
A Systems-Level Interrogation Identifies Regulators of Blood Cell Number and Survival
Signaling networks that drive cell survival and proliferation regulate cell number in development and disease. We use a simple Drosophila model of cell number control, which centers on PDGF/VEGF receptor signaling. Performing a genome-wide RNAi screen under Pvr-sensitized conditions, we identify regulators of cell number that have not been found in conventional screens. Validation by in vivo genetics reveals previously unrecognized roles for EcR and InR in the balance of cell survival in the Drosophila embryo. Phosphoproteomic analysis demonstrates distinct mechanisms of cell survival regulation by EcR and receptor tyrosine kinase signaling. It further identifies common phosphorylation targets of Pvr and InR including regulators of cell survival, and receptor-specific phosphorylation targets mediating unique functions of Pvr and InR. Importantly, the study provides precedence that the selection of phosphorylation targets by signaling receptors can change with the signaling status of the cell, which may have wide-reaching implications for other cell regulatory systems.
Published in the journal:
. PLoS Genet 11(3): e32767. doi:10.1371/journal.pgen.1005056
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005056
Summary
Signaling networks that drive cell survival and proliferation regulate cell number in development and disease. We use a simple Drosophila model of cell number control, which centers on PDGF/VEGF receptor signaling. Performing a genome-wide RNAi screen under Pvr-sensitized conditions, we identify regulators of cell number that have not been found in conventional screens. Validation by in vivo genetics reveals previously unrecognized roles for EcR and InR in the balance of cell survival in the Drosophila embryo. Phosphoproteomic analysis demonstrates distinct mechanisms of cell survival regulation by EcR and receptor tyrosine kinase signaling. It further identifies common phosphorylation targets of Pvr and InR including regulators of cell survival, and receptor-specific phosphorylation targets mediating unique functions of Pvr and InR. Importantly, the study provides precedence that the selection of phosphorylation targets by signaling receptors can change with the signaling status of the cell, which may have wide-reaching implications for other cell regulatory systems.
Introduction
The regulation of cell number varies greatly and typically depends on developmental and environmental stimuli that determine the intracellular balance of pro - and anti-death, and proliferative signals [1–3]. Proto-oncogenes and tumor suppressors play roles as regulators of cell number and the pathological extension of cell survival is a major hallmark of tumorigenesis [4]. Accordingly, understanding the complex signaling networks that regulate cell survival is an important yet incompletely accomplished goal [4,5], which can be facilitated by studying a simple model organism.
Blood cells in the fruitfly Drosophila melanogaster have been instrumental in the discovery of fundamental concepts in immunity, hematopoiesis and wound healing [6–11], but they are also a convenient model to study mechanisms that regulate cell number. In particular, the Drosophila PDGF/VEGF Receptor (Pvr), a member of the Receptor Tyrosine Kinase (RTK) family, controls anti-apoptotic survival signaling in Drosophila blood cells (hemocytes) in vivo and in the embryonic cell line Kc in culture [12]. In other instances, Pvr has been reported to regulate cell proliferation [13,14], differentiation [15,16], cell size [17,18], cytoskeletal architecture [19] and cell migration [20–22]. Drosophila Pvr therefore parallels roles of the vertebrate family of PDGF/VEGF Receptors in development and disease [12,21,23–26].
Here, we took advantage of the role of Pvr in embryonic blood cell survival and performed a systematic RNAi screen to identify regulators of cell number, using the Drosophila cell line Kc under sensitized conditions of Pvr knockdown. The screen identified enhancers and suppressors of the Pvr RNAi phenotype, many of which were not found in conventional RNAi screens examining cell growth and viability. In particular, we found that knockdown of InR enhanced the Pvr RNAi phenotype while knockdown of the Ecdysone receptor (EcR) [27] and its co-receptor ultraspiracle (usp) [28] suppressed the Pvr RNAi phenotype. We confirmed functional roles for these genes related to Pvr both in cell culture and in vivo. Phosphoproteomic analyses revealed major differences in the signaling signature of Pvr deficient cells rescued by activation of InR as compared to inactivation of EcR. Further, our analysis identified distinct sets of phosphorylation targets, common to both Pvr and InR, and unique to each receptor. Most importantly, we provide precedence that the selection of phosphorylation targets by signaling receptors can depend on the signaling status of the cell, which may have wide-reaching implications for cell regulatory systems in animal development, disease, and the experimental and therapeutic manipulation of signaling pathways.
Results
Pvr signaling in the embryonic hemocyte cell line Kc
Previously, we demonstrated that the Drosophila PDGF/VEGF Receptor, Pvr, is essential for anti-apoptotic survival in embryonic hemocytes and in the related cell line Kc, which maintains autocrine Pvr signaling [12,29]. Taking advantage of these systems, we sought to examine the signaling networks that mediate anti-apoptotic survival and regulate cell number.
First, we confirmed that RNAi-mediated knockdown of Pvr induces apoptotic cell death in Kc cells. RNAi silencing of the Drosophila inhibitor of apoptosis DIAP1, or thread (th), served as positive control (Fig. 1A). Expression of the baculovirus inhibitor of apoptosis p35 [30] rescued hemocyte survival, leading us to establish a selected pool of Kcp35 cells (Kcp35 cells, Fig. 1A). Immunoblotting confirmed that Pvr knockdown was equally efficient in Kc and Kcp35 cells (S1 Fig). Closer examination by incorporation of the thymidine nucleoside analog EdU (5-ethynyl-2’deoxyuridine) in Kc versus Kcp35 cells revealed that Pvr also moderately contributes toward cell proliferation in this system (Fig. 1B), an effect that could not be distinguished in a previous study employing cell cycle profiling [12]. Reduction in proliferation was also suggested by immunoblotting, where lysates of equal numbers of cells showed a decrease in the proliferation marker phospho-histone H3 (pHH3) in Pvr knockdown samples (S1 Fig).

Using Kc cells, we queried signaling pathways that might be involved in Pvr-dependent cell survival and proliferation. Examining activity of the Akt/TOR and Mek/Erk pathways by using antibodies to phosphorylated forms of S6Kinase (S6K, an Akt pathway target), Mek and Erk, we found that both pathways are active in Kc cells. Pvr RNAi led to a significant reduction in the phosphorylation levels of these proteins, indicating that Pvr is a major activator of these pathways in Kc cells. Single signaling mediator knockdowns of Akt, the TOR-associated Raptor, S6K, Mek and Erk served as controls (Fig. 1C). Phosphorylation signals were also quantified and displayed as a ratio with the amount of unphosphorylated signaling mediator (Fig. 1C). These findings suggest that Pvr triggers activation of the Akt/TOR and Mek/Erk and pathways, thereby supporting anti-apoptotic cell survival and proliferation.
Next, we asked whether silencing of either or both of these pathways is sufficient to affect cell viability and mimic loss of Pvr function. Combining dsRNAs targeting various mediators of the Akt/Tor and Mek/Erk pathways, we found that, despite efficient knockdown of the genes (S2 Fig), neither single nor simultaneous inhibition of both pathways caused a significant reduction of cell numbers, as quantified by CellTiterGlo assay based on ATP content (Fig. 1D), and cell counting (Fig. 1E). In contrast, Pvr RNAi, showed significant decreases in cell number (Fig. 1D, E). This predicted the presence of one or more additional, redundant cell survival/proliferation pathway(s) downstream of Pvr (‘X’, Fig. 1F), and/or parallel signaling pathways that contribute to the overall survival and proliferation of the cell (‘Y’, Fig. 1F).
A genome-wide RNAi screen for modifiers of Pvr
Based on our prediction, we sought to identify other signaling pathways that contribute to the anti-apoptotic survival of Kc cells. We hypothesized that re-activation of just one survival or proliferation pathway would be sufficient to rescue cell numbers in Pvr deficient cells (Fig. 1F). Indeed, silencing of negative regulators of the Akt/Tor and Erk pathways rescued the Pvr RNAi phenotype, validating our screening approach. For these experiments, we ruled out that silencing of downstream signaling mediators such as Akt would result in upregulation of Pvf2 expression, the major Pvr ligand in Kc cells that mediates autocrine signaling (S3 Fig). Expanding our approach, we screened the DRSC Genome-Wide RNAi library 1.0 (Drosophila RNAi Screening Center, Harvard Medical School) for modifiers of cell number, specifically under conditions of Pvr RNAi-mediated silencing compared to a control background (Fig. 2A). The DRSC 1.0 set targets 22,914 distinct amplicons based on Flybase release 5.51 of the Drosophila genome, corresponding to 13,777 unique genes [31], 6944 of which are expressed in Kc cells [29]. Screening was performed in 384-well format, quantifying ATP content as a readout of cell number (CellTiterGlo). To determine an increase or decrease over the average value of ATP content, Z scores were calculated for each well. Focusing on those dsRNAs that show differential effects in Pvr knockdown (Pvr RNAi) versus control cells (GFP RNAi), we calculated the difference of each of the Z scores (ZDiff = Z[Pvr]-Z[GFP]), and selected amplicons with ZDiff> = 2 and ZDiff< = -2 as primary screen hits (S1 Table). Cluster analysis of the values Z[Pvr], Z[GFP], and ZDiff for each amplicon revealed three distinct classes of signatures, i.e. Pvr Suppressors, Pvr Enhancers, and Pvr ‘Upstream Genes’ (Fig. 2B). By our cutoff criteria, 64 amplicons scored as suppressors of the Pvr knockdown phenotype, rescuing cell numbers more effectively in the Pvr RNAi background compared to control cells. 65 amplicons scored as Pvr Enhancers, exacerbating the Pvr knockdown phenotype. We classified 290 amplicons as Pvr ‘Upstream Genes’, reducing cell numbers in control cells, but having rather minor effects in the Pvr silenced background. Among this group we found amplicons targeting Pvr itself and many ribosomal proteins, suggesting that many of the targeted genes play a role in the production or activation of Pvr (S1 Table).

Subsequent secondary testing of screen hits was carried out for Pvr Suppressors and Pvr Enhancers. We selected 47 suppressor genes and 47 enhancer genes based on a cutoff of ZDiff> = 2.2 and ZDiff< = -2.2 (S2 Table) and synthesized non-overlapping alternative amplicons that were free of 19bp or larger overlaps with other genes, in order to avoid off-target effects [32,33]. As in the primary screen, amplicons were tested for their ability to modify cell number, specifically comparing Pvr knockdown cells relative to control cells (S2 Table). To identify promising ‘high confidence candidates’ for further analysis, we calculated the average of the ZDiff scores among all amplicons of a gene from the primary and secondary screens (ZDiffFinal) (S3 Table). Based on ZDiffFinal values of > = 1.6 and < = -1.2, we report 30 high-confidence Pvr Suppressors and 14 high-confidence Pvr Enhancers (S3 Table). Z value cutoffs were guided by the scores of predicted genes within the set, such as members of the Akt/Tor and Mek/Erk pathways. Candidates of specific interest were confirmed by live/dead cell counting, omitting genes with obvious roles in RNA interference, such as AGO2 (S4 Fig).
Relatively few genes scored as Pvr Enhancers. Among those, we identified the RTK InR [34], and cropped (crp) encoding the helix-loop-helix transcription factor that is a homolog of the mammalian transcription factor AP-4 [35]. The screen also identified tonalli (tna), encoding a protein similar to mammalian ZMIZ1 and ZMIZ2 involved in sumoylation [36] that interacts genetically with the Brahma ATP-dependent chromatin remodeling complex in Drosophila [37].
Among the Pvr Suppressors, the screen yielded all known tumor suppressors and negative regulators of the Akt/TOR pathway, including Phosphatase and Tensin Homolog (Pten), Tuberous Sclerosis Protein 1 (Tsc1), gigas (gig)/Tuberous Sclerosis Protein 2 (Tsc2), SNF4A–a and -γ, also known as AMP-Activated Protein Kinase subunits a and γ(AMPK–α and AMPK–γ), Forkhead Box Protein (foxo), and Lobe (L), a protein with similarities to the vertebrate Proline-rich Akt substrate of 40 kDa (PRAS40) [38–41]. We further identified negative regulators of the Ras/Erk pathway Mitogen-activated protein kinase phosphatase 3 (Mkp3), and microtubule star (mts) and widerborst (wdb), which encode components of the protein phosphatase PP2A complex [42–44]. We calculated which protein complexes were over-represented with respect to the frequency of their components among the high confidence hits in the RNAi screen, and found, besides the PP2A complex, two other major protein complexes among the high confidence hits in the RNAi screen (Fig. 2C): the ecdysone receptor complex, consisting of the nuclear hormone receptors EcR and usp [27,45], and the Brahma SWI2/SNF2 family ATPase chromatin-remodeling complex, comprising osa and dalao [46,47]. Other Pvr Suppressors were CG6182, an ortholog of mammalian TBC1 domain member 7 (TBC7), and GckIII, and CG31635, an ortholog of mammalian LRRC68. Given the reported interplay between ecdysone and insulin signaling during development [48], we wanted to dissect whether common and/or distinct downstream mechanisms mediate Pvr suppression, and therefore chose InR and EcR /usp for in vivo validation.
Pro-apoptotic effects of EcR/Usp signaling and anti-apoptotic effects of insulin signaling in cell culture
Using Kc cells, we examined the functional roles of InR and EcR/Usp in more detail. EcR and Usp form a heterodimer and are induced by binding of the steroid hormone 20-hydroxyecdysone (20E) [45,49]. Signaling by the EcR complex plays a major role during molting and metamorphosis [50], yet a role in embryonic cell death and cell number control has not been established [51]. We confirmed the effects of silencing or stimulating InR, or silencing EcR or usp, on Pvr RNAi-induced apoptosis using TUNEL assays, and we quantified effects on proliferation using EdU incorporation in Kcp35 cells (Fig. 3A-C). Consistent with the results from the screen, we found that, in combination with Pvr knockdown, silencing of InR exacerbated apoptosis. Further, silencing of EcR or usp, or stimulation of InR with insulin rescued apoptosis (Fig. 3A). In contrast, when examining proliferation, only insulin stimulation or a Tsc2/gigas (gig) RNAi Akt pathway control significantly suppressed proliferation defects, suggesting that EcR and Usp mainly function in the regulation of cell death, rather than proliferation (Fig. 3B, C). InR knockdown seemed to enhance the reduction of EdU incorporation in Pvr knockdown cells, but differences were not statistically significant based on three independent biological replicate experiments (Fig. 3C).
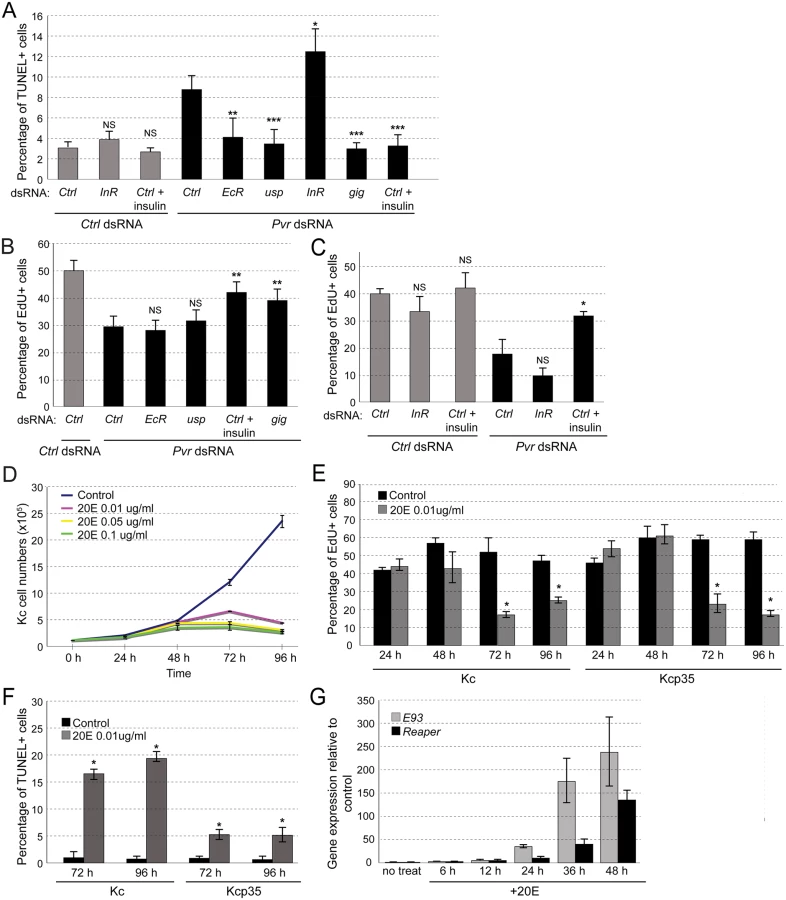
Next we examined the effects of ecdysone stimulation. Anti-proliferative effects of ecdysone in Kc cells have been reported previously [52–54], but whether ecdysone also has direct pro-apoptotic effects in embryonic cells not been determined. To test this, we stimulated Kc cells with 20E at concentrations close to physiological levels (0.01ug/ml) [54,55]. Overall, 20E induced a marked reduction in cell number at stimulation times of >48h (Fig. 3D). As expected, it resulted in a reduction of cell proliferation as measured by EdU incorporation, both in Kc and in apoptosis-resistant Kcp35 cells (Fig. 3E). However, TUNEL analysis showed a substantial increase in apoptotic cells upon 20E stimulation, which was largely suppressed in Kcp35 cells (Fig. 3F). 20E did not cause a decrease of Pvr protein levels (S5 Fig), suggesting that molecular mechanisms other than Pvr downregulation account for the observed increase in apoptosis. During metamorphosis-associated programmed cell death (PCD), several genes have been described as ecdysone-induced pro-death targets, in particular Ecdysone-induced protein 93F (E93), broad (br), Ecdysone-induced protein 74EF (E74A), and reaper (rpr) [56–58]. When we examined the expression levels of these genes during ecdysone stimulation of Kc cells we found that, indeed, rpr and E93 levels increased from the first day of 20E stimulation (Fig. 3G), consistent with the induction of apoptosis. We also examined whether Pvr knockdown would have an effect on the expression of rpr and E93 but found no significant difference relative to controls (S6 Fig).
In summary, we conclude that the EcR complex has pro-apoptotic functions in the cell line Kc, which become apparent under sensitized conditions of Pvr loss of function, or experimental addition of 20E.
EcR and InR are opposing modifiers of Pvr in vivo
To test the role of InR and EcR in the suppression and enhancement of apoptosis in vivo, we examined the function of these genes in the survival of hemocytes in the Drosophila embryo. Drosophila embryos typically show a developmentally fixed number of ~600 hemocytes post stage 11 until early stage 17, and loss of Pvr signaling causes a rapid decline in hemocytes due to their apoptotic death and phagocytic clearance by the small number of remaining live hemocytes [12]. Based on our findings in Kc cells, we predicted that inhibition of EcR would rescue, and inhibition of InR would enhance, Pvr loss-of-function in embryonic blood cells [12]. Indeed, hemocyte-specific suppression of EcR signaling by expression of dominant-negative forms of EcR [59,60] partially rescued hemocyte counts in Pvr1 mutant embryos (Fig. 4), resembling rescue by the baculovirus inhibitor of apoptosis, p35 [12] (see also Fig. 4A). Conversely, expression of dominant-negative InR in hemocytes enhanced the Pvr phenotype, further reducing embryonic hemocyte numbers (Fig. 4). Consistently, we previously demonstrated that activated PI3K, a positive mediator of the Akt/TOR pathway downstream of InR, can partially rescue the Pvr mutant in vivo phenotype [12]. To confirm hemocyte autonomous effects of EcR and InR, we induced embryonic hemocyte death by hemocyte-specific expression of dominant-negative PvrΔC [12], and examined the effects of co-expressed dominant-negative versions of EcR or InR. Again, we found that dominant-negative EcR rescued apoptotic loss of hemocytes, while dominant-negative InR exacerbated the cell death phenotype (Fig. 4). Expression of the transgenes in the wild type background had no significant effects (Fig. 4A).

Intrigued by the mild increase of hemocyte numbers upon overexpression of dominant-negative EcRdn (Fig. 4A), we asked whether blocking EcR signaling alone would have a positive effect on hemocyte numbers at a later point during development, for example at the transition from the embryo to the larval stage. A time course of total hemocyte counts in live animals illustrates that, compared to stage 16 embryos, hemocyte numbers in young 1st instar larvae decline to about 60%, suggesting a putative connection with the embryonic ecdysone peak in mid-embryogenesis [61] (S7 Fig). However, comparing live hemocyte counts of controls to animals with hemocyte-specific expression of EcRdn, we did not see a significant rescue in the total number of hemocytes, despite a mild increase in EcRdn overexpressing larvae (S7 Fig).
Taken together, our findings suggest that EcR signaling accounts for a basic level of pro-death signaling in embryonic hemocytes, which however is revealed only under sensitized conditions such as Pvr loss of function. Conversely, signaling by InR contributes to the trophic survival of embryonic hemocytes, which acts redundantly with Pvr signaling, and therefore again is only evident in conjunction with loss of Pvr signaling (Fig. 4G).
Signaling networks downstream of Pvr, InR and EcR
Based on our findings, we sought to further dissect the relationship between Pvr, InR and EcR signaling. First, we asked whether signaling by the EcR complex acts epistatically or in parallel with RTK-triggered signaling pathways such as Akt/Tor. When comparing the effects of silencing of the EcR/Usp and Akt/Tor pathways separately and in combination, we found that simultaneous knockdown of genes from both pathways resulted in increased cell number rescue (e.g. EcR and Pten), which in many cases was significant when compared to knockdown of two genes from the same pathway (i.e. EcR and usp, or Pten and gig). This suggested a parallel, rather than epistatic relationship (Fig. 5A). Biochemically, insulin stimulation of Pvr deficient cells restored, albeit to distinctive levels, phosphorylation of downstream signaling mediators of the Akt/Tor and Mek/Erk pathways, while EcR knockdown did not show such effects (Fig. 5B). This suggested similar but not identical signaling profiles for the RTKs Pvr and InR, and distinct mechanisms for the EcR complex.

To compare the signaling profiles of Pvr, InR and EcR in a more systematic manner, we chose a phosphoproteomics approach. We utilized mass spectrometry and an isobaric labeling strategy that enables multiplexing and relative quantification between samples [62,63]. For this analysis, we surveyed the phosphoproteome by formally comparing conditions of (1) ‘high Pvr’ signaling (+ control dsRNA; taking advantage of the high endocrine Pvr activity in Kc cells); (2) ‘low Pvr’ signaling (+ Pvr dsRNA); (3) ‘high InR’ signaling (+ insulin, to stimulate endogenous InR in Kc cells); (4) ‘low InR’ signaling (+ control dsRNA; taking advantage of the low InR activity in Kc cells under standard culture conditions presumably due to low levels of dIlp expression [29], see also Fig. 5B); (5) ‘high EcR’ (endogenous EcR in Kc cells); and (6) ‘low EcR’ (+ EcR dsRNA).
First, we assessed which phosphoproteins were up - or downregulated in the rescue of Pvr silenced Kc cells. We surveyed the phosphoproteome under conditions of high and low Pvr activity (Fig. 6A and B, respectively), and analyzed separately for the two conditions the effects of EcR silencing or InR activation, assessing biological duplicates (S4 Table and S5 Table). Under ‘high Pvr’ conditions, approximately 10% of the detected phosphorylation was altered more than 1.5-fold under conditions of InR stimulation, which we refer to as the ‘InR-specific set’ (Fig. 6A and S4 Table). This percentage nearly doubled under ‘low Pvr’ conditions (Fig. 6B and S5 Table). Although some of these phosphorylations could be attributed to the fact the InR may phosphorylate Pvr targets in the absence of Pvr, this finding also suggested the emergence of new sets of up and down-regulated phosphosites that were not observed upon InR activation under ‘high Pvr’ conditions (below).

Secondly, we sought to directly measure the degree to which phosphosites were altered by InR under conditions of ‘high’ versus ‘low’ Pvr signaling, hypothesizing a ‘sensitization’ of InR signaling by the absence of Pvr. We repeated our phosphoproteomic analysis, this time directly comparing the six experimental signaling conditions among each other (Fig. 7A and S6 Table). While nearly three-quarters of the ‘InR-specific set’ of phosphopeptides remained upregulated following InR activation in the absence of Pvr, the ‘InR-specific set’ showed qualitative differences in the absence and presence of Pvr signaling. For instance, InR stimulation elevated levels of phosphorylation of fifteen phosphoproteins specifically under ‘low Pvr’ activity as compared to ‘high Pvr’ signaling. These included Chromosome-associated protein (Cap), lava lamp (lva), Enhancer of decapping 3 (Edc3), Bicaudal D (BicD), lethal(2)03709, eukaryotic translation Initiation Factor 2α (eIF-2 α) and several uncharacterized gene products. InR activation restored phosphorylation to nearly all sites downregulated in Pvr deficient cells, (Fig. 7C). These phosphorylations likely account for the ability of insulin to rescue Pvr deficiency. EcR knockdown, meanwhile, had very little effect on the phosphoproteome, both in low and high Pvr conditions (Fig. 6A, B), despite efficient knockdown (S8 Fig). Similar findings were made from the comparative analysis of all six experimental conditions (Fig. 7A, B). This is consistent with an alternative mode of action, such as the transcriptional modulation of EcR/Usp target genes (see Fig. 3G).

Lastly, we identified a pool of common phosphoproteins induced by both Pvr and InR, which comprise signaling mediators for common functions in cell survival and proliferation. At the same time, we distinguished Pvr - or InR-associated targets that may mediate receptor-specific functions. A common set of phosphorylation targets for Pvr and InR, either direct or indirect, can be inferred from the reciprocal effects of Pvr knockdown and InR stimulation, comparing ‘low Pvr’ and ‘high InR’ conditions (Fig. 7B; 153 phosphosites: S7 Table). Examples include phosphorylation of Structure specific recognition protein (Ssrp), La related protein (Larp), eukaryotic translation initiation factor 4G (eIF4G), Lamin, NAT1, Claspin, Gartenzwerg (Garz), Nedd4, Nopp140, Lk6, Yorkie (Yki), Stat92E, and Moleskin (Msk). Many of these common signaling mediators function in cell survival and cell proliferation. For example, the transcription factor Yki coordinates cell proliferation and apoptosis by directing the expression of cell cycle and cell death regulators [64]. Stat92E loss-of-function has been reported to inhibit hemocyte proliferation [65,66], while the importin Msk localizes MAP kinase to the nucleus to promote cell proliferation and survival [67]. We found enrichment for the regulation of phosphorylation of components of specific complexes by both InR and Pvr, including the Chs5p/Arf1-binding protein complex, the chromatin remodeling FACT complex, the translation initiation factor 2 complex, the cohesion-Sa complex, TRAPP complex and splicing associated factor complex (Fig. 7D, E). While we do not expect that all components of an individual complex require an alteration in phosphorylation in order for complex activity to change, more confidence for implication of that complex downstream of Pvr or InR is gleaned from multiple components exhibiting altered phosphorylation. As such, we expect that these complexes play key roles downstream of both InR and Pvr.
To distinguish Pvr - or InR-specific targets that may mediate receptor-specific functions we compared phosphoproteomes under ‘high Pvr, low InR’ and ‘low Pvr, high InR’ conditions (S8 Table). Among the Pvr-specific phosphorylations, we identified phosphoproteins involved in cell migration, cytoskeleton, and regulation of cell shape such as CIN85 and CD2AP ortholog (Cindr), Tenascin major (Ten-m), Vacuolar protein sorting 4 (Vps4), Rab7, Rho GTPase activating protein at 15B (RhoGAP15B), and Sprouty (Sty). Cindr is a recognized component of the CIN85 complex, one of three complexes for which multiple components exhibited a dependence on Pvr for specific phosphorylation (Fig. 7F). With respect to InR-specific phosphorylations, we detected phosphoproteins associated with the Gene Ontology Consortium terms growth regulation (i.e. Gp150, Foxo, L, Chico), glycogen metabolism (i.e. Glycogen Synthase), and the innate immune response (i.e. G protein-coupled receptor kinase interacting ArfGAP and Mustard). These differential phosphorylations likely provide receptor specificity and function to modulate the activity of specific complexes such as those over-represented in terms of the number of components modulated by InR activity (Fig. 7G).
Discussion
Here, we present a genome - and proteome-wide survey in Drosophila to identify signaling networks and cellular regulators that control cell survival and cell number. Starting from a genome-wide RNAi screen for modifiers of cell number under Pvr sensitized conditions, we established a new proapoptotic role for the EcR complex, and an anti-apoptotic function for InR, in the balance of blood cell number in the Drosophila embryo. Phosphoproteomic analyses of Pvr deficient cells under low and high InR signaling states enabled the identification of common Pvr and InR phosphorylation targets regulating cell survival, and receptor-specific phosphorylation targets that mediate unique functions of Pvr and InR (model, Fig. 8). Our study further highlights the ability of signaling receptors to modulate their targets depending on cellular context, in our case, specifically based on the activity of other RTKs. These observations are important in light of mechanisms of acquired RTK inhibitor resistance that were recently described [68].

A sensitized screen to identify novel regulators of cell number
Previously, we demonstrated that Pvr mediates cell survival in the Drosophila embryonic hematopoietic system and in Drosophila Kc cells in culture [12]. Similar roles for Pvr in other cell populations such as glia were subsequently reported [69]. Here, we find that Pvr also contributes to the proliferation of Kc cells, which is revealed when Pvr-dependent cell death is suppressed. These Pvr functions are well conserved with mammalian systems, where PDGF/VEGF Receptors mediate cell survival and proliferation during normal development [70,71] and in pathologies such as leukemias and other forms of cancer [26,72,73].
Our findings encompassing the role of Pvr in the activation of the Mek/Erk and Akt/Tor pathways are consistent with previous reports of Pvr-dependent phosphorylation of Erk [21,23], the activation of the TOR1 Complex and Erk by Pvr [14], and the physical interaction of Pvr with PVRAP, Grb2, Shc, and the regulatory subunit of PI3K in cell culture [14,74]. Since our screen was designed to eliminate general regulators of cell number and instead focus on those genes that show differential effects under sensitized conditions, it predominantly revealed genes with tumor suppressor-like activities (Pvr Suppressors), many of which were not detected in conventional RNAi screens for cell proliferation or survival previously [17,75–78]. Several of the identified Pvr Enhancers (5/14) and half of the Pvr Suppressors scored as hits in other genome-wide RNAi screens examining RTK signaling, specifically InR and EGFR signaling using the same screening platform and dsRNA libraries [79]; see S3 Table for specific overlap).
Many genes identified in the screen regulate redundant pro-survival pathways downstream of Pvr (Fig. 8), as was predicted by our initial screening hypothesis (Fig. 1F), and which is also supported by others [14]. However, some regulators identified in the screen instead act in pathways parallel to Pvr signaling, as we demonstrated for InR and EcR signaling. Among the RNAi screen hits, we distinguished three major classes of modifiers. First, we identified a large group of ‘Upstream Genes’ that specifically affect cell number only in signaling competent, but not Pvr depleted cells. Among these, we found a large number of ribosomal protein genes. Interestingly, a recent Drosophila in vivo study identified ribosomal protein RpS8 as functional upstream regulator of Pvr in hemocytes of the lymph gland, proposing it may exert its function by interaction with Bip1 (bric à brac interacting protein 1), which shows similar phenotypes [16]. While our screen did not identify Bip1, it revealed RpS8 as putative Pvr ‘Upstream Gene’. Ribosomal subunits may promote Pvr expression also as part of the general translation machinery, or may play more specialized roles in translation regulation, according to previous reports on target-specific ribosomal activities that may influence the cellular signaling makeup in development and tumorigenesis [80–82]
Second, inherent to our system, our screen yielded relatively few Pvr Enhancers. From this group we chose InR for verification analysis by in vivo genetics, which we further complemented with a phosphoproteomic survey that illuminated synergy between Pvr and InR. Analogous synergistic relationships between InR with other RTKs have been reported in Drosophila development [69,83,84], and vertebrate signaling [85]. The specificity of redundant RTK signaling pathways is of major interest is the fields of cell signaling and cancer research and subject of ongoing intense study [68,86].
Third, the screen yielded a group of Pvr Suppressors, which function as tumor suppressor-like genes whose loss rescues cell survival under sensitized conditions. This group contains all negative regulators of the Akt/Tor pathway, many of which are known tumor suppressors in mammalian systems [39–41], and several negative regulators of the Mek/Erk pathway such as mts and wdb, encoding for components of the PP2a complex [42,43], and Mkp3, which encodes for a phosphatase known to negatively regulate Erk [44]. As expected, several genes identified in the Pvr modifier screen also scored in previous screens for signaling mediators of the Pvr, Akt/Tor and RTK/Erk pathways [14,83,87]. The screen also revealed novel, or only recently characterized, genes. CG6182 is an ortholog of the mammalian TBC7, that interacts physically with Tsc1 [88]; GckIII is a counterpart of mammalian Serine Threonine Kinase 25 (STK25), also known as SOK1, that localizes to the Golgi [89] and induces cell death upon overexpression in mammalian cell culture [90]. Some of the identified genes have been characterized in Drosophila, yet no role in cell number control in the embryo has been described. For example, we identified multiple members of the Brahma SWI2/SNF2 family ATPase chromatin-remodeling complex [46,47], with osa and dalao scoring as Pvr Suppressors, and Brahma associated protein 60kD (Bap60) and moira (mor) scoring in mixed categories. Two of the strongest hits among the Pvr Suppressors were genes encoding the nuclear hormone receptors EcR and Usp [27,45], which we followed up with subsequent analyses.
Role of the EcR complex in embryonic cell death
EcR and Usp have previously been studied for their roles in proliferation, differentiation and cell death during larval molting and metamorphosis [50,61,91]. In Kc cells, the EcR/Usp ligand ecdysone has been known to arrest the cell cycle and trigger a cell differentiation program [52–54]. However, neither in the embryo nor in Kc cells has ecdysone signaling been previously associated with cell death [51,92]. Here, we describe a role for ecdysone signaling in embryonic cell death, a function revealed only under sensitized conditions or when directly stimulating EcR pathway activity. When treating Kc cells with 20E, we find that the EcR targets E93 and rpr are transcriptionally upregulated, consistent with previous reports describing these genes as transcriptional targets of EcR [93]. E93 and Rpr drive apoptosis [57,94–96] and are required for ecdysone-induced death of the larval midgut and salivary glands during metamorphosis and in the larval cell line l(2)mbn [56,57,97–100]. As for the mechanism of cell death rescue by EcR silencing, we were unable to detected measurable levels of Halloween gene expression, which is required for biosynthetic maturation of 20E [101]. We therefore propose that Kc cultures produce low levels of 20E through low-level expression of Halloween genes, or the EcR complex may have residual pro-death functions even in the absence of ligand. Previous publications have suggested that the unligated EcR complex has an active role and can bind to ecdysone response elements [102,103].
Vertebrate counterparts of EcR and Usp are the liver X receptors (LXRs), and retinoid X receptor (RXR), respectively [104,105]. RXR plays central roles in cell proliferation, apoptosis, and differentiation [106–108] during development and in pathologies such as cancer and metabolic disease [109,110]. Lack of activation of the RXR/Retinoic acid receptor (RAR) pathways causes Acute Promyelocytic Leukemia (APL) and other malignancies due to impairments in cell differentiation and increased cell survival [109,111,112] and treatment with synthetic retinoids or rexinoids has proven promising in reverting malignant phenotypes [109,111]. Interestingly, dependence on additional anti-apoptotic pathways has been reported in RxR-dependent APL. In particular, Akt/Tor signaling contributes to the increased cell survival in APL, and, consequently, dual therapy with PI3K inhibitors and retinoids has shown great therapeutic promise [113].
Signaling by InR and the EcR complex
Drosophila InR and Akt/TOR signaling were recently reported in several studies for their multifaceted roles in the regulation of lymph gland hemocytes, an independent blood cell lineage in Drosophila [114–116]. Drosophila InR further promotes the trophic survival of germline stem cells in the Drosophila ovary [117], which relies on its downstream mediator Tor [118]. Besides this, Drosophila InR is known mostly for its role in cell growth and regulation of body and organ size [119]. In contrast, the related mammalian Insulin-like Growth Factors 1 and 2 (IGF1 and IGF2) play important roles in the trophic survival of various cell types [120]. Our study now demonstrates that Drosophila InR also exerts trophic function in embryonic blood cells, which is revealed once redundant receptor activity such as Pvr activity is suppressed.
Several mammalian orthologs of InR phosphorylation targets uncovered from our phosphoproteomic analyses have been previously reported to be regulated by insulin, such as Ssrp, Larp, eIF4G, Lamin, NAT1, Claspin, Garz, Nedd4, Nopp140, Yki and Lk6 [121,122]. These phosphoproteins are also regulated by Pvr and contribute to the roster of common RTK targets that likely account for Pvr/InR-induced cell survival. An additional example of a commonly targeted phosphosite is the activating phosphorylation of Stat92E [123], a proposed target of the insulin receptor [124]. These examples highlight the success of our phosphoproteomic approach to uncover bona fide targets shared by InR and PVR. The approach also unveiled novel downstream effectors. For example, the requirement of Pvr for Ssrp phosphorylation hints to a relationship between Pvr and the chromatin remodeling FACT complex, a heterodimer comprised of Ssrp and Dre4 [125]. This hypothesis is reinforced by 1) the suppression of Pvr deficient cell proliferation by dre4 knockdown; and 2) a reported two-hybrid interaction between Pvr and Spt6 [126], a component of an elongation complex that includes FACT [127]. The possibility that FACT functions downstream of these RTKs to regulate transcriptional initiation and elongation as a cell survival mechanism will be an interesting area of future investigation.
Our phosphoproteomics experiments additionally uncovered InR-specific phosphorylations: e.g. phosphosites on Foxo, Unkempt (Unk), Chico, Tsc1, Spaghetti (Spag), L, Ajuba (Jub), and Git. Notably, many of these proteins were identified by affinity purification and mass spectrometry as components of an InR/Tor protein interaction network [128], supporting our proposition that indeed these phosphoproteins serve InR-specific functions. Remarkably, four of the thirty high confidence Pvr Suppressors and two of the fourteen high confidence Pvr Enhancers exhibited altered phosphorylation specifically under InR activation indicating these localized phosphorylation events may be critical for the rescue of Pvr deficient cells provided by InR stimulation.
Our analysis identified very few EcR-dependent phosphoproteins, however, we cannot rule out that these few may indeed regulate cell number. For example, phosphorylation of the methionine sulfoxide reductase Ecdysone-induced protein 28/29kD (Eip71CD) was upregulated by EcR knockdown. Eip71CD confers protection to oxidative stress, increases cell size and number, and promotes longevity [129,130]. Additionally, phosphorylation of the diacylglycerol O-acyltransferase Midway (Mdy) was upregulated upon EcR knockdown. mdy mutant egg chambers exhibit premature nurse cell death and degeneration during mid-oogenesis [131] comparable to EcR and Eip75B germline clones [132]. We observed an upregulation of phosphorylated Transforming acidic coiled-coil (Tacc) in Pvr deficient cells subjected to either EcR knockdown. Vertebrate TACC proteins interact with RxRβ to regulate specific gene expression [133]. Phosphorylation could potentially influence Tacc interaction with Usp, the Drosophila ortholog of RxR, and consequently impact Usp-dependent gene expression, thereby permitting cell survival. We cannot exclude the possibility that, due to incomplete coverage, our phosphoproteomics analyses may have failed to capture critical phosphorylation changes induced by EcR knockdown that account for Pvr deficient cell survival. Our analyses did, however, generate a list of candidates for future study and highlight the substantially different responses by insulin and EcR knockdown to rescue Pvr loss.
Signaling networks in mammalian development and disease
The dual dependence of Drosophila Kc cells and embryonic hemocytes on the Akt/TOR and Mek/Erk pathways (this study and [12,14] echoes the dependence of many mammalian cells, in particular tumor cells, on these two signaling pathways. In addition to concomitant activation by upstream receptors, Akt/Tor and Mek/Erk signaling further show a substantial degree of crosstalk between each other [134]. Dual inhibition of these two pathways has therefore become a promising approach in targeted cancer therapies [135]. However, many molecularly targeted approaches remain challenging due to the plasticity of signaling, the involvement of additional undefined redundant signaling pathways, and the variation of signaling networks downstream of even closely related receptors [68,135]. Findings from the Drosophila model provide insight into the pools of common and unique signaling targets of the related RTKs Pvr and InR. Further, this study suggests that the qualitative signaling specificity of receptors can be switched in response to the signaling status of the cell. This notion may be of wide-reaching consequences for many cellular processes, and requires careful consideration when aiming for the experimental or therapeutic manipulation of signaling systems.
Materials and Methods
Fly stocks and crosses
Fly lines used were: Pvr1/CyO [12], srpHemoGAL4 [12], Pxn-GAL4 [136], UAS-PvrΔC [12], UAS-p35 [30], UAS-srcEGFP (E. Spana), UAS-lacZnls (E. Spana), UAS-mCD8::GFP [137], UAS-Stinger [138], UAS-EcRA, UAS-EcRB1, UAS-EcRB2 [57], UAS-EcRB1 W650A (dominant-negative) [59] and UAS-EcR A W650A (dominant-negative) [60], UAS-InR-dn [139].
For in vivo quantification of hemocytes in Drosophila embryos, the srp-Hemo-GAL4 driver was used to express UAS-lacZnls and UAS-srcEGFP in hemocytes. Genotypes of Pvr1 mutant rescue/enhancement experiments were: Pvr1,UAS-srcEGFP/ Pvr1,srpHemoGAL4; UAS-p35/UAS-lacZnls and Pvr1,UAS-EcR (A or B1) W650A or UAS-InR-dn/Pvr1,srpHemoGAL4; UAS-mCD8::GFP/UAS-lacZnls. Genotype for the alternative rescue/enhancement of Pvr dominant-negative expressing hemocytes were: srpHemoGAL4, UAS-srcEGFP/+; UAS-PvrΔC, UAS-EcRA W650A or UAS-InR-dn/ UAS-lacZnls.
Embryo staining, stimulation, and microscopy
Embryos were collected on apple juice agar plates and fixed and stained as described previously [12]. Antibodies used were goat anti-GFP (1 : 1500) (Molecular Probes) and mouse anti-β-Gal (1 : 750) (Promega), and Alexa Fluor secondary antibodies (Invitrogen) Imaging was done on Leica DMI 4000B and Leica SP5 microscopes. Hemocyte counts were conducted under fluorescent microscopy at 40X, assessing 10 independent embryos per genotype and stage. Standard deviations and p values by Student’s t-test were calculated.
Live hemocyte counting
To examine hemocyte numbers at the embryo-larva transition, hemocytes were marked by Pxn-GAL4 driven expression of UAS-Stinger. The transgenic driver UAS-Stinger; Pxn-GAL4 was crossed to w1118 (control), or UAS-EcRA dn, respectively. Dechorionated embryos or larvae from 2 hour timed collections were mounted under glass slides and subjected to visual/manual counting under a fluorescence microscope. At least ten embryos or larvae per time point and genotype were assessed. Standard deviations and p values by Student’s t-test were calculated.
Cell maintenance and stimulation
Kc167 cells [140], here labeled Kc, were cultured in Schneider’s Drosophila Medium (Millipore, Gibco) supplemented with 10% Fetal Bovine Serum (FBS) and 1000 units/ml Penicillin and 1000mcg/ml Streptomycin. Insulin was supplemented to a final concentration of 5 μg/ml for InR stimulation experiments.
20E experiments: 20-Hydroxyecdysone (Sigma-Aldrich), 20E, was dissolved in ethanol to make a 5mg/ml stock. A subsequent stock of 1μg/ml stock was made by diluting in water. 1x105 cells were seeded into each well of a 24 well plate and 20E was added to achieve the indicated final concentrations.
p35 stable cell line
All cell experiments were based on Kc167 cells, in short Kc. Effectene Transfection Reagent (Qiagen) was utilized for transfection. Kc cells were co-transfected with driver Actin-GAL4, UAS-puromycin, UAS-GFP and UAS-p35 plasmid constructs. Three days after transfection, cells were selected with puromycin 10ug/ml. After 2 weeks, surviving cells were harvested and sorted by Fluorescence-activated cell sorting (FACS) to isolate the highest 20 percentile of GFP-expressing cells. To further select cells that are resistant to apoptosis, thread RNAi knockdown was used to eliminate cells with weak resistance to caspase-dependent apoptosis. The surviving cells were expanded for experimental use. The presence of p35 transgene in the p35 stable cell pool was confirmed by PCR verification.
Cell-based RNAi
RNAi knockdown was performed as described previously [141]. Briefly, Kc167 cells were re-suspended and diluted in serum-free medium before seeding. dsRNAs targeting each specific gene were added and incubated for 45 minutes before supplementing with complete medium with FBS to adjust to a final concentration of 10% FBS.
Genome-wide RNAi screening
We screened a set of 62 384-well plates that were pre-arrayed with dsRNAs, corresponding to 22,914 distinct amplicons based on Flybase release 5.51 of the Drosophila genome corresponding to 13,777 unique genes [31], and 7463 Sanger predictions [142] (DRSC). To determine differential effects between Pvr silenced and control cells, we screened each plate under two conditions, dsRNA-mediated knockdown of Pvr, or knockdown of a control (GFP). All experiments were performed in duplicate. Each well contained 0.25ug of pre-arrayed dsRNA. Before seeding, Kc cell suspensions were pre-mixed with Pvr or control (GFP) dsRNAs in batch, corresponding to a final concentration of 0.3ug per well. Cells were seeded at a density of 7,000 cells/well and incubated for 4 days. CellTiter-Glo assay (Promega) was performed according to the instructions of the manufacturer, and luminescence was read using Analyst GT or SpectraMax plate readers (Molecular Devices). Liquid handling was performed using WellMate (Matrix), MicroFill (BioTek), or MultiDrop (Thermo), high-throughput dispensers. Z scores [z = (χ-μ)/σ] were calculated as follows: μ = Mean of readings from controls wells (i.e. wells without pre-arrayed candidate dsRNAs), σ = Standard deviation from readings of the control wells. χ = Reading of candidate gene well. Z score for Pvr knockdown condition (Z[Pvr]) and for control knockdown condition (Z[GFP]) were generated and the differential effects in Pvr knockdown condition and control knockdown were calculated by the difference of each Z scores (i.e. Zdiff = Z[Pvr] - Z[GFP]). Cluster analysis of primary screen data was performed of amplicons scoring ZDiff> = 2.0 and ZDiff< = -2.0, using Z[Pvr], Z[GFP], and Zdiff values for each amplicon. Analysis included hierarchical clustering by centered correlation, and complete linkage, and results were displayed using TreeView [143]. For verification screening, genes were selected at cutoffs of ZDiff> = 2.2 and ZDiff< = -2.2, and one or two amplicons per gene, non-overlapping with the primary screen amplicons, and devoid of 19bp off-target overlaps, were utilized (DRSC). Secondary screening involved differential screening of Pvr RNAi and GFP RNAi cells as outlined above. Assays were performed in replicate and repeated in two independent duplicates. Final, ‘high confidence’ Pvr modifiers were determined by calculation of final average ZDiff scores determined from the averaged ZDiff scores of all amplicons targeting specific genes that were evaluated in both primary and secondary screening. Regarding the error rate of the screen, we generated false positive and false negative rates as follows: To evaluate false positives we i) assembled a list of 355 protein-coding genes that are not expressed across Drosophila tissue/stage/cell lines based on both modEncode RNA-Seq data as well as FlyAtlas data; ii) compared this list with genes scoring in the primary screen (there is only 1 gene overlapping and the relevant amplicon has >5 predicted off targets); and iii) estimated a false positive rate: 1/355 = <1%. To evaluate false negatives we i) assembled a list of 38 high confidence genes based on secondary screening hits, which are the genes that scored with at least 2 independent amplicons and each amplicon was consistently scored among replicates; ii) identified all amplicons relevant to these 38 genes from the genome library and found 80 of them. 41 scored in the primary screen while 39 failed to score; and iii) used these numbers to calculate a false negative rate: 39/80 = 49%.
Cell counting, EdU and TUNEL assays
To obtain cell counts, 1x105 cells were seeded into each well of a 24-well plate followed by treatment with dsRNAs or 20E. 3.3ug of dsRNA targeting each specific gene knockdown was added. After culturing for the indicated period of time, cells were re-suspended and diluted 1 : 1 with 0.4% Trypan Blue. Numbers of viable/dead cells were assessed by hemocytometer counting based on Trypan Blue exclusion/staining.
For EdU and TUNEL assays, 20,000 Kc cells were seeded into each well of 96-well black clear bottom plate and immediately treated with dsRNAs or 20E. 0.825ug of dsRNA was used to target each specific gene knockdown.
To assess cell proliferation, cells were incubated for 4 hours with 10uM of Click-iT EdU (Invitrogen) one or several days after the dsRNAs or 20E treatment. The Click-iT EdU cell proliferation assay was conducted according to manufacturer's instructions.
For assessing cell death, cells were processed by TUNEL assay according to manufacturer's instructions (Invitrogen).
Stained cells were counted visually/manually and by ImageJ automated cell quantification. In brief, for ImageJ analysis, still images were converted to 8 bit images and cells were selected by setting a threshold against the background. Highlighted cells were then counted by the ‘Analyze Particles’ function. At least three still images for each sample were taken at random sites using a 40X objective. Percentages of EdU or TUNEL positive cells were calculated as follows: (# of EdU or TUNEL positive cells/ total # of cells) * 100. Cell culture figures show compilations of three independent biological replicate experiments. Error bars indicate standard deviation. Student’s t-test as indicated. * for p < 0.05; ** for p < 0.01; *** for p < 0.001; NS for not significant.
dsRNAs design and generation
In most cases, dsRNA amplicon sequences were selected by the Drosophila RNAi Screening Center (DRSC), as indicated by DRSC amplicon numbers. Primers used for generating the amplicon template contained a 5' T7 RNA polymerase-binding site (TAATACGACTCACTATAGG) following by the amplicon specific sequences. dsRNAs were generated by in vitro transcription using Megascript T7 transcription kit (Ambion). dsRNAs were purified with a RNeasy Mini Kit (Qiagen) and product size confirmed by agarose gel electrophoresis. dsRNA concentrations were measured with a Nanodrop 2000C spectrophotometer (Thermo Scientific).
Real time PCR
Total RNA was extracted using a RNeasy mini kit (Qiagen), according to manufacturer's instructions. Total purified RNA were measured with a Nanodrop 2000C spectrophotometer (Thermo Scientific). 1ug—0.1ug of purified RNA was reversed transcribed into cDNA using an iScript cDNA synthesis kit (Biorad). Real time PCR reactions were carried out using iQ SYBR Green Supermix (Bio-Rad) on a Bio-Rad CFX96 Real Time System and gene expression levels were analyzed with CFX Manager Software (Bio-Rad). Primers for real time PCR assays were designed using web-based software ProbeFinder (Roche Applied Science Universal ProbeLibrary Assay Design Center) or by the author. Primer sequences for real-time PCR assessment will be made available upon request.
Protein lysates and immunoblotting
Kc cells were lysed using Triton lysis buffer (50mM Tris-HCl (pH 7.5), 150mM NaCl, 1% Trition X-100, 30mM NaF) freshly supplemented with 1mM Na3VO4 and protease inhibitors (Complete, Roche) and immunoblot analysis was performed as described previously [12]. Primary antibodies were obtained from Cell Signaling Technology except monoclonal anti-β-tubulin (Sigma T5168), anti-Pvr [12], anti-EcR (Developmental Studies Hybridoma Bank, DSHB) and anti-histone H3 (Abcam 39950); signal was detected by HRP conjugated secondary antibodies (Amersham NA934V/NXA931 and Jackson ImmunoResearch 706–035–148) and ECL.
Sample preparation for mass spectrometric analysis
Kc cells were serum starved for 1 hr, incubated with dsRNA for 30 minutes and then diluted either in Schneider’s Drosophila Medium (Gibco) supplemented with Fetal Bovine Serum (FBS) (final concentration of 10%), Penicillin (50 units/ml final concentration), and Streptomycin (50 ug/ml final concentration), with or without insulin (5ug/ml final concentration). After two days cells were lysed in: 8M urea, 75mM NaCl, 50mM Tris-HCl pH 8.2, 1mM NaF, 1mM β-glycerophosphate, 1mM sodium orthovanadate, 10mM sodium pyrophosphate, 1mM PMSF, EDTA-free Protease Inhibitor Cocktail Tablet (Roche). One milligram of protein from each sample was reduced with 5mM dithiothreitol at 56°C for 25 minutes. Cysteines were alkylated with 14mM iodoacetamide for 30 minutes at room temperature in the dark. Unreacted iodoacetamide was quenched by incubation with additional dithiothreitol to 5mM for 15 minutes at room temperature in the dark. Lysates were diluted 1 : 5 with 25mM Tris-HCl, pH 8.2 and CaCl2 added to 1mM. Digestion with 5ug sequencing grade trypsin (Promega) was overnight at 37°C with agitation. Peptides were acidified with 10% trifluoroacetic acid and desalted using 1cc Sep-Pak tC18 solid-phase extraction cartridges (Waters). Eluted peptides were lyophilized, resuspended in 200mM Na-HEPES pH8.2, and labeled with TMT reagent (Thermo Scientific) in anhydrous acetonitrile (2mg TMT reagent per sample) for 1 hour at room temperature. TMT labeling was as follows:
Experiment 1. control dsRNA, biological replicate #1 : 126 (high Pvr, low InR); control dsRNA, biological replicate #2 : 127 (high Pvr, low InR); control dsRNA + insulin, biological replicate #1 : 128 (high Pvr, high InR); control dsRNA + insulin, biological replicate #2 : 129 (high Pvr, high InR); control dsRNA + EcR dsRNA, biological replicate #1 : 130 (high Pvr, low EcR); control dsRNA + EcR dsRNA, biological replicate #2 : 131 (high Pvr, low EcR).
Experiment 2. Pvr dsRNA + control dsRNA, biological replicate #1 : 126 (low Pvr, low InR); Pvr dsRNA + control dsRNA, biological replicate #2 : 127 (low Pvr, low InR); Pvr dsRNA + control dsRNA + insulin, biological replicate #1 : 128 (low Pvr, high InR); Pvr dsRNA + control dsRNA + insulin, biological replicate #2 : 129 (low Pvr, high InR); Pvr dsRNA + EcR dsRNA, biological replicate #1 : 130 (low Pvr, low EcR); Pvr dsRNA + EcR dsRNA, biological replicate #2 : 131 (low Pvr, low EcR).
Experiment 3. Pvr dsRNA + control dsRNA: 126 (low Pvr, low InR); Pvr dsRNA + control dsRNA + insulin: 127 (low Pvr, high InR); Pvr dsRNA + EcR dsRNA: 128 (low Pvr, low EcR); control dsRNA: 129 (high Pvr, low InR); control dsRNA + insulin: 130 (high Pvr, high InR); control dsRNA + EcR dsRNA: 131 (high Pvr, low EcR).
Reactions were quenched by the addition of hydroxylamine to 0.3% and incubation at room temperature for 15 min. Labeled peptides were combined, lyophilized, and stored at -80°C until further processing. Samples were acidified with 10% trifluoroacetic acid and desalted using a 3cc Sep-Pak tC18 solid-phase extraction cartridge (Waters). Phosphopeptides were enriched by strong cation exchange chromatography (SCX; [144]). Lyophilized peptides were resuspended in 400 ul SCX buffer A (7 mM KH2PO4, pH 2.65, 30% acetonitrile) and injected onto a SCX column (Polysulfoethyl aspartamide, 9.4 mm×250mm, 5 uM particle size, 200 Ǻ pore size, PolyLC). A gradient was developed over 35 min from 0% to 30% buffer B (7 mM KH2PO4, pH 2.65, 30% acetonitrile, 350 mM KCl) at a flow rate of 2.5 ml/min. 12 fractions were collected and lyophilized. Peptides were then desalted with 1cc Waters Sep-Pak tC18 solid-phase extraction cartridges and subjected to TiO2 based phosphopeptide enrichment [145] using 0.5mg titanium dioxide microspheres per mg protein. Eluates were further desalted using STAGE tips [146] and lyophilized. Samples were reconstituted in 4ul 5% formic acid / 5% acetonitrile.
Mass spectrometric analysis
In most signaling systems, the major gatekeeper of signal transduction is protein phosphorylation, which can be adjusted rapidly according to the needs of a cell. A caveat of solely measuring phosphorylation is that a change in phosphopeptide levels for any particular peptide can result from a change in phosphorylation of the peptide, or from a change in levels of that protein. We expect that roughly a quarter of altered phosphorylation we observe would be explained by the latter mechanism, based on previous reports (Bodenmiller et al. Science Signaling 2010 and Wu et al. Mol. Cell Proteomics, 2011; Sopko et al. Dev Cell 2014). Given that coverage of the Drosophila proteome is not yet comprehensive in a single mass spec run, normalization of phosphorylation to protein amounts can only be estimated. Further, lowly expressed proteins are often missed. For these reasons, we chose to focus exclusively on phosphorylation in our study. Samples were subjected to LC-MS/MS with an Orbitrap Velos Pro mass spectrometer (Thermo Scientific) using higher-energy collision dissociation (HCD; [147]) and a top ten method [148]. MS/MS spectra were searched against a composite database of Drosophila melanogaster proteins derived from Flybase version 5.23 in both the forward and reverse orientation using the Sequest algorithm [149]. Search parameters included: a precursor mass tolerance of 20 ppm; up to two missed cleavages; static modification of TMT tags on lysine residues and peptide N termini (+229.162932 Da) and +57.021464 Da accounting for carbamidomethylation on Cys; dynamic modification of phosphorylation (+79.966330 Da) on Ser, Thr and Tyr and oxidation (+15.994915 Da) on Met. A target-decoy database search strategy [150] enabled thresholding of the false discovery rate (FDR) for MS/MS spectral assignment at 1%. Correct spectral matches were distinguished from incorrect matches using linear discriminant analysis based on parameters including Xcorr, ΔCn, precursor mass error, peptide length, and charge state [151]. The localizations of individual phosphorylations were assigned using the probability-based AScore algorithm [152] and only phosphosites with AScores greater than 13 (p < 0.05) were considered in our analysis. Moreover, only phosphopeptides with isolation specificity greater than 0.75 were considered for further analysis. Further filtering of the dataset resulted in a final protein FDR of ~2% and a peptide FDR near 0.15%. TMT labeling was >98% efficient. For TMT reporter ion quantification, a 0.03 Da window centered on the expected mass of each reporter ion was monitored and the intensity of the signal closest to the expected mass was recorded. Reporter ion signals were further adjusted to correct for impurities associated with each TMT label, as described elsewhere [153]. Raw TMT reporter ion intensities for individual phosphopeptides were normalized to the summed reporter ion intensity for each TMT label. Adjusted reporter ion intensities were averaged between replicates. Peptides for which only one replicate TMT labeled sample generated detectable reporter ions were excluded from further analysis.
Database information
Complete information on Pvr modifier screen data, and DRSC library dsRNA amplicons can be accessed at http://www.flyrnai.org/.
Supporting Information
Zdroje
1. Opferman JT (2007) Life and death during hematopoietic differentiation. Curr Opin Immunol 19 : 497–502. 17662585
2. Wright KM, Deshmukh M (2006) Restricting apoptosis for postmitotic cell survival and its relevance to cancer. Cell Cycle 5 : 1616–1620. 16880745
3. Parihar A, Eubank TD, Doseff AI Monocytes and macrophages regulate immunity through dynamic networks of survival and cell death. J Innate Immun 2 : 204–215. doi: 10.1159/000296507 20375558
4. Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144 : 646–674. doi: 10.1016/j.cell.2011.02.013 21376230
5. Green DR, Evan GI (2002) A matter of life and death. Cancer Cell 1 : 19–30. 12086884
6. Hoffmann JA, Reichhart JM (2002) Drosophila innate immunity: an evolutionary perspective. Nat Immunol 3 : 121–126. 11812988
7. Martinez-Agosto JA, Mikkola HK, Hartenstein V, Banerjee U (2007) The hematopoietic stem cell and its niche: a comparative view. Genes Dev 21 : 3044–3060. 18056420
8. Hartenstein V (2006) Blood cells and blood cell development in the animal kingdom. Annu Rev Cell Dev Biol 22 : 677–712. 16824014
9. Lemaitre B, Hoffmann J (2007) The host defense of Drosophila melanogaster. Annu Rev Immunol 25 : 697–743. 17201680
10. Evans CJ, Hartenstein V, Banerjee U (2003) Thicker than blood: conserved mechanisms in Drosophila and vertebrate hematopoiesis. Dev Cell 5 : 673–690. 14602069
11. Gold KS, Brückner K (2014) Drosophila as a model for the two myeloid blood cell systems in vertebrates. Experimental hematology 42 : 717–727. doi: 10.1016/j.exphem.2014.06.002 24946019
12. Brückner K, Kockel L, Duchek P, Luque CM, Rørth P, et al. (2004) The PDGF/VEGF Receptor controls blood cell survival in Drosophila. Dev Cell 7.
13. Munier AI, Doucet D, Perrodou E, Zachary D, Meister M, et al. (2002) PVF2, a PDGF/VEGF-like growth factor, induces hemocyte proliferation in Drosophila larvae. EMBO Rep 3 : 1195–1200. 12446570
14. Tran TA, Kinch L, Pena-Llopis S, Kockel L, Grishin N, et al. (2013) Platelet-derived growth factor/vascular endothelial growth factor receptor inactivation by sunitinib results in Tsc1/Tsc2-dependent inhibition of TORC1. Molecular and cellular biology 33 : 3762–3779. doi: 10.1128/MCB.01570-12 23878397
15. Mondal BC, Mukherjee T, Mandal L, Evans CJ, Sinenko SA, et al. (2011) Interaction between differentiating cell - and niche-derived signals in hematopoietic progenitor maintenance. Cell 147 : 1589–1600. doi: 10.1016/j.cell.2011.11.041 22196733
16. Mondal BC, Shim J, Evans CJ, Banerjee U (2014) Pvr expression regulators in equilibrium signal control and maintenance of Drosophila blood progenitors. eLife 3: e03626. doi: 10.7554/eLife.03626 25201876
17. Bjorklund M, Taipale M, Varjosalo M, Saharinen J, Lahdenpera J, et al. (2006) Identification of pathways regulating cell size and cell-cycle progression by RNAi. Nature 439 : 1009–1013. 16496002
18. Sims D, Duchek P, Baum B (2009) PDGF/VEGF signaling controls cell size in Drosophila. Genome Biol 10: R20. doi: 10.1186/gb-2009-10-2-r20 19216764
19. Rosin D, Schejter E, Volk T, Shilo B (2004) Apical accumulation of the Drosophila PDGF/VEGF receptor ligands provides a mechanism for triggering localized actin polymerization. Development: in press.
20. Wu Y, Brock AR, Wang Y, Fujitani K, Ueda R, et al. (2009) A blood-borne PDGF/VEGF-like ligand initiates wound-induced epidermal cell migration in Drosophila larvae. Curr Biol 19 : 1473–1477. doi: 10.1016/j.cub.2009.07.019 19646875
21. Duchek P, Somogyi K, Jekely G, Beccari S, Rørth P (2001) Guidance of cell migration by the Drosophila PDGF/VEGF receptor. Cell 107 : 17–26. 11595182
22. McDonald JA, Pinheiro EM, Montell DJ (2003) PVF1, a PDGF/VEGF homolog, is sufficient to guide border cells and interacts genetically with Taiman. Development 130 : 3469–3478. 12810594
23. Cho NK, Keyes L, Johnson E, Heller J, Ryner L, et al. (2002) Developmental control of blood cell migration by the Drosophila VEGF pathway. Cell 108 : 865–876. 11955438
24. Ashman LK (1999) The biology of stem cell factor and its receptor C-kit. Int J Biochem Cell Biol 31 : 1037–1051. 10582338
25. Ferrara N, Gerber HP, LeCouter J (2003) The biology of VEGF and its receptors. Nat Med 9 : 669–676. 12778165
26. Kelly LM, Gilliland DG (2002) Genetics of myeloid leukemias. Annu Rev Genomics Hum Genet 3 : 179–198. 12194988
27. Koelle MR, Talbot WS, Segraves WA, Bender MT, Cherbas P, et al. (1991) The Drosophila EcR gene encodes an ecdysone receptor, a new member of the steroid receptor superfamily. Cell 67 : 59–77. 1913820
28. Thomas HE, Stunnenberg HG, Stewart AF (1993) Heterodimerization of the Drosophila ecdysone receptor with retinoid X receptor and ultraspiracle. Nature 362 : 471–475. 8385270
29. Cherbas L, Willingham A, Zhang D, Yang L, Zou Y, et al. (2011) The transcriptional diversity of 25 Drosophila cell lines. Genome research 21 : 301–314. doi: 10.1101/gr.112961.110 21177962
30. Hay BA, Wolff T, Rubin GM (1994) Expression of baculovirus P35 prevents cell death in Drosophila. Development 120 : 2121–2129. 7925015
31. Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, et al. (2000) The genome sequence of Drosophila melanogaster. Science 287 : 2185–2195. 10731132
32. Perrimon N, Mathey-Prevot B (2007) Matter arising: off-targets and genome-scale RNAi screens in Drosophila. Fly (Austin) 1 : 1–5.
33. Kulkarni MM, Booker M, Silver SJ, Friedman A, Hong P, et al. (2006) Evidence of off-target effects associated with long dsRNAs in Drosophila melanogaster cell-based assays. Nat Methods 3 : 833–838. 16964256
34. Goberdhan DC, Wilson C (2003) The functions of insulin signaling: size isn't everything, even in Drosophila. Differentiation; research in biological diversity 71 : 375–397. 12969331
35. King-Jones K, Korge G, Lehmann M (1999) The helix-loop-helix proteins dAP-4 and daughterless bind both in vitro and in vivo to SEBP3 sites required for transcriptional activation of the Drosophila gene Sgs-4. J Mol Biol 291 : 71–82. 10438607
36. Rodriguez-Magadan H, Merino E, Schnabel D, Ramirez L, Lomeli H (2008) Spatial and temporal expression of Zimp7 and Zimp10 PIAS-like proteins in the developing mouse embryo. Gene Expr Patterns 8 : 206–213. 18053775
37. Gutierrez L, Zurita M, Kennison JA, Vazquez M (2003) The Drosophila trithorax group gene tonalli (tna) interacts genetically with the Brahma remodeling complex and encodes an SP-RING finger protein. Development 130 : 343–354. 12466201
38. Wang YH, Huang ML (2009) Reduction of Lobe leads to TORC1 hypoactivation that induces ectopic Jak/STAT signaling to impair Drosophila eye development. Mechanisms of development 126 : 781–790. doi: 10.1016/j.mod.2009.08.005 19733656
39. Zoncu R, Efeyan A, Sabatini DM (2011) mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 12 : 21–35. doi: 10.1038/nrm3025 21157483
40. Sarbassov DD, Ali SM, Sabatini DM (2005) Growing roles for the mTOR pathway. Curr Opin Cell Biol 17 : 596–603. 16226444
41. Manning BD, Cantley LC (2007) AKT/PKB signaling: navigating downstream. Cell 129 : 1261–1274. 17604717
42. Janssens V, Goris J (2001) Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J 353 : 417–439. 11171037
43. Van Hoof C, Goris J (2003) Phosphatases in apoptosis: to be or not to be, PP2A is in the heart of the question. Biochim Biophys Acta 1640 : 97–104. 12729918
44. Kim M, Cha GH, Kim S, Lee JH, Park J, et al. (2004) MKP-3 has essential roles as a negative regulator of the Ras/mitogen-activated protein kinase pathway during Drosophila development. Mol Cell Biol 24 : 573–583. 14701731
45. Yao TP, Segraves WA, Oro AE, McKeown M, Evans RM (1992) Drosophila ultraspiracle modulates ecdysone receptor function via heterodimer formation. Cell 71 : 63–72. 1327536
46. Papoulas O, Beek SJ, Moseley SL, McCallum CM, Sarte M, et al. (1998) The Drosophila trithorax group proteins BRM, ASH1 and ASH2 are subunits of distinct protein complexes. Development 125 : 3955–3966. 9735357
47. Tamkun JW (1995) The role of brahma and related proteins in transcription and development. Curr Opin Genet Dev 5 : 473–477. 7580139
48. Colombani J, Bianchini L, Layalle S, Pondeville E, Dauphin-Villemant C, et al. (2005) Antagonistic actions of ecdysone and insulins determine final size in Drosophila. Science 310 : 667–670. 16179433
49. Oro AE, McKeown M, Evans RM (1990) Relationship between the product of the Drosophila ultraspiracle locus and the vertebrate retinoid X receptor. Nature 347 : 298–301. 2169594
50. Kozlova T, Thummel CS (2000) Steroid regulation of postembryonic development and reproduction in Drosophila. Trends Endocrinol Metab 11 : 276–280. 10920384
51. Chavez VM, Marques G, Delbecque JP, Kobayashi K, Hollingsworth M, et al. (2000) The Drosophila disembodied gene controls late embryonic morphogenesis and codes for a cytochrome P450 enzyme that regulates embryonic ecdysone levels. Development 127 : 4115–4126. 10976044
52. Cherbas L, Koehler MM, Cherbas P (1989) Effects of juvenile hormone on the ecdysone response of Drosophila Kc cells. Dev Genet 10 : 177–188. 2500282
53. Andres AJ, Fletcher JC, Karim FD, Thummel CS (1993) Molecular analysis of the initiation of insect metamorphosis: a comparative study of Drosophila ecdysteroid-regulated transcription. Dev Biol 160 : 388–404. 8253272
54. Stevens B, Alvarez CM, Bohman R, O'Connor JD (1980) An ecdysteroid-induced alteration in the cell cycle of cultured Drosophila cells. Cell 22 : 675–682. 6780197
55. Rosset R (1978) Effects of ecdysone on a Drosophila cell line. Exp Cell Res 111 : 31–36. 620695
56. Jiang C, Baehrecke EH, Thummel CS (1997) Steroid regulated programmed cell death during Drosophila metamorphosis. Development 124 : 4673–4683. 9409683
57. Lee CY, Wendel DP, Reid P, Lam G, Thummel CS, et al. (2000) E93 directs steroid-triggered programmed cell death in Drosophila. Mol Cell 6 : 433–443. 10983989
58. Lee CY, Simon CR, Woodard CT, Baehrecke EH (2002) Genetic mechanism for the stage - and tissue-specific regulation of steroid triggered programmed cell death in Drosophila. Developmental biology 252 : 138–148. 12453466
59. Cherbas L, Hu X, Zhimulev I, Belyaeva E, Cherbas P (2003) EcR isoforms in Drosophila: testing tissue-specific requirements by targeted blockade and rescue. Development 130 : 271–284. 12466195
60. Brown HL, Cherbas L, Cherbas P, Truman JW (2006) Use of time-lapse imaging and dominant negative receptors to dissect the steroid receptor control of neuronal remodeling in Drosophila. Development 133 : 275–285. 16354717
61. Thummel CS (1995) From embryogenesis to metamorphosis: the regulation and function of Drosophila nuclear receptor superfamily members. Cell 83 : 871–877. 8521511
62. Dayon L, Hainard A, Licker V, Turck N, Kuhn K, et al. (2008) Relative quantification of proteins in human cerebrospinal fluids by MS/MS using 6-plex isobaric tags. Analytical chemistry 80 : 2921–2931. doi: 10.1021/ac702422x 18312001
63. Thompson A, Schafer J, Kuhn K, Kienle S, Schwarz J, et al. (2003) Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Analytical chemistry 75 : 1895–1904. 12713048
64. Huang J, Wu S, Barrera J, Matthews K, Pan D (2005) The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell 122 : 421–434. 16096061
65. Remillieux-Leschelle N, Santamaria P, Randsholt NB (2002) Regulation of larval hematopoiesis in Drosophila melanogaster: a role for the multi sex combs gene. Genetics 162 : 1259–1274. 12454071
66. Luo H, Rose PE, Roberts TM, Dearolf CR (2002) The Hopscotch Jak kinase requires the Raf pathway to promote blood cell activation and differentiation in Drosophila. Mol Genet Genomics 267 : 57–63. 11919715
67. Vrailas AD, Marenda DR, Cook SE, Powers MA, Lorenzen JA, et al. (2006) smoothened and thickveins regulate Moleskin/Importin 7-mediated MAP kinase signaling in the developing Drosophila eye. Development 133 : 1485–1494. 16540506
68. Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, et al. (2012) Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 487 : 505–509. doi: 10.1038/nature11249 22763448
69. Learte AR, Forero MG, Hidalgo A (2008) Gliatrophic and gliatropic roles of PVF/PVR signaling during axon guidance. Glia 56 : 164–176. 18000865
70. Hoch RV, Soriano P (2003) Roles of PDGF in animal development. Development 130 : 4769–4784. 12952899
71. Breen EC (2007) VEGF in biological control. J Cell Biochem 102 : 1358–1367. 17979153
72. Scheijen B, Griffin JD (2002) Tyrosine kinase oncogenes in normal hematopoiesis and hematological disease. Oncogene 21 : 3314–3333. 12032772
73. Gerber HP, Ferrara N (2003) The role of VEGF in normal and neoplastic hematopoiesis. J Mol Med 81 : 20–31. 12545246
74. Ishimaru S, Ueda R, Hinohara Y, Ohtani M, Hanafusa H (2004) PVR plays a critical role via JNK activation in thorax closure during Drosophila metamorphosis. Embo J 23 : 3984–3994. 15457211
75. Boutros M, Kiger AA, Armknecht S, Kerr K, Hild M, et al. (2004) Genome-wide RNAi analysis of growth and viability in Drosophila cells. Science 303 : 832–835. 14764878
76. Bettencourt-Dias M, Giet R, Sinka R, Mazumdar A, Lock WG, et al. (2004) Genome-wide survey of protein kinases required for cell cycle progression. Nature 432 : 980–987. 15616552
77. Chew SK, Chen P, Link N, Galindo KA, Pogue K, et al. (2009) Genome-wide silencing in Drosophila captures conserved apoptotic effectors. Nature 460 : 123–127. doi: 10.1038/nature08087 19483676
78. Yi CH, Sogah DK, Boyce M, Degterev A, Christofferson DE, et al. (2007) A genome-wide RNAi screen reveals multiple regulators of caspase activation. J Cell Biol 179 : 619–626. 17998402
79. Friedman AA, Tucker G, Singh R, Yan D, Vinayagam A, et al. (2011) Proteomic and functional genomic landscape of receptor tyrosine kinase and ras to extracellular signal-regulated kinase signaling. Sci Signal 4: rs10. doi: 10.1126/scisignal.2002029 22028469
80. Silvera D, Formenti SC, Schneider RJ (2010) Translational control in cancer. Nat Rev Cancer 10 : 254–266. doi: 10.1038/nrc2824 20332778
81. Stumpf CR, Ruggero D (2011) The cancerous translation apparatus. Curr Opin Genet Dev.
82. Xue S, Barna M (2012) Specialized ribosomes: a new frontier in gene regulation and organismal biology. Nature reviews Molecular cell biology 13 : 355–369. doi: 10.1038/nrm3359 22617470
83. Friedman A, Perrimon N (2006) A functional RNAi screen for regulators of receptor tyrosine kinase and ERK signalling. Nature 444 : 230–234. 17086199
84. Marin-Hincapie M, Garofalo RS (1999) The carboxyl terminal extension of the Drosophila insulin receptor homologue binds IRS-1 and influences cell survival. The Journal of biological chemistry 274 : 24987–24994. 10455177
85. van der Geer P, Hunter T, Lindberg RA (1994) Receptor protein-tyrosine kinases and their signal transduction pathways. Annu Rev Cell Biol 10 : 251–337. 7888178
86. McCubrey JA, Steelman LS, Chappell WH, Sun L, Davis NM, et al. (2012) Advances in targeting signal transduction pathways. Oncotarget 3 : 1505–1521. 23455493
87. Kockel L, Kerr KS, Melnick M, Bruckner K, Hebrok M, et al. (2010) Dynamic Switch of Negative Feedback Regulation in Drosophila Akt-TOR Signaling. PLoS Genet 6: e1000990. doi: 10.1371/journal.pgen.1000990 20585550
88. Nakashima A, Yoshino K, Miyamoto T, Eguchi S, Oshiro N, et al. (2007) Identification of TBC7 having TBC domain as a novel binding protein to TSC1-TSC2 complex. Biochem Biophys Res Commun 361 : 218–223. 17658474
89. Matsuki T, Matthews RT, Cooper JA, van der Brug MP, Cookson MR, et al. (2011) Reelin and stk25 have opposing roles in neuronal polarization and dendritic Golgi deployment. Cell 143 : 826–836.
90. Nogueira E, Fidalgo M, Molnar A, Kyriakis J, Force T, et al. (2008) SOK1 translocates from the Golgi to the nucleus upon chemical anoxia and induces apoptotic cell death. J Biol Chem 283 : 16248–16258. doi: 10.1074/jbc.M709724200 18364353
91. Sorrentino RP, Carton Y, Govind S (2002) Cellular immune response to parasite infection in the Drosophila lymph gland is developmentally regulated. Dev Biol 243 : 65–80. 11846478
92. Kozlova T, Thummel CS (2003) Essential roles for ecdysone signaling during Drosophila mid-embryonic development. Science 301 : 1911–1914. 12958367
93. Gauhar Z, Sun LV, Hua S, Mason CE, Fuchs F, et al. (2009) Genomic mapping of binding regions for the Ecdysone receptor protein complex. Genome Res 19 : 1006–1013. doi: 10.1101/gr.081349.108 19237466
94. Baehrecke EH, Thummel CS (1995) The Drosophila E93 gene from the 93F early puff displays stage - and tissue-specific regulation by 20-hydroxyecdysone. Dev Biol 171 : 85–97. 7556910
95. Goyal L, McCall K, Agapite J, Hartwieg E, Steller H (2000) Induction of apoptosis by Drosophila reaper, hid and grim through inhibition of IAP function. Embo J 19 : 589–597. 10675328
96. Meier P, Silke J, Leevers SJ, Evan GI (2000) The Drosophila caspase DRONC is regulated by DIAP1. EMBO J 19 : 598–611. 10675329
97. Lee CY, Cooksey BA, Baehrecke EH (2002) Steroid regulation of midgut cell death during Drosophila development. Dev Biol 250 : 101–111. 12297099
98. Choi YJ, Lee G, Park JH (2006) Programmed cell death mechanisms of identifiable peptidergic neurons in Drosophila melanogaster. Development 133 : 2223–2232. 16672345
99. Kilpatrick ZE, Cakouros D, Kumar S (2005) Ecdysone-mediated up-regulation of the effector caspase DRICE is required for hormone-dependent apoptosis in Drosophila cells. J Biol Chem 280 : 11981–11986. 15657059
100. Chittaranjan S, McConechy M, Hou YC, Freeman JD, Devorkin L, et al. (2009) Steroid hormone control of cell death and cell survival: molecular insights using RNAi. PLoS Genet 5: e1000379. doi: 10.1371/journal.pgen.1000379 19214204
101. Rewitz KF, Rybczynski R, Warren JT, Gilbert LI (2006) The Halloween genes code for cytochrome P450 enzymes mediating synthesis of the insect moulting hormone. Biochem Soc Trans 34 : 1256–1260. 17073797
102. Cherbas L, Lee K, Cherbas P (1991) Identification of ecdysone response elements by analysis of the Drosophila Eip28/29 gene. Genes Dev 5 : 120–131. 1899227
103. Schubiger M, Truman JW (2000) The RXR ortholog USP suppresses early metamorphic processes in Drosophila in the absence of ecdysteroids. Development 127 : 1151–1159. 10683169
104. Owen GI, Zelent A (2000) Origins and evolutionary diversification of the nuclear receptor superfamily. Cell Mol Life Sci 57 : 809–827. 10892345
105. King-Jones K, Thummel CS (2005) Nuclear receptors—a perspective from Drosophila. Nat Rev Genet 6 : 311–323. 15803199
106. Ross SA, McCaffery PJ, Drager UC, De Luca LM (2000) Retinoids in embryonal development. Physiol Rev 80 : 1021–1054. 10893430
107. Altucci L, Gronemeyer H (2001) Nuclear receptors in cell life and death. Trends Endocrinol Metab 12 : 460–468. 11701345
108. Bastien J, Rochette-Egly C (2004) Nuclear retinoid receptors and the transcription of retinoid-target genes. Gene 328 : 1–16. 15019979
109. Altucci L, Leibowitz MD, Ogilvie KM, de Lera AR, Gronemeyer H (2007) RAR and RXR modulation in cancer and metabolic disease. Nat Rev Drug Discov 6 : 793–810. 17906642
110. Means AL, Gudas LJ (1995) The roles of retinoids in vertebrate development. Annu Rev Biochem 64 : 201–233. 7574480
111. Bushue N, Wan YJ (2010) Retinoid pathway and cancer therapeutics. Adv Drug Deliv Rev 62 : 1285–1298. doi: 10.1016/j.addr.2010.07.003 20654663
112. Lin RJ, Sternsdorf T, Tini M, Evans RM (2001) Transcriptional regulation in acute promyelocytic leukemia. Oncogene 20 : 7204–7215. 11704848
113. Billottet C, Banerjee L, Vanhaesebroeck B, Khwaja A (2009) Inhibition of class I phosphoinositide 3-kinase activity impairs proliferation and triggers apoptosis in acute promyelocytic leukemia without affecting atra-induced differentiation. Cancer research 69 : 1027–1036. doi: 10.1158/0008-5472.CAN-08-2608 19176369
114. Shim J, Gururaja-Rao S, Banerjee U (2013) Nutritional regulation of stem and progenitor cells in Drosophila. Development 140 : 4647–4656. doi: 10.1242/dev.079087 24255094
115. Dragojlovic-Munther M, Martinez-Agosto JA (2012) Multifaceted roles of PTEN and TSC orchestrate growth and differentiation of Drosophila blood progenitors. Development 139 : 3752–3763. 22951642
116. Benmimoun B, Polesello C, Waltzer L, Haenlin M (2012) Dual role for Insulin/TOR signaling in the control of hematopoietic progenitor maintenance in Drosophila. Development 139 : 1713–1717. doi: 10.1242/dev.080259 22510984
117. Hsu HJ, Drummond-Barbosa D (2009) Insulin levels control female germline stem cell maintenance via the niche in Drosophila. Proceedings of the National Academy of Sciences of the United States of America 106 : 1117–1121. doi: 10.1073/pnas.0809144106 19136634
118. Pritchett TL, McCall K (2012) Role of the insulin/Tor signaling network in starvation-induced programmed cell death in Drosophila oogenesis. Cell Death Differ 19 : 1069–1079. doi: 10.1038/cdd.2011.200 22240900
119. Brogiolo W, Stocker H, Ikeya T, Rintelen F, Fernandez R, et al. (2001) An evolutionarily conserved function of the Drosophila insulin receptor and insulin-like peptides in growth control. Current biology: CB 11 : 213–221. 11250149
120. Kurmasheva RT, Houghton PJ (2006) IGF-I mediated survival pathways in normal and malignant cells. Biochimica et biophysica acta 1766 : 1–22. 16844299
121. Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, et al. (2011) The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 332 : 1317–1322. doi: 10.1126/science.1199498 21659604
122. Yu Y, Yoon SO, Poulogiannis G, Yang Q, Ma XM, et al. (2011) Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science 332 : 1322–1326. doi: 10.1126/science.1199484 21659605
123. Zhang Y, Liu Y, Li X, Gao W, Zhang W, et al. (2013) Effects of insulin and IGF-I on growth hormone - induced STAT5 activation in 3T3-F442A adipocytes. Lipids in health and disease 12 : 56. doi: 10.1186/1476-511X-12-56 23631823
124. Coffer PJ, van Puijenbroek A, Burgering BM, Klop-de Jonge M, Koenderman L, et al. (1997) Insulin activates Stat3 independently of p21ras-ERK and PI-3K signal transduction. Oncogene 15 : 2529–2539. 9399641
125. Orphanides G, Lagrange T, Reinberg D (1996) The general transcription factors of RNA polymerase II. Genes & development 10 : 2657–2683.
126. Bianco A, Poukkula M, Cliffe A, Mathieu J, Luque CM, et al. (2007) Two distinct modes of guidance signalling during collective migration of border cells. Nature 448 : 362–365. 17637670
127. Lindstrom DL, Squazzo SL, Muster N, Burckin TA, Wachter KC, et al. (2003) Dual roles for Spt5 in pre-mRNA processing and transcription elongation revealed by identification of Spt5-associated proteins. Molecular and cellular biology 23 : 1368–1378. 12556496
128. Glatter T, Schittenhelm RB, Rinner O, Roguska K, Wepf A, et al. (2011) Modularity and hormone sensitivity of the Drosophila melanogaster insulin receptor/target of rapamycin interaction proteome. Molecular systems biology 7 : 547. doi: 10.1038/msb.2011.79 22068330
129. Chung H, Kim AK, Jung SA, Kim SW, Yu K, et al. (2010) The Drosophila homolog of methionine sulfoxide reductase A extends lifespan and increases nuclear localization of FOXO. FEBS letters 584 : 3609–3614. doi: 10.1016/j.febslet.2010.07.033 20655917
130. Roesijadi G, Rezvankhah S, Binninger DM, Weissbach H (2007) Ecdysone induction of MsrA protects against oxidative stress in Drosophila. Biochem Biophys Res Commun 354 : 511–516. 17239346
131. Buszczak M, Lu X, Segraves WA, Chang TY, Cooley L (2002) Mutations in the midway gene disrupt a Drosophila acyl coenzyme A: diacylglycerol acyltransferase. Genetics 160 : 1511–1518. 11973306
132. Buszczak M, Freeman MR, Carlson JR, Bender M, Cooley L, et al. (1999) Ecdysone response genes govern egg chamber development during mid-oogenesis in Drosophila. Development 126 : 4581–4589. 10498692
133. Vettaikkorumakankauv AK, Lauffart B, Gangisetty O, Cincotta MA, Hawthorne LA, et al. (2008) The TACC proteins are coregulators of the Retinoid X Receptor beta. Cancer Therapy 6. 19129927
134. Mendoza MC, Er EE, Blenis J (2011) The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends in biochemical sciences 36 : 320–328. doi: 10.1016/j.tibs.2011.03.006 21531565
135. Britten CD (2013) PI3K and MEK inhibitor combinations: examining the evidence in selected tumor types. Cancer chemotherapy and pharmacology 71 : 1395–1409. doi: 10.1007/s00280-013-2121-1 23443307
136. Stramer B, Wood W, Galko MJ, Redd MJ, Jacinto A, et al. (2005) Live imaging of wound inflammation in Drosophila embryos reveals key roles for small GTPases during in vivo cell migration. J Cell Biol 168 : 567–573. 15699212
137. Lee T, Luo L (1999) Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron 22 : 451–461. 10197526
138. Barolo S, Castro B, Posakony JW (2004) New Drosophila transgenic reporters: insulated P-element vectors expressing fast-maturing RFP. Biotechniques 36 : 436–440, 442. 15038159
139. Parks AL, Cook KR, Belvin M, Dompe NA, Fawcett R, et al. (2004) Systematic generation of high-resolution deletion coverage of the Drosophila melanogaster genome. Nat Genet 36 : 288–292. 14981519
140. Echalier G, Ohanessian A (1969) [Isolation, in tissue culture, of Drosophila melangaster cell lines]. C R Acad Sci Hebd Seances Acad Sci D 268 : 1771–1773. 4976834
141. Clemens JC, Worby CA, Simonson-Leff N, Muda M, Maehama T, et al. (2000) Use of double-stranded RNA interference in Drosophila cell lines to dissect signal transduction pathways. Proc Natl Acad Sci U S A 97 : 6499–6503. 10823906
142. Hild M, Beckmann B, Haas SA, Koch B, Solovyev V, et al. (2003) An integrated gene annotation and transcriptional profiling approach towards the full gene content of the Drosophila genome. Genome Biol 5: R3. 14709175
143. Eisen MB, Spellman PT, Brown PO, Botstein D (1998) Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A 95 : 14863–14868. 9843981
144. Villen J, Gygi SP (2008) The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry. Nature protocols 3 : 1630–1638. doi: 10.1038/nprot.2008.150 18833199
145. Kettenbach AN, Gerber SA (2011) Rapid and reproducible single-stage phosphopeptide enrichment of complex peptide mixtures: application to general and phosphotyrosine-specific phosphoproteomics experiments. Anal Chem 83 : 7635–7644. doi: 10.1021/ac201894j 21899308
146. Rappsilber J, Ishihama Y, Mann M (2003) Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Analytical chemistry 75 : 663–670. 12585499
147. Olsen JV, Macek B, Lange O, Makarov A, Horning S, et al. (2007) Higher-energy C-trap dissociation for peptide modification analysis. Nature methods 4 : 709–712. 17721543
148. Dephoure N, Zhou C, Villen J, Beausoleil SA, Bakalarski CE, et al. (2008) A quantitative atlas of mitotic phosphorylation. Proceedings of the National Academy of Sciences of the United States of America 105 : 10762–10767. doi: 10.1073/pnas.0805139105 18669648
149. Eng JKM, A.L., Yates J.R. 3rd (1994) An Approach to Correlate Tandem Mass Spectral Data of Peptides with Amino Acid Sequences in a Protein Database. J Am Soc Mass Spectrom 5 : 976–989. doi: 10.1016/1044-0305(94)80016-2 24226387
150. Elias JE, Gygi SP (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nature methods 4 : 207–214. 17327847
151. Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, et al. (2010) A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143 : 1174–1189. doi: 10.1016/j.cell.2010.12.001 21183079
152. Beausoleil SA, Villen J, Gerber SA, Rush J, Gygi SP (2006) A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nature biotechnology 24 : 1285–1292. 16964243
153. McAlister GC, Huttlin EL, Haas W, Ting L, Jedrychowski MP, et al. (2012) Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Analytical chemistry 84 : 7469–7478. doi: 10.1021/ac301572t 22880955
154. Vinayagam A, Hu Y, Kulkarni M, Roesel C, Sopko R, et al. (2013) Protein complex-based analysis framework for high-throughput data sets. Sci Signal 6: rs5. doi: 10.1126/scisignal.2003629 23443684
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 3
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Clonality and Evolutionary History of Rhabdomyosarcoma
- Morphological Mutations: Lessons from the Cockscomb
- Maternal Filaggrin Mutations Increase the Risk of Atopic Dermatitis in Children: An Effect Independent of Mutation Inheritance
- Transcriptomic Profiling of Reveals Reprogramming of the Crp Regulon by Temperature and Uncovers Crp as a Master Regulator of Small RNAs
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy