
Glycosyl Phosphatidylinositol Anchor Biosynthesis Is Essential for Maintaining Epithelial Integrity during Embryogenesis
Cell surface proteins, such as receptors, either integrate into the plasma membrane through a transmembrane domain or are tethered to it by an accessory glycosylated phospholipid (GPI) anchor that is attached to them after they are made. The GPI-anchor biosynthesis pathway is highly conserved from yeast to humans and null mutations in any of the key enzymes are lethal at early developmental stages. Point mutations in several genes encoding for GPI-anchor biosynthesis enzymes have been linked to human disease. Specifically, mutations in PIGV are associated with multiple congenital malformations, including renal and anorectal malformation and mental retardation. It is currently not known how the mutations in PIGV lead to these diseases. Here we describe a point mutation in the PIGV ortholog of the nematode Caenorhabditis elegans, pigv-1, which is found to cause a high degree of embryonic lethality. We documented a substantial reduction in the level of GPI-anchors in the mutant. Importantly, following its development using 4D microscopy and employing tissue-specific rescue, we identified loss of epithelial integrity as the primary cause of developmental arrest. Our results highlight the importance of GPI-anchored proteins for epithelial integrity in vivo and suggest a possible etiology for human diseases associated with PIGV mutations.
Published in the journal:
. PLoS Genet 11(3): e32767. doi:10.1371/journal.pgen.1005082
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005082
Summary
Cell surface proteins, such as receptors, either integrate into the plasma membrane through a transmembrane domain or are tethered to it by an accessory glycosylated phospholipid (GPI) anchor that is attached to them after they are made. The GPI-anchor biosynthesis pathway is highly conserved from yeast to humans and null mutations in any of the key enzymes are lethal at early developmental stages. Point mutations in several genes encoding for GPI-anchor biosynthesis enzymes have been linked to human disease. Specifically, mutations in PIGV are associated with multiple congenital malformations, including renal and anorectal malformation and mental retardation. It is currently not known how the mutations in PIGV lead to these diseases. Here we describe a point mutation in the PIGV ortholog of the nematode Caenorhabditis elegans, pigv-1, which is found to cause a high degree of embryonic lethality. We documented a substantial reduction in the level of GPI-anchors in the mutant. Importantly, following its development using 4D microscopy and employing tissue-specific rescue, we identified loss of epithelial integrity as the primary cause of developmental arrest. Our results highlight the importance of GPI-anchored proteins for epithelial integrity in vivo and suggest a possible etiology for human diseases associated with PIGV mutations.
Introduction
Proteins can attach to the plasma membrane by intrinsic transmembrane domains or by post-translational modifications with lipid moieties. One such lipid modification, tethering proteins to the outer leaflet of the plasma membrane (PM), is a glycosylphospatidylinositol (GPI) anchor, whose synthesis and attachment to proteins in the endoplasmic reticulum (ER) is a multi-step process involving >30 enzymes [1]. Proteins subjected to GPI anchor modification harbor two signal peptides, an N terminal signal peptide that targets them to the ER and a C terminal signal peptide that serves as a marker for GPI attachment [2]. The basic structure of a GPI anchor consists of a phosphoethanolamine linker, a glycan core and a phospholipid tail. The glycan core of GPI can be modified by other phosphoethanolamine or other sugar groups, giving rise to diverse GPI anchor structures [3].
From the ER, GPI-anchor proteins (GPI-APs) are transported to the Golgi, where the phospholipid tail of GPI anchor undergoes lipid remodeling to increase the efficiency of membrane binding. GPI-AP are then sorted and subsequently delivered to the outer leaflet of the PM through the trans-Golgi network [4]. GPI-APs are mostly localized to the apical membrane of polarized cells and are enriched in domains known as lipid rafts [5,6]. Extraction of lipid rafts using weak non-ionic detergent pulls down GPI-APs along with the rafts [7]. Apical polarization of GPI-APs has also been observed in vivo in epithelial cells of pancreas, intestine and urinary bladder in GFP-GPI-expressing mice. GPI-APs are also present in non-polarized tissues with equal distribution across the membrane [8].
GPI-APs have very diverse functions in various cells across species. They are required for viability and cell wall biosynthesis in yeast, act as defense against host immune system in trypanosome, mediate cell-cell interactions, signal transduction, and perform enzymatic activity in mammalian cells [1,3,9]. At the tissue level, GPI-APs were shown to be important for germline and oocyte development in the nematode Caenorhabditis elegans (C. elegans). Mutation in piga-1 (ortholog of mammalian PIGA), the catalytic subunit of phosphatidylinositol N-acetylglucosaminyltransferase complex, the first enzyme playing a role in GPI biosynthesis, decreases the number of germline mitotic cells and compromises oocyte formation and maturation [10]. At the organism level, GPI-APs were shown to be essential for mouse and human embryogenesis. A complete PIGA knockout mouse could never be obtained and this may be explained by the fact that mouse embryonic stem cells depleted of PIGA form embryoid bodies that are arrested at an early stage of differentiation [11,12].
While each individual GPI-AP has a unique function that depends on the protein itself, there is evidence to suggest that GPI anchors themselves, independent of the proteins they anchor, play a role in organizing the PM. Moreover, despite the fact that GPI-anchors are positioned in the outer leaflet of the PM, they have been shown to be affected by the organization of the actin cortex underlying the PM [13,14].
Mounting evidence supporting essential roles for GPI-APs during human embryogenesis comes from human genetic studies conducted in the past decade. Missense mutations in genes encoding enzymes catalyzing various steps of GPI anchor biosynthesis, such as PIGW that catalyzes attachment of acyl group to phosphatidylinositiol [15,16], PIGV that catalyzes transfer of second mannose to GPI intermediate [17,18], PIGT that attaches GPI to proteins [19] and PGAP2 that modifies the phospholipid tail of PI [20] result in congenital diseases known as hyperphosphatasia mental retardation syndrome (HPMRS), Hirschprung disease, morphological malformation and renal anomalies [21,22,23,24].
Studies on the roles of GPI-APs during embryogenesis have been hindered by the difficulties in obtaining viable mutants for GPI biosynthesis enzymes. In this study, we exploit a hypomorphic temperature-sensitive allele of pigv-1 (human PIGV ortholog) in C. elegans to investigate the role of GPI-APs during embryogenesis. We found that GPI-APs are vital for the integrity of epithelial tissues during morphogenesis, suggesting an essential role for GPI-APs in stabilizing the apical membrane of epithelial tissues under stress.
Results
A temperature sensitive missense mutation in pigv-1(qm34) leads to defects in embryonic elongation
In the course of our whole genome sequencing of maternal-effect morphologically abnormal (mal) mutants isolated by Hekimi et al. [25] in an ethyl methanesulfonate (EMS) mutagenesis screen, we discovered that mal-3(qm34) (Fig. 1A) has a missense mutation at amino acid 361 of previously unassigned gene T09B4.1, converting glycine to glutamate (Fig. 1B). BLAST analysis of the T09B4.1 protein sequence suggested that it is an ortholog of human GPI mannosyltransferase 2, which is known as PIGV (S1A–B Fig.). The mutation was verified by conventional sequencing, and from this point onwards we refer to mal-3(qm34) as pigv-1(qm34).

Sometimes, EMS mutagenesis results in hypomorphic alleles that are temperature-sensitive. We investigated this possibility by growing pigv-1(qm34) worms at 15, 20 and 25°C and measuring their viability at each temperature. We found that pigv-1(qm34) is a heat-sensitive allele, with more than 80% embryonic lethality at 25°C (Fig. 1C, S1 Table). We followed embryogenesis by time-lapse differential interference contrast (DIC) microscopy and observed phenotypes resulting from pigv-1 inactivation at 25°C. At this temperature C. elegans embryogenesis takes 10.5 hours from the first division till hatching (S2 Fig.). The first 3 hours of embryogenesis are characterized by formation of the founder cells, rapid cell division and gastrulation. At around 3 hours the epidermis is born on the dorsal side of the embryo and the next 3.5 hours are dominated by epidermal morphogenesis, a three step process made up of intercalation, enclosure, and elongation [26]. Loss of pigv-1 resulted in defects appearing during elongation with cysts forming inside the embryo and/or cells leaking out from the embryo body, resulting in elongation arrest and embryonic lethality (Fig. 1D). Quantification of 176 pigv-1(qm34) embryos showed that over 80% displayed ruptures and/or cyst formation and arrested in elongation (Fig. 1E). Few escapers hatched and became L1 larva with body shape defects (Fig. 1A).
Utilizing the heat sensitivity of pigv-1(qm34), we determined the developmental period when pigv-1 activity is required through reciprocal temperature shift experiments. We found that pigv-1 activity is essential from the one cell embryo stage until elongation. Once elongation was underway inactivation of PIGV-1 had less effect on embryogenesis (Fig. 1F, S3 Table).
We confirmed that these phenotypes are caused by the mutation in pigv-1 by rescue experiments. Transformation of pigv-1(qm34) worms with a fosmid that contains a wild-type allele of the pigv-1 gene significantly rescued embryonic lethality (P<0.01) (Fig. 1G, S4 Table). Expression of a gfp-tagged pigv-1 under the control of 2.4 kb upstream of the pigv-1 start codon failed to rescue embryonic lethality in pigv-1(qm34) mutant worms, most likely due to low expression. On the other hand, expression of pigv-1 under the control of the erm-1 promoter, which resulted in 3 fold stronger expression, successfully rescued embryonic lethality of pigv-1(qm34) (Fig. 1G, S4 Table), confirming that the mutated gene causing lethality in the pigv-1(qm34) strain is pigv-1.
GPI-APs are present throughout embryogenesis and are enriched in apical membranes of epithelial cells after differentiation
To visualize GPI-AP distribution during embryogenesis, we used Alexa-488 labeled proaerolysin (FLAER), a bacterial toxin that binds specifically to GPI-AP [27], to label embryos at different stages of development (Fig. 2). In the one-cell embryo, GPI-APs accumulated at perinuclear areas and were enriched in the anterior cytoplasm (Fig. 2, first row). As soon as new membrane was delivered to the cell surface, during cell division, GPI-APs accumulated on the plasma membrane (Fig. 2, second row). During gastrulation we observed GPI-APs accumulated at the membrane of all cells (Fig. 2, fourth row). While being uniformly localized on membrane of all cells in the early embryo, non-uniform GPI-AP distribution was observed upon tissue differentiation. For example, during dorsal intercalation, GPI-APs were highly enriched on pharyngeal cell membranes in a non-polarized manner, whereas later on, during elongation, they became apically enriched (Fig. 2, fifth to seventh rows).

PIGV-1 is expressed in all embryonic epithelial tissues where it is required for GPI-anchor biosynthesis
To gain insight into the spatial and temporal activity of the PIGV-1 enzyme during embryogenesis, we visualized an N-terminally GFP-tagged PIGV-1 driven by its endogenous promoter in an extrachromosomal array. We could not detect GFP::PIGV-1 in the early embryo, possibly due to silencing of the transgenes in the germline. Later in development the expression level was low. Nevertheless, we observed GFP::PIGV-1 to be prominent in the epidermis, pharynx, intestine, rectum and excretory cell (Fig. 3A), all tissues with epithelial character. At the subcellular level, PIGV-1 localized to intracellular structures that appear to be ER [28,29].
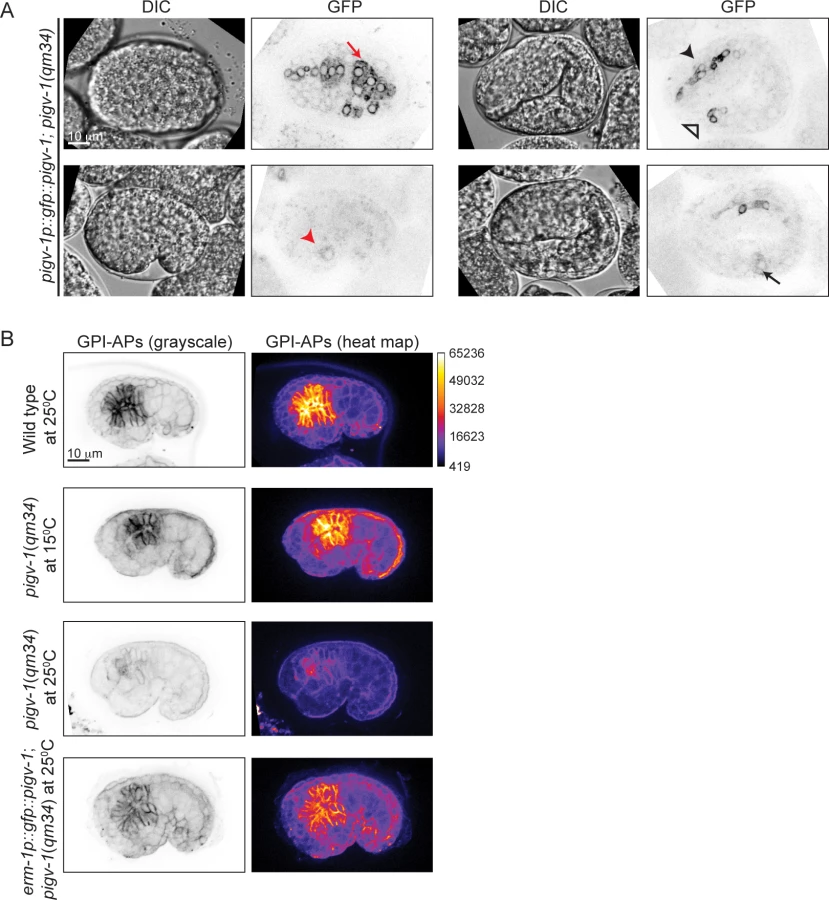
Using FLAER staining as readout for GPI-anchor biosynthesis, we compared the intensity of FLAER staining in pigv-1(qm34) at the permissive (15°C) and restrictive (25°C) temperatures. At 15°C, pigv-1(qm34) embryos exhibited FLAER levels similar to wild type, whereas at 25°C, abrogation of PIGV-1 activity led to a 4-fold reduction in the FLAER signal (Fig. 3B, second and third rows). FLAER staining was restored to wild-type levels in pigv-1(qm34) embryos at 25°C when pigv-1 was expressed in all epithelial tissues by the erm-1 promoter (Fig. 3B, fourth row). Taken together these data show that GPI-APs are enriched in epithelial tissues and the abundance of GPI-APs at the cell membrane is dependent on the activity of PIGV-1.
Loss of function of piga-1 exhibits phenotypes similar to pigv-1(qm34) mutant embryos
In mammalian cells, more than 30 enzymes are known to regulate the GPI anchor biosynthesis pathway. Many of these enzymes have orthologs in C. elegans (S1B Fig.). A previous study has shown that RNAi-mediated knockdown of most C. elegans GPI anchor biosynthesis enzymes does not lead to any phenotype and two of them, namely pigk and pigo resulted in sterility [10]. We scanned a range of feeding RNAi conditions for pigk and pigo, with the rationale that partial loss of function might bypass their requirement for germline development, and expose a possible role in embryogenesis. However, we observed either sterility or no phenotype when each of the two enzymes was depleted. Thus, we turned our attention towards piga-1(tm2939) mutant worms characterized in the previous study [10]. Progeny of homozygous piga-1(tm2939) worms are embryonic lethal, and they display a deformed eggshell due to increased osmotic sensitivity during germline development. To uncouple the functions of PIGA-1 during germline development and embryogenesis, we used piga-1(tm2939) worms rescued by piga-1 expression under the control of lag-2, a distal tip cell promoter. First, we examined whether lag-2 drives piga-1 expression during embryogenesis and found piga-1 expressed ubiquitously during embryogenesis (Fig. 4A). Since the plag-2::piga-1::gfp construct is expressed as an extrachromosomal array, some embryos lose piga-1 expression during embryogenesis. We identified which embryos lost the extrachromosomal array and followed their embryonic phenotypes. While all the embryos retaining piga-1 expression during embryogenesis hatched, ~50% of the embryos devoid of piga-1 expression were arrested during elongation. In one-third of arrested embryos, internal cells leaked out from the embryo body (Fig. 4B), a phenotype reminiscent of pigv-1(qm34) embryos, suggesting that weakening of epithelial tissue integrity is not a specific phenotype of pigv-1 loss of function, but rather a general consequence of disruption of the GPI biosynthesis pathway.

Loss of GPI-APs leads to breaches in epithelial tissue in the epidermis, embryonic intestine and excretory canal
The elongation phase of C. elegans embryogenesis is characterized by the formation of circumferential actin bundles (CFB) in the dorsal and ventral epidermal cells and actomyosin contraction in the lateral epidermal cells [30]. Contractility of the muscle tissues is known to be required for elongation beyond the 2-fold length [31]. We tested whether the elongation arrest occurring upon pigv-1 inactivation is caused by defective CFB and/or muscle organization. We examined CFB in pigv-1(qm34) embryos using an F-actin reporter (VAB-10 actin binding domain tagged with GFP) and found that CFB structure is indistinguishable from that of wild type embryo (S3A Fig.). Myotactin antibodies were utilized to examine muscle organization and we observed no difference between muscle organization in wild type and in pigv-1(qm34) embryos (S3B Fig.). Moreover, some pigv-1(qm34) embryos elongated beyond two-fold stage. These results suggest that elongation arrest in pigv-1(qm34) embryos is not caused by defects in CFB or muscle structure.
We then set out to characterize the embryonic phenotypes resulting from pigv-1 inactivation in more detail. We employed several cell junction and membrane markers expressed in specific tissues to pinpoint the location of the defects. Using AJM-1::GFP and HMP-1::GFP as a marker for epidermal apical junctions, we observed gaps between epidermal cells through which internal cells leaked out, most often from the embryo anterior (Fig. 5A and S1–S4 Movies). In some embryos, the gap is created by misalignment of leading ventral epidermal cells coming from opposite ends to enclose the embryo at the ventral midline (Fig. 5A). Using a plasma membrane marker specifically expressed in the pharynx and intestine, we identified cysts to be located at the basal side of the intestine, and using CED-10::GFP, which highlights plasma membrane of all cells, we observed cysts to be located between the intestine and its surrounding basal lamina (Fig. 5B-C, S4 Fig.). Using AJM-1::GFP to highlight the apical junctions of intestinal cells we observed widening of the lumen in pigv-1(qm34) embryos (Fig. 5D). We measured intestinal lumen width of wild type and pigv-1(qm34) embryos in early (2–2.5 fold) and later (3–3.5 fold) stages of elongation and found that the lumen width of pigv-1(qm34) embryos was significantly wider (P<0.05) than that of wild type embryos at late elongation (Fig. 5D). Furthermore, we noticed that the intestine in pigv-1(qm34) embryos was often twisted (S6C Fig.).

Using mCherry-tagged AQP-8, a water channel specifically localized to the excretory canal, expressed at a low level which maintains a normal translumenal flux, we found the excretory canal to be another location where cysts formed in pigv-1(qm34) embryos (Fig. 5E). In contrast with the intestinal cysts that formed in extracellular space the excretory canal cysts formed within the cell. Few embryos that survived embryogenesis hatched with excretory canal cysts (S5 Fig.). The excretory canals in larvae with excretory canal cysts were usually very short. Not only the length, but the branching of the excretory canal is also affected in pigv-1(qm34) embryo. In wild-type worms the excretory canal extends four tubules shaped like an H: a pair towards anterior and another pair towards posterior from the cell body. However, the excretory canal in pigv-1(qm34) embryo often has one or two more tubules extending from the cell body or branching from the original tubules (S7 Fig.).
GPI-APs are known to be targeted to apical membranes in polarized epithelial cells [5,6]. We therefore examined whether the apicobasal polarity of epithelial cells is affected in pigv-1(qm34) embryos. We observed that the apical markers PAR-6 and PKC-3 were correctly localized on the apical intestinal and excretory cell membranes in pigv-1(qm34) embryos (S6A Fig.), and AJM-1 was localized at the apical side of epidermal and intestinal cell-cell junctions (S6B–C Fig.). Conversely, the basolateral marker LET-413 was localized to the basolateral membranes in epidermal and intestinal cells in pigv-1(qm34) embryos, indistinguishable from wild type (S6B Fig.). Similarly, the intermediate filament IFB-2 was correctly localized beneath the apical membrane in intestinal tissue in pigv-1(qm34) embryos (S6C Fig). Altogether, these results rule out a polarity defect as the underlying cause for the pigv-1(qm34) mutant phenotypes.
GPI-AP biosynthesis in all epithelial tissues is required to rescue embryonic lethality of pigv-1(qm34)
We observed pigv-1 loss of function to affect the integrity of three epithelial tissues: epidermis, intestine and excretory canal. However, it was not immediately evident which defective tissue was responsible for the embryonic lethality. To address this question we restored pigv-1 expression specifically in each epithelial tissue or in all epithelial tissues of pigv-1(qm34) worms and determined their embryonic viability. We employed the lin-26 promoter to drive expression in the epidermis, the pha-4 promoter to drive expression in the pharynx and intestine, the aqp-8 promoter to drive expression in the excretory canal, and the erm-1 promoter to drive expression in all epithelial tissues (Fig. 6A). All promoters drove pigv-1 expression at comparable levels.

While restoring pigv-1 expression in the pharynx-intestine or in the excretory canal partially reduced pigv-1(qm34) embryonic lethality, restoring pigv-1 expression in the epidermis did not significantly reduce pigv-1(qm34) embryonic lethality (Fig. 6B, S4 Table). The most significant rescue of embryonic lethality achieved by expression of pigv-1 in a single tissue was a 17% reduction in lethality. In contrast, expressing pigv-1 in all epithelial tissues using the erm-1 promoter sharply decreased pigv-1(qm34) embryonic lethality down by 62%, comparable to the sum of the embryonic rescue of each epithelial tissue (Fig. 6B, S4 Table). Thus, it appears that pigv-1 function is required in all epithelial tissues for embryonic viability.
Overexpression of ERM-1::GFP rescues embryonic lethality of pigv-1(qm34)
The cytoskeletal cortex underlying the plasma membrane provides it with structural support and protects the membrane from mechanical stress. Depletion of spectrin in erythrocytes changes membrane rigidity and subsequently leads to cell fragmentation [32]. Thus, we hypothesized that strengthening the actin cortex in pigv-1(qm34) embryos might positively affect membrane integrity. First, we examined whether providing more actin has any impact on epithelial membrane integrity. We examined pigv-1(qm34) embryos overexpressing YFP::ACT-5 in the epidermis and intestine and found that embryonic lethality in these worms is indistinguishable from that of pigv-1(qm34) (Fig. 7A, S5 Table). We then explored whether strengthening the link between the actin cortex and the cell membrane might influence membrane integrity. We chose worms overexpressing ERM-1::GFP at a level which does not cause any phenotypic defect since a previous study showed that at high levels of expression ERM-1 leads to formation of excretory canal cysts [33]. We crossed the ERM-1::GFP-overexpressing worms with pigv-1(qm34) and found that embryonic lethality was significantly reduced in the pigv-1(qm34);ERM-1::GFP strain. Depletion of overexpressed ERM-1 by gfp(RNAi) in this stain reverted embryonic lethality back to pigv-1(qm34) level, confirming ERM-1 overexpression is responsible for rescuing pigv-1-associated embryonic lethality (Fig. 7A, S5 Table). Careful examination of embryogenesis in pigv-1(qm34) embryos overexpressing ERM-1::GFP revealed strong suppression of pigv-1 phenotypes, and a significant portion of embryos (36%) hatched without any visible defects (Fig. 7B, S2 Table). Considering ERM-1 localization at intestine and excretory canal apical membranes, we reasoned that ERM-1 overexpression could rescue apical-associated phenotypes in these tissues. Measuring the width of intestinal lumen we found that it was reduced to the wild type dimension (Fig. 7C).

To gain insight into the mechanism of pigv-1-phenotype rescue by ERM-1 overexpression, we examined whether endogenous ERM-1 distribution and level were altered in pigv-1(qm34) embryos. Immunolabeling with ERM-1 antibodies showed no difference in ERM-1 distribution or level between wild type and pigv-1(qm34) embryos (Fig. 8A). We then asked whether ERM-1 might rescue pigv-1 mutant by enhancing the residual pigv-1 activity and restoring the level of GPI-APs. Using FLAER as the probe for GPI-APs, we found that GPI-AP level in pigv-1(qm34) embryos overexpressing ERM-1 is similar to pigv-1(qm34) embryos alone, suggesting that ERM-1 overexpression does not rescue pigv-1 embryonic lethality by restoring GPI-APs (Fig. 8B).

Discussion
GPI anchor is an important post-translational protein modification whose functions and mechanisms have been widely studied using unicellular organisms and mammalian cells in culture [1,6,9]. However, the role of GPI biosynthesis in animals remains poorly understood. In humans, somatic mutations in PIGA gene loci lead to paroxysmal nocturnal hemaglobinuria, a disease characterized by increased susceptibility of erythrocytes to lysis by the complement immune system [34]. No heritable mutation in piga gene in human has been identified, suggesting that PIGA function is required during embryogenesis. Indeed, deletion of PIGA gene in mice, which completely abrogates GPI biosynthesis, resulted in early embryogenesis defects [11,35]. However, this condition precludes the study of GPI function throughout embryogenesis.
Also in C. elegans, a null mutation in piga-1 results in germline defects and early embryonic lethality. In this study, we circumvented the early requirements for GPI-APs by using a temperature sensitive hypomorphic allele of pigv-1. The amount of GPI-APs remaining upon pigv-1 inactivation was sufficient for normal germline development, thus enabling us to uncover their requirement during embryogenesis. We showed that GPI-APs are present and function throughout embryogenesis. Interestingly, the phenotypes of pigv-1 inactivation, i.e., weakened epithelial tissues, are manifested only late in embryogenesis during the elongation stage of epidermal morphogenesis. This may be due to increased mechanical forces generated by actomyosin in muscle and epidermis tissues at that stage.
Although present at the membrane of all cells in the C. elegans embryo, pigv-1 loss of function exhibits no defect in early development events, such as gastrulation or tissue differentiation. This is in contrast to mammalian embryogenesis, in which complete PIGA depletion results in defects in tissue differentiation [35]. One possible explanation for this difference is that the residual GPI-APs in pigv-1 animals are sufficient for normal differentiation. Another reason could be differences in the proteins regulating differentiation. Tissue differentiation in mammals is regulated by BMP/ TGF-β signaling whose activation requires GPI-anchored co-receptors, Dragon and Cripto-1 [35,36]. Although present in C. elegans, BMP/TGF-β signaling is not required during embryogenesis, but operates during postembryonic development, regulating body size [37,38].
Inactivation of pigv-1 in C. elegans embryos resulted mainly in epithelial defects. Failures in epidermal enclosure and intestinal cyst formation are consistent with weaker cell-cell adhesion. In the epidermis, improper cell-cell adhesion creates gaps between ventral epidermal cells from which internal tissues leak out during elongation. Compromised cell-cell junctions in the intestine, which has higher osmotic pressure than the surrounding tissues, would allow passage of low molecular weight substances, such as water molecules, from the intestinal lumen to the intestine basal side. The presence of a basal lamina separating the intestine from the pseudocoelom results in accumulation of these substances in the form of cysts.
One possible explanation of these results is that the reduction in the amount of one or more specific cell adhesion proteins that are GPI-anchored causes the observed defects. However, amongst the GPI-APs that have been experimentally identified in C. elegans none are known to mediate cell-cell adhesion [7,10]. While we do not rule out the involvement of yet unknown GPI-anchored adhesion proteins, we propose another mechanism to explain the observed epithelial phenotypes that does not depend on a specific protein, but rather on the GPI anchors themselves. Goswami et. al. have shown that cortical actin affects the organization of GPI-AP in the membrane [13]. We propose that GPI-APs are enriched in apical membranes of polarized epithelial cells where they play a role in organizing the membrane into domains that interact with the actin cortex within the cell and through these interactions stabilize the apical membrane. According to this model, a decrease in GPI-APs will lead to fewer membrane-cortex connections and thus to a weaker apical membrane. In support of this idea, we observed a widening of the intestinal lumen in pigv-1 mutant embryos, as would be expected if the apical membrane of the intestine is weakened and thereby cannot resist as well the osmotic pressure from inside the lumen. Further support for this model comes from the finding that overexpression of ERM-1 rescues lumen width and overall embryonic lethality of pigv-1 mutant embryos. ERM-1, the sole C. elegans ortholog of ezrin, radixin and moesin, is a linker protein that has an actin-binding domain and attaches to the PM through its FERM domain, serving to connect the PM with the actin cortex [39,40]. ERM-1 overexpression did not increase the levels of GPI-APs in pigv-1 embryos and hence the reduction in lethality associated with it is most likely due to its membrane-cortex cross-linking function. From this we deduce that loss of GPI-APs leads to a weakening of apical membranes in epithelial cells, irrespective of the proteins attached to the GPI-anchor.
Another epithelial tissue affected by pigv-1 loss of function is the excretory canal. Down regulation of GPI-APs in the excretory canal leads to cysts formation and this cystic excretory canal is usually short, consistent with apical membrane weakening upon the loss of GPI-APs. Unlike the intestine, which is a multicellular tubule, the excretory canal is a unicellular tubule that extends actively during embryo elongation. It has been demonstrated that a balance between membrane-actin cortex recruitment and translumenal flux is essential for the excretory canal extension [33]. Weakened apical membrane upon down regulation of GPI-APs in pigv-1 mutant embryo may prevent further recruitment of membrane components and actin undercoat to extend the canal, creating an imbalance between the two forces. Consequently, the dominant force, the translumenal flux is transmitted to enlarge the canal diameter, resulting in cysts formation. Another phenotype we observed in the excretory canal of pigv-1 mutants is ectopic branching. To our knowledge such a phenotype has not been associated with loss of function of any gene so far, opening a new avenue to study the regulation of tubular branching.
Besides epithelial tissues, loss of GPI-APs in C. elegans may also affects neuronal and/or muscle tissues, suggested by the lethargic phenotype of pigv-1(qm34) worms, although the GPI-APs responsible for this phenotype remains to be identified. The short excretory canal that has been observed occasionally in pigv-1(qm34) could also result from the loss of GPI-APs from neuronal membrane. The neuronal cell adhesion molecules (NCAM) that are essential for axon outgrowth and pathfinding have been demonstrated to regulate excretory canal extension [41,42,43]. In the absence of NCAM, the excretory canal does not grow to full extent. Supporting this view, the in vitro and the in silico experiments found several NCAMs (rig-3, rig-6, rig-7 and wrk-1) to be potentially GPI-modified [7,44].
Not all epithelial tissues displayed abnormal phenotypes in pigv-1 mutants. The pharynx and rectum are two epithelial tissues that do not seem to be affected by down regulation of GPI-APs. High enrichment of GPI-APs at pharyngeal membranes compared to other tissues could provide an explanation for the absence of weakened membrane phenotypes. However, this reason does not hold for the rectum, as GPI-APs at rectal membranes are not more abundant than other tissues that display weakened membrane. Since both pharynx and rectum are covered by a cuticle, the most likely explanation for the absence of visible phenotypes is that the cuticle protects both tissues from potential damage resulting from weakened membranes.
Restoring PIGV-1 expression individually in the epidermis, intestine and excretory canal in pigv-1(qm34) embryos revealed that the defects in these epithelial tissues do not contribute equally to the embryonic lethality. The intestine and the excretory canal defects have higher contribution to embryonic lethality as compared to the epidermal defects. This is somewhat unexpected because 56% of pigv-1(qm34) embryos die due to tissues leakage from the embryo interior, indicating that a gap between epidermal cells is present from where the tissues pass through. However, uncontained high pressure built in the intestinal and excretory canal due to cell adhesion defects and membrane weakening may be sufficient to open epidermal junction and push the internal tissues out of embryo interior.
Mutations in the human ortholog of C. elegans pigv-1, PIGV, have been associated in genetic studies with hyperphosphatasia-mental retardation syndrome (a.k.a Mabry syndrome). This autosomal recessive syndrome has a wide spectrum of phenotypes including intellectual disabilities, facial anomalies, hyperphosphatasia, vesicoureteral and renal anomalies, and anorectal anomalies [21]. With the exception of hyperphosphatasia, which is known to be the result of loss of GPI-anchored complement inhibitors in blood cells [34], the proteins and cellular functions that are affected in humans with PIGV mutations are unknown. Although our findings in C. elegans cannot possibly fully explain the cellular physiology of the human disease, it does point to a basic mechanism, i.e., weakening of apical membranes in epithelial cells, that may be playing a role in some of the manifestations of the disease. Furthermore, if it will be discovered that epithelial membrane integrity is affected in human patients then our work also suggests a promising avenue for therapy, i.e., strengthening of the membrane-cortex connection, based on our ERM-1 overexpression results.
Materials and Methods
Strains and alleles
Strains were grown and maintained at 20°C under standard conditions [45]. Wild type strain N2 was used as a control. The pigv-1(qm34) was retrieved from an EMS screening conducted by Hekimi et al. [25]. For analysis using GFP fusions, F2 progeny exhibiting pigv-1 phenotypes and carrying the markers were selected from crosses between pigv-1(qm34) and the following strains: SU93 jcIs1[ajm-1::gfp, unc-29(+), rol-6p::rol-6(su1006)] [46], SU265 jcIs17[hmp-1p::hmp-1::gfp, dlg-1p::dlg-1::dsRed, rol-6p::rol-6(su1006)] [47], SU467 pIs7[pha-4p::pm::gfp, rol-6p::rol-6(su1006)] [48], FT17 xnIs3[par-6p::par-6::gfp, unc-119(+)]; unc-119(ed3) III, MOT63 temIs59[pIC26::pkc-3]; unc-119(ed3) III, WS4918 opIS310[ced-1p::yfp::act-5::let-858 3'UTR, unc-119(+)] [49], VJ402 fgEx1[erm-1p::erm-1::gfp, rol-6p::rol-6(su1006)] [33], ML1735 mcIs50[lin-26p::vab-10(actin-binding domain)::gfp, myo-2p::GFP] [50], plag-2p::piga-1::egfp-expressing strain was generated by Murata et al [10].
Plasmid construction
All plasmids generated in this study were constructed in a modified pPD95.75 backbone. For tissue-specific rescue of pigv-1 loss of function, GFP position was changed to be at the N terminal instead of at the C terminal of the multiple cloning sites (MCS), whereas for AQP-8-expressing plasmid, GFP at C terminal was replaced with mCherry. To construct pigv-1p::gfp::pigv-1 plasmid, pigv-1 promoter (2.4 kb sequence upstream of pigv-1 start codon) and coding sequence were amplified and inserted into SbfI and AgeI sites upstream of gfp in original pPD95.75 vector. Circular PCR was performed to amplify the whole plasmid, but the gfp region using a pair of primers harboring XhoI sites at their 5’ ends. PCR product was then ligated to produce a circular plasmid containing pigv-1 promoter and coding sequence, but without gfp. Second circular PCR was conducted to insert two new restriction sites, i.e.: NotI and BglII between pigv-1 promoter and coding sequence. PCR product was then subjected to digestion using NotI and BglII. gfp coding sequence was amplified from original pPD95.75 and subcloned into pJET (Thermo Scientific). The recombinant plasmid was digested using NotI and BglII and gfp sequence-containing product was ligated to pigv-1-containing pPD95.75, resulting in a plasmid expressing gfp::pigv-1 driven by pigv-1 promoter. Four different promoters were used to rescue pigv-1(qm34) in different tissues: 4.1 kb sequence of lin-26 promoter to drive pigv-1 expression in epidermis, 7.1 kb of pha-4 promoter for expression in pharynx and intestine, 2.2 kb of aqp-8 promoter for expression in excretory canal and 3 kb of erm-1 promoter for expression in all epithelial tissues. They are inserted into modified pPD95.75 at SbfI/NotI sites replacing pigv-1 promoter. Transgenic animals generated by injecting the constructs into the gonad of hermaphrodite animals resulted in the following strains: RZB40 (pigv-1(qm34); msnEx40[lin-26p::gfp::pigv-1; rol-6(su1006)]), RZB41 (pigv-1(qm34); msnEx41[pha-4p::gfp::pigv-1; rol-6(su1006)]), RZB129 (pigv-1(qm34); msnEx129[aqp-8p::gfp::pigv-1; rol-6(su1006)]) and RZB128 (pigv-1(qm34); msnEx128[erm-1p::gfp::pigv-1; rol-6(su1006)]).
To construct aqp-8::mCherry-expressing plasmid, mCherry coding sequence was amplified from pAA64 and ligated to circularly amplified pPD95.75 devoid of gfp sequence using Gibson assembly (NEB). Subsequently, 2.2 kb aqp-8 promoter together with aqp-8 genomic sequence were inserted at SbfI/BamHI sites in modified pPD95.75. Injection of this construct resulted in strain RZB221 (pigv-1(qm34); msnEx221[aqp-8p::aqp-8::mCherry; rol-6(su1006)]).
Microinjection
Microinjection was performed as described by Mello and Fire [51]. Injection mix includes 100 μg/μl salmon sperm DNA digested with PvuII, 20 μg/μl rol-6(su1006) digested with SbfI and 5–10 μg/μl each construct digested with SbfI.
Whole genome sequencing and mutation validation
Genomic DNA was extracted from pigv-1(qm34) mutant worms using standard method and subjected to whole genome sequencing using Illumina platform and annotated using MAQGene [52]. The whole genome sequencing and its annotation were performed by Hobert lab (Columbia University). Candidate genes altered in pigv-1(qm34) were narrowed down using genetic mapping results done by Hekimi et al. [25]. Point mutation in pigv-1 gene was confirmed by amplification of pigv-1 gene in pigv-1(qm34) mutant worms, subcloning into pJET vector (Thermo Scientific) and followed by conventional sequencing (First Base). Further validation of pigv-1 missense mutation as the phenotype-causing gene in pigv-1(qm34) worms was done by injection of 100 μg/μl fosmid WRM063BcC08, which contains pigv-1 gene, together with the co-transformation marker rol-6(su1006) into the gonad of pigv-1(qm34) hermaphrodites. F2 rollers were upshifted to 25°C and examined for embryonic lethality.
Quantification of embryonic lethality
Ten to fifteen gravid hermaphrodites were placed on the plate and incubated for several hours to lay more than 100 eggs. Hermaphrodites were then removed and the number of eggs laid was counted. Twenty-four hours later, the number of larvae hatched was determined. Each experiment was done in duplicate and repeated five times. Beside experiments determining temperature sensitivity that are conducted at three different temperatures (15°C, 20°C and 25°C), the remaining experiments were conducted solely at 25°C to get the highest extent of pigv-1 inactivation. In this case, L4 larvae were upshifted from 20°C to 25°C for 20 to 24 hours prior to the test.
Temperature shift experiments
For upshift experiment, embryos were dissected from gravid pigv-1(qm34) worms grown at 15°C and incubated at 25°C for the duration of embryogenesis. Each embryo was staged and scored for hatching. For downshift experiment, similar procedure was performed, except that pigv-1(qm34) worms were kept at 25°C for 24 hours before downshifted to 15°C. Embryos that do not hatch at the end of embryogenesis were considered as lethal.
4D microscopy and Pigv-1 phenotype classification
Larvae or embryos collected from gravid hermaphrodite, mounted onto 3% agarose padded-glass slide, closed with coverslip and sealed with wax. Normaski images shown in Fig. 1A, B and S2B were captured using a Nikon Ti Eclipse widefield microscope equipped with DIC 1.40NA oil condenser and a charged-coupled device camera Cool Snap HQ2 (Photometrics). All other imaging were done using spinning disk confocal system composed of a Nikon Ti Eclipse microscope with a CSU-X1 spinning disk confocal head (Yokogawa), DPSS-Laser (Roper Scientific) at 491 and 568 nm excitation wavelength and an Evolve Rapid-Cal electron multiplying charged-coupled device camera (Photometrics). For both microscopes, Metamorph software (Molecular Devices) was used to control acquisition. Projected images were created using Fiji.
After 24 hour DIC recording, wild type, pigv-1(qm34) and pigv-1(qm34) embryos expressing ERM-1::GFP were scored as viable or lethal and each category is further classified into four subcategories; i.e.: without visible defects, with cysts and rupture, with cyst only and with rupture only.
GFP knockdown by RNAi
IPTG plate used for gfp(RNAi) feeding was prepared as described [53]. Wild type and pigv-1(qm34) L1 larvae expressing ERM-1::GFP were fed using bacterial-feeding strain of gfp for 3 days at 15°C till they become L4 and then upshifted to 25°C for overnight. The absence of GFP signal was verified by using fluorescent stereomicroscope and only those devoid of the signal were subjected for embryonic lethality test.
FLAER staining and immunolabeling
Fixation and indirect immunofluorescence were performed essentially as described [54]. The following primary mouse antibodies were used: ERM-1 (DSHB; 1/20), AJM-1 (MH27, DSHB; 1/10), myotactin (MH46, DSHB; 1/5) and LET-413 (DSHB; 1/2) and IFB-2 (MH33, DSHB; 1/5). Donkey anti-mouse coupled to Alexa 647 (1/500) (Life technologies) was used as secondary antibodies and proaerolysin coupled to Alexa 488 (FLAER, Protox Biotech) was used to detect GPI-APs. Images were taken on a Nikon Ti Eclipse spinning disk microscope with 100x objective and processed further using Fiji. To measure lumen width in wild type and pigv-1(qm34) mutant embryos, N2 and pigv-1(qm34) embryos expressing AJM-1::GFP were fixed, maximum intensity projection of embryonic intestine in GFP channel was constructed and the widest section of intestinal lumen was determined. The same procedure was done to measure lumen width in pigv-1(qm34) embryos expressing ERM-1::GFP, except that AJM-1 antibodies were used instead of AJM-1::GFP expression.
Statistical analysis
Statistical analyses were done using Microsoft Excel. Two-tailed Student’s t-test was applied to compare the values.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Ferguson MA (1999) The structure, biosynthesis and functions of glycosylphosphatidylinositol anchors, and the contributions of trypanosome research. J Cell Sci 112 (Pt 17): 2799–2809. 10444375
2. Fujita M, Jigami Y (2008) Lipid remodeling of GPI-anchored proteins and its function. Biochim Biophys Acta 1780 : 410–420. 17913366
3. Paulick MG, Bertozzi CR (2008) The glycosylphosphatidylinositol anchor: a complex membrane-anchoring structure for proteins. Biochemistry 47 : 6991–7000. doi: 10.1021/bi8006324 18557633
4. Maeda Y, Kinoshita T (2011) Structural remodeling, trafficking and functions of glycosylphosphatidylinositol-anchored proteins. Prog Lipid Res 50 : 411–424. doi: 10.1016/j.plipres.2011.05.002 21658410
5. Brown DA, Rose JK (1992) Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell 68 : 533–544. 1531449
6. Paladino S, Pocard T, Catino MA, Zurzolo C (2006) GPI-anchored proteins are directly targeted to the apical surface in fully polarized MDCK cells. J Cell Biol 172 : 1023–1034. 16549497
7. Rao W, Isaac RE, Keen JN (2011) An analysis of the Caenorhabditis elegans lipid raft proteome using geLC-MS/MS. J Proteomics 74 : 242–253. doi: 10.1016/j.jprot.2010.11.001 21070894
8. Kondoh G, Gao XH, Nakano Y, Koike H, Yamada S, et al. (1999) Tissue-inherent fate of GPI revealed by GPI-anchored GFP transgenesis. FEBS Lett 458 : 299–303. 10570928
9. Leidich SD, Drapp DA, Orlean P (1994) A conditionally lethal yeast mutant blocked at the first step in glycosyl phosphatidylinositol anchor synthesis. J Biol Chem 269 : 10193–10196. 8144596
10. Murata D, Nomura KH, Dejima K, Mizuguchi S, Kawasaki N, et al. (2012) GPI-anchor synthesis is indispensable for the germline development of the nematode Caenorhabditis elegans. Mol Biol Cell 23 : 982–995. doi: 10.1091/mbc.E10-10-0855 22298425
11. Dunn DE, Yu J, Nagarajan S, Devetten M, Weichold FF, et al. (1996) A knock-out model of paroxysmal nocturnal hemoglobinuria: Pig-a(-) hematopoiesis is reconstituted following intercellular transfer of GPI-anchored proteins. Proc Natl Acad Sci U S A 93 : 7938–7943. 8755581
12. Keller P, Tremml G, Rosti V, Bessler M (1999) X inactivation and somatic cell selection rescue female mice carrying a Piga-null mutation. Proc Natl Acad Sci U S A 96 : 7479–7483. 10377440
13. Goswami D, Gowrishankar K, Bilgrami S, Ghosh S, Raghupathy R, et al. (2008) Nanoclusters of GPI-anchored proteins are formed by cortical actin-driven activity. Cell 135 : 1085–1097. doi: 10.1016/j.cell.2008.11.032 19070578
14. Gowrishankar K, Ghosh S, Saha S, C R, Mayor S, et al. (2012) Active remodeling of cortical actin regulates spatiotemporal organization of cell surface molecules. Cell 149 : 1353–1367. doi: 10.1016/j.cell.2012.05.008 22682254
15. Murakami Y, Siripanyapinyo U, Hong Y, Kang JY, Ishihara S, et al. (2003) PIG-W is critical for inositol acylation but not for flipping of glycosylphosphatidylinositol-anchor. Mol Biol Cell 14 : 4285–4295. 14517336
16. Umemura M, Okamoto M, Nakayama K, Sagane K, Tsukahara K, et al. (2003) GWT1 gene is required for inositol acylation of glycosylphosphatidylinositol anchors in yeast. J Biol Chem 278 : 23639–23647. 12714589
17. Kang JY, Hong Y, Ashida H, Shishioh N, Murakami Y, et al. (2005) PIG-V involved in transferring the second mannose in glycosylphosphatidylinositol. J Biol Chem 280 : 9489–9497. 15623507
18. Fabre AL, Orlean P, Taron CH (2005) Saccharomyces cerevisiae Ybr004c and its human homologue are required for addition of the second mannose during glycosylphosphatidylinositol precursor assembly. FEBS J 272 : 1160–1168. 15720390
19. Ohishi K, Inoue N, Kinoshita T (2001) PIG-S and PIG-T, essential for GPI anchor attachment to proteins, form a complex with GAA1 and GPI8. EMBO J 20 : 4088–4098. 11483512
20. Tashima Y, Taguchi R, Murata C, Ashida H, Kinoshita T, et al. (2006) PGAP2 is essential for correct processing and stable expression of GPI-anchored proteins. Mol Biol Cell 17 : 1410–1420. 16407401
21. Horn D, Wieczorek D, Metcalfe K, Baric I, Palezac L, et al. (2014) Delineation of PIGV mutation spectrum and associated phenotypes in hyperphosphatasia with mental retardation syndrome. Eur J Hum Genet 22 : 762–767. doi: 10.1038/ejhg.2013.241 24129430
22. Kvarnung M, Nilsson D, Lindstrand A, Korenke GC, Chiang SC, et al. (2013) A novel intellectual disability syndrome caused by GPI anchor deficiency due to homozygous mutations in PIGT. J Med Genet 50 : 521–528. doi: 10.1136/jmedgenet-2013-101654 23636107
23. Krawitz PM, Murakami Y, Riess A, Hietala M, Kruger U, et al. (2013) PGAP2 mutations, affecting the GPI-anchor-synthesis pathway, cause hyperphosphatasia with mental retardation syndrome. Am J Hum Genet 92 : 584–589. doi: 10.1016/j.ajhg.2013.03.011 23561847
24. Chiyonobu T, Inoue N, Morimoto M, Kinoshita T, Murakami Y (2014) Glycosylphosphatidylinositol (GPI) anchor deficiency caused by mutations in PIGW is associated with West syndrome and hyperphosphatasia with mental retardation syndrome. J Med Genet 51 : 203–207. doi: 10.1136/jmedgenet-2013-102156 24367057
25. Hekimi S, Boutis P, Lakowski B (1995) Viable maternal-effect mutations that affect the development of the nematode Caenorhabditis elegans. Genetics 141 : 1351–1364. 8601479
26. Chisholm AD, Hardin J (2005) Epidermal morphogenesis. WormBook: 1–22.
27. Diep DB, Nelson KL, Raja SM, Pleshak EN, Buckley JT (1998) Glycosylphosphatidylinositol anchors of membrane glycoproteins are binding determinants for the channel-forming toxin aerolysin. J Biol Chem 273 : 2355–2360. 9442081
28. Audhya A, Desai A, Oegema K (2007) A role for Rab5 in structuring the endoplasmic reticulum. J Cell Biol 178 : 43–56. 17591921
29. Orlean P, Menon AK (2007) Thematic review series: lipid posttranslational modifications. GPI anchoring of protein in yeast and mammalian cells, or: how we learned to stop worrying and love glycophospholipids. J Lipid Res 48 : 993–1011. 17361015
30. Priess JR, Hirsh DI (1986) Caenorhabditis elegans morphogenesis: the role of the cytoskeleton in elongation of the embryo. Dev Biol 117 : 156–173. 3743895
31. Barstead RJ, Waterston RH (1991) Vinculin is essential for muscle function in the nematode. J Cell Biol 114 : 715–724. 1907975
32. Mohandas N, Chasis JA, Shohet SB (1983) The influence of membrane skeleton on red cell deformability, membrane material properties, and shape. Semin Hematol 20 : 225–242. 6353591
33. Khan LA, Zhang H, Abraham N, Sun L, Fleming JT, et al. (2013) Intracellular lumen extension requires ERM-1-dependent apical membrane expansion and AQP-8-mediated flux. Nat Cell Biol 15 : 143–156. doi: 10.1038/ncb2656 23334498
34. Takeda J, Miyata T, Kawagoe K, Iida Y, Endo Y, et al. (1993) Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell 73 : 703–711. 8500164
35. Chen G, Ye Z, Yu X, Zou J, Mali P, et al. (2008) Trophoblast differentiation defect in human embryonic stem cells lacking PIG-A and GPI-anchored cell-surface proteins. Cell Stem Cell 2 : 345–355. doi: 10.1016/j.stem.2008.02.004 18397754
36. Klauzinska M, Castro NP, Rangel MC, Spike BT, Gray PC, et al. (2014) The multifaceted role of the embryonic gene Cripto-1 in cancer, stem cells and epithelial-mesenchymal transition. Semin Cancer Biol 29C: 51–58.
37. Taneja-Bageshwar S, Gumienny TL (2013) Regulation of TGFbeta superfamily signaling by two separable domains of glypican LON-2 in C. elegans. Worm 2: e23843. doi: 10.4161/worm.23843 24778932
38. Schultz RD, Bennett EE, Ellis EA, Gumienny TL (2014) Regulation of extracellular matrix organization by BMP signaling in Caenorhabditis elegans. PLoS One 9: e101929. doi: 10.1371/journal.pone.0101929 25013968
39. Fehon RG, McClatchey AI, Bretscher A (2010) Organizing the cell cortex: the role of ERM proteins. Nat Rev Mol Cell Biol 11 : 276–287. doi: 10.1038/nrm2866 20308985
40. McClatchey AI (2014) ERM proteins at a glance. J Cell Sci 127 : 3199–3204. doi: 10.1242/jcs.098343 24951115
41. Schwarz V, Pan J, Voltmer-Irsch S, Hutter H (2009) IgCAMs redundantly control axon navigation in Caenorhabditis elegans. Neural Dev 4 : 13. doi: 10.1186/1749-8104-4-13 19341471
42. Katidou M, Tavernarakis N, Karagogeos D (2013) The contactin RIG-6 mediates neuronal and non-neuronal cell migration in Caenorhabditis elegans. Dev Biol 373 : 184–195. doi: 10.1016/j.ydbio.2012.10.027 23123963
43. Zallen JA, Kirch SA, Bargmann CI (1999) Genes required for axon pathfinding and extension in the C. elegans nerve ring. Development 126 : 3679–3692. 10409513
44. Eisenhaber F, Eisenhaber B, Kubina W, Maurer-Stroh S, Neuberger G, et al. (2003) Prediction of lipid posttranslational modifications and localization signals from protein sequences: big-Pi, NMT and PTS1. Nucleic Acids Res 31 : 3631–3634. 12824382
45. Brenner S (1974) The genetics of Caenorhabditis elegans. Genetics 77 : 71–94. 4366476
46. Koppen M, Simske JS, Sims PA, Firestein BL, Hall DH, et al. (2001) Cooperative regulation of AJM-1 controls junctional integrity in Caenorhabditis elegans epithelia. Nat Cell Biol 3 : 983–991. 11715019
47. Zaidel-Bar R, Joyce MJ, Lynch AM, Witte K, Audhya A, et al. (2010) The F-BAR domain of SRGP-1 facilitates cell-cell adhesion during C. elegans morphogenesis. J Cell Biol 191 : 761–769. doi: 10.1083/jcb.201005082 21059849
48. Praitis V, Simske J, Kniss S, Mandt R, Imlay L, et al. (2013) The secretory pathway calcium ATPase PMR-1/SPCA1 has essential roles in cell migration during Caenorhabditis elegans embryonic development. PLoS Genet 9: e1003506. doi: 10.1371/journal.pgen.1003506 23696750
49. Neukomm LJ, Frei AP, Cabello J, Kinchen JM, Zaidel-Bar R, et al. (2011) Loss of the RhoGAP SRGP-1 promotes the clearance of dead and injured cells in Caenorhabditis elegans. Nat Cell Biol 13 : 79–86. doi: 10.1038/ncb2138 21170032
50. Gally C, Wissler F, Zahreddine H, Quintin S, Landmann F, et al. (2009) Myosin II regulation during C. elegans embryonic elongation: LET-502/ROCK, MRCK-1 and PAK-1, three kinases with different roles. Development 136 : 3109–3119. doi: 10.1242/dev.039412 19675126
51. Mello C, Fire A (1995) DNA transformation. Methods Cell Biol 48 : 451–482. 8531738
52. Bigelow H, Doitsidou M, Sarin S, Hobert O (2009) MAQGene: software to facilitate C. elegans mutant genome sequence analysis. Nat Methods 6 : 549. doi: 10.1038/nmeth.f.260 19620971
53. Kamath RS, Martinez-Campos M, Zipperlen P, Fraser AG, Ahringer J (2001) Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol 2: RESEARCH0002.
54. Gonczy P, Schnabel H, Kaletta T, Amores AD, Hyman T, et al. (1999) Dissection of cell division processes in the one cell stage Caenorhabditis elegans embryo by mutational analysis. J Cell Biol 144 : 927–946. 10085292
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 3
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Clonality and Evolutionary History of Rhabdomyosarcoma
- Morphological Mutations: Lessons from the Cockscomb
- Maternal Filaggrin Mutations Increase the Risk of Atopic Dermatitis in Children: An Effect Independent of Mutation Inheritance
- Transcriptomic Profiling of Reveals Reprogramming of the Crp Regulon by Temperature and Uncovers Crp as a Master Regulator of Small RNAs
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy