi: 10.1038/npp.2009.75 19606083
19. Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S, et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science. 2003;301(5634):805–9. 12907793
20. Schloesser RJ, Manji HK, Martinowich K. Suppression of adult neurogenesis leads to an increased hypothalamo-pituitary-adrenal axis response. Neuroreport. 2009;20(6):553–7. doi: 10.1097/WNR.0b013e3283293e59 19322118
21. Snyder JS, Soumier A, Brewer M, Pickel J, Cameron HA. Adult hippocampal neurogenesis buffers stress responses and depressive behaviour. Nature. 2011;476(7361):458–61. doi: 10.1038/nature10287 21814201
22. Waterhouse EG, An JJ, Orefice LL, Baydyuk M, Liao GY, Zheng K, et al. BDNF Promotes Differentiation and Maturation of Adult-born Neurons through GABAergic Transmission. J Neurosci. 2012;32(41):14318–30. doi: 10.1523/JNEUROSCI.0709-12.2012 23055503
23. Brezun JM, Daszuta A. Depletion in serotonin decreases neurogenesis in the dentate gyrus and the subventricular zone of adult rats. Neuroscience. 1999;89(4):999–1002. 10362289
24. Brummelte S, Pawluski JL, Galea LA. High post-partum levels of corticosterone given to dams influence postnatal hippocampal cell proliferation and behavior of offspring: A model of post-partum stress and possible depression. Horm Behav. 2006;50(3):370–82. 16780843
25. Lajud N, Roque A, Cajero M, Gutierrez-Ospina G, Torner L. Periodic maternal separation decreases hippocampal neurogenesis without affecting basal corticosterone during the stress hyporesponsive period, but alters HPA axis and coping behavior in adulthood. Psychoneuroendocrinology. 2012;37(3):410–20. doi: 10.1016/j.psyneuen.2011.07.011 21862224
26. Lajud N, Gonzalez-Zapien R, Roque A, Tinajero E, Valdez JJ, Clapp C, et al. Prolactin administration during early postnatal life decreases hippocampal and olfactory bulb neurogenesis and results in depressive-like behavior in adulthood. Horm Behav. 2013;64(5):781–9. doi: 10.1016/j.yhbeh.2013.10.005 24144492
27. Hayashi F, Takashima N, Murayama A, Inokuchi K. Decreased postnatal neurogenesis in the hippocampus combined with stress experience during adolescence is accompanied by an enhanced incidence of behavioral pathologies in adult mice. Mol Brain. 2008;1 : 22. doi: 10.1186/1756-6606-1-22 19091092
28. Li XJ, Li SH, Sharp AH, Nucifora FC Jr., Schilling G, Lanahan A, et al. A huntingtin-associated protein enriched in brain with implications for pathology. Nature. 1995;378(6555):398–402. 7477378
29. Sheng G, Xu X, Lin YF, Wang CE, Rong J, Cheng D, et al. Huntingtin-associated protein 1 interacts with Ahi1 to regulate cerebellar and brainstem development in mice. J Clin Invest. 2008;118(8):2785–95. doi: 10.1172/JCI35339 18636121
30. Li SH, Yu ZX, Li CL, Nguyen HP, Zhou YX, Deng C, et al. Lack of huntingtin-associated protein-1 causes neuronal death resembling hypothalamic degeneration in Huntington's disease. J Neurosci. 2003;23(17):6956–64. 12890790.
31. Rong J, McGuire JR, Fang ZH, Sheng G, Shin JY, Li SH, et al. Regulation of intracellular trafficking of huntingtin-associated protein-1 is critical for TrkA protein levels and neurite outgrowth. J Neurosci. 2006;26(22):6019–30. 16738245
32. Xiang J, Yang H, Zhao T, Sun M, Xu X, Zhou XF, et al. Huntingtin-associated protein 1 regulates postnatal neurogenesis and neurotrophin receptor sorting. J Clin Invest. 2014;124(1):85–98. doi: 10.1172/JCI69206 24355921
33. Gauthier LR, Charrin BC, Borrell-Pages M, Dompierre JP, Rangone H, Cordelieres FP, et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118(1):127–38. 15242649
34. Wu LLY, Fan YJ, Li SH, Li XJ, Zhou XF. Huntingtin-associated Protein-1 Interacts with Pro-brain-derived Neurotrophic Factor and Mediates Its Transport and Release. J Biol Chem. 2010;285(8):5614–23. doi: 10.1074/jbc.M109.073197 19996106
35. Yang M, Lim Y, Li XJ, Zhong JH, Zhou XF. Precursor of Brain-derived Neurotrophic Factor (proBDNF) Forms a Complex with Huntingtin-associated Protein-1 (HAP1) and Sortilin That Modulates proBDNF Trafficking, Degradation, and Processing. J Biol Chem. 2011;286(18). doi: 10.1074/jbc.M111.294355 22069308
36. Pardo R, Molina-Calavita M, Poizat G, Keryer G, Humbert S, Saudou F. pARIS-htt: an optimised expression platform to study huntingtin reveals functional domains required for vesicular trafficking. Mol Brain. 2010;3. doi: 10.1186/1756-6606-3-3 20205763
37. Liot G, Zala D, Pla P, Mottet G, Piel M, Saudou F. Mutant Huntingtin Alters Retrograde Transport of TrkB Receptors in Striatal Dendrites. J Neurosci. 2013;33(15):6298–309. doi: 10.1523/JNEUROSCI.2033-12.2013 23575829
38. Xu X, Yang H, Lin YF, Li X, Cape A, Ressler KJ, et al. Neuronal Abelson helper integration site-1 (Ahi1) deficiency in mice alters TrkB signaling with a depressive phenotype. Proc Natl Acad Sci U S A. 2010;107(44):19126–31. doi: 10.1073/pnas.1013032107 20956301
39. Erlandsson A, Larsson J, Forsberg-Nilsson K. Stem cell factor is a chemoattractant and a survival factor for CNS stem cells. Exp Cell Res. 2004;301(2):201–10. 15530856
40. Jin K, Mao XO, Sun Y, Xie L, Greenberg DA. Stem cell factor stimulates neurogenesis in vitro and in vivo. J Clin Invest. 2002;110(3):311–9. 12163450
41. Sun L, Lee J, Fine HA. Neuronally expressed stem cell factor induces neural stem cell migration to areas of brain injury. J Clin Invest. 2004;113(9):1364–74. 15124028
42. Parker G, Hadzi-Pavlovic D, Austin MP, Mitchell P, Wilhelm K, Hickie I, et al. Sub-typing depression, I. Is psychomotor disturbance necessary and sufficient to the definition of melancholia? Psychol Med. 1995;25(4):815–23. 7480459.
43. Teicher MH. Actigraphy and motion analysis: new tools for psychiatry. Harv Rev Psychiatry. 1995;3(1):18–35. 9384925.
44. Teicher MH, Glod CA, Magnus E, Harper D, Benson G, Krueger K, et al. Circadian rest-activity disturbances in seasonal affective disorder. Arch Gen Psychiatry. 1997;54(2):124–30. 9040280.
45. Nakamura T, Kiyono K, Yoshiuchi K, Nakahara R, Struzik ZR, Yamamoto Y. Universal scaling law in human behavioral organization. Phys Rev Lett. 2007;99(13):138103. 17930642.
46. Sheng G, Chang GQ, Lin JY, Yu ZX, Fang ZH, Rong J, et al. Hypothalamic huntingtin-associated protein 1 as a mediator of feeding behavior. Nat Med. 2006;12(5):526–33. 16604089
47. Saarelainen T, Hendolin P, Lucas G, Koponen E, Sairanen M, MacDonald E, et al. Activation of the TrkB neurotrophin receptor is induced by antidepressant drugs and is required for antidepressant-induced behavioral effects. J Neurosci. 2003;23(1):349–57. 12514234.
48. Der-Avakian A, Markou A. The neurobiology of anhedonia and other reward-related deficits. Trends Neurosci. 2012;35(1):68–77. doi: 10.1016/j.tins.2011.11.005 22177980
49. Amsterdam JD, Settle RG, Doty RL, Abelman E, Winokur A. Taste and smell perception in depression. Biol Psychiatry. 1987;22(12):1481–5. 3676376.
50. Dragatsis I, Levine MS, Zeitlin S. Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat Genet. 2000;26(3):300–6. 11062468
51. Dragatsis I, Zeitlin S. CaMKIIalpha-Cre transgene expression and recombination patterns in the mouse brain. Genesis. 2000;26(2):133–5. 10686608.
52. Li XJ, Sharp AH, Li SH, Dawson TM, Snyder SH, Ross CA. Huntingtin-associated protein (HAP1): discrete neuronal localizations in the brain resemble those of neuronal nitric oxide synthase. Proc Natl Acad Sci U S A. 1996;93(10):4839–44. 8643490; PubMed Central PMCID: PMC39366.
53. Gutekunst CA, Li SH, Yi H, Ferrante RJ, Li XJ, Hersch SM. The cellular and subcellular localization of huntingtin-associated protein 1 (HAP1): comparison with huntingtin in rat and human. J Neurosci. 1998;18(19):7674–86. 9742138.
54. Pham K, Nacher J, Hof PR, McEwen BS. Repeated restraint stress suppresses neurogenesis and induces biphasic PSA-NCAM expression in the adult rat dentate gyrus. Eur J Neurosci. 2003;17(4):879–86. 12603278.
55. Gregus A, Wintink AJ, Davis AC, Kalynchuk LE. Effect of repeated corticosterone injections and restraint stress on anxiety and depression-like behavior in male rats. Behav Brain Res. 2005;156(1):105–14. 15474655
56. Luo C, Xu H, Li XM. Quetiapine reverses the suppression of hippocampal neurogenesis caused by repeated restraint stress. Brain Res. 2005;1063(1):32–9. 16271709
57. Dunn AJ, Swiergiel AH. Effects of acute and chronic stressors and CRF in rat and mouse tests for depression. Ann N Y Acad Sci. 2008;1148 : 118–26. doi: 10.1196/annals.1410.022 19120099
58. Earnheart JC, Schweizer C, Crestani F, Iwasato T, Itohara S, Mohler H, et al. GABAergic control of adult hippocampal neurogenesis in relation to behavior indicative of trait anxiety and depression states. J Neurosci. 2007;27(14):3845–54. 17409249
59. Li G, Bien-Ly N, Andrews-Zwilling Y, Xu Q, Bernardo A, Ring K, et al. GABAergic interneuron dysfunction impairs hippocampal neurogenesis in adult apolipoprotein E4 knockin mice. Cell stem cell. 2009;5(6):634–45. doi: 10.1016/j.stem.2009.10.015 19951691; PubMed Central PMCID: PMC2992822.
60. Song J, Sun J, Moss J, Wen Z, Sun GJ, Hsu D, et al. Parvalbumin interneurons mediate neuronal circuitry-neurogenesis coupling in the adult hippocampus. Nat Neurosci. 2013;16(12):1728–30. doi: 10.1038/nn.3572 24212671
61. Masiulis I, Yun S, Eisch AJ. The interesting interplay between interneurons and adult hippocampal neurogenesis. Mol Neurobiol. 2011;44(3):287–302. doi: 10.1007/s12035-011-8207-z 21956642
62. Islam MN, Fujinaga R, Yanai A, Jahan MR, Takeshita Y, Kokubu K, et al. Characterization of the "sporadically lurking HAP1-immunoreactive (SLH) cells" in the hippocampus, with special reference to the expression of steroid receptors, GABA, and progenitor cell markers. Neuroscience. 2012;210 : 67–81. doi: 10.1016/j.neuroscience.2012.02.029 22421101
63. Li Y, Chin LS, Levey AI, Li L. Huntingtin-associated protein 1 interacts with hepatocyte growth factor-regulated tyrosine kinase substrate and functions in endosomal trafficking. J Biol Chem. 2002;277(31):28212–21. 12021262
64. Kittler JT, Thomas P, Tretter V, Bogdanov YD, Haucke V, Smart TG, et al. Huntingtin-associated protein 1 regulates inhibitory synaptic transmission by modulating gamma-aminobutyric acid type A receptor membrane trafficking. Proc Natl Acad Sci U S A. 2004;101(34):12736–41. 15310851
65. Twelvetrees AE, Yuen EY, Arancibia-Carcamo IL, MacAskill AF, Rostaing P, Lumb MJ, et al. Delivery of GABAARs to synapses is mediated by HAP1-KIF5 and disrupted by mutant huntingtin. Neuron. 2010;65(1):53–65. doi: 10.1016/j.neuron.2009.12.007 20152113
66. Chan EY, Nasir J, Gutekunst CA, Coleman S, Maclean A, Maas A, et al. Targeted disruption of Huntingtin-associated protein-1 (Hap1) results in postnatal death due to depressed feeding behavior. Hum Mol Genet. 2002;11(8):945–59. 11971876.
67. Vos T, Flaxman AD, Naghavi M, Lozano R, Michaud C, Ezzati M, et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380(9859):2163–96. doi: 10.1016/S0140-6736(12)61729-2 23245607
68. Jane Costello E, Erkanli A, Angold A. Is there an epidemic of child or adolescent depression? J Child Psychol Psychiatry. 2006;47(12):1263–71. 17176381
69. Springer KW, Sheridan J, Kuo D, Carnes M. The long-term health outcomes of childhood abuse. An overview and a call to action. J Gen Intern Med. 2003;18(10):864–70. 14521650; PubMed Central PMCID: PMC1494926.
70. Surget A, Saxe M, Leman S, Ibarguen-Vargas Y, Chalon S, Griebel G, et al. Drug-dependent requirement of hippocampal neurogenesis in a model of depression and of antidepressant reversal. Biol Psychiatry. 2008;64(4):293–301. doi: 10.1016/j.biopsych.2008.02.022 18406399
71. Jayatissa MN, Henningsen K, Nikolajsen G, West MJ, Wiborg O. A reduced number of hippocampal granule cells does not associate with an anhedonia-like phenotype in a rat chronic mild stress model of depression. Stress. 2010;13(2):95–105. doi: 10.3109/10253890902951786 19929309
72. Mirescu C, Gould E. Stress and adult neurogenesis. Hippocampus. 2006;16(3):233–8. 16411244
73. Katafuchi T, Li AJ, Hirota S, Kitamura Y, Hori T. Impairment of spatial learning and hippocampal synaptic potentiation in c-kit mutant rats. Learn Mem. 2000;7(6):383–92. doi: 10.1101/Lm.33900 WOS:000166134800003.
74. Donahue CP, Jensen RV, Ochiishi T, Eisenstein I, Zhao MR, Shors T, et al. Transcriptional profiling reveals regulated genes in the hippocampus during memory formation. Hippocampus. 2002;12(6):821–33. 12542233
75. Masson K, Heiss E, Band H, Ronnstrand L. Direct binding of Cbl to Tyr568 and Tyr936 of the stem cell factor receptor/c-Kit is required for ligand-induced ubiquitination, internalization and degradation. Biochem J. 2006;399(1):59–67. 16780420
76. Jahn T, Seipel P, Coutinho S, Urschel S, Schwarz K, Miething C, et al. Analysing c-kit internalization using a functional c-kit-EGFP chimera containing the fluorochrome within the extracellular domain. Oncogene. 2002;21(29):4508–20. 12085229
77. van Praag H, Shubert T, Zhao CM, Gage FH. Exercise enhances learning and hippocampal neurogenesis in aged mice. J Neurosci. 2005;25(38):8680–5. 16177036
78. Kempermann G, Gast D, Gage FH. Neuroplasticity in old age: Sustained fivefold induction of hippocampal neurogenesis by long-term environmental enrichment. Ann Neurol. 2002;52(2):135–43. doi: 10.1002/Ana.10262 WOS:000177140000002.
79. Lin YF, Xu X, Cape A, Li S, Li XJ. Huntingtin-associated protein-1 deficiency in orexin-producing neurons impairs neuronal process extension and leads to abnormal behavior in mice. J Biol Chem. 2010;285(21):15941–9. doi: 10.1074/jbc.M110.107318 20304926
80. Aakalu G, Smith WB, Nguyen N, Jiang CG, Schuman EM. Dynamic visualization of local protein synthesis in hippocampal neurons. Neuron. 2001;30(2):489–502. 11395009
81. McGuire JR, Rong J, Li SH, Li XJ. Interaction of Huntingtin-associated protein-1 with kinesin light chain: implications in intracellular trafficking in neurons. J Biol Chem. 2006;281(6):3552–9. 16339760
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 4
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Lack of GDAP1 Induces Neuronal Calcium and Mitochondrial Defects in a Knockout Mouse Model of Charcot-Marie-Tooth Neuropathy
- Proteolysis of Virulence Regulator ToxR Is Associated with Entry of into a Dormant State
- Frameshift Variant Associated with Novel Hoof Specific Phenotype in Connemara Ponies
- Ataxin-2 Regulates Translation in a New BAC-SCA2 Transgenic Mouse Model
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy