
Inhibits Neuromuscular Junction Growth by Downregulating the BMP Receptor Thickveins
Bi-directional signaling between neurons and their target cells is critical for synapse formation, growth, and plasticity, as well as for neuronal survival. Bone morphogenetic protein (BMP) acts as a retrograde signal promoting synaptic growth at the Drosophila neuromuscular junction (NMJ), but little is known about proteins that regulate BMP signaling by controlling BMP release, receptor expression, and signal transduction. We report here that a previously uncharacterized and evolutionally conserved member of the S6 kinase (S6K) family S6K like (S6KL) inhibits BMP signaling by interacting with and promoting proteasome-mediated degradation of the BMP receptor Thickveins (Tkv). In S6KL mutants, there was an elevated level of Tkv protein, together with overgrown NMJs characterized by excess satellite boutons. Reducing the gene dose of tkv by half in S6KL null background restored normal NMJ morphology, suggesting that S6KL normally serves to suppress Tkv-mediated BMP signaling. Biochemically, S6KL interacted with Tkv. Overexpression of S6KL down-regulated Tkv and this effect was inhibited by blocking the proteasomal degradation pathway. Collectively, our data demonstrate that S6KL regulates NMJ synapse development by promoting the proteasomal degradation of Tkv. Thus, we have identified a novel negative regulator of BMP signaling in the Drosophila nervous system.
Published in the journal:
. PLoS Genet 11(3): e32767. doi:10.1371/journal.pgen.1004984
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004984
Summary
Bi-directional signaling between neurons and their target cells is critical for synapse formation, growth, and plasticity, as well as for neuronal survival. Bone morphogenetic protein (BMP) acts as a retrograde signal promoting synaptic growth at the Drosophila neuromuscular junction (NMJ), but little is known about proteins that regulate BMP signaling by controlling BMP release, receptor expression, and signal transduction. We report here that a previously uncharacterized and evolutionally conserved member of the S6 kinase (S6K) family S6K like (S6KL) inhibits BMP signaling by interacting with and promoting proteasome-mediated degradation of the BMP receptor Thickveins (Tkv). In S6KL mutants, there was an elevated level of Tkv protein, together with overgrown NMJs characterized by excess satellite boutons. Reducing the gene dose of tkv by half in S6KL null background restored normal NMJ morphology, suggesting that S6KL normally serves to suppress Tkv-mediated BMP signaling. Biochemically, S6KL interacted with Tkv. Overexpression of S6KL down-regulated Tkv and this effect was inhibited by blocking the proteasomal degradation pathway. Collectively, our data demonstrate that S6KL regulates NMJ synapse development by promoting the proteasomal degradation of Tkv. Thus, we have identified a novel negative regulator of BMP signaling in the Drosophila nervous system.
Introduction
Reliable and effective communication between neurons and their targets across the synaptic cleft through cell adhesion molecules and signaling pathways is critical for the formation, growth, and plasticity of synapses [1–3]. For example, the retrograde bone morphogenetic protein (BMP) signaling from postsynaptic muscles to presynaptic motoneurons is crucial for synaptic development and plasticity at the Drosophila larval neuromuscular junctions (NMJs) [4–8]. A similar role of BMP signaling has also been identified at the central synapses in vertebrates [9].
At the Drosophila NMJ, the BMP homolog Glass bottom boat (Gbb) is secreted from muscles and binds to the constitutively active presynaptic BMP type II receptor wishful thinking (Wit), which in turn leads to the recruitment of the type I receptors thickveins (Tkv) and saxophone (Sax). Wit phosphorylates and activates Tkv and Sax, which in turn phosphorylate mothers against decapentaplegic (Mad). Phosphorylated Mad (pMad) forms a complex with co-Smad Medea that translocates to the nucleus and regulates the transcription of target genes required for NMJ growth. Mutation of any member of this cascade leads to a drastic reduction in the number of synaptic boutons and the amount of neurotransmitter released at the NMJ [10–13]. Conversely, up-regulation of BMP signaling leads to NMJ overgrowth [8,14–17]. Thus BMP pathway is both required and sufficient for Drosophila NMJ growth.
At the Drosophila NMJ, BMP signaling is tightly regulated at multiple levels. For example, BMP signaling is attenuated by endocytosis and endosomal trafficking of BMP receptors Wit and Tkv [14–19]. Recently, we uncovered that brain tumor (Brat), a translational suppressor, suppresses Mad expression in motoneurons [8]. We report here that an evolutionally conserved and previously uncharacterized kinase, namely a protein kinase encoded by a gene mapped at chromosome band 17E (PK17E), acts as a negative regulator of Tkv in regulating NMJ synapse growth. Based on sequence homology and kinase activity assays, we renamed PK17E as S6KL, short for ribosomal protein S6 kinase like.
We report here that S6KL null mutation causes NMJ overgrowth characterized by excess satellite boutons. This NMJ overgrowth is caused by up-regulation of BMP signaling based on genetic and immunochemical analyses. We further show that S6KL physically interacts with Tkv and promotes its proteasomal degradation, resulting in downregulation of BMP signaling and inhibition of NMJ growth. Thus, our study identifies a novel regulatory mechanism of BMP signaling in the Drosophila nervous system.
Results
Isolation and characterization of S6KL mutants
To better understand the neuronal functions of Drosophila fragile X mental retardation protein (dFMRP) in Drosophila, we screened a Drosophila embryo cDNA library with the yeast two-hybrid system using the N-terminal 220 amino acids peptide of dFMRP as a bait. We identified three positive genes including PK17E represented by one clone encoding the 98 amino acid residues of PK17E C-terminal followed by 272 bp 3’-UTR sequence. Since dFMRP regulates NMJ development, we examined the synaptic role of PK17E for which there were mutants surviving to the third instar larvae. Though the candidate clone for S6KL turned out to be a false positive as the clone alone without bait expression was sufficient to activate the reporter expression, we found that multiple S6KL mutants, including trans-allelic EY06723 and EY10051 with a P element inserted in the genomic S6KL gene (Fig. 1D) or hemizygotes of each insertion, showed obvious NMJ overgrowth phenotype. On the contrary, overexpression of S6KL was reported exhibiting fewer synaptic boutons from a gain-of-function screen for genes controlling motor axon guidance and synaptogenesis in Drosophila [20]. Thus, we suspected that S6KL played a role in synapse development and characterized its synaptic function in the present study.

Sequence comparison showed that PK17E is a member of the S6K family and contains a single serine/threonine protein kinase (STK) domain with 36% identity and 52% similarity, and 33% identity and 48% similarity to the STK domain of human S6K1 and Drosophila dS6K, respectively. Given that the previously characterized Drosophila S6K (dS6K) is highly homologous to human S6K1 and S6K2 within the STK domain (83% identity), we renamed PK17E “S6K like” (S6KL). S6KL is well conserved from C. elegans to humans. Specifically, the STK domain of the Drosophila S6KL is 45% identical and 63% similar to that of the uncharacterized human homolog SGK494 (http://www.kinase.com/cgi-bin) (Fig. 1A). So far, the physiological functions of S6KL and its homologs have not been characterized in any organisms.
Sequence analysis revealed that S6KL contains a catalytic domain shared by the serine/threonine protein kinase A, G, and C (AGC) families categorized based on their tendency to phosphorylate sites surrounded by basic amino acids. To verify if S6KL has an intrinsic kinase activity, S6KL was tagged with a Flag epitope and expressed in S2 cells using an actin promoter. To control for nonspecific kinase activity, a putative kinase-dead mutant of S6KL, Flag-S6KLK193Q, in which lysine 193 was mutated to glutamine disrupting ATP binding and hence the kinase activity based on previous studies on S6 kinases [21,22] (Fig. 1A), and a vector control Flag-GFP were similarly expressed in S2 cells. Following transfection, S2 cells were lysed, and the cell lysates were subjected to immunoprecipitation with anti-Flag antibody (Fig. 1B). The immunoprecipitates were then assayed for kinase activity by detecting γ-32P incorporation from [γ-32P] ATP into a generic kinase substrate myelin basic protein (MBP). While GFP control and S6KLK193Q showed no kinase activity, wild-type S6KL effectively phosphorylated MBP (Fig. 1C), demonstrating an intrinsic kinase activity for S6KL.
To investigate the in vivo functions of S6KL, we generated multiple S6KL mutant alleles by imprecise excision of EY10051. The S6KL158 allele had a deletion of 2376 base pairs (bp), whereas S6KL140 had a deletion of 2553 bp spanning the first to the fifth exon (Fig. 1D). Full-length S6KL mRNA was not detected in homozygous S6KL140 adults (Fig. 1E), whereas mRNAs of the flanking CG6961 and the intragenic CG7053 were expressed normally (Fig. 1E). Western analysis with a rabbit polyclonal antibody against a peptide spanning amino acid residues 66–82 of S6KL (Fig. 1D) detected no target protein expression in homozygous S6KL140 adult heads (Fig. 1F). S6KL140 appeared to be a null allele, as it had an intragenic deletion removing the N-terminal 261 amino acid residues including part of the kinase domain and behaved as a null phenotypically (see below). Immunostaining showed no apparent expression of the endogenous S6KL in the ventral nerve cord (Fig. 2A and 2B), wing disc, and eye disc (S1 Fig.), but appreciable expression in a small subset of cells in the leg disc (S1 Fig.). When overexpressed driven by the motoneuron specific OK6-Gal4, S6KL was present primarily in the cytoplasm (Fig. 2C). Endogenous S6KL was not detectable at the NMJs, but overexpressed S6KL can localize to both presynapse and postsynapse when UAS-S6KL is under the control of the motor neuron specific OK6-Gal4 and the muscular C57-Gal4, respectively (Fig. 2D–2G’). Homozygous S6KL140 and S6KL158 mutants were fully viable, fertile in both males and females, and showed no obvious developmental and behavioral abnormalities. In this study, we focused on characterizing the synaptic phenotypes of S6KL140 mutants at NMJ terminals.
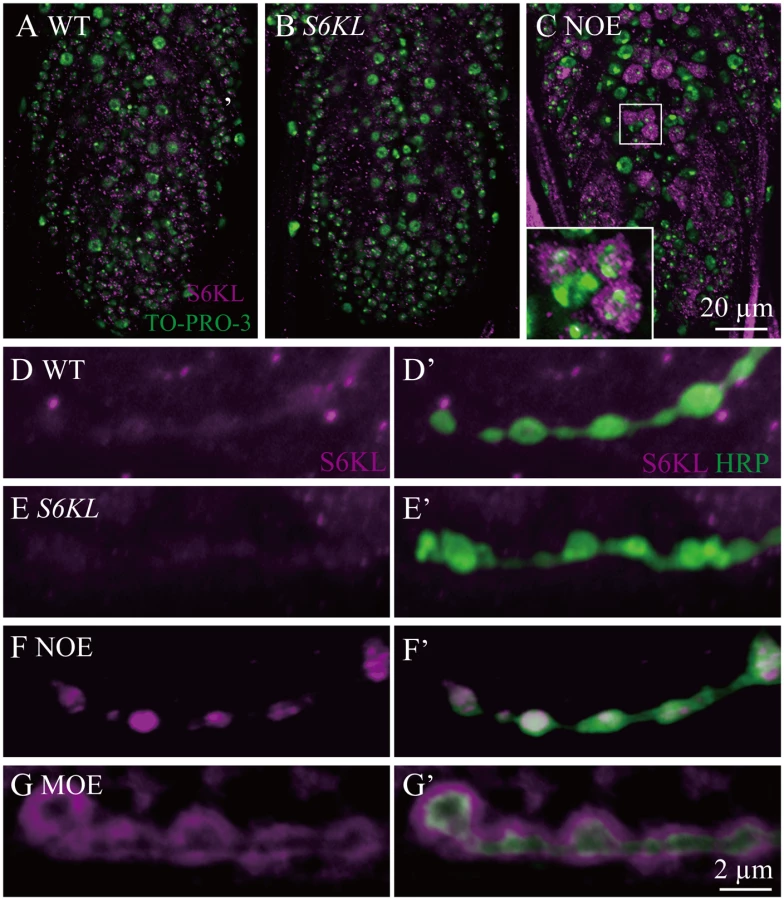
Overgrown NMJ terminals in S6KL mutants
The Drosophila NMJ is an effective model system for studying synaptic development and function. To elucidate a possible role for S6KL at synapses, we first examined the NMJ morphology of S6KL mutants. The wild type muscle 4 NMJ (NMJ4) showed the stereotypical “beads-on-a-chain” morphology when co-stained with an antibody against cysteine string protein (CSP), a synaptic vesicle-associated protein, and anti-horse radish peroxidase (HRP) which labels neuronal membrane. The NMJ synapses in S6KL140 mutants were obviously overgrown with extra boutons (Fig. 3A and 3B). The mean total number of synaptic boutons per NMJ4 was 39.47±1.14 in S6KL140 mutants, significantly higher than 25.24±1.10 in wild types (p<0.001; Fig. 3A, 3B, and 3G). Satellite boutons are ectopic boutons that emerge from the main nerve terminal or bud from primary boutons [14,23]. Compared with wild type, the mean satellite bouton number was more than four-fold higher in S6KL140 mutants (p<0.001; Fig. 3A, 3B, and 3H). The NMJ phenotype of hemizygous S6KL140/Df(1)ED7413 mutants (Df(1)ED7413 deletes the S6KL locus completely) was similar to that of homozygous S6KL140 mutants. Presynaptic expression of S6KL driven by the motoneuron-specific elav-Gal4 in S6KL140 background restored the overgrown NMJ to wild-type level (Fig. 3D, 3G and 3H), whereas Mhc-Gal4-driven postsynaptic expression of S6KL did not (Fig. 3E, 3G and 3H), suggesting a specific role in presynaptic neurons.

To investigate whether the kinase activity of S6KL is required for NMJ growth regulation, we carried out rescue experiments by expressing wild-type and the kinase-dead K193Q mutant S6KL in S6KL140 background. The overgrown NMJs in S6KL mutants were fully rescued by presynaptic expression of wild-type but not S6KLK193Q under the control of elav-Gal4 (Fig. 3F–3H), though S6KLK193Q was expressed at a substantial level as the wild-type S6KL (insets of Fig. 3D and 3F). These results demonstrate that the kinase activity of S6KL is required for inhibiting NMJ growth.
S6KL restrains NMJ growth presynaptically
The above-mentioned tissue-specific rescue results suggest a presynaptic function of S6KL. To further verify this notion, we carried out tissue-specific alterations of S6KL expression and examined their effects on NMJ growth. Presynaptic overexpression of S6KL by the pan-neuronal elav-Gal4 resulted in a significant decrease in the number of total boutons (26.67±1.32 boutons for WT and 16.43±0.95 boutons for elav-Gal4 > UAS-S6KL; p<0.001; Fig. 4A, 4B and 4F), consistent with a previous report [20]. Whereas overexpression of S6KL in postsynaptic muscles using C57-Gal4 had no effect on bouton number (26.67±1.32 boutons for WT and 24.86±1.13 boutons for C57-Gal4 > UAS-S6KL; Fig. 4A, 4C, and 4F). Targeted knockdown of S6KL in presynaptic neurons by elav-Gal4-driven RNAi (VDRC line 102179) resulted in a significant increase in the number of total and satellite boutons compared with wild type (26.67±1.32 boutons for WT and 38.22±1.41 boutons for elav-Gal4 > RNAi; p<0.001; Fig. 4A, 4D, 4F and 4G), whereas RNAi knockdown of S6KL in postsynaptic muscles by C57-Gal4 showed no effect on bouton number (26.67±1.32 boutons for WT and 27.06±1.19 boutons for C57-Gal4 > RNAi; Fig. 4A, 4E–4G). Similar results were observed using a second VDRC RNAi line 47326 targeting an independent sequence (26.67±1.32 boutons for WT and 41.04±1.33 boutons for elav-Gal4 > RNAi 47326; p<0.001). These findings demonstrate that S6KL inhibits NMJ growth presynaptically.

Fewer but larger synaptic vesicles in S6KL mutant boutons
To investigate further the effect of S6KL on the ultrastructure of NMJs, we examined NMJ morphology by transmission electron microscopy. Presynaptic structures essential for neurotransmitter release at NMJ terminals include synaptic vesicles (SVs) and active zones with T bars (arrows in Fig. 5A and 5B), while the most prominent postsynaptic structure is the sub-synaptic reticulum (SSR) composed of a meshwork of convoluted muscle plasma membrane (Fig. 5A and 5B). The postsynaptic SSR appeared largely normal in S6KL140 mutants (compare Fig. 5B with 5A). However, the SV density within the whole presynaptic bouton was 71.85±3.96 SVs/μm2 in S6KL140 mutants, moderately but significantly reduced than 99.39±4.14 SVs/μm2 in wild type (p<0.001; Fig. 5A, 5B, and 5E). A small subpopulation of vesicles were larger in S6KL140 mutants (asterisks in Fig. 5D). Statistical analysis of the density and size of SVs within a 200 nm radius around active zones showed that there were 16.09±0.57 SVs in S6KL140 mutants, fewer than 22.03±0.74 SVs in wild type (p<0.001; Fig. 5C, 5D, and 5F), whereas the mean SV diameter was 44.75±0.53 nm in S6KL140 mutants, significantly larger than 34.23±0.22 nm in wild type (p<0.001; Fig. 5G). A cumulative probability plot showed that 80% of wild-type SVs were < 40 nm, compared with only 35% of SVs < 40 nm in diameter in S6KL140 mutants (Fig. 5H). Hence, loss of S6KL led to reduced vesicle density, and fewer and larger SVs at the neurotransmitter release sites in NMJ terminals, phenotypes similar to that observed in many endocytic mutants such as brat, endophilin, synaptojanin, and AP180/lap [8,24–26].

Normal evoked neurotransmission but increased quantal size in S6KL mutants
To examine the functional consequences of these altered NMJ synapses in S6KL mutants, we recorded both evoked excitatory junction potentials (EJPs) and spontaneous miniature EJPs (mEJPs) at muscle 6 using intracellular electrodes. The results showed that the mean EJP amplitude was not significantly altered in S6KL140 mutants (39.32±1.586 mV for wild type and 41.66±1.249 mV for S6KL140 mutants; p = 0.25). However, the mean mEJP amplitude, also known as quantal size, was 1.677±0.033 mV in S6KL140 mutants, significantly higher than 1.123±0.021 mV in wild type (p<0.001; Fig. 6A, 6B, and 6D). A cumulative probability plot showed that 80% of mEJP amplitudes were < 1.5 mV in wild types, while only 47% of mEJP amplitudes were <1.5 mV in S6KL140 mutants; the difference was significant by Kolmogorov-Smirnov test (D = 0.33; p<0.001; Fig. 6E). As with the restoration of normal NMJ morphology (Fig. 3), elav-Gal4-driven presynaptic expression of S6KL in S6KL140 mutant background also fully rescued the increased mean mEJP amplitude (p>0.05 compared with wild type; Fig. 6C, 6D and 6E), indicating that the increased quantal size is specifically caused by S6KL mutations.

Defective endocytosis at S6KL mutant NMJ synapses
As with S6KL mutants, several endocytic protein mutants also show more satellite boutons, fewer and larger SVs, and increased mEJP amplitudes at NMJs [23,26–28]. To assess if endocytosis was affected by the S6KL mutation, we first performed FM1–43 dye uptake assays at the NMJs of third instar larvae. The lipophilic FM1–43 is non-fluorescent in the aqueous extracellular environment, but quantum yield is increased more than 40-fold when bound to lipid membrane. Hence, newly endocytosed vesicles in the presence of FM1–43 will be fluorescently labeled, providing a quantitative measure of vesicle endocytosis [8,29]. Compared with wild-type controls, which displayed intense labeling of FM1–43, dye uptake was reduced by 30.95% in S6KL140 mutant boutons (p<0.001; Fig. 7A, 7B, and 7E). Similarly, hemizygous S6KL mutants (S6KL140/Df(1)ED7413) also showed significantly reduced FM1–43 uptake compared with wild type (p<0.001; Fig. 7A, 7C, and 7E). The decrease in FM1–43 dye loading in S6KL mutants was completely reversed by presynaptic expression of S6KL driven by OK6-Gal4 (Fig. 7A, 7D and 7E). These results suggest that S6KL is required for synaptic endocytosis.

Efficient endocytosis is required to sustain neurotransmitter release during intense stimulation. As shown in Fig. 7F, EJP amplitudes in S6KL mutants declined to about 72.13% of the initial response during 10 min tetanic stimulation at 10 Hz in 1 mM Ca2+. In contrast, wild-type controls maintained release at about 92.27% of the initial response after the same stimulus train. Again, this phenotype was fully rescued by presynaptic expression of S6KL in S6KL140 mutants (Fig. 7F). The inability of S6KL mutants to maintain normal levels of transmission during intense activity is consistent with a defect in synaptic endocytosis or vesicle trafficking.
S6KL interacts with BMP pathway components in regulating NMJ growth
Up-regulation of BMP signaling leads to NMJ overgrowth characterized by more boutons including satellite boutons [14,16,17], similar to what we observed in S6KL mutants, suggesting that loss of S6KL may lead to upregulated BMP signaling. To examine possible involvement of BMP signaling in mediating the overgrowth of synaptic boutons in S6KL mutants, genetic interactions between S6KL and the BMP receptor tkv and other components of the pathway were examined. The NMJs in tkv7/tkvk16713 mutants were undergrown (25.24±1.11 boutons for WT and 16.13±0.66 boutons for tkv7/tkvk16713; p<0.001; Fig. 8A, 8D, and 8M). Interestingly, the number of total boutons in S6KL140; tkv7/tkvk16713 double mutants were not different from those in tkv single mutants (p>0.05; Fig. 8D, 8E, and 8M), suggesting that synaptic overgrowth induced by S6KL mutation requires BMP signaling through Tkv. Importantly, mutating one copy of the BMP type I receptor tkv in S6KL140 background (S6KL140; tkv7/+) completely reversed the increase in total bouton number in S6KL140 mutants (24.80±1.46 boutons for S6KL140; tkv7/+ and 39.47±1.14 boutons for S6KL140 mutants; p<0.001; Fig. 8A–8C and 8M), whereas the number of total boutons in heterozygous tkv7 mutants (tkv7/+) was normal as wild type (p>0.05; Fig. 8M). Similarly, mutating one copy of mad, which encodes the BMP effector mothers against decapentaplegic (25.20±0.92 boutons for S6KL140; mad237/+; p<0.001 compared with S6KL140 mutants), or wit, which encodes the type II BMP receptor Wit also suppressed the overgrown NMJs of S6KL mutants (23.47±1.22 boutons for S6KL140; witA12/+; p<0.001 compared with S6KL140 mutants).
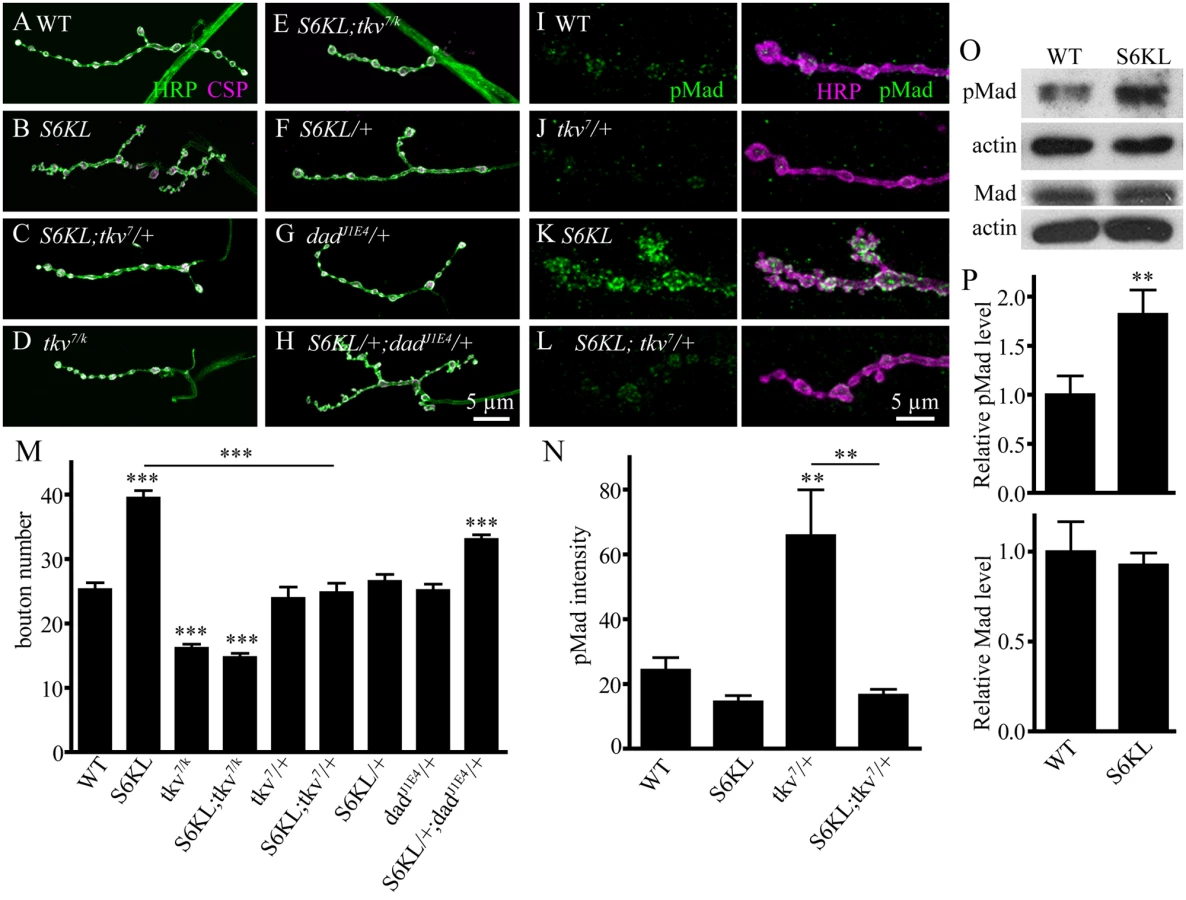
We then examined genetic interactions between S6KL and dad. Dad is a BMP signaling inhibitor and dad mutants show overgrown NMJ terminals [14,16]. Synaptic growth was normal in S6KL140/+ and dadJ1E4/+ single heterozygous mutants. However, the total bouton number was 33.05±0.73 in S6KL140/+; dadJ1E4/+ transheterozygous mutants, significantly higher than that in the single heterozygotes (26.53±1.08 for S6KL140/+ and 25.13±0.97 for dadJ1E4/+; p<0.001; Fig. 8F–8H and 8M). These results together indicate that the excessive synaptic growth in S6KL mutants may be caused by up-regulation of BMP signaling.
Activation of BMP receptors by ligand binding leads to Mad phosphorylation (pMad), and pMad is the major transducer of BMP signaling in Drosophila motoneurons. Thus, pMad level serves as a molecular readout of BMP signaling. The intensity of pMad staining in the presynaptic terminals of S6KL140 mutants was 2.71-fold higher than wild type (Fig. 8I, 8K, and 8N). Consistently, western analysis of larval brains showed an elevated level of pMad (1.82-fold higher than wild type) but a normal level of Mad protein in S6KL140 mutants (Fig. 8O and 8P). Furthermore, the staining intensity of β–galactosidase encoded by dad-lacZ which has been widely used to monitor the transcriptional output of BMP pathway [30,31] was apparently increased in the motoneuron nuclei of S6KL mutants compared with wild type (S2 Fig.). As with the NMJ overgrown phenotype, pMad upregulation was reversed to wild-type level by mutating one copy of tkv in S6KL140 background (Fig. 8I–8L and 8N). Taken together, these results indicate that the synaptic overgrowth in S6KL mutants requires BMP signaling and that S6KL normally serves to restrain NMJ growth by inhibiting BMP signaling.
S6KL physically interacts with Tkv and negatively regulates its protein level
To uncover the molecular mechanism underlying the up-regulation of BMP signaling in S6KL mutants, we examined the protein levels of two BMP receptors Tkv and Wit for which there are tags or antibodies are available. As a quality Tkv antibody was not available to us, we took advantage of the commercial GFP antibodies to detect the expression of GFP - or YFP-tagged Tkv. Immunostaining showed that the level of Tkv-GFP driven by the pan-neuronal elav-Gal4 at NMJ terminals was higher in S6KL140 mutants than wild types (Figs. 9A–9B’). Western analysis of larval brain extracts showed a 2.46 fold increase of Tkv-GFP protein level in S6KL140 mutants than in wild types (Fig. 9C). Consistently, a 2.35 fold increase of Tkv protein level was detected by western analysis in S6KL140 mutants expressing an YFP protein tag which labels the endogenous Tkv (Fig. 9C). In contrast, the protein level of Wit was not altered in S6KL140 mutants compared with wild type (Fig. 9C). Quantitative PCR revealed that the level of tkv mRNA was normal in the larval brains of S6KL140 mutants (S3 Fig.), indicating a negative regulation of Tkv by S6KL at the post-transcriptional level.

To further understand the specific effects of S6KL on Tkv protein expression, we first assessed Tkv-GFP protein levels in S2 cells expressing altered levels of S6KL by western analysis. The level of Tkv-GFP protein was higher in S6KL knockdown cells, whereas a decreased level of Tkv-GFP was observed in S6KL—overexpressing cells compared with control S2 cells expressing the endogenous level of S6KL (Fig. 9D and 9E). Furthermore, the reduction of the Tkv-GFP protein level in S6KL—overexpressing S2 cells was blocked by the potent 26S proteasome inhibitor MG132 (Fig. 9E), suggesting that S6KL down-regulates Tkv by facilitating proteasome-dependent degradation. To further confirm that S6KL affects Tkv protein level by proteasome pathway, we examined Tkv ubiquitination in S2 cells co-transfected with Myc-Tkv and double-stranded RNAs (dsRNA) against S6KL. In S6KL—knockdown cells with distinct dsRNAs, Tkv poly-ubiquitination was apparently decreased to different extents (Fig. 9G). We then performed immunoprecipitation assays from S2 cells co-expressing Flag-tagged S6KL and Myc-tagged Tkv or GFP to determine if S6KL physically interacted with Tkv. Western blotting of the immunoprecipitates by anti-Flag and anti-Myc antibodies revealed robust, specific co-immunoprecipitation of S6KL and Tkv (Fig. 9F), suggesting that S6KL forms a complex with Tkv in S2 cells, though an in vivo interaction between S6KL and Tkv remains to be established. The interaction was not dependent on the kinase activity, as the kinase dead S6KLK193Q mutant still associated with Tkv (Fig. 9F), in support of a dominant negative effect of the point mutation [21,22]. Co-localization of overexpressed S6KL and Tkv-GFP in the soma of motor neurons in the ventral nerve cord (Fig. 9H) further supports the physical interaction between S6KL and Tkv.
Discussion
In this study, we uncover that S6KL, a previously uncharacterized and evolutionally conserved serine-threonine kinase of the S6K family, inhibits NMJ growth by suppressing BMP signaling. We further demonstrate that S6KL suppresses BMP signaling by promoting proteasome-mediated degradation of the BMP receptor Tkv. Thus our results shed new light on the in vivo functions of S6KL and the regulation of BMP signaling in synapse development.
S6KL plays a role distinct from S6K
S6K1 and S6K2 are downstream effectors of the mammalian target of rapamycin (mTOR) pathways [32]. S6K1 regulates cell growth by controlling the biosynthesis of translational components, most notably ribosomal proteins [32]. S6K1 mutant mice are small in body size and exhibit glucose intolerance due to a selective decrease in β-cell size [33,34]. In Drosophila, there is only one S6K homologue named dS6K. Drosophila dS6K mutants show extreme delay in development and severe reduction in adult body size due to a reduced cell size of multiple cell types [35]. Several S6K substrates have been identified, the most extensively studied of which is ribosomal protein S6. In addition to S6, the eukaryotic translation initiation factor eIF4B is also a physiologically relevant target of S6K1 that could explain its effect on translation and cell growth; eIF4B is required for efficient recruitment of ribosomes to mRNA [36].
Different from Drosophila dS6K mutants, S6KL mutants showed normal body size and no delay in development, indicating that S6KL plays a role distinct from the cell growth regulation by S6K. At the NMJ terminals, S6KL also acts differently from dS6K. dS6K regulates bouton size, active zone number, and synaptic function without influencing bouton number [37,38], whereas S6KL inhibits NMJ growth by attenuating BMP signaling. The in vivo functions of S6KL in other organisms such as C. elegans and humans remain to be characterized.
S6KL restrains NMJ growth by inhibiting BMP signaling
Bone morphogenetic protein signaling is the prevalent retrograde pathway promoting Drosophila NMJ growth [2,4,5,8]. In this study, we present genetic and immunochemical data demonstrating that S6KL restrains synapse growth by antagonizing BMP signaling activity. First, the NMJ terminals were overgrown with more synaptic boutons and satellite boutons in S6KL mutants (Fig. 3), consistent with enhanced BMP signaling. Second, the NMJ overgrowth phenotype was reversed by removing one copy of the BMP signaling pathway components such as tkv (Fig. 8C and 8M). Third, S6KL and dad mutants displayed dosage-sensitive genetic interactions; trans-heterozygotes of S6KL and dad had significantly more boutons compared with that of heterozygous mutants for either gene alone (Fig. 8H and 8M). This type of genetic interaction is rarely observed for gene products that do not directly influence each other. Finally, the immunostaining intensity of pMad and dad-LacZ, two distinct reporters of BMP signaling strength, was increased in S6KL mutant NMJs (Fig. 8K and 8N; S2 Fig.). Together, our data demonstrate that S6KL normally inhibits NMJ growth by antagonizing BMP signaling.
In addition to the up-regulation of BMP signaling in S6KL mutants, synaptic endocytosis is impaired in S6KL mutants. As endocytosis attenuates BMP signaling [14], one simple explanation for the elevated BMP signaling in S6KL mutants is defective endocytosis. However, our results suggest that S6KL may not directly affect endocytosis in regulating NMJ growth. First, synaptic proteins, such as Endophilin, Dynamin, and Eps15 are normally expressed at S6KL mutant NMJs (S4 Fig.), indicating that S6KL does not affect the protein level and localization of these proteins at NMJ synapses. In contrast, many endocytic proteins such as Dynamin, Synaptojanin, AP180, and Endophilin are reduced or mislocalized at endocytic mutant NMJs [24,28,39]. Second, the protein level of Tkv but not Wit was specifically increased in S6KL mutants compared with wild type, while both Tkv and Wit protein level was normal in endoA mutants (S5 Fig.). Immunostaining further confirmed an elevated level of Tkv in S6KL mutants but a normal level of Tkv in endocytic mutants (Fig. 8 and S6 Fig.). Similarly, a normal level of BMP receptors was reported in spichthyin and nervous wreck mutants with endocytic trafficking defects [14,17]. Thus, although S6KL mutants share key synaptic phenotypes with endocytic mutants such as satellite boutons, reduced FM1–43 uptake, more large SVs, increased amplitudes of mEJPs, and faster decline of EJP amplitudes (Figs. 3–7) [8,19,23,26,27,39,40], S6KL negatively regulates Tkv protein level whereas endocytic proteins do not.
There are multiple possibilities, not mutually exclusive, for the observed endocytic defects in S6KL mutants. First, the increased level of Tkv proteins in S6KL mutants may impinge on endocytosis through endocytic proteins, as Tkv interacts with Dap160 and Dynamin via nervous wreck [14]. Second, while endocytosis inhibits BMP signaling [14], BMP signaling may in turn suppress endocytosis. For example, increased BMP signaling in dad mutants results in endocytosis defects [8]. In addition, the endocytic defect detected by FM1–43 uptake in both S6KL and Brat mutants could be rescued by reducing the dose of BMP signaling components (S7 Fig. and [8]). Trio, a Rac GTPase-specific guanine nucleotide exchange factor, is a downstream target of BMP signaling in motoneurons [41]. Thus, the compromised endocytosis in S6KL mutants could be attributable, at least partially, to elevated BMP signaling and subsequently enhanced Trio-Rac-mediated actin reorganization, as actin cytoskeleton is closely involved in endocytosis [19]. Last, but not least, S6KL may affect endocytosis by directly regulating the activity, localization, or both of an endocytic protein not examined in this study. The exact mechanism by which loss of S6KL leads to endocytic defects remains to be characterized.
S6KL facilitates proteasome-mediated degradation of Tkv
Multiple lines of evidence support that S6KL physically interacts with Tkv, and facilitates its proteasomal degradation. First, Tkv functions downstream of S6KL and is required for synaptic overgrowth in S6KL mutants (Fig. 8). Second, Tkv protein level was elevated in S6KL mutants and S2 cells expressing reduced levels of S6KL by RNAi knockdown (Fig. 9C and 9D). Conversely, Tkv protein level was decreased in S6KL—overexpressing S2 cells, and this decrease was blocked by MG132, a potent 26S proteasome inhibitor (Fig. 9E). The negative regulation of Tkv by S6KL is specific, as the protein levels of Wit and Mad, two components of the BMP signaling, remain unaltered in S6KL mutants (Figs. 8 and 9). Third, S6KL can be co-immunoprecipitated with Tkv from S2 cells and co-localizes with Tkv in the motoneuron soma (Fig. 9F and 9H), suggesting that they interact physically, though an in vivo interaction between the two remains to be determined. Together, our results show that in the absence of S6KL, Tkv accumulates, leading to upregulated BMP signaling and consequently overgrowth of NMJ terminals and defective endocytosis. It is worth noting that the negative regulation of BMP signaling by S6KL also applies to vein development in the wing (S8 Fig.).
How might S6KL target Tkv for degradation? The strong biochemical and genetic interactions between S6KL and Tkv raised several possible models for S6KL to inhibit Tkv degradation. The simplest model is that S6KL directly phosphorylates Tkv and facilitates its proteasomal degradation. However, we found no phosphorylation of Tkv by S6KL in an in vitro kinase assay. Alternatively, S6KL could act as an adaptor to recruit critical proteins, e.g., unidentified kinases and ubiquitin E3 ligases, initiating proteasomal degradation of Tkv, as has been reported for transmembrane receptors in different processes [31,42,43]. For example, Fused, a serine/threonine kinase, functions in concert with the E3 ligase Smurf to regulate ubiquitination and proteolysis of Tkv in the cystoblasts of the Drosophila female germline [31]. Fused likely acts on Tkv by phosphorylating the Ser238 site of Tkv, leading to Tkv ubiquitination by Smurf and subsequent proteasome-mediated degradation, though experimental evidence demonstrating Tkv is a direct phosphorylation target of Fused is still lacking [31]. In the Drosophila nervous system, S6KL may exert a function similar to Fused in the ovary, inhibiting BMP signaling by promoting Tkv degradation. As Smurf null mutants showed normal NMJ terminals (S9 Fig.), the identity of the ubiquitin ligase that cooperates with S6KL in motoneurons to promote Tkv degradation remains to be determined. In addition, as membrane receptors can be degraded by lysosomes [43,44], S6KL facilitated degradation of Tkv by lysosomes is also possible.
Regardless of detailed mechanisms, our study has established a close connection between S6KL and Tkv, and uncovered a critical role of S6KL in regulating synaptic growth and function by inhibiting BMP signaling activity through promoting Tkv degradation. Because of the broad roles of tightly regulated BMP signaling in development and dysregulation of BMP signaling in many diseases, including cancer and nervous system diseases, it will be of great interest to determine if mammalian homologs of S6KL act similarly on BMP signaling.
Materials and Methods
Drosophila strains and genetics
Fly stocks were maintained on standard cornmeal food at 25°C unless specified. The w1118 strain was used as the wild type control in all experiments. P element-mediated excisions were used to generate deletions in S6KL following a standard protocol. The original stock EY10051 from the Bloomington Stock Center has a P element insertion in the first intron of S6KL (Fig. 1D). Before mobilizing EY10051 by P transposase Δ2–3, we isogenized the original stock. The w+ deletion lines with the P insertion excised imprecisely (S6KL158 and S6KL140) were verified by PCR, DNA sequencing, and western blotting.
Transgenic lines carrying UAS-S6KL or UAS-S6KLK193Q (a critical ATP binding residue lysine at 193 mutated to glutamine) were generated by inserting the full-length open reading frame of wild-type or mutant S6KL into the pUAST vector, followed by standard germline transformation. The tissue-specific Gal4 drivers elav-Gal4 (pan-neuronal), OK6-Gal4 (motor neuron specific), Mhc-Gal4 (muscle specific), and C57-Gal4 (muscle specific) were used for tissue-specific expression experiments. The UAS-Tkv-GFP transgenic line was supplied by Marcos Gonzalez-Gaitan (University of Geneva, Geneva, Switzerland). A yellow fluorescent protein (YFP) genetrap line labeling the endogenous Tkv with YFP was obtained from the Drosophila Genome Research Center (stock number 115298; Kyoto, Japan). The S6KL RNAi lines 102179 and 47326 were from the Vienna Drosophila Stock Center (VDRC). The following stocks were obtained from the Bloomington Stock Center: EP438, Df(1)ED7413, witA12, witB11, tkvk16713, tkv7, dadj1E4, and mad237.
Information on additional stocks, as well as antibodies and quantitative PCR analysis, can be found in S1 Text.
Generation of rabbit antibodies against S6KL
Polyclonal antibodies against S6KL were raised by immunizing rabbits with peptides containing amino acid residues 66–82 (TNQPTQSHPPGDEEQPV) of S6KL. The antibodies were purified using an affinity column coupled with the S6KL peptide according to the manufacturer’s instructions and used at 1 : 1000 for both staining and western analysis.
Immunoprecipitation and in vitro kinase activity assay
Coding sequences for GFP, S6KL, and S6KLK193Q were subcloned into pAC5.1 plasmids to produce N-terminally Flag tagged fusion proteins. Recombinant plasmids were transfected into S2 cells by Cellfectin II reagent (Invitrogen). After 48 h, transfected cells were lysed in RIPA buffer (50 mM Tris-HCl at pH 7.4, 150 mM NaCl, 0.1% SDS, 1% NP-40) for 1 h at 4°C. Cell lysates were precleared with protein G-Sepharose (GE) for 1 h and then incubated with anti-Flag monoclonal antibody (Sigma) for 3 h at 4°C. The immuno-complexes were precipitated with protein G-sepharose (GE), washed five times with RIPA buffer, and then washed two times with kinase buffer containing 25 mM Tris-HCl (pH 7.5), 5 mM β-glycerophosphate, 2 mM dithiothreitol (DTT), 0.1 mM Na3VO4, and 10 mM MgCl2. To perform the kinase activity assay, the anti-Flag immunoprecipitates were incubated with 50 μM ATP (Sigma), 10 μCi [γ-32P] ATP (PerkinElmer) and 5 μg myelin basic protein (Sigma) as substrates for 40 min at 30°C. The reaction products were separated on 12% SDS-polyacrylamide gels and autoradiographed to detect γ-32P incorporation.
Immunohistochemistry and confocal microscopy
Immunostaining of larval fillets was performed as described with minor modifications [7,8]. Late third instar larvae were dissected in Ca2+-free standard saline, fixed in fresh 4% paraformaldehyde for 40 min, and then immunostained with the following primary antibodies: FITC-conjugated goat anti-HRP at 1 : 100 (Jackson ImmunoResearch), rabbit anti-S6KL at 1 : 1000, rabbit anti-phosphorylated Mad at 1 : 500 (from P. ten Dijke, Leiden University, Leiden, the Netherlands) [45], and anti-CSP at 1 : 500 (6D6, Developmental Studies Hybridoma Bank, DSHB). Alexa 488 - or 568-conjugated anti-mouse or anti-rabbit secondary antibodies (Invitrogen) were used at 1 : 1000 to visualize primary antibody staining. All images were acquired using a Leica SP5 or Olympus BX51 laser scanning confocal microscope and processed with Adobe Photoshop.
Morphological analysis of NMJs was described previously [7,8]. Briefly, serial confocal images of NMJ 4 in the abdominal segments A2 and A3 were acquired and individual boutons were identified by immunostaining with the synaptic vesicle marker anti-CSP.
Electron microscopy
Electron microscopy of NMJ synapses was performed as previously described [8,19]. Briefly, dissected larvae were fixed overnight at 4°C in a mixture of 2% glutaraldehyde and 2% paraformaldehyde in 0.1 M cacodylate buffer (pH 7.4), rinsed in cacodylate buffer, and post-fixed in 0.5% OsO4/0.8% K3Fe(CN)6 cacodylate buffer for 90 min at room temperature. Samples were washed and stained in saturated aqueous uranyl acetate for 2 h on ice, dehydrated in a graded acetone series, and embedded in Spurr resin (Electron Microscopy Sciences). Ultrathin (70–80 nm) longitudinal sections of NMJ6/7 were cut with a Leica UC6 ultramicrotome. Grids were post-stained with saturated uranyl acetate for 20 min, followed by staining with Reynold’s lead citrate for 2–3 min. Processed samples were observed under a JEOL JEM-1400 transmission electron microscope. Statistical analysis of synaptic vesicles was performed as previously described [8,19].
Electrophysiology
Intracellular recordings from Drosophila muscle were performed essentially as described [8,19]. Briefly, wandering third instar larvae were dissected in calcium free HL3 saline [46] (in mM, 70 NaCl, 5 KCl, 20 MgCl2, 10 NaHCO3, 5 trehalose, 5 HEPES, 115 sucrose; pH 7.3). Excitatory junctional potentials (EJPs) were evoked at 0.3 Hz by a suction electrode using a depolarizing pulse delivered by a Grass S48 stimulator (Astro-Grass Inc.). Both EJPs and miniature EJPs (mEJPs) were recorded from muscle 6 of abdominal segment A2 or A3 in HL3 saline containing 0.5 mM Ca2+ using intracellular microelectrodes (10–20 MΩ) filled with 3 M KCl, and processed with Clampfit 10.2 software. To examine synaptic transmission upon high-frequency stimulation, presynaptic axons were stimulated at 10 Hz for 10 min in HL3 saline containing 1 mM extracellular Ca2+ [19,47]. All electrophysiological responses were recorded using Axoclamp 2B amplifier (Axon Instruments) in Bridge mode and analyzed using Clampfit 10.2 software. Only recordings from cells with resting potentials ≤ -60 mV and input resistances > 6 MΩ were analyzed.
FM1–43 uptake assay
Fluorescent lipophilic FM dyes are widely used to follow endocytosis, vesicle trafficking, and exocytosis. The FM1–43 uptake assay for measurement of endocytosis at NMJ synapses was described previously [8,29]. Briefly, late third instar larvae were dissected in low Ca2+ (0.2 mM) saline while keeping the central nervous system intact and then perfused with normal medium containing (in mM) 128 NaCl, 2 KCl, 4 MgCl2, 1.8 CaCl2, 5 HEPES and 35.5 sucrose at pH 7.3 [48]. Endocytosis of FM1–43 was induced by high K+ (90 mM) in the normal medium containing 10 μM FM1–43 (Molecular Probes) and reduced NaCl by an equivalent amount for 3 min. Depolarized and FM1–43-loaded preparations were then vigorously washed three times with Ca2+-free saline (normal medium with 0 Ca2+ and 0.5 mM EGTA) for 2 min and imaged using a Leica SP5 confocal microscope with a 40× water-immersion lens for multiple Z stack sections.
Cell culture, co-immunoprecipitation, and western analysis
Culture of Drosophila S2 cells and RNAi-mediated knockdown of gene expression were performed using protocols previously described [49]. S2 cells were maintained at 25°C in optimized serum-free medium (SF-900 II, Gibco). Cells were transfected using Cellfectin II reagent (Invitrogen) and expression was induced with 0.5 mM CuSO4 for 48 h. For dsRNA synthesis, the DNA template was PCR-amplified from a plasmid containing the full-length S6KL ORF using the following S6KL—specific primer pairs, 5’-TAATACGACTCACTATAGGGGCAGCAGGAGAATCCAGTTC-3’ and 5’-TAATACGACTCACTATAGGGTTGTGGAGAAAATCCAAGGC-3’ for S6KL dsRNA1, and 5’-TAATACGACTCACTATAGGGGAAGCAAATAGATCTGTGGCAAAAC-3’ and 5’-TAATACGACTCACTATAGGGGCTGCTTGGAGCTCAGGTTATAGTC-3’ for S6KL dsRNA2. The PCR products were used as templates for synthesis of double-stranded RNA using the MEGAscript T7 kit (Ambion). For RNAi-induced gene silencing, cultured cells were resuspended at a final concentration of 1×106 cells per ml medium and plated at 1 ml per well in six-well culture plates. After 24 h incubation, 10 μg dsRNA was added and mixed by gently swaying the plate in a circular motion six to eight times. Forty eight hours later, transfected cells were lysed in RIPA buffer for 1 h at 4°C. Protein expression levels were analyzed using the following antibodies: rabbit anti-S6KL at 1 : 1000, anti-GFP at 1 : 1000 (Clontech), anti-Flag at 1 : 1000 (Sigma), and anti-actin at 1 : 50,000 (Millipore). For immunoprecipitation, S6KL—Flag and Tkv-Myc were co-expressed in S2 cells. Cell lysates were pre-cleared with protein G-Sepharose (GE) for 1 h and then incubated with anti-Flag antibody at 1 : 100 (Sigma) or anti-Myc antibody at 1 : 100 (Millipore) for 3 h at 4°C. The immune complexes were precipitated with protein G-sepharose (GE), which were then washed five times with ice-cold RIPA buffer and boiled for 5 min in 30 μl sample buffer. Freed proteins were then separated by SDS-PAGE and subjected to western analysis using anti-Flag at 1 : 1000 and anti-Myc at 1 : 1000. The secondary antibody was HRP-coupled anti-mouse IgG (Sigma) or anti-rabbit IgG (Sigma) used at 1 : 50000. The protein bands were visualized using a chemiluminescent HRP substrate (Millipore).
To determine whether the degradation of Tkv was dependent on proteasome, S2 cells were transfected with plasmids co-expressing Flag-S6KL and Tkv-GFP. After 48 h culture, cells were treated with or without 50 μM MG132 (Sigma) for additional 4 h, followed by western analysis.
For western blotting of different genotypes, 60 adult heads from each genotype were homogenized in ice-cold 120 μl RIPA lysis buffer containing 2% SDS. The immunoblots (proteins from ten heads of each genotype per lane) were incubated overnight at 4°C with anti-S6KL at 1 : 1000, anti-GFP at 1 : 1000, and anti-Wit at 1 : 50 (DSHB). Actin was used as a loading control.
Statistical analysis
Quantification of pMad staining and endocytosed FM1–43 intensities was performed using Image J. Anti-HRP staining was used as an internal control for pMad intensity quantification. For each channel, an arbitrary threshold was set and used for all relevant images. Each staining experiment was repeated at least three times. To quantify the expression levels of target proteins, positive signals on western blots from multiple independent repeats were calculated using ImageJ and normalized to the actin control. All the data are expressed as mean ± standard error of the mean (SEM). Statistical significance between paired group means was determined by Student’s t-tests (Fig. 5). Multiple group means were evaluated by one-way ANOVA with Tukey’s post hoc tests for pair-wise comparisons. The cumulative probabilities of quantal size between wild type and S6KL mutants in Fig. 6E were compared by Kolmogorov-Smirnov test. All comparisons were between a specific genotype and the control unless otherwise indicated. No asterisk denotes p>0.05, * p<0.05, ** p<0.01, and *** p<0.001.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Giagtzoglou N, Ly CV, Bellen HJ (2009) Cell adhesion, the backbone of the synapse: “vertebrate” and “invertebrate” perspectives. Cold Spring Harb Perspect Biol 1: a003079. doi: 10.1101/cshperspect.a003079 20066100
2. Melom JE, Littleton JT (2011) Synapse development in health and disease. Curr Opin Genet Dev 21 : 256–261. doi: 10.1016/j.gde.2011.01.002 21277192
3. Packard M, Mathew D, Budnik V (2003) Wnts and TGF beta in synaptogenesis: old friends signalling at new places. Nat Rev Neurosci 4 : 113–120. 12563282
4. Bayat V, Jaiswal M, Bellen HJ (2011) The BMP signaling pathway at the Drosophila neuromuscular junction and its links to neurodegenerative diseases. Curr Opin Neurobiol 21 : 182–188. doi: 10.1016/j.conb.2010.08.014 20832291
5. Collins CA, DiAntonio A (2007) Synaptic development: insights from Drosophila. Curr Opin Neurobiol 17 : 35–42. 17229568
6. Fuentes-Medel Y, Budnik V (2010) Menage a Trio during BMP-Mediated Retrograde Signaling at the NMJ. Neuron 66 : 473–475. doi: 10.1016/j.neuron.2010.05.016 20510851
7. Liu Z, Huang Y, Hu W, Huang S, Wang Q, et al. (2014) dAcsl, the Drosophila ortholog of acyl-CoA synthetase long-chain family member 3 and 4, inhibits synapse growth by attenuating bone morphogenetic protein signaling via endocytic recycling. J Neurosci 34 : 2785–2796. doi: 10.1523/JNEUROSCI.3547-13.2014 24553921
8. Shi W, Chen Y, Gan G, Wang D, Ren J, et al. (2013) Brain tumor regulates neuromuscular synapse growth and endocytosis in Drosophila by suppressing mad expression. J Neurosci 33 : 12352–12363. doi: 10.1523/JNEUROSCI.0386-13.2013 23884941
9. Xiao L, Michalski N, Kronander E, Gjoni E, Genoud C, et al. (2013) BMP signaling specifies the development of a large and fast CNS synapse. Nat Neurosci 16 : 856–864. doi: 10.1038/nn.3414 23708139
10. Aberle H, Haghighi AP, Fetter RD, McCabe BD, Magalhaes TR, et al. (2002) wishful thinking encodes a BMP type II receptor that regulates synaptic growth in Drosophila. Neuron 33 : 545–558. 11856529
11. Marques G, Bao H, Haerry TE, Shimell MJ, Duchek P, et al. (2002) The Drosophila BMP type II receptor Wishful Thinking regulates neuromuscular synapse morphology and function. Neuron 33 : 529–543. 11856528
12. McCabe BD, Marques G, Haghighi AP, Fetter RD, Crotty ML, et al. (2003) The BMP homolog Gbb provides a retrograde signal that regulates synaptic growth at the Drosophila neuromuscular junction. Neuron 39 : 241–254. 12873382
13. McCabe BD, Hom S, Aberle H, Fetter RD, Marques G, et al. (2004) Highwire regulates presynaptic BMP signaling essential for synaptic growth. Neuron 41 : 891–905. 15046722
14. O’Connor-Giles KM, Ho LL, Ganetzky B (2008) Nervous wreck interacts with thickveins and the endocytic machinery to attenuate retrograde BMP signaling during synaptic growth. Neuron 58 : 507–518. doi: 10.1016/j.neuron.2008.03.007 18498733
15. Rodal AA, Blunk AD, Akbergenova Y, Jorquera RA, Buhl LK, et al. (2011) A presynaptic endosomal trafficking pathway controls synaptic growth signaling. J Cell Biol 193 : 201–217. doi: 10.1083/jcb.201009052 21464232
16. Sweeney ST, Davis GW (2002) Unrestricted synaptic growth in spinster-a late endosomal protein implicated in TGF-beta-mediated synaptic growth regulation. Neuron 36 : 403–416. 12408844
17. Wang X, Shaw WR, Tsang HT, Reid E, O’Kane CJ (2007) Drosophila spichthyin inhibits BMP signaling and regulates synaptic growth and axonal microtubules. Nat Neurosci 10 : 177–185. 17220882
18. Nahm M, Lee MJ, Parkinson W, Lee M, Kim H, et al. (2013) Spartin regulates synaptic growth and neuronal survival by inhibiting BMP-mediated microtubule stabilization. Neuron 77 : 680–695. doi: 10.1016/j.neuron.2012.12.015 23439121
19. Zhao L, Wang D, Wang Q, Rodal AA, Zhang YQ (2013) Drosophila cyfip Regulates Synaptic Development and Endocytosis by Suppressing Filamentous Actin Assembly. PLoS Genet 9: e1003450. doi: 10.1371/journal.pgen.1003450 23593037
20. Kraut R, Menon K, Zinn K (2001) A gain-of-function screen for genes controlling motor axon guidance and synaptogenesis in Drosophila. Curr Biol 11 : 417–430. 11301252
21. Barcelo H, Stewart MJ (2002) Altering Drosophila S6 kinase activity is consistent with a role for S6 kinase in growth. Genesis 34 : 83–85. 12324955
22. von Manteuffel SR, Dennis PB, Pullen N, Gingras AC, Sonenberg N, et al. (1997) The insulin-induced signalling pathway leading to S6 and initiation factor 4E binding protein 1 phosphorylation bifurcates at a rapamycin-sensitive point immediately upstream of p70s6k. Mol Cell Biol 17 : 5426–5436. 9271419
23. Dickman DK, Lu Z, Meinertzhagen IA, Schwarz TL (2006) Altered synaptic development and active zone spacing in endocytosis mutants. Curr Biol 16 : 591–598. 16546084
24. Verstreken P, Koh TW, Schulze KL, Zhai RG, Hiesinger PR, et al. (2003) Synaptojanin is recruited by endophilin to promote synaptic vesicle uncoating. Neuron 40 : 733–748. 14622578
25. Verstreken P, Kjaerulff O, Lloyd TE, Atkinson R, Zhou Y, et al. (2002) Endophilin mutations block clathrin-mediated endocytosis but not neurotransmitter release. Cell 109 : 101–112. 11955450
26. Zhang B, Koh YH, Beckstead RB, Budnik V, Ganetzky B, et al. (1998) Synaptic vesicle size and number are regulated by a clathrin adaptor protein required for endocytosis. Neuron 21 : 1465–1475. 9883738
27. Koh TW, Verstreken P, Bellen HJ (2004) Dap160/intersectin acts as a stabilizing scaffold required for synaptic development and vesicle endocytosis. Neuron 43 : 193–205. 15260956
28. Verstreken P, Ohyama T, Haueter C, Habets RL, Lin YQ, et al. (2009) Tweek, an evolutionarily conserved protein, is required for synaptic vesicle recycling. Neuron 63 : 203–215. doi: 10.1016/j.neuron.2009.06.017 19640479
29. Verstreken P, Ohyama T, Bellen HJ (2008) FM 1–43 labeling of synaptic vesicle pools at the Drosophila neuromuscular junction. Methods Mol Biol 440 : 349–369. doi: 10.1007/978-1-59745-178-9_26 18369958
30. Chang YJ, Pi H, Hsieh CC, Fuller MT (2013) Smurf-mediated differential proteolysis generates dynamic BMP signaling in germline stem cells during Drosophila testis development. Dev Biol 383 : 106–120. doi: 10.1016/j.ydbio.2013.08.014 23988579
31. Xia L, Jia S, Huang S, Wang H, Zhu Y, et al. (2010) The Fused/Smurf complex controls the fate of Drosophila germline stem cells by generating a gradient BMP response. Cell 143 : 978–990. doi: 10.1016/j.cell.2010.11.022 21145463
32. Um SH D’Alessio D, Thomas G (2006) Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cell Metab 3 : 393–402. 16753575
33. Shima H, Pende M, Chen Y, Fumagalli S, Thomas G, et al. (1998) Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. EMBO J 17 : 6649–6659. 9822608
34. Pende M, Kozma SC, Jaquet M, Oorschot V, Burcelin R, et al. (2000) Hypoinsulinaemia, glucose intolerance and diminished beta-cell size in S6K1–deficient mice. Nature 408 : 994–997. 11140689
35. Montagne J, Stewart MJ, Stocker H, Hafen E, Kozma SC, et al. (1999) Drosophila S6 kinase: a regulator of cell size. Science 285 : 2126–2129. 10497130
36. Raught B, Peiretti F, Gingras AC, Livingstone M, Shahbazian D, et al. (2004) Phosphorylation of eucaryotic translation initiation factor 4B Ser422 is modulated by S6 kinases. EMBO J 23 : 1761–1769. 15071500
37. Cheng L, Locke C, Davis GW (2011) S6 kinase localizes to the presynaptic active zone and functions with PDK1 to control synapse development. J Cell Biol 194 : 921–935. doi: 10.1083/jcb.201101042 21930778
38. Shen W, Ganetzky B (2009) Autophagy promotes synapse development in Drosophila. J Cell Biol 187 : 71–79. doi: 10.1083/jcb.200907109 19786572
39. Koh TW, Korolchuk VI, Wairkar YP, Jiao W, Evergren E, et al. (2007) Eps15 and Dap160 control synaptic vesicle membrane retrieval and synapse development. J Cell Biol 178 : 309–322. 17620409
40. Marie B, Sweeney ST, Poskanzer KE, Roos J, Kelly RB, et al. (2004) Dap160/intersectin scaffolds the periactive zone to achieve high-fidelity endocytosis and normal synaptic growth. Neuron 43 : 207–219. 15260957
41. Ball RW, Warren-Paquin M, Tsurudome K, Liao EH, Elazzouzi F, et al. (2010) Retrograde BMP signaling controls synaptic growth at the NMJ by regulating trio expression in motor neurons. Neuron 66 : 536–549. doi: 10.1016/j.neuron.2010.04.011 20510858
42. Lu D, Lin W, Gao X, Wu S, Cheng C, et al. (2011) Direct ubiquitination of pattern recognition receptor FLS2 attenuates plant innate immunity. Science 332 : 1439–1442. doi: 10.1126/science.1204903 21680842
43. Kowanetz M, Lonn P, Vanlandewijck M, Kowanetz K, Heldin CH, et al. (2008) TGFbeta induces SIK to negatively regulate type I receptor kinase signaling. J Cell Biol 182 : 655–662. doi: 10.1083/jcb.200804107 18725536
44. Itoh S, ten Dijke P (2007) Negative regulation of TGF-beta receptor/Smad signal transduction. Curr Opin Cell Biol 19 : 176–184. 17317136
45. Persson U, Izumi H, Souchelnytskyi S, Itoh S, Grimsby S, et al. (1998) The L45 loop in type I receptors for TGF-beta family members is a critical determinant in specifying Smad isoform activation. FEBS Lett 434 : 83–87. 9738456
46. Stewart BA, Atwood HL, Renger JJ, Wang J, Wu CF (1994) Improved stability of Drosophila larval neuromuscular preparations in haemolymph-like physiological solutions. J Comp Physiol A 175 : 179–191. 8071894
47. Yao CK, Lin YQ, Ly CV, Ohyama T, Haueter CM, et al. (2009) A synaptic vesicle-associated Ca2+ channel promotes endocytosis and couples exocytosis to endocytosis. Cell 138 : 947–960. doi: 10.1016/j.cell.2009.06.033 19737521
48. Jan LY, Jan YN (1976) Properties of the larval neuromuscular junction in Drosophila melanogaster. J Physiol 262 : 189–214. 11339
49. Rogers SL, Rogers GC (2008) Culture of Drosophila S2 cells and their use for RNAi-mediated loss-of-function studies and immunofluorescence microscopy. Nat Protoc 3 : 606–611. doi: 10.1038/nprot.2008.18 18388942
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 3
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Clonality and Evolutionary History of Rhabdomyosarcoma
- Morphological Mutations: Lessons from the Cockscomb
- Maternal Filaggrin Mutations Increase the Risk of Atopic Dermatitis in Children: An Effect Independent of Mutation Inheritance
- Transcriptomic Profiling of Reveals Reprogramming of the Crp Regulon by Temperature and Uncovers Crp as a Master Regulator of Small RNAs
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy