
Opposite Phenotypes of Muscle Strength and Locomotor Function in Mouse Models of Partial Trisomy and Monosomy 21 for the Proximal Region
Down syndrome is the most common genetic cause of intellectual disabilities, and marked hypotonia is among the constant diagnostic traits. Here we observe the opposite changes in locomotion, muscle strength, and energetic balance in new mouse models of DS and M21 for the Hspa13-App proximal region of human chromosome 21. The differential expression analysis revealed downregulation of skeletal muscle genes controlling energetic metabolism, mitochondrial activity, and biogenesis in Ts3Yah, while upregulation of similar set of genes was found in Ms3Yah mice. This phenomenon correlates with the changes in mitochondrial proliferation with increased membrane permeability of Ts3Yah mitochondria and decreased mitochondrial ROS production in Ms3Yah mice. Our results demonstrate the opposite phenotypic effect of trisomy and monosomy of the Hspa13-App syntenic region of human chromosome 21, highlighting new physiological mechanisms for hypotonia in DS individuals.
Published in the journal:
. PLoS Genet 11(3): e32767. doi:10.1371/journal.pgen.1005062
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005062
Summary
Down syndrome is the most common genetic cause of intellectual disabilities, and marked hypotonia is among the constant diagnostic traits. Here we observe the opposite changes in locomotion, muscle strength, and energetic balance in new mouse models of DS and M21 for the Hspa13-App proximal region of human chromosome 21. The differential expression analysis revealed downregulation of skeletal muscle genes controlling energetic metabolism, mitochondrial activity, and biogenesis in Ts3Yah, while upregulation of similar set of genes was found in Ms3Yah mice. This phenomenon correlates with the changes in mitochondrial proliferation with increased membrane permeability of Ts3Yah mitochondria and decreased mitochondrial ROS production in Ms3Yah mice. Our results demonstrate the opposite phenotypic effect of trisomy and monosomy of the Hspa13-App syntenic region of human chromosome 21, highlighting new physiological mechanisms for hypotonia in DS individuals.
Introduction
DS and M21 are complex genetic conditions that arise from an altered dosage of genes on human chromosome 21 (Hsa21). Full M21 is rare and generally not compatible with life, with only a few cases reported in literature, and the oldest case survived for only a few days after birth [1–5]. The remaining M21 cases are partial deletion or mosaics. The phenotypes of M21 are variable among individuals and depend on the specific deleted region of Hsa21. Some patients have only mild to moderate intellectual disabilities [6, 7], while others are more severely affected and present multiple dysmorphic craniofacial, skeletal, and cardiac features, as well as muscular, ocular, pulmonary, renal, and genitourinary abnormalities [8–12].
DS, with an incidence of one in 700 live births [13], is still the most common viable cause of chromosomal aneuploidy in humans and the most frequent genetic cause of intellectual disability. Besides the characteristic of facial dysmorphology, hypotonia is one of the major traits observable at birth in this disorder. More than 80 features occur with various degrees of expression and frequency in DS, and some have been associated with regions of Hsa21 [12, 14]. However, the causes of delayed motor performance affecting both gross and fine motor skills, weak muscle strength, and exercise performance are not known. Motor impairments are prominent throughout life and may contribute to the delay in cognitive skills [15, 16]. Poor locomotor skills have been attributed to impaired coordinated input due to cerebellar dysfunction [17, 18] but might have a more complex origin combining deficit in balance and postural control, stiffness, reduced muscular strength, and hypotonia.
Hypotonia is a state of low muscular tonicity due to decreased springlike properties of the striated muscle fibres, which suffer from a lack of energy to keep working at a normal level for a long time and a slower speed of response. Hypotonia may also be accompanied by reduced muscular strength. It can be caused by abnormal muscle function, abnormal neurological input, or metabolic diseases. Impaired exercise endurance is observed in DS individuals and could arise from endocrinal or metabolic perturbations [19, 20]. However, little is known about the origin of hypotonia in DS.
Mouse models provide a powerful tool to study the physiological, cellular, and molecular aspects of human diseases. Moreover, the possibility to manipulate large genomic regions in the mouse genome has offered a chance to develop mouse models of aneuploidies and investigate the relationship between phenotype and genotype. Hsa21 genes are found in three syntenic regions localised on mouse chromosomes 16 (Mmu16, 23.3 Mb, 115 Hsa21 orthologous genes), 17 (Mmu17, 1.1 Mb, 19 Hsa21 orthologous genes), and 10 (Mmu10, 2.3 Mb, 41 Hsa21 orthologous genes) (http://www.ensembl.org).
Several mouse models have been generated over the years that display many features of DS [21–25], but locomotor activity has not previously been fully investigated in these models. There is little in literature about the locomotor phenotype of the trisomic models for genes located on the Mmu16. Moreover, conflicting results have been reported on the motor abilities of Ts65Dn mice that are trisomic for 102 orthologous of Hsa21 genes (http://www.ensembl.org) located between Mprl39 and Zfp295 [26, 27]. Additionally, this model is trisomic for 50 genes and 10 pseudogenes present in the centromeric part of Mmu17 that is irrelevant to DS [28]. Studies from Costa and collaborators reported lower maximal treadmill running speeds, deficits in balance and motor coordination assessed by the rotarod test, and reduction in grip force [29, 30], whereas Hyde and collaborators found no impairment in motor learning in Ts65Dn mice [31, 32]. Several analyses have described abnormal electrical and biochemical properties in motor neurons and muscle cells both in human and DS mouse models [33–35]. The analysis of the distal part of the Hsa21 did not reveal locomotor deficit in a mouse model trisomic for the Abcg1-U2af1 segment on Mmu17 [25] and demonstrated that the Cstb-Prmt2 segment is also not necessary, as going back to two copies of this region in the Tc1 transchromosomic model does not rescue the locomotor deficit observed in this model (although five genes in this region were not tested here as they are not trisomic in Tc1) [36].
To assess the contribution of the DS phenotype associated with the 9.4 Mb Hspa13-App Hsa21 syntenic region on Mmu16 that is not trisomic in the widely used Ts65Dn model, we generated through chromosome engineering a trisomic mouse model (Ts3Yah) and the corresponding monosomic model (Ms3Yah) for this region. Those mice were evaluated for different behavioural phenotypes associated with DS and displayed opposite locomotor phenotypes. While Ts3Yah mice have impaired locomotion that is associated with increased grip strength and muscle mass, the opposite phenotype was observed in Ms3Yah animals. The transcription profile analysis of skeletal muscles revealed coordinate changes in muscle metabolic activity and mitochondrial biogenesis. This finding was confirmed by alteration in the oxidative capacity and mitochondrial content of the skeletal muscles. This study unravels new insight into the metabolic changes that result in defects of skeletal muscle strength and performance that are associated with Hsa21 aneuploidy and suggest new candidate genes involved in DS and M21.
Results
Generating models of aneuploidy for the Hspa13-App region on mouse chromosome 16
Chromosomal engineering based on the MICER [37] and long-range Cre/loxP-mediated recombination [38] were used in embryonic stem (ES) cells to produce the segmental duplication (Dp[16Hspa13-App]2Yah; MGI5519056) and the corresponding deletion (Del[16Hspa13-App]3Yah; MGI5519049) of the Hspa13-App 9.4 Mb interval located on Mmu16, as described in Materials and Methods and Fig. 1. This mouse syntenic region contains 15 Hsa21 orthologous genes (Fig. 1A, S1 Table). As a result of the insertion points of the targeting vectors, the Hspa13 gene at the proximal end of the 9.5 Mb region was in two functional copies in the duplication and lost in the deletion, whereas the App gene at the distal end was only in one functional copy in the duplication and lost in the deletion. The derived aneuploid mouse models corresponding to the tandem duplication and the reciprocal deletion were respectively named Dp(16)(Hspa13-App)2Yah for the trisomy (abbreviated as Ts3Yah) and Del(16)(Hspa13-App)3Yah for the monosomy (abbreviated as Ms3Yah). The two models were validated by the Southern blot analysis (Fig. 1B), fluorescent in situ hybridisation (FISH) analysis (Fig. 1E), and array-based comparative genomic hybridisation (Fig. 1F) as described in Materials and Methods. Ts3Yah and Ms3Yah mice are viable and fertile, with normal life spans, and exhibit no obvious gross anatomical anomalies on the C57BL/6J genetic background. Ts3Yah show no weight difference (weight at 10 weeks, n = 12 male mice per genotype: control 26.4±0.9g vs Ts3Yah 26.5±0.7g, Student’s t-test p = 0.97). While their weight is not different at birth from their littermates (weight taken at J2, n = 9 male mice per genotype: control 1.8g±0.1g vs Ms3Yah 1.9g±0.1 g, Student’s t-test p = 0.78), adult Ms3Yah mice weight approximately 10% less than their wild-type littermates (weight at 10 weeks, n = 10 male mice per genotype: control 28.6±0.6 g vs Ms3Yah 24.6g±1.1g, Student’s t-test p = 0.005).

Locomotor performances are oppositely affected in mice trisomic and monosomic for the Hspa13-App region
Motor function was assessed first using two rotarod motor performance tests: one with five-minute sessions of incremental fixed speeds and the second consisting of two five-minute trials with accelerating speed. Ts3Yah mice showed performances that were significantly lower than their diploid littermates in both tests at age 8 to 12 months, and Ms3Yah mice performed significantly lower than their diploid littermates at the same age as well (Fig. 2A). Looking at both Ts3Yah and Ms3Yah mice, the weight of the animal was not correlated with its performance, excluding that the increased performance in Ms3Yah animals was due to mice being leaner. The effects of the trisomy and monosomy on rotarod test performance were however not observed in the young mice aged 3 months (S1A Fig). In order to assess muscle strength, the maximal grip force was measured and normalised to the body weight of the animal. Muscle strength was increased in Ts3Yah and decreased in Ms3Yah mice compared to diploid littermates at 3 to 4 months and 8 to 12 months of age (Fig. 2B, at 8 to 12 months of age). Muscle endurance capacity was tested by having the mice run on a treadmill and measuring the maximal distance travelled. Ts3Yah mice were only able to run about half of the distance covered by diploid control mice, indicating that they were more susceptible to fatigue (Fig. 2C), whereas Ms3Yah mice showed performances comparable to diploid controls (S1B Fig). Spontaneous locomotor activity and habituation were measured in an open field by recording the distance travelled by the mouse during the first 15 minutes and the last 15 minutes of a 30 minute session. No difference was found in Ts3Yah mice (S1C Fig), while Ms3Yah mice had a slight overall increase in the distance travelled and did not reduce their exploratory activity as much as the controls animals during the last fifteen minutes of the session, suggesting a slight hyperactivity (Fig. 2D). The mice were tested for hind limb coordination by walking across a notched bar. No significant difference was observed for the percentage of errors in hind limb coordination in Ts3Yah mice (S1D Fig), while Ms3Yah mice made significantly less errors than their control littermates (Fig. 2E).

Copy number variation of the Hspa13-App region triggers opposite expression changes of pathways involved in mitochondrial function and biogenesis in the muscle
Muscle strength and locomotion variation suggest that the observed motor phenotypes at least partly result from a peripheral muscular phenotype. Hence, we decided to focus our attention on the skeletal muscles. To identify molecular changes in the skeletal muscles of Ts3Yah and Ms3Yah mice, we studied the transcriptional profiles of gastrocnemius muscles isolated from adult mice. From the 45,281 probe sets sampled on the microarray, 43,058 and 43,632 were expressed in the Ts3Yah and the Ms3Yah, respectively. There were 2,302 (corresponding to 1,614 genes) and 2,246 (corresponding to 1,631 genes) probes differentially expressed in Ts3Yah and Ms3Yah muscles, respectively, compared to their euploid littermate controls, with a significant p-value <0.05 and resampling p-values of 0.081 (Ts3Yah) and 0.073 (Ms3Yah) (see Materials and Methods for details of the methods used). About half of them (1,129 and 1,169, respectively) were upregulated and half of them (1,173 and 1,077, respectively) downregulated. Fold changes (FC) observed varied between 0.5 and 2.1 for both Ts3Yah and Ms3Yah, with only about 5% of the deregulated genes having a FC>1.2 or <0.8 (Fig. 3A), indicating that the trisomy and the monosomy of the Hspa13-App region do not have a dramatic effect on gene expression. The final lists of the most deregulated probes (FC>|1.2|) for Ts3Yah and Ms3Yah are presented along with their annotation, FC, and p-value in Supplementary Tables (S2–S3 Tables), respectively, and the clustering of those deregulated genes are presented in Fig. 3B. Three genes from the Hspa13-App region (Chodl, Atp5j, and D16Ertd472e [C21orf91 in human]) were among the most deregulated in Ts3Yah muscles. In addition, several protein kinases (Ttn, Nmrk2, Csnk2a1-rs3, Pctk1, Pacsin3, Prkaa2 Raf1, Cdc2l2, Smg1, Hk2, Ckmt2, Dapk2, Mpp3, and Atp1b1), three members of translation initiation factors (Eif2c2, Eif4e3, and Eif2b5), and a binding protein of a translation initiation factor (Eif4ebp2) were overexpressed, and a few mitochondrial-located proteins and enzymes were underexpressed (Atp5a1, Ckmt2, Mgst1, Stard7, Abhd10, Cyb5b, Pdpr, Hk2, Tmem65, Tomm22, Hsd3b2, and Tmem160). In Ms3Yah muscles, only 26 genes were downregulated at FC<0.8, and six of them (App, Atp5j, Chodl, D16Ertd472e, Usp25, and Jam2) corresponded to genes mapping within the Hspa13-App monosomic segment. Interestingly, among the 84 genes upregulated in Ms3Yah muscles, 29 were mitochondrial related (Hadhb, Alas1, Ndufa8, Brp17, Aifm1, Acadl, Gbas, Mfn2, Acaa2, As3mt, Mrpl47, Adh1, Tmem143, Akr1b10, Slc25a11, Pptc7, Cyc1, Uqcc, LOC433224, Slc25a20, Mpc2, Rmnd1, Coq5, Slc25a3, Slc25a4, Idh3g, Hmgcl, Akr1b10, Coq6, Ppif, and Ndufaf4). By comparing the lists of the most deregulated probes in Ts3Yah and Ms3Yah muscles, we found eight different genes displaying opposite deregulation in the two conditions: Chodl, Atp5j, and D16Ertd472e in the Hspa13-App region and Wfs1, Ffar3, mt-Nd4l, Gpihbp1, and Hist1h4h located elsewhere (see genes in bold in S2–S3 Tables).

We selected the genes located in the Hspa13-App interval and at its borders for the analysis of their expression profile by quantitative PCR analysis (QRT-PCR) (Fig. 3C). Only three genes within the Hspa13-App interval, namely, Ncam2, Samsn1, and Prss7 (also known as Tmprss15), were not detected by QRT-PCR analysis in the striated muscle as reported in the literature. Nevertheless, most of the genes present within this interval and were not detected by the microarray experiment were found expressed in skeletal muscles by QRT-PCR analysis. The genes present in three copies in Ts3Yah muscles were upregulated at around 1.5 fold, at the exception of Cxadr and Chodl. As expected, the expression of App that is not duplicated in the Ts3Yah chromosome was unchanged in Ts3Yah muscle. In Ms3Yah animals, all the monosomic genes were globally 0.5 fold underexpressed compared to their disomic controls. In addition, we measured the expression levels of five genes at the border of the rearrangement, namely, Adamts1, Adamts5, Usp16, Cct8, and Bach1 (Fig. 3C). No significant change of expression was observed for border genes present in two copies in the genome of Ts3Yah and Ms3Yah mice. Finally, genes found differentially expressed in Ts3Yah and Ms3Yah were tested by QRT-PCR. Most of the genes were validated and found to be significantly deregulated in Ms3Yah muscle as in the microarray analysis, although deregulation was only observed for Wnt4 in Ts3Yah animals (Fig. 3C).
We further analysed our microarray data with the software tool ‘Gene Set Enrichment Analysis’ (GSEA; http://www.broad.mit.edu/gsea) [39, 40]. This analysis helps find correlation of gene expression. The genes belonging to a defined pathway are ranked together according to their change in expression in Ts3Yah and Ms3Yah muscles compared to their diploid controls and calculating a maximum enrichment score (ES) for each gene set. More than 20 informative gene sets were significantly downregulated in Ts3Yah and upregulated in Ms3Yah muscles compared to diploid muscles (S4–S5 Tables), whereas no category was found for upregulated in Ts3Yah or downregulated in Ms3Yah muscles. Most of the deregulated gene sets were associated with mitochondrial energy metabolism (electron transport chain and oxidative phosphorylation or ‘VOXPHOS’), fatty acid metabolism (fatty acid degradation), and carbohydrate metabolism (TCA cycle, glycolysis and gluconeogenesis, and butanoate metabolism). About half of the genes assigned to mitochondrial gene sets overlapped with those categories. The complementary GSEA ([40]) revealed a major overlap in the leading edge subset of genes (genes in the gene set that contribute most to the ES) between Ts3Yah and Ms3Yah enriched gene sets (see S4–S5 Tables for leading edge genes in each gene set) (see among the top-ranked genes sets, one called ‘PGC,’ for peroxisome proliferator-activated receptor-gamma coactivator) and corresponding to a set of genes involved in oxidative phosphorylation and whose expression is coordinately decreased in human diabetic muscles and activated upon expression of Pgc-1α (Ppargc-1α) in C2C12 cells [41]. Leading edge genes from this set show a 25% to 50% overlap with leading edge genes from genes sets implicated in energy and metabolic pathways identified in our analysis. Fig. 4 shows GSEA-scoring plots and their corresponding heat maps for ‘PGC’ and the four gene sets, including ‘electron transport chain,’ ‘TCA cycle,’ ‘fatty acid degradation,’ and ‘glycolysis/gluconeogenesis,’ that show the most overlap with gene set ‘PGC.’ Together, these findings suggest a shift in energy metabolism to a more oxidative state with increased mitochondrial biogenesis and/or activity in Ms3Yah skeletal muscles and an opposite suppression of the oxidative state in Ts3Yah muscles that might be triggered by signaling pathway(s) involving transcriptional coactivators of the PGC family or other cooperating partners involved in this regulatory pathway.

The Hspa13-App region is implicated in the regulation of skeletal muscle oxidative capacity
Changes in locomotor capacity, muscle strength, and deregulation of the transcriptional control of energy metabolism led us to explore the muscle properties in Ts3Yah and Ms3Yah animals. We found an increased muscle mass in Ts3Yah animals and decreased muscle mass in Ms3Yah animals (Fig. 5A) that could account for the change in muscle strength observed in the animals. Gastrocnemius muscles are composed predominantly of type II glycolytic fibres and are poor in oxidative fast-twitch fibres that contain high amounts of mitochondria. We estimated the mitochondrial content of the gastrocnemius muscle by quantifying mtDNA and comparing the copy number of mtCOX2 normalised by nuclear DNA in transgenic animals versus their diploid control littermates. We observed a significant increase in mtDNA content in Ms3Yah muscles and close to significant decrease in Ts3Yah muscles (Student’s t-test Ts3Yah vs diploid, p = 0.06; Fig. 5B).

Mitochondrial activity was assessed by staining histological muscles sections for the presence of the mitochondrial enzyme succinate dehydrogenase (SDH). In tibialis anterioris (TA), a skeletal muscle rich in glycolytic fibres, the SDH-stained fibres versus nonstained fibres were counted. Ms3Yah muscles revealed significant increase in SDH-stained fibres compared to diploid and Ts3Yah muscles (Fig. 5C, top), but no change in the percentage of slow-twitch oxidative type I myofibers using MyHCI staining was observed (Fig. 5D, top). In the soleus, mainly composed of oxidative fibres, differences between highly oxidative and less oxidative fibres were determined by scoring darkly SDH-stained fibres from lightly stained fibres. A significant increase in the number of oxidative fibres was found in Ms3Yah compared to diploid and Ts3Yah muscles (Fig. 5C, bottom). An increased proportion of type I fibres was found in Ms3Yah compared to both diploid and Ts3Yah muscles. A decreased proportion of type I myofibers was also found in Ts3Yah soleus compared to diploid (Fig. 5D, bottom). Hence, we found that monosomy increased the oxidative state of the muscle while trisomy decreased it. We analysed the TA in electron microscopy. In skeletal muscle fibres, mitochondria are usually localised between myofibrils, aligned parallel to the myofibrils, and in pairs at the Z-disc of the sarcomere and packed strands underneath the sarcolemma. In Ts3Yah TA, some fibres (estimated <10%) were found having few small-sized mitochondria compared to normal fibres (Fig. 5E). In contrast, we could find areas with increased mitochondrial mass present between adjacent fibres and containing few degenerative mitochondria in Ms3Yah muscles (Fig. 5E). Overall, these results indicate increased muscle oxidative capacity in Ms3Yah muscles and decreased oxidative capacity in Ts3Yah muscles due to change in mitochondrial content.
Analysis of mitochondrial function in skeletal muscles
To determine if mitochondrial function is altered in the skeletal muscles of Ts3Yah and Ms3Yah mice, we assessed mitochondrial functional capacity in isolated equal amounts of mitochondrial preparations from hind limb muscles. We measured mitochondrial respiration using pyruvate/malate (PM), whose breakdown products generate NADH and lead to the reduction of mitochondrial complex I and the citric acid intermediate succinate (S), which donates electrons to FAD-containing complex II of the electron transport chain. While Ts3Yah mitochondrial respiration rates during state 3 respiration with ADP was comparable to the respiratory rates of diploid mitochondria (S2A Fig), they were significantly higher compared to controls in both states 2 (basal respiration in the presence of substrate alone) (ANOVA ‘genotype,’ ‘substrate’; F[1, 13] = 3.674, p = 0.078) (Fig. 6A) and 4o (respiration in presence of oligomycin, an inhibitor of ATPase) (ANOVA ‘genotype,’ ‘substrate’; F[1, 13] = 56.681, p = 0.026) (Fig. 6B). The fact that the respiratory flux is increased in both states 2 and 4o in Ts3Yah mitochondria suggests an increase in the membrane permeability. The respiratory control ratio, defined as the respiration in state 3 (maximal ADP-stimulated respiration) divided by that in state 4o, remained unchanged, showing intact mitochondria (S2B Fig). The activity of cytochrome oxidase (COX), the terminal enzyme in the mitochondrial electron transport chain, was investigated by isolating its respiratory activity using antimycin and was not changed in Ts3Yah mitochondria (S2C Fig). To further explore proton leak in Ts3Yah mitochondria, we used the batch of mitochondria subjected to respiration with succinate and measured the Δψm in parallel with respiration in the absence of net ATP synthesis (presence of oligomycin) to calculate the basal proton conductance of the membrane [42]. A voltage plot for the conductance over a range of potentials was generated by starting in state 4o and progressively limiting electron transport by titrating malonate and simultaneously determining respiration and Δψm. The curve of the relationship between oxygen consumption and membrane potential in Ts3Yah mitochondria was shifted to the left compared to diploid controls indicating that the rate of oxygen consumed to counteract proton leak was slightly increased (Fig. 6C). In addition, reporting the kinetic response of substrate oxidation activity to mitochondrial potential during states 2, 3, and 4o revealed increased respiratory activity in Ts3Yah compared to diploid controls as seen by the shift of the slope to the right (Fig. 6D). Finally, in non-phosphorylating conditions (state 4o), the mitochondrial potential in Ts3Yah mitochondria (186.7±1.9 mV) was the same as in diploid controls (185.0±2.2 mV), indicating that the higher rate of oxygen consumption observed in Ts3Yah was not due to an increase in oxidation of succinate. Together, those results suggest a slight change in the inner membrane proton conductance in Ts3Yah mitochondria. The mitochondrial respiration and COX activity in Ms3Yah mitochondria were similar to that of diploid controls (S2D–S2G Fig). However, the mitochondrial protein yield calculated from the reported amount of mitochondria extracted to the weight of muscle used for the extraction was significantly increased (37%; Student’s t-test, p = 0.024; Fig. 6E), suggesting that the increased oxidative capacity observed in Ms3Yah muscle is due solely to increased mitochondrial proliferation and not to increased mitochondrial respiration.
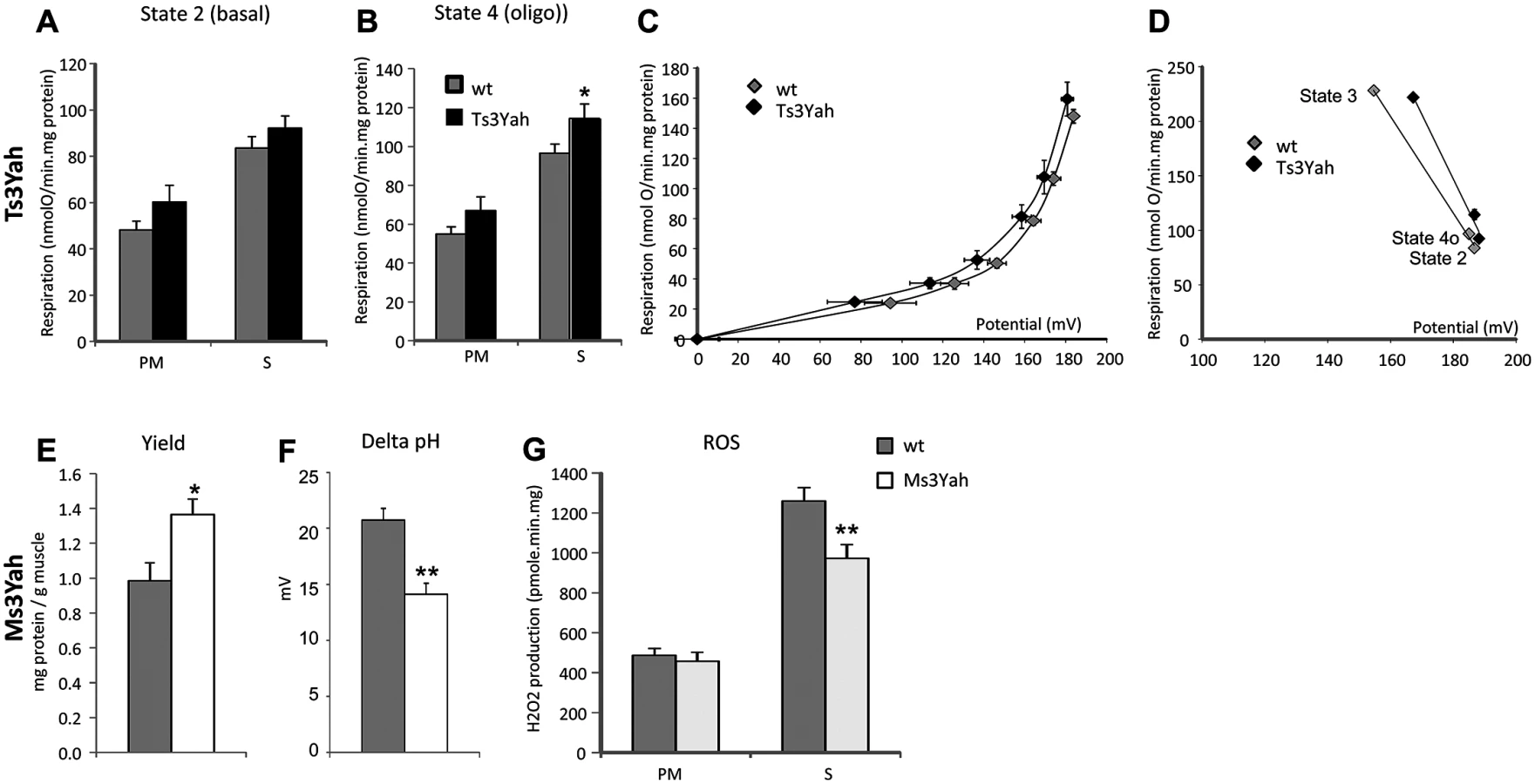
The kinetic analysis between oxygen consumption and membrane potential in Ms3Yah mitochondria was similar to that of diploid controls indicating a similar basal proton leak, as expected by the absolute respiratory rates observed in Ms3Yah mitochondria. However, although the basal proton motive force values were not significantly different between genotypes (Ms3Yah: 182 ± 3 mV; diploid control: 191±4 mV), the chemical component of the proton motive force ΔpH was significantly lower (~30%; t-test, p = 0.001) in Ms3Yah mice (14±1 mV) than in diploid controls (21±1 mV) (Fig. 6F). We also observed a 26% decrease in Ms3Yah mitochondrial ROS production (Fig. 6G, decrease in H2O2 release as a marker of ROS production) under basal respiration with S, which corresponds to the respiratory state that produces most ROS (ANOVA ‘genotype,’ ‘substrate’; F[1,31] = 6.068, p = 0.02, and post hoc tests with all pairwise multiple comparison procedure using Holm-Sidak method: Ms3Yah vs diploid with succinate as substrate p = 0.001). ROS production from S comes from a reverse electron flux from complex II to complex I (NAD) that produces superoxide at a very high rate and is highly dependent on the pH gradient (ΔpH) across the mitochondrial inner membrane [43]. Overall, our analysis revealed that there is a propensity of increased membrane permeability in Ts3Yah mitochondria and of decreased ROS production by Ms3Yah mitochondria that is due to a decrease of pH across the mitochondrial inner membrane.
Discussion
The phenotypic characterisation of two new mouse models trisomic and monosomic for the 9.4 Mb Mmu16 Hspa13-App region syntenic to the centromeric 21q11.2-q11.3 region revealed the contribution of gene dosage to locomotion, muscle strength, and mass and energy balance. The locomotor deficits observed in DS were attributed to impaired coordination input due to cerebellar or more general central dysfunction. Structural anomalies have been observed in the cerebellum of DS patients [44–46], but the presence of other motor-related phenotypes such as increased time for feedback adjustments in grip force, higher static grip forces, and difficulties to maintain constant muscle power suggests a more complex origin [47, 48]. Studies on motor control have described specific preferential muscle coactivation patterns in people who have DS during voluntary movements or postural adjustments. This coactivation of the agonist-antagonist muscle pair might produce less resistance to motion and might contribute to hypotonia [49, 50].
The locomotor deficit in Ts3Yah is accompanied by increased grip strength but decreased muscle endurance. Functional characterisation of the soleus muscle of Ts65Dn mice revealed that the muscle weakness observed in vivo was not the result of a deficit in force-generating capacity of the muscle in the basal state but was rather due to post-fatigue muscle weakness [51]. Hence, hypotonia and motor deficits observed in DS might be associated more with the fatigability of the muscle due to a lower energetic state. Indeed, hypotonia corresponds to a state of low muscular tonicity as the consequence of lack of energy to keep working at a normal level for a long time (and of a slower speed of response [52, 53]. Lower exercise endurance capacity with diminished basal metabolism and aerobic capacity has been observed in DS persons [54–56]. Accordingly, walking anomalies observed in DS children are associated with difficulties to maintain constant muscle power [57]. Interestingly, our Ts3Yah mouse model shows locomotor deficits as a consequence of energy decrease and lack of endurance.
The contribution of skeletal muscles to DS locomotor phenotypes and their physiological and biochemical characterisation have been poorly explored. A recent analysis of the soleus muscle in Ts65Dn mice failed to highlight a clear change in mitochondrial content or in muscle fibre type composition in the muscle although it identified alterations in pathways involved in glucose and fat metabolism as well as ATP biosynthesis [51]. The examination of hormonal and metabolic responses after a maximal effort in a treadmill test in DS and age-matched control people revealed that exercise limitation in DS people was caused by limited aerobic capacity [56]. This limitation was attributed to the disruption of the hormonal response (cortisol and catecholamines) leading to an incapacity to control glycolysis, neoglucogenesis, and lipolysis when muscle exercise is pursued [56]. Altered mitochondrial activity and increased oxidative stress would play a similar role in muscular function as found in the central nervous system, with DS accelerated aging and neurodegeneration [58–61].
Ms3Yah mitochondria revealed a decreased ROS production under respiration with succinate that could be linked to a lower pH gradient (ΔpH) of the mitochondrial inner membrane, whereas no change was observed for the membrane electrical potential (Δψ). ROS production with succinate is mainly generated by reverse electron flux from complex II to complex I [62–65] and is at least threefold more sensitive to the pH gradient (ΔpH) across the mitochondrial inner membrane than to the membrane potential (Δψ) [43]. PH gradient can be generated by proton pumping during substrate oxidation or by the ATP synthase pumping protons into the mitochondrial matrix during ATP synthesis. Interestingly, Atp5J, which encodes the F6 subunit of the mitochondrial ATP synthase [66], is found in the Hspa13-App region and shows opposite deregulation in Ts3Yah and Ms3Yah mice. Change in the dosage of this subunit might lead to a change in ATP synthase efficiency and thus to a change in pH gradient across the mitochondrial inner membrane. Hence, transgenic mice overexpressing Atp5J have sustained a decrease in intracellular pH in tissues leading to acidosis [67]. A deficit of mitochondrial ATP production due to alteration of the catalytic OXPHOS capacity and associated to a compensatory enhancement of glycolysis and mitochondrial mass was seen in fibroblasts from DS subjects [68]. Increased respiration during non-phosphorylating conditions, an indicator of nonoptimal OXPHOS efficiency, was also observed in the same study and suggests a greater proton leak across the mitochondrial inner membrane of DS fibroblasts. Interestingly, as seen by Valenti and collaborators [68], we did not observe major changes in mitochondrial respiratory capacity in Ts3Yah and Ms3Yah mitochondria, but Ts3Yah mitochondria showed a slight increase in respiration during non-phosphorylating conditions and a shift in the relationship between oxygen consumption and mitochondrial membrane potential indicative of a greater proton leak. This finding, together with the presence in the Hspa13-App region of the gene coding for a subunit of the ATP synthase, suggest that ATP synthesis might be affected in Ts3Yah mitochondria. It would be interesting to further measure the ATPase activity as well as the ratio of ATP synthesis over oxygen consumption in Ts3Yah mitochondria.
The transcriptome analysis of Ts3Yah and Ms3Yah skeletal muscles identifies the deregulation of genes involved in energy metabolic pathways. These results are consistent with previous findings in the hearts of DS fetuses and in DS amniocytes [69, 70]. Moreover, our analysis suggests that Pgc-1α or members of the Pgc-1α pathway are important to the molecular changes observed in Ts3Yah and Ms3Yah muscles. Consistent with this hypothesis, we increased expression of Pgc-1α in the transcriptome of Ms3Yah muscles (FC = 1.25, p = 0.0027) and increased expression in Ms3Yah (FC = 1.18, p = 0.03) and decrease expression in Ts3Yah (FC = 0.81, p = 0.01) muscles of ERRα (ESRRA), encoding the orphan nuclear receptor ERRα (NR3B1) that is known to have co-expression with Pgc-1α in tissues with high energy demand and whose transcriptional activity has been extensively linked to members of the PGC-1 family of coactivators [71]. Pgc-1α is a transcriptional coactivator of steroid and nuclear receptors and was found to play a central role as a modulator of oxidative metabolism in response to external physiological stimuli by activating the expression of a wide array of genes involved in fuel intake, gluconeogenesis, and fatty acid oxidation [72]. Most importantly, Pgc-1α controls mitochondrial biogenesis through the regulation of the synthesis of mitochondrial proteins involved not only in oxidative phosphorylation [73, 74] but also in the mitochondrial ribosomal machinery, protein transport, and dynamic [75]. Interestingly, the gene encoding the mitochondrial enzyme citrate synthase (Cs) needed for pyruvate-derived acetyl-CoA entry to the TCA cycle and whose activity is a marker of increased mitochondrial biogenesis is found in the leading edge genes of the mitochondrial gene set identified in the GSEA analysis. In addition, genes coding for proteins involved in the transcription and translation of mitochondrial genes (Tfam, Tufm, Tfb1m, Tfb2m, Lars2, Wars2, Mtif2, and Mtrf1) and for proteins participating in mitochondrial protein import and translocation across the outer and inner mitochondrial membrane (Mipep,Surf1, Tomm70a, Timm17b, Timm9, Timm44, and Tomm22) were found in mitochondrial leading edge genes in either Ts3Yah or Ms3Yah or both (see leading edge gene set ‘HUMAN_MITODB_6_2002,’ S4–S5 Tables). Hence, molecular changes observed in Ts3Yah and Ms3Yah muscles point at gene(s) within the Hspa13-App region affecting the Pgc1a coactivator network regulating energy metabolism and mitochondrial biogenesis.
Eight of the 15 genes conserved between human and mouse in the Hspa13-App region, namely, Hspa13, Nrip1, Usp25, Btg3, D16Ertd472e, Mrpl39, Jam2, Atp5J, and Gabpa, were globally 0.5 fold downregulated in Ms3Yah muscles and 1.5 fold upregulated in Ts3Yah muscles following gene dosage. Among those genes, two (Gabpa and Nrip1) are strong candidates for the muscular phenotypes as they are known to be part of the network of proteins involved in the transcriptional control of cellular energy homeostasis. Gabpa (GA-binding protein subunit α) encodes the ETS DNA-binding subunit of the NRF2 (nuclear respiratory factor) transcription factor that was found to interact with PGC-1α and ERRα to drive the expression of mitochondrial genes [76, 77]. Genetic disruption and knockdown of mouse Gabpa caused early embryonic lethality [78, 79] with mitochondrial dysfunction and have been proposed as a possible explanation for embryonic loss. Yang and collaborators (2014) recently deleted Gabpa in cultured primary mouse fibroblasts and found out that this resulted in a reduction of mitochondrial mass, ATP production, and mitochondrial methyltransferase Tfb1m, which is essential for mitochondrial protein translation and mitochondrial biogenesis [80]. However, the Ms3Yah mice with only one copy of Gabpa have increased mitochondrial biogenesis, and the Gabpα heterozygous knockout mice display no phenotype and express similar levels of GABPα protein as the wild-type mice in their muscles [79]. Moreover, no increase in protein expression of GABPα could be seen in human DS fibroblasts [81].
The receptor-interacting protein (RIP140 or NRIP1) identified as a corepressor of nuclear receptors [82–84] was found to affect oxidative metabolism and mitochondrial biogenesis by negatively controlling mitochondrial pathways regulated by PGC-1α [85]. Nrip1 null mice displayed an increase in the number of oxidative fibres as measured by SDH and MyHC isoforms staining and an increase in the quantity of mitochondria, whereas overexpression of NRIP1 reduced oxidative activity and mitochondrial biogenesis in skeletal muscle [86]. In addition, the absence of NRIP1 resulted in the upregulation of the same gene sets (oxidative phosphorylation, fatty acid oxidation, TCA cycle, and glycolysis) that we found deregulated in our models [86]. A siRNA experiment to decrease NRIP1 expression in trisomic human fetal fibroblasts enabled to recover normal levels of nuclear-encoded mitochondrial genes and normal mitochondrial function in those fibroblasts [87]. As such Nrip1 changes in copy number might explain most of the phenotypes observed in Ts3Yah and Ms3Yah mice, but we cannot exclude the contribution of other genes from the interval. Indeed, the change in the steady-state level of a subunit of mitochondrial ribosomal protein (MRP) encoded by the Mrpl39 gene within the Hspa13-App region may also trigger perturbation in the translation of mitochondrial-encoded proteins of the OXPHOS system, leading to variations of mitochondrial function observed in Ts3Yah and Ms3Yah mice. Translational deficiencies due to mutations in genes encoding MRPs have been described for four of the more than 70 MRPs, leading to growth retardation, cardiomyopathy, hypotonia, and brain anomalies [88–91] but are expected to range from lethality to slightly impaired energy metabolic deficiencies and give rise to various tissue-specific defects such as neuromuscular disorders or metabolic acidosis [92, 93]. In addition, deficit in the OXPHOS machinery can trigger the mitochondrial membrane permeability transition (MPT) leading to mitochondrial apoptosis or necrosis [94] as observed in Ms3Yah muscles.
DS and M21 are complex genetic diseases, and mouse models represent invaluable tools to decipher Hsa21 dosage-sensitive genes and associated deregulated molecular pathways. Our new models show that subtle deregulations contribute to the complexity of the DS-related pathology and indicate the need to further study the contribution of different Hsa21 regions to the modulation of DS. In particular, this study contributes to understanding the motor deficits in people who have DS by providing new insight into the effect of trisomy on muscle dysfunction and proposing new deregulated pathways that might represent novel therapeutic targets for DS therapy.
Materials and Methods
Ethics statement
All animals were treated in compliance with animal welfare policies from the French Ministry of Agriculture (Law 87 848 and YH accreditation 45–31 and 67–369). Mice were bred on C57BL/6J background for at least five generations prior to the functional analysis. The animals were bred under SPF conditions and were treated in compliance with animal welfare policies. In all the experiments, disomic littermates matched for age and gender were used as controls. A series of behavioural experiments were conducted in order to evaluate motor and muscular functions. All experimental procedures have been described previously in [36]. Behavioural protocols (rotarod, grip strength, treadmill, notched bar, and open field) were approved by the local Ethics Committee for Animal Experimentation under the accreditation number (2012–069). For all these tests, mice were kept in SPF conditions with free access to food and water. The light-dark cycle was 12 : 12, with lights on at 7 : 00 a.m. All the tests were done between 9 : 00 a.m. and 4 : 00 p.m.
After weaning, male mice were gathered by litters in the same cage. The different apparatus used were placed in a dimly lit testing room (approximatively 60 lux). To produce experimental groups, only animals coming from litters containing a minimum of two male pups were selected. Groups of animals were transferred to the experimental room 30 minutes before each experimental test. No invasive procedure was used, and the mice were euthanized at the end of the analysis.
Generating the Ts3Yah and Ms3Yah mouse strains
The Ms3Yah monosomic mice, Del(16Stch-App)3Yah, and the Ts3Yah trisomic mice, Dp(16Stch-App)2Yah, were generated by chromosomal engineering in vivo as described in more details in S1 Text. Briefly, Hspa13 and App targeting vectors containing loxP sites and isolated from 5’Hprt and 3’Hprt libraries [95] were inserted by homologous recombination in HM1–1 Hprt-deficient ES cells in a trans configuration [96] (Fig. 1B–C). Transient expression of the Cre recombinase generated clones with the tandem duplication and the deletion of the Hspa13-App fragment (Fig. 1D) that were subsequently injected to C57BL/6J blastocysts to generate chimeras. Chimeras were mated with C57Bl6/J animals and pups carrying either the Mmu16 with the deletion or the Mmu16 with the duplication that were identified by Southern blot analysis. Those animals were subsequently bred with C57BL/6J mice to obtain the Ms3Yah and Ts3Yah lines.
Behavioural analyses
Groups (n = 10–15) of transgenic and diploid littermate males with matched age and genetic background (B6 from N5 backcross level) were used for the different behavioural tests, following the recommendations from the standard operating procedures developed by the Eumorphia network (http://www.eumorphia.org). A series of behavioural experiments were conducted in order to evaluate the motor conditions in mice with group of 10 to 15 males per genotype. For all these tests, mice were kept in SPF conditions with free access to food and water. Several tests were carried out, including open-field, rotarod, notched bar,and grip strength at ages 3 to 4 months and rotarod, grip strength, treadmill endurance, and notched bar at ages 8 to 12 months. All procedures, except for the treadmill exercise, are presented in S1 text. The treadmill system consists of a belt, which is enclosed in a Plexiglas chamber, and a stimulus device with a metal shock grid attached to the rear of the belt. The speed and slope of the belt are electronically adjusted. The animals were acclimatised with a 20-minute run at 25 cm/s and a 5-degree incline the day before the running test. For the actual test, the experiment was started at 25 cm/s and a 5-degree incline. Every 20 minutes, a progressive increase of 5 cm/s in speed is applied. The distance run and the time spent running were recorded, and a mouse was considered exhausted and removed from the experiment if it received approximately 100 shocks (1 mA) in a period of 5 minutes or if it was spending more than 20 seconds on the shock grid. The duration of running and the total distance covered evaluate the performance of the mice.
Gene expression analysis
RNA extraction
Total RNA was extracted from frozen gastrocnemius muscles from Ms3Yah and Ts3Yah and their respective control diploid littermates (n = 6) using TRIzol Reagent (Invitrogen) followed by RNase-Free DNaseI (Qiagen) treatment.
Microarray hybridisation and data analysis
Biotinylated cDNA were prepared from total RNAs and hybridised to Illumina’s MouseWG-6 v2.0 Expression BeadChips. Raw data was normalised by the quantile method [97], and values were log transformed. A principal component analysis was done using the GeneSpring software (Agilent Technologies) and showed that one diploid sample corresponding to the Ts3Yah control group had a completely different profile. This sample was excluded from the analysis. The data were analysed separately for Ms3Yah and Ts3Yah, comparing each set to its own littermate diploid group. The genes expressed above the background level were determined by selecting the probe sets having a signal value above the 35th percentile of all expression values in at least one array. This corresponded to a threshold value above 90 in raw data (6.49 in log scale). The genes differentially expressed were selected by statistical analysis (Student’s t-test p<0.05 between transgenic mice and their respective diploid littermates). Verification was done to ensure that selected probes have acceptable false discovery rate (<10%, [98]), and resampling analysis was used to make 1,000 permutations of the samples for each probe in order to calculate the false discovery rate p-value (Zoe software; http://www-microarrays.u-strasbg.fr/base.php?page=analysisExpressionFilterZoeE.php). FC was calculated as a ratio of averages from transgenic and diploid signals with values <1 representing probes that are underexpressed in the transgenic (Ts3Yah or Ms3Yah) compared to diploid controls and values >1 representing probes that are overexpressed in the transgenic compared to the diploid controls.
Hierarchical clustering was carried out on significantly deregulated genes (p<0.05) with Cluster 3.0 software [99] using Euclidian distances to calculate the distances between the genes and between the samples. The calculated distances were then clustered by complete linkage clustering. The red-to-green colour scale represents the mean-adjusted expression values, where red corresponds to higher expression and green to lower expression.
Gene Set Enrichment Analysis (GSEA) was used to test sets of related genes that might be coordinately deregulated in Ts3Yah and Ms3Yah gastrocnemius muscles [40, 41]. The whole transcriptome output lists of Ts3Yah and Ms3Yah differentially expressed genes, together with their diploid controls, were submitted to the GSEA tool, and the functional gene sets containing curated biological pathways from the MSigDB C2 curated database were tested [40]. A number of 1,000 permutations were specified as recommended in order to assess the statistical significance of the enrichment score (ES). The GSEA analysis report highlights enrichment gene sets with an FDR<5%. All data are accessible under the GEO accession number (GSE58463) [NCBI tracking system #17057227].
QRT-PCR
cDNA synthesis was performed using the SuperScript III First-Strand Synthesis SuperMix for QRT-PCR (Invitrogen). A series of primer pairs (available upon request) were designed to span intron-exon junctions. Efficiencies of the TaqMan assays were checked using a cDNA dilution series from the extracts of gastrocnemius muscle samples. The QPCR was performed with 300 nM of each primer and 100 nM of HPLC purified FAM-TAMRA-labelled double-dye TaqMan probes in a final reaction of 15 μl with a standard amplification procedure. Normalisation was performed by carrying out in parallel the amplification of five housekeeping genes (Actb, Pgk1, Hprt1, Ppia, and Gnas) and by using the GeNorm procedure [100] in order to correct the variations of the amount of source RNA in the starting material. All the tested samples were performed in triplicate, and the results were reported as the mean ± sem.
mtDNA quantification by quantitative real-time PCR
DNA was isolated from muscle tissue, and quantitative PCR using the TaqMan technology was performed in triplicate to determine the relative quantity of the mitochondrial DNA marker cyclooxygenase 2 (Cox2) and the genomic DNA marker myxovirus resistance 1 (Mx1) gene. mtCOX2 and Mx1 primers were purchased from Sigma-Aldrich and TaqMan MGB probes for mtCOX2 and Mx1 from Applied Biosystem. PCR conditions were as follows: (1) 50°C for 2 minutes, (2) 95°C for 10 minutes, (3) 95°C for 15 seconds, and (4) 60°C for 1 minute (steps 3 and 4 were repeated 50 times). The results were calculated from the difference in threshold cycle (ΔCT) values for mtDNA and nuclear-specific amplification. Data were expressed as mtDNA in transgenic mice relative to diploid control groups.
SDH and immunofluorescence staining on frozen muscle sections
Tibialis anterioris muscles and soleus tissues were collected from 5 to 7 mice aged 4 to 6 months per genotype and immediately frozen in 5 minutes and 3 minutes, respectively, in isopentane cooled in liquid nitrogen (-190°C). A total of 10 μm thick serial sections were obtained using a cryostat (Leica CM3050) at -25°C. The sections were processed for succinate dehydrogenase (SDH) staining (S1 Text). Alternatively, type I fibres in tibialis and soleus were characterised by MyHC-I immunostaining using the primary monoclonal mouse antibody against type I (M844, Sigma) as described in S1 Text. Quantification was done by counting muscle fibres using the ImageJ software (W. Rasband, NIH; http://rsb.info.nih.gov/ij/) on at least three consecutive sections.
Electron microscopy
TA samples (n = 3–5) of 3-month and 12-month old mice were fixed in 2.5% glutaraldehyde and 2.5% paraformaldehyde in cacodylate buffer (0.1 M, pH 7.4), postfixed in 1% osmium tetroxide in 0.1M cacodylate buffer, and dehydrated through graded alcohol (50%, 70%, 90%, and 100%) and propylene oxide. The samples were oriented longitudinally and embedded in Epon 812. Semithin sections were cut at 2 μm, and ultrathin sections were cut at 70 nm and contrasted with uranyl acetate and lead citrate and examined at 70 kv with a Morgagni 268D electron microscope. The images were captured digitally by a MegaView III camera (Soft Imaging System).
Analysis of mitochondrial function
Mitochondria were isolated from hind limb skeletal muscles from one or two adult mice per mitochondrial preparation as described in S1 Text. The protein concentration of mitochondrial suspensions was determined in duplicate by a biuret method with bovine serum albumin to normalise the samples. Oxygen consumption was measured with a Clark oxygen electrode (Rank Brothers Ltd) in a stirred and closed chamber as described in S1 text. The cytochrome-c oxidase (COX) activity was determined by the mitochondrial respiration rate in the mitochondrial suspension, which includes 10 mM antimycin, 2 mM ascorbate, and 0.5 mM TMPD (N,N,N’,N’-TetraMethyl-p-Phenylene-Diamine). Measurements of COX activity were done in duplicate on n = 5 per genotype.
Mitochondrial membrane potential was measured using an electrode sensitive to the lipophilic cation triphenylmethylphosphonium (TPMP+). The protocol described in S1 Text allows us to calculate the ΔpH value as the difference between membrane potential values in the presence (ΔpH + Δψ) and in the absence (Δψ) of nigericin.
Respiration and membrane potential were then progressively inhibited through successive steady states induced by the addition of malonate. After each run, 2 μM FCCP was added to release TPMP+ back into the medium for baseline correction. Membrane potentials were calculated as described previously [101], assuming a TPMP+ binding correction of 0.35 mg protein/μl for skeletal muscle mitochondria [102].
Radical oxygen species (ROS) production by isolated mitochondria was evaluated by measuring the rate of H2O2 released using an SFM-25 fluorometer (Kontron) at excitation and emission wavelengths of 560 nm and 584 nm, respectively. The respiratory buffer (1 ml) was supplemented with 5 U/ml horseradish peroxidase, 1 μM Amplex Red reagent, and mitochondria (15–20 μg/ml). Respiratory substrates (5 mM succinate or 5 mM pyruvate/2.5 malate or 40 μM palmitoyl-L-carnitine/2.5 malate) were added to start the reaction. The fluorescent signal was calibrated using a standard curve obtained after successive addition of H2O2 (20 to 80 pmoles).
Statistics
Unless otherwise stated, statistical analyses were performed comparing two groups, the Ts3Yah or Ms3Yah and their diploid control littermates, using Student’s t-test when appropriate or the nonparametric Mann-Whitney rank sum test. For tests requiring serial recordings, data were analysed by two-way ANOVA using the SigmaPlot software. Data are presented as mean ± sem. Significant threshold was set for p<0.05.
Supplemental information
Supplemental information includes additional experimental procedures, two figures, and five tables and can be found online.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. Joosten AMS, DeVos S, VanOpstal D, Brandenburg H, Gaillard JLJ, VermeijKeers C. Full monosomy 21, prenatally diagnosed by fluorescent in situ hybridization. Prenatal Diagnosis. 1997;17(3):271–5. doi: 10.1002/(sici)1097-0223(199703)17 : 3<271::aid-pd51>3.0.co;2-p 9110372
2. Kulharya AS, Tonk VS, Lovell C, Flannery DB. Complete Monosomy 21 Confirmed by FISH and Array-CGH. American Journal of Medical Genetics Part A. 2012;158A(4):935–7. doi: 10.1002/ajmg.a.35251 22407893
3. Manolakos E, Peitsidis P, Eleftheriades M, Dedoulis E, Ziegler M, Orru S, et al. Prenatal detection of full monosomy 21 in a fetus with increased nuchal translucency: Molecular cytogenetic analysis and review of the literature. Journal of Obstetrics and Gynaecology Research. 2010;36(2):435–40. doi: 10.1111/j.1447-0756.2009.01140.x 20492403
4. Mori MA, Lapunzina P, Delicado A, Nunez G, Rodriguez JI, de Torres ML, et al. A prenatally diagnosed patient with full monosomy 21: Ultrasound, cytogenetic, clinical, molecular, and necropsy findings. American Journal of Medical Genetics Part A. 2004;127A(1):69–73. doi: 10.1002/ajmg.a.20622 15103721
5. Fisher D, DiPietro A, Murdison KA, Lemieux CA. Full Monosomy 21: Echocardiographic Findings in the Third Molecularly Confirmed Case. Pediatric Cardiology. 2013;34(3):733–5. doi: 10.1007/s00246-012-0334-4 22562777
6. Wakui K, Toyoda A, Kubota T, Hidaka E, Ishikawa M, Katsuyama T, et al. Familial 14-Mb deletion at 21q11.2-q21.3 and variable phenotypic expression. Journal of Human Genetics. 2002;47(10):511–6. doi: 10.1007/s100380200076 12376739
7. Tinkel-Vernon H, Finkernagel S, Desposito F, Pittore C, Reynolds K, Sciorra L. Patient with a deletion of chromosome 21q and minimal phenotype. American Journal of Medical Genetics Part A. 2003;120A(1):142–3. doi: 10.1002/ajmg.a.10210 12794708
8. Chettouh Z, Croquette MF, Delobel B, Gilgenkrants S, Leonard C, Maunoury C, et al. MOLECULAR MAPPING OF 21 FEATURES ASSOCIATED WITH PARTIAL MONOSOMY-21—INVOLVEMENT OF THE APP-SOD1 REGION. American Journal of Human Genetics. 1995;57(1):62–71. 7611297
9. Lindstrand A, Malmgren H, Sahlen S, Schoumans J, Nordgren A, Ergander U, et al. Detailed molecular and clinical characterization of three patients with 21q deletions. Clinical Genetics. 2010;77(2):145–54. doi: 10.1111/j.1399-0004.2009.01289.x 19863549
10. Roberson EDO, Wohler ES, Hoover-Fong JE, Lisi E, Stevens EL, Thomas GH, et al. Genomic analysis of partial 21q monosomies with variable phenotypes. European Journal of Human Genetics. 2011;19(2):235–8. doi: 10.1038/ejhg.2010.150 20823914
11. Korenberg JR, Kalousek DK, Anneren G, Pulst SM, Hall JG, Epstein CJ, et al. DELETION OF CHROMOSOME-21 AND NORMAL INTELLIGENCE—MOLECULAR DEFINITION OF THE LESION. Human Genetics. 1991;87(2):112–8. doi: 10.1007/bf00204163 2066097
12. Lyle R, Béna F, Gagos S, Gehrig C, Lopez G, Schinzel A, et al. Genotype-phenotype correlations in Down syndrome identified by array CGH in 30 cases of partial trisomy and partial monosomy chromosome 21. Eur J Hum Genet. 2009;17(4):454–66. doi: 10.1038/ejhg.2008.214 19002211
13. Parker SE, Mai CT, Canfield MA, Rickard R, Wang Y, Meyer RE, et al. Updated National Birth Prevalence Estimates for Selected Birth Defects in the United States, 2004–2006. Birth Defects Research Part a-Clinical and Molecular Teratology. 2010;88(12):1008–16. doi: 10.1002/bdra.20735
14. Korbel J, Tirosh-Wagner T, Urban A, Chen X, Kasowski M, Dai L, et al. The genetic architecture of Down syndrome phenotypes revealed by high-resolution analysis of human segmental trisomies. Proc Natl Acad Sci U S A. 2009;106(29):12031–6. doi: 10.1073/pnas.0813248106 19597142
15. Anderson DI, Campos JJ, Witherington DC, Dahl A, Rivera M, He MX, et al. The role of locomotion in psychological development. Frontiers in Psychology. 2013;4. doi: 10.3389/fpsyg.2013.00440
16. Campos JJ, Anderson DI, Barbu-Roth MA, Hubbard EM, Hertenstein MJ, Witherington D. Travel Broadens the Mind. Infancy. 2000;1(2):149–219. doi: 10.1207/s15327078in0102_1
17. Frith U, Frith CD. SPECIFIC MOTOR DISABILITIES IN DOWNS-SYNDROME. Journal of Child Psychology and Psychiatry and Allied Disciplines. 1974;15(4):293–301.
18. Shumwaycook A, Woollacott MH. DYNAMICS OF POSTURAL CONTROL IN THE CHILD WITH DOWN SYNDROME. Physical Therapy. 1985;65(9):1315–22. 3162178
19. Hestnes A, Stovner LJ, Husoy O, Folling I, Fougner KJ, Sjaastad O. HORMONAL AND BIOCHEMICAL DISTURBANCES IN DOWNS-SYNDROME. Journal of Mental Deficiency Research. 1991;35 : 179–93. 1833549
20. Gonzalez-Aguero A, Ara I, Moreno LA, Vicente-Rodriguez G, Casajus JA. Fat and lean masses in youths with Down syndrome: Gender differences. Research in Developmental Disabilities. 2011;32(5):1685–93. doi: 10.1016/j.ridd.2011.02.023 21435834
21. O'Doherty A, Ruf S, Mulligan C, Hildreth V, Errington ML, Cooke S, et al. An aneuploid mouse strain carrying human chromosome 21 with Down syndrome phenotypes. Science. 2005;309(5743):2033–7. doi: 10.1126/science.1114535 16179473
22. Yu T, Li ZY, Jia ZP, Clapcote SJ, Liu CH, Li SM, et al. A mouse model of Down syndrome trisomic for all human chromosome 21 syntenic regions. Human Molecular Genetics. 2010;19(14):2780–91. doi: 10.1093/hmg/ddq179 20442137
23. Yu T, Liu CH, Belichenko P, Clapcote SJ, Li SM, Pao AN, et al. Effects of individual segmental trisomies of human chromosome 21 syntenic regions on hippocampal long-term potentiation and cognitive behaviors in mice. Brain Research. 2010;1366 : 162–71. doi: 10.1016/j.brainres.2010.09.107 20932954
24. Li ZY, Yu T, Morishima M, Pao A, LaDuca J, Conroy J, et al. Duplication of the entire 22.9 Mb human chromosome 21 syntenic region on mouse chromosome 16 causes cardiovascular and gastrointestinal abnormalities. Human Molecular Genetics. 2007;16(11):1359–66. doi: 10.1093/hmg/ddm086 17412756
25. Pereira PL, Magnol L, Sahun I, Brault V, Duchon A, Prandini P, et al. A new mouse model for the trisomy of the Abcg1-U2af1 region reveals the complexity of the combinatorial genetic code of down syndrome. Human Molecular Genetics. 2009;18(24):4756–69. doi: 10.1093/hmg/ddp438 19783846
26. Reeves RH, Irving NG, Moran TH, Wohn A, Kitt C, Sisodia SS, et al. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat Genet. 1995;11(2):177–84. 7550346
27. Duchon A, Raveau M, Chevalier C, Nalesso V, Sharp AJ, Herault Y. Identification of the translocation breakpoints in the Ts65Dn and Ts1Cje mouse lines: relevance for modeling down syndrome. Mamm Genome. 2011. doi: 10.1007/s00335-011-9356-0
28. Duchon A, Raveau M, Chevalier C, Nalesso V, Sharp AJ, Herault Y. Identification of the translocation breakpoints in the Ts65Dn and Ts1Cje mouse lines: relevance for modeling down syndrome. Mammalian Genome. 2011;22(11–12):674–84. doi: 10.1007/s00335-011-9356-0 21761260
29. Costa ACS, Walsh K, Davisson MT. Motor dysfunction in a mouse model for Down syndrome. Physiology & Behavior. 1999;68(1–2):211–20.
30. Costa ACS, Stasko MAP, Schmidt C, Davisson MT. Behavioral validation of the Ts65Dn mouse model for Down syndrome of a genetic background free of the retinal degeneration mutation Pde6b(rd1). Behavioural Brain Research. 2010;206(1):52–62. doi: 10.1016/j.bbr.2009.08.034 19720087
31. Hyde LA, Crnic LS, Pollock A, Bickford PC. Motor learning in Ts65Dn mice, a model for Down syndrome. Developmental Psychobiology. 2001;38(1):33–45. doi: 10.1002/1098-2302(2001)38 : 1<33::aid-dev3>3.0.co;2-0 11150059
32. Vidal V, Garcia S, Martinez P, Corrales A, Florez J, Rueda N, et al. LACK OF BEHAVIORAL AND COGNITIVE EFFECTS OF CHRONIC ETHOSUXIMIDE AND GABAPENTIN TREATMENT IN THE TS65DN MOUSE MODEL OF DOWN SYNDROME. Neuroscience. 2012;220 : 158–68. doi: 10.1016/j.neuroscience.2012.06.031 22728103
33. Ault B, Caviedes P, Hidalgo J, Epstein CJ, Rapoport SI. ELECTROPHYSIOLOGICAL ANALYSIS OF CULTURED FETAL MOUSE DORSAL-ROOT GANGLION NEURONS TRANSGENIC FOR HUMAN SUPEROXIDE DISMUTASE-1, A GENE IN THE DOWN SYNDROME REGION OF CHROMOSOME-21. Brain Research. 1989;497(1):191–4. 2529019
34. Nieminen K, Suarezisla BA, Rapoport SI. ELECTRICAL-PROPERTIES OF CULTURED DORSAL-ROOT GANGLION NEURONS FROM NORMAL AND TRISOMY-21 HUMAN-FETAL TISSUE. Brain Research. 1988;474(2):246–54. 2974749
35. Peng S, Rapoport SI, Pearce RJ, Galdzicki Z. Abnormal chloride and potassium conductances in cultured embryonic tongue muscle from trisomy 16 mouse. Developmental Brain Research. 2000;122(2):193–7. 10960688
36. Duchon A, Pothion S, Brault V, Sharp AJ, Tybulewicz VLJ, Fisher EMC, et al. The telomeric part of the human chromosome 21 from Cstb to Prmt2 is not necessary for the locomotor and short-term memory deficits observed in the Tc1 mouse model of Down syndrome. Behavioural Brain Research. 2011;217(2):271–81. doi: 10.1016/j.bbr.2010.10.023 21047530
37. Adams DJ, Biggs PJ, Cox T, Davies R, van der Weyden L, Jonkers J, et al. Mutagenic insertion and chromosome engineering resource (MICER). Nature Genetics. 2004;36(8):867–71. doi: 10.1038/ng1388 15235602
38. Brault V, Besson V, Magnol L, Duchon A, Herault Y. Cre/loxP-mediated chromosome engineering of the mouse genome. Handb Exp Pharmacol. 2007;178 : 29–48. 17203650
39. Mootha VK, Lepage P, Miller K, Bunkenborg J, Reich M, Hjerrild M, et al. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(2):605–10. doi: 10.1073/pnas.242716699 12529507
40. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(43):15545–50. doi: 10.1073/pnas.0506580102 16199517
41. Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1 alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nature Genetics. 2003;34(3):267–73. doi: 10.1038/ng1180 12808457
42. Hafner RP, Brown GC, Brand MD. ANALYSIS OF THE CONTROL OF RESPIRATION RATE, PHOSPHORYLATION RATE, PROTON LEAK RATE AND PROTONMOTIVE FORCE IN ISOLATED-MITOCHONDRIA USING THE TOP-DOWN APPROACH OF METABOLIC CONTROL-THEORY. European Journal of Biochemistry. 1990;188(2):313–9. doi: 10.1111/j.1432-1033.1990.tb15405.x 2156698
43. Lambert AJ, Brand MD. Superoxide production by NADH: ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochemical Journal. 2004;382 : 511–7. doi: 10.1042/bj20040485 15175007
44. Pinter JD, Eliez S, Schmitt JE, Capone GT, Reiss AL. Neuroanatomy of Down's syndrome: A high-resolution MRI study. American Journal of Psychiatry. 2001;158(10):1659–65. doi: 10.1176/appi.ajp.158.10.1659 11578999
45. Raz N, Torres IJ, Briggs SD, Spencer WD, Thornton AE, Loken WJ, et al. SELECTIVE NEUROANATOMICAL ABNORMALITIES IN DOWNS-SYNDROME AND THEIR COGNITIVE CORRELATES—EVIDENCE FROM MRI MORPHOMETRY. Neurology. 1995;45(2):356–66. 7854539
46. Weis S, Weber G, Neuhold A, Rett A. DOWN-SYNDROME—MR QUANTIFICATION OF BRAIN STRUCTURES AND COMPARISON WITH NORMAL CONTROL SUBJECTS. American Journal of Neuroradiology. 1991;12(6):1207–11. 1837203
47. Cole KJ, Abbs JH, Turner GS. DEFICITS IN THE PRODUCTION OF GRIP FORCES IN DOWN SYNDROME. Developmental Medicine and Child Neurology. 1988;30(6):752–8. 2976689
48. Ringenbach SD, Chua R, Maraj BKV, Kao JC, Weeks DJ. Bimanual coordination dynamics in adults with Down syndrome. Motor Control. 2002;6(4):388–407. 12429892
49. Aruin AS, Almeida GL, Latash ML. Organization of a simple two-joint synergy in individuals with Down syndrome. American Journal on Mental Retardation. 1996;101(3):256–68. 8933900
50. Latash ML. Learning motor synergies by persons with Down syndrome. Journal of Intellectual Disability Research. 2007;51 : 962–71. doi: 10.1111/j.1365-2788.2007.01008.x 17991003
51. Cowley PM, Keslacy S, Middleton FA, DeRuisseau LR, Fernhall B, Kanaley JA, et al. Functional and biochemical characterization of soleus muscle in Down syndrome mice: insight into the muscle dysfunction seen in the human condition. American Journal of Physiology-Regulatory Integrative and Comparative Physiology. 2012;303(12):R1251–R60. doi: 10.1152/ajpregu.00312.2012
52. Davis WE, Kelso JAS. ANALYSIS OF INVARIANT CHARACTERISTICS IN THE MOTOR CONTROL OF DOWNS-SYNDROME AND NORMAL SUBJECTS. Journal of Motor Behavior. 1982;14(3):194–212. 15153410
53. Morris AF, Vaughan SE, Vaccaro P. MEASUREMENTS OF NEUROMUSCULAR TONE AND STRENGTH IN DOWNS-SYNDROME CHILDREN. Journal of Mental Deficiency Research. 1982;26(MAR):41–6.
54. Allison DB, Gomez JE, Heshka S, Babbitt RL, Geliebter A, Kreibich K, et al. DECREASED RESTING METABOLIC-RATE AMONG PERSONS WITH DOWN-SYNDROME. International Journal of Obesity. 1995;19(12):858–61. 8963352
55. Luke A, Roizen NJ, Sutton M, Schoeller DA. ENERGY-EXPENDITURE IN CHILDREN WITH DOWN-SYNDROME—CORRECTING METABOLIC-RATE FOR MOVEMENT. Journal of Pediatrics. 1994;125(5):829–38. 7965444
56. Bricout VA, Flore P, Guinot M, Faure P, Garnier P, Eberhard Y, et al. Hormonal responses of Down's syndrome subjects to exercise. Science & Sports. 2007;22(6):293–6. doi: 10.1016/j.scispo.2007.09.002
57. Mercer VS, Lewis CL. Hip Abductor and Knee Extensor Muscle Strength of Children with and without Down Syndrome. Pediatr Phys Ther. 2001;13(1):18–26. 17053646
58. Cenini G, Dowling ALS, Beckett TL, Barone E, Mancuso C, Murphy MP, et al. Association between frontal cortex oxidative damage and beta-amyloid as a function of age in Down syndrome. Biochimica Et Biophysica Acta-Molecular Basis of Disease. 2012;1822(2):130–8. doi: 10.1016/j.bbadis.2011.10.001
59. Busciglio J, Pelsman A, Wong C, Pigino G, Yuan ML, Mori H, et al. Altered metabolism of the amyloid beta precursor protein is associated with mitochondrial dysfunction in Down's syndrome. Neuron. 2002;33(5):677–88. 11879646
60. Iannello RC, Crack PJ, de Haan JB, Kola I. Oxidative stress and neural dysfunction in Down Syndrome. Journal of Neural Transmission-Supplement. 1999;(57):257–67. 10666681
61. Zana M, Janka Z, Kalman J. Oxidative stress: A bridge between Down's syndrome and Alzheimer's disease. Neurobiology of Aging. 2007;28(5):648–76. doi: 10.1016/j.neurobiolaging.2006.03.008 16624449
62. Genova MW, Pich MM, Biondi A, Bernacchia A, Falasca A, Bovina C, et al. Mitochondrial production of oxygen radical species and the role of coenzyme Q as an antioxidant. Experimental Biology and Medicine. 2003;228(5):506–13. 12709577
63. Batandier C, Guigas B, Detaille D, El-Mir MY, Fontaine E, Rigoulet M, et al. The ROS production induced by a reverse-electron flux at respiratory-chain complex 1 is hampered by metformin. Journal of Bioenergetics and Biomembranes. 2006;38(1):33–42. doi: 10.1007/s10863-006-9003-8 16732470
64. Liu YB, Fiskum G, Schubert D. Generation of reactive oxygen species by the mitochondrial electron transport chain. Journal of Neurochemistry. 2002;80(5):780–7. doi: 10.1046/j.0022-3042.2002.00744.x 11948241
65. Hansford RG, Hogue BA, Mildaziene V. Dependence of H2O2 formation by rat heart mitochondria on substrate availability and donor age. Journal of Bioenergetics and Biomembranes. 1997;29(1):89–95. doi: 10.1023/a:1022420007908 9067806
66. Johnson JA, Ogbi M. Targeting the F1Fo ATP Synthase: Modulation of the Body's Powerhouse and Its Implications for Human Disease. Current Medicinal Chemistry. 2011;18(30):4684–714. 21864274
67. Osanai T, Tanaka M, Magota K, Tomita H, Okumura K. Coupling factor 6-induced activation of ecto-F1Fo complex induces insulin resistance, mild glucose intolerance and elevated blood pressure in mice. Diabetologia. 2012;55(2):520–9. doi: 10.1007/s00125-011-2341-z 22038518
68. Valenti D, Tullo A, Caratozzolo MF, Merafina RS, Scartezzini P, Marra E, et al. Impairment of F1F0-ATPase, adenine nucleotide translocator and adenylate kinase causes mitochondrial energy deficit in human skin fibroblasts with chromosome 21 trisomy. Biochemical Journal. 2010;431 : 299–310. doi: 10.1042/bj20100581 20698827
69. Conti A, Fabbrini F, D'Agostino P, Negri R, Greco D, Genesio R, et al. Altered expression of mitochondrial and extracellular matrix genes in the heart of human fetuses with chromosome 21 trisomy. Bmc Genomics. 2007;8. doi: 268 10.1186/1471-2164-8-268
70. Lee SH, Lee S, Jun HS, Jeong HJ, Cha WT, Cho YS, et al. Expression of the mitochondrial ATPase6 gene and Tfam in Down syndrome. Molecules and Cells. 2003;15(2):181–5. 12803480
71. Hock MB, Kralli A. Transcriptional Control of Mitochondrial Biogenesis and Function. Annual Review of Physiology. 2009;71 : 177–203. doi: 10.1146/annurev.physiol.010908.163119 19575678
72. Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): Transcriptional coactivator and metabolic regulator. Endocrine Reviews. 2003;24(1):78–90. doi: 10.1210/er.2002-0012 12588810
73. Scarpulla RC. Nuclear activators and coactivators in mammalian mitochondrial biogenesis. Biochimica Et Biophysica Acta-Gene Structure and Expression. 2002;1576(1–2):1–14. doi: 10.1016/s0167-4781(02)00343-3
74. Scarpulla RC. Transcriptional activators and coactivators in the nuclear control of mitochondrial function in mammalian cells. Gene. 2002;286(1):81–9. doi: 10.1016/s0378-1119(01)00809-5 11943463
75. Dominy JE, Puigserver P. Mitochondrial Biogenesis through Activation of Nuclear Signaling Proteins. Cold Spring Harbor Perspectives in Biology. 2013;5(7). doi: 10.1101/cshperspect.a015008
76. Mootha VK, Handschin C, Arlow D, Xie XH, St Pierre J, Sihag S, et al. Err alpha and Gabpa/b specify PGC-1 alpha-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle (vol 101, pg 6570, 2004). Proceedings of the National Academy of Sciences of the United States of America. 2005;102(29):10405-. doi: 10.1073/pnas.0505194102
77. Villena JA, Vinas O, Mampel T, Iglesias R, Giralt M, Villarroya F. Regulation of mitochondrial biogenesis in brown adipose tissue: nuclear respiratory factor-2/GA-binding protein is responsible for the transcriptional regulation of the gene for the mitochondrial ATP synthase beta subunit. Biochemical Journal. 1998;331 : 121–7. 9512469
78. Yang ZF, Mott S, Rosmarin AG. The Ets transcription factor GABP is required for cell-cycle progression. Nature Cell Biology. 2007;9(3):339–U164. doi: 10.1038/ncb1548 17277770
79. Ristevski S, O'Leary DA, Thornell AP, Owen MJ, Kola I, Hertzog PJ. The ETS transcription factor GABP alpha is essential for early embryogenesis. Molecular and Cellular Biology. 2004;24(13):5844–9. doi: 10.1128/mcb.24.13.5844-5849.2004 15199140
80. Yang ZF, Drumea K, Mott S, Wang JL, Rosmarin AG. GABP Transcription Factor (Nuclear Respiratory Factor 2) Is Required for Mitochondrial Biogenesis. Molecular and Cellular Biology. 2014;34(17):3194–201. doi: 10.1128/mcb.00492-12 24958105
81. O'Leary DA, Pritchard MA, Xu DK, Kola I, Hertzog PJ, Ristevski S. Tissue-specific overexpression of the HSA21 gene GABP alpha: implications for DS. Biochimica Et Biophysica Acta-Molecular Basis of Disease. 2004;1739(1):81–7. doi: 10.1016/j.bbadis.2004.09.005
82. Cavailles V. Fine tuning of transcription by nuclear hormone receptors cofactors. M S-Medecine Sciences. 1998;14(10):1127–8.
83. Lee CH, Chinpaisal C, Wei LN. Cloning and characterization of mouse RIP140, a corepressor for nuclear orphan receptor TR2. Molecular and Cellular Biology. 1998;18(11):6745–55. 9774688
84. Cavailles V, Dauvois S, Danielian PS, Parker MG. INTERACTION OF PROTEINS WITH TRANSCRIPTIONALLY ACTIVE ESTROGEN-RECEPTORS. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(21):10009–13. doi: 10.1073/pnas.91.21.10009 7937828
85. White R, Morganstein D, Christian M, Seth A, Herzog B, Parker MG. Role of RIP140 in metabolic tissues: Connections to disease. Febs Letters. 2008;582(1):39–45. doi: 10.1016/j.febslet.2007.11.017 18023280
86. Seth A, Steel JH, Nichol D, Pocock V, Kumaran MK, Fritah A, et al. The transcriptional corepressor RIP140 regulates oxidative metabolism in skeletal muscle. Cell Metabolism. 2007;6(3):236–45. doi: 10.1016/j.cmet.2007.08.004 17767910
87. Izzo A, Manco R, Bonfiglio F, Cali G, de Cristofaro T, Patergnani S, et al. NRIP1/RIP140 siRNA-mediated Attenuation Counteracts Mitochondrial Dysfunction In Down Syndrome. Human Molecular Genetics. 2014;23(16):43. doi: 10.1093/hmg/ddu157
88. Galmiche L, Serre V, Beinat M, Assouline Z, Lebre AS, Chretien D, et al. Exome Sequencing Identifies MRPL3 Mutation in Mitochondrial Cardiomyopathy. Human Mutation. 2011;32(11):1225–31. doi: 10.1002/humu.21562 21786366
89. Miller C, Saada A, Shaul N, Shabtai N, Ben-Shalom E, Shaag A, et al. Defective mitochondrial translation caused by a ribosomal protein (MRPS16) mutation. Annals of Neurology. 2004;56(5):734–8. doi: 10.1002/ana.20282 15505824
90. Saada A, Shaag A, Amon S, Dolfin T, Miller C, Fuchs-Telem D, et al. Antenatal mitochondrial disease caused by mitochondrial ribosomal protein (MRPS22) mutation. Journal of Medical Genetics. 2007;44(12):784–6. doi: 10.1136/jmg.2007.053116 17873122
91. Serre V, Rozanska A, Beinat M, Chretien D, Boddaert N, Munnich A, et al. Mutations in mitochondrial ribosomal protein MRPL12 leads to growth retardation, neurological deterioration and mitochondrial translation deficiency. Biochimica Et Biophysica Acta-Molecular Basis of Disease. 2013;1832(8):1304–12. doi: 10.1016/j.bbadis.2013.04.014 23603806
92. O'Brien TW, O'Brien BJ, Norman RA. Nuclear MRP genes and mitochondrial disease. Gene. 2005;354 : 147–51. doi: 10.1016/j.gene.2005.03.026 15908146
93. Rotig A. Human diseases with impaired mitochondrial protein synthesis. Biochimica Et Biophysica Acta-Bioenergetics. 2011;1807(9):1198–205. doi: 10.1016/j.bbabio.2011.06.010 21708121
94. Kim JS, He LH, Lemasters JJ. Mitochondrial permeability transition: a common pathway to necrosis and apoptosis. Biochemical and Biophysical Research Communications. 2003;304(3):463–70. doi: 10.1016/s0006-291x(03)00618-1 12729580
95. Zheng BH, Mills AA, Bradley A. A system for rapid generation of coat color-tagged knockouts and defined chromosomal rearrangements in mice. Nucleic Acids Research. 1999;27(11):2354–60. 10325425
96. Magin T, McWhir J, Melton D. A new mouse embryonic stem cell line with good germ line contribution and gene targeting frequency. Nucleic Acids Res. 1992;20(14):3795–6. 1641353
97. Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19(2):185–93. 12538238
98. Westfall PH, Young SS. P-VALUE ADJUSTMENTS FOR MULTIPLE TESTS IN MULTIVARIATE BINOMIAL MODELS. Journal of the American Statistical Association. 1989;84(407):780–6. doi: 10.2307/2289666 12155379
99. de Hoon MJL, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinformatics. 2004;20(9):1453–4. doi: 10.1093/bioinformatics/bth078 14871861
100. Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biology. 2002;3(7). doi: 0034.1 10.1186/gb-2002-3-7-research0034 12184816
101. MD B. Measurement of mitochondrial proton motive force. In: BGaC CE, editor. Bioenergetics—A practical approach. Oxford, UK: IRL; 1995. p. 39–62.
102. Rolfe DFS, Hulbert AJ, Brand MD. CHARACTERISTICS OF MITOCHONDRIAL PROTON LEAK AND CONTROL OF OXIDATIVE-PHOSPHORYLATION IN THE MAJOR OXYGEN-CONSUMING TISSUES OF THE RAT. Biochimica Et Biophysica Acta-Bioenergetics. 1994;1188(3):405–16. doi: 10.1016/0005-2728(94)90062-0
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 3
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Clonality and Evolutionary History of Rhabdomyosarcoma
- Morphological Mutations: Lessons from the Cockscomb
- Maternal Filaggrin Mutations Increase the Risk of Atopic Dermatitis in Children: An Effect Independent of Mutation Inheritance
- Transcriptomic Profiling of Reveals Reprogramming of the Crp Regulon by Temperature and Uncovers Crp as a Master Regulator of Small RNAs
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy