
The RNA-Binding Protein QKI Suppresses Cancer-Associated Aberrant Splicing
Alternative pre-mRNA splicing is a key mechanism for increasing proteomic diversity and modulating gene expression. Emerging evidence indicates that splicing program is frequently deregulated during tumorigenesis, and cancer cells favor to produce protein isoforms that can promote growth and survival. Lung cancer is one of the most common cancers and the leading cause of cancer-related death worldwide. Although a number of lung cancer-related splicing events have been detected in several genome-wide analyses, much less is known about how aberrant splicing takes place in lung cancer and how it contributes to tumor development. In this study, we characterized the RNA-binding protein QKI as a new critical regulator of alternative splicing in lung cancer and as a potential marker for prognosis. Genome-wide analysis of QKI-dependent splicing by RNA-Seq identified some cancer-associated splicing changes as its targets. Our results demonstrate that QKI-5 inhibits cancer cell proliferation and prevents inappropriate activation of the Notch signaling pathway by regulating its key target, NUMB. We further showed that QKI-5 represses the inclusion of NUMB alternative exon through competing with a core splicing factor SF1. In summary, our data indicate that down-regulation of QKI causes aberrant splicing in lung cancer and suggest a novel tumor suppression mechanism involving QKI-mediated repression of Notch signaling.
Published in the journal:
. PLoS Genet 10(4): e32767. doi:10.1371/journal.pgen.1004289
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004289
Summary
Alternative pre-mRNA splicing is a key mechanism for increasing proteomic diversity and modulating gene expression. Emerging evidence indicates that splicing program is frequently deregulated during tumorigenesis, and cancer cells favor to produce protein isoforms that can promote growth and survival. Lung cancer is one of the most common cancers and the leading cause of cancer-related death worldwide. Although a number of lung cancer-related splicing events have been detected in several genome-wide analyses, much less is known about how aberrant splicing takes place in lung cancer and how it contributes to tumor development. In this study, we characterized the RNA-binding protein QKI as a new critical regulator of alternative splicing in lung cancer and as a potential marker for prognosis. Genome-wide analysis of QKI-dependent splicing by RNA-Seq identified some cancer-associated splicing changes as its targets. Our results demonstrate that QKI-5 inhibits cancer cell proliferation and prevents inappropriate activation of the Notch signaling pathway by regulating its key target, NUMB. We further showed that QKI-5 represses the inclusion of NUMB alternative exon through competing with a core splicing factor SF1. In summary, our data indicate that down-regulation of QKI causes aberrant splicing in lung cancer and suggest a novel tumor suppression mechanism involving QKI-mediated repression of Notch signaling.
Introduction
Lung cancer is one of the most common cancers and the leading cause of cancer-related death worldwide [1]. Due to the lack of detectable early-stage symptoms and limited treatment options, the 5-year survival rates remain poor for most patients [2]. Thus, more comprehensive investigations of gene expression alterations are needed for understanding the molecular mechanisms of lung tumorigenesis with a goal of identifying reliable earlier markers and effective therapeutic targets.
Alternative pre-mRNA splicing, the process by which multiple mRNA variants can be produced from a single gene, is a key mechanism for increasing proteomic diversity and modulating gene expression [3]. Misregulation of splicing underlies many human diseases, including cancer [4]–[6]. During the initiation and progression of cancer, the splicing program together with other layers of gene expression programs is subject to substantial alterations. A large fraction of RNA-binding proteins can function as splicing regulators to affect splice site selection through recognizing regulatory elements, located in either exons or introns, and interacting with spliceosomal factors or other splicing regulators [7]–[9]. The role of splicing regulators in cancer was best demonstrated by an SR protein, SRSF1, which functions as a proto-oncogene [10], [11]. Overexpression of SRSF1 resulted in the transformation of immortal fibroblasts and mammary epithelial cells partly through regulating alternative splicing of candidate genes involved in signal transduction and apoptosis. Other splicing regulators such as RBFOX2, hnRNP H, PTB have been shown to control cancer-associated splicing alterations that affect gene products participating in key cellular programs [12]–[14]. Many splicing factors exhibit differential expression between normal and tumor tissues [15], but their functions and targets during cancer development remain elusive.
Aberrant splicing has been implicated in lung tumorigenesis [16]. Indeed, a number of lung cancer-related splicing events have been detected in several genome-wide analyses using splicing sensitive microarray or deep sequencing technologies [17]–[21]. However, our understanding of the functionally important splicing events that contribute to tumorigenesis and the mechanisms that lead to aberrant splicing in lung cancer is very limited.
To search for splicing regulators that control lung-cancer associated splicing changes, we surveyed the expression of 59 known splicing regulators in microarray data collected from Gene Expression Omnibus (GEO) database [22]–[24] and found that the RNA-binding protein Quaking (QKI) is the most frequently down-regulated splicing factor in lung cancer tissues. QKI is a conserved STAR (signal transduction and activation of RNA) family protein that plays an essential role during embryonic and postnatal development [25]. QKI contains a STAR domain, which is composed of a maxi-KH RNA binding domain and two flanking QUA domains (QUA1 and QUA2), several SH3-binding sites, and a tyrosine-rich tail. The QKI gene encodes at least three protein isoforms (QKI-5, -6, and -7) that are generated by alternative splicing in mouse and human. QKI-5 isoform is a nucleus-cytoplasm shuttling protein, but found mostly in the nucleus [26]. QKI-6 and -7 isoforms are localized to the cytoplasm exclusively [27]. The expression of QKI proteins is developmentally regulated with high expression in adult lung, brain, heart, and testes [28], [29]. QKI proteins are involved in diverse aspects of RNA metabolism including pre-mRNA splicing [30]–[33], mRNA localization and transport [34], [35], mRNA and miRNA stability [35]–[40], translation [41], [42], and miRNA processing [43]. Based on SELEX and PAR-CLIP analyses, QKI has been shown to specifically bind an ACUAAY (Y = C or U) motif [44]–[46]. Recently, Hall et al showed that QKI-5 enhances Capzb exon 9 inclusion through recognizing ACUAA motifs downstream of exon 9 [32]. QKI-6 can also regulate alternative splicing in oligodendrocyte indirectly via controlling the translation or mRNA stability of a well-known splicing regulator, hnRNP A1 [39], [42]. However, the molecular mechanism underlying the direct role of QKI-5 in splicing regulation remains elusive.
In this study, we investigated the role of QKI during lung tumorigenesis. We found that QKI is frequently reduced in non-small cell lung cancer (NSCLC) and that its down-regulation is associated with shortened survival of patients. Through RNA-Seq analysis, we identified QKI as a master regulator of alternative splicing in lung cancer cells. Our results demonstrate that QKI inhibits lung cancer cell growth at least in part through regulating the alternative splicing of the Notch pathway regulator NUMB. Moreover, we show that QKI can inhibit splicing by selectively competing with a core splicing factor SF1 for targeting an authentic splicing signal, the branchpoint. Together, our data establish QKI as a critical regulator of splicing in lung cancer and present a new QKI/NUMB/Notch pathway in the regulation of cell proliferation.
Results
QKI is a prognostic marker that is frequently down-regulated in NSCLC
To identify splicing factors that control lung cancer-associated splicing changes, we analyzed mRNA expression levels of 59 known splicing factors in microarray data collected from 80 normal and 104 adenocarcinoma patient samples [22]–[24] and found that QKI is one of the splicing factors frequently down-regulated (Figure S1). By surveying public gene expression databases (http://www.oncomine.org), we also observed that the expression levels of QKI mRNA are dramatically down-regulated in three major NSCLC subtypes, including adenocarcinomas, squamous cell carcinomas, and large cell carcinomas (Figure 1A). To validate it, we first examined the protein levels of QKI in primary lung cancer samples. In 8 out of 10 paired adenocarcinoma patient tissues, the expression level of QKI-5, the dominant isoform in lung cells, is significantly reduced in tumor tissues, compared to adjacent normal tissues (Figure 1B). We next found that both QKI-5 mRNA and protein levels are down-regulated in a panel of human lung cancer cell lines, including A549, H1373, H520, and H358, compared with a bronchial epithelial cell, BEAS2B (Figure 1C and D). Importantly, using an online survival analysis tool (http://www.kmplot.com) [47], we found that QKI expression positively correlates with the overall survival of patients (Figure 1E). In particular, down-regulation of QKI in patients at stage I is significantly associated with a shortened survival rate (Figure 1F). These findings suggest that QKI may serve as an early-stage prognostic marker in the management of NSCLC.

QKI-5 inhibits lung cancer cell proliferation and transformation
To assess the role of QKI-5 in the pathogenesis of lung cancer, we made stable A549 and H520 cell lines overexpressing FLAG-tagged QKI-5 (Figure 2A and Figure S2A). Ectopic expression of QKI-5 in A549 and H520 cells showed a significantly slower proliferation than control cells as determined by the MTT assay (Figure 2B and Figure S2B). Furthermore, by performing anchorage-independent colony formation assay, we observed a dramatic decrease in the colony numbers from A549 and H520 cells overexpressing QKI-5 (Figure 2C and Figure S2C). These data indicate that QKI-5 is capable of inhibiting both cell proliferation and transformation in vitro.

To determine whether QKI-5 suppresses tumor growth in vivo, we injected A549 cells stably expressing FLAG-tagged QKI-5 into nude mice (n = 10, each group). Six weeks after injection, 100% of mice injected with the control A549 cells formed tumors. In contrast, only 30% of mice injected with A549 cells expressing exogenous QKI-5 generated tumors. Additionally, these tumors were much smaller in volume (Figure 2D and E). We conclude that QKI-5 inhibits cancer cell growth in vitro and in vivo.
QKI is a key regulator of alternative splicing in lung cancer cells
To understand the molecular basis for QKI function in lung cancer, we performed a genome-wide search for mRNA targets of QKI. To do this, QKI was depleted from BEAS2B cells using retroviruses expressing shRNAs targeting all three QKI isoforms (Figure 3A). Total RNAs were then isolated from control - (sh-Luc) or QKI-knockdown (sh-Q3) cells followed by sequencing using an Illumina GAII sequencer. In total, 78.4 and 76.5 million reads were generated for control - and QKI-knockdown samples, respectively; and ∼73% of reads were mapped to the human genome (hg19 version) uniquely. Using this method, we identified 213 genes whose mRNAs exhibited an over 2-fold change in abundance after QKI knockdown (RPKM> = 0.5). We also detected 799 events of splicing changes in 629 genes (Benjamini-Hochberg adjusted P-value<0.05, Figure 3B and Table S1). Among them, only 3 alternatively spliced genes had an over 2-fold change in the mRNA level, suggesting that the mRNA abundance and alternative splicing of most QKI targets are not co-regulated. To validate the RNA-Seq data, we performed semi-quantitative RT-PCR analyses for a subset of cassette exon-type splicing events. Among the 90 tested cassette exon events, 81 events (90%) displayed changes in splicing upon QKI depletion in BEAS2B cells, confirming our RNA-Seq results (Table S2). As representatives, we show 10 validated targets for which QKI functions either as a splicing activator or as a splicing repressor (Figure 3C).

As described above, we have found that QKI is down-regulated in NSCLC, which correlates with a poorer prognosis. To investigate the significance of QKI-mediated splicing regulation in cancer, we examined approximately 40 splicing events from Table S2 in tumor tissues in comparison with matching normal tissues from the same patients. RT-PCR analyses detected at least 25 alternative splicing events that underwent changes in the same direction as shown in BEAS2B cells upon QKI knockdown (Table S3). For example, the splicing patterns of ADD3, EXOC1, MYO18A, NFYA, and NUMB were changed in 4 pairs of adenocarcinoma (A1-4#) and 4 pairs of squamous cell carcinoma tissues (S1-4#), in which the expression levels of QKI are significantly reduced in tumor tissues compared to the adjacent normal tissues. However, the splicing patterns of these genes were not changed in 1 pair of adenocarcinoma tissues (A5#) in which comparable amount of QKI was detected between tumor and normal tissues (Figure 3D). These results suggest that down-regulation of QKI in NSCLC causes cancer-related splicing events.
QKI suppresses the Notch pathway through regulating the alternative splicing of NUMB
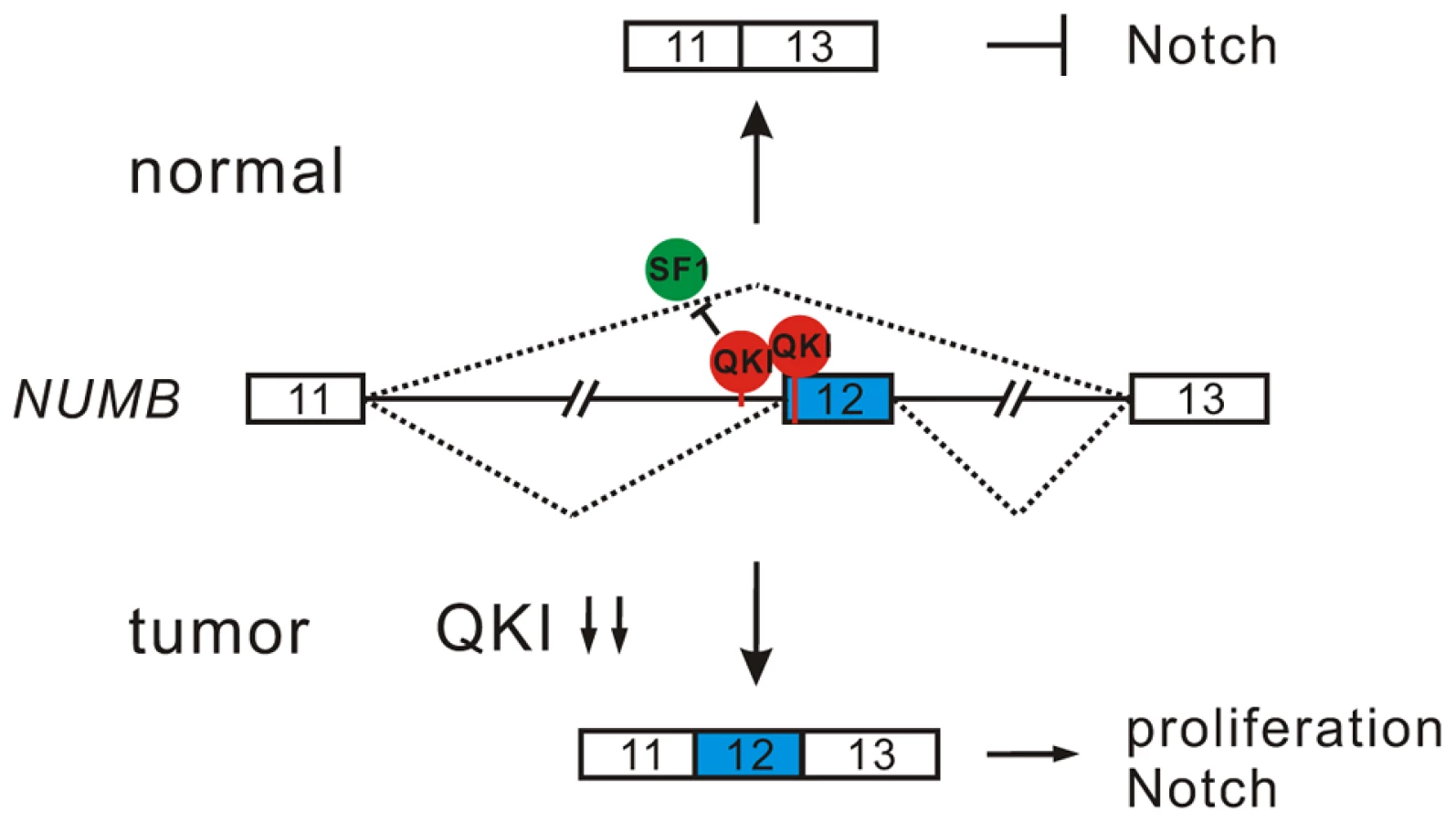
NUMB is an evolutionarily conserved signaling adaptor protein that plays a critical role in cell fate determination and functions as a negative regulator of the Notch signaling pathway [48]–[50]. The human NUMB gene generates four isoforms (p65, p66, p71, and p72) that result from the alternative splicing of two variable exons [51]. Exon 6 encodes 11 amino acids within the phosphotyrosine binding (PTB) domain, while exon 12 encodes 48 amino acids within the C-terminal proline-rich region (PRR). Interestingly, we found that NUMB is a target of QKI. Knockdown of QKI in BEAS2B cells stimulated exon 12 inclusion, and re-introduction of QKI-5 in QKI-depleted cells repressed exon 12 inclusion (Figure 4A and B). In contrast, QKI-5 did not regulate the splicing of exon 6 in NUMB (Figure S3).

To investigate the significance of QKI regulation on NUMB alternative splicing, we designed two shRNAs (sh-N1 and sh-N2) targeting different regions of NUMB exon 12. Knockdown of the exon 12-containing isoforms of NUMB in A549 cells significantly decreased cell proliferation (Figure 4C and D). Furthermore, introduction of a NUMB isoform containing exon 6 and exon 12 (p72) into A549 cells ectopically expressing QKI-5 partially abrogated the anti-proliferation activity of QKI-5. In contrast, expression of a NUMB isoform with exon 6 but without exon 12 (p66) augmented the inhibitory effect of QKI-5 on cell proliferation (Figure 4E and F). Thus, our data strongly suggest that QKI-5 suppresses cell proliferation, at least in part, through controlling the alternative splicing of NUMB.
To determine whether alternative splicing of NUMB regulates the Notch pathway, we examined the expression of the Notch targets, HES1, Hey1, and Hey2, by RT-qPCR analysis after isoform-specific knockdown of NUMB in A549 cells. Removal of the NUMB isoforms containing exon 12 resulted in a 2 - and 5-fold decrease of HES1 and Hey2 mRNA levels, respectively. However, the same treatment had little effect on Hey1 expression (Figure 4G). We next tested whether QKI also regulates the expression of these Notch targets through changing the splicing pattern of NUMB. Overexpression of QKI-5 in A549 cells inhibited HES1 and Hey2 expression, but did not affect Hey1 expression. Importantly, re-expression of the NUMB isoform p72 in QKI-5-overexpressing cells fully rescued Hey2 expression, while it slightly increased HES1 expression (Figure 4H). In contrast, introduction of the NUMB isoform p66 in QKI-5-overexpressing cells did not increase, but slightly decreased the expression of HES1 and Hey2. Together, these results indicate that QKI represses the expression of Notch targets through inhibiting the inclusion of NUMB exon 12.
To understand how the alternative splicing of NUMB regulates the Notch pathway, we investigated the ability of the two NUMB isoforms p72 and p66 to activate the Notch receptor in C2C12 cells, which express endogenous Notch1 receptor at a relatively high level. To do this, the released intracellular fragment of Notch (NICD) was detected after transiently transfecting C2C12 cells with the NUMB isoforms followed by EDTA treatment. As previously reported, production of the active form of Notch receptor (NICD) was stimulated in C2C12 upon EDTA treatment [52] (Figure 4I, lanes 1 and 2). Remarkably, NICD was not detected when the NUMB isoform p66 was overexpressed in C2C12 cells. In contrast, overexpression of the NUMB isoform p72 enhanced NICD release (Figure 4I, lanes 3 and 4). These data indicate that the two isoforms of NUMB have opposite effects on the Notch signaling activation, i.e. the NUMB isoform without exon 12 inhibits NICD activation, whereas the NUMB isoform carrying exon 12 promotes NICD activation. Taken together, our data demonstrate that QKI-5 suppresses the activation of the Notch pathway through regulating the alternative splicing of NUMB.
QKI-5 inhibits the inclusion of NUMB exon 12 by recognizing two QKI binding sites located upstream of and within exon 12
To understand how QKI regulates NUMB exon 12 splicing, we first checked whether QKI is associated with NUMB pre-mRNA in vivo by performing RNA immunoprecipitation (RIP) assay. Our data showed that QKI antibody specifically immunoprecipitated NUMB pre-mRNA but not the β-actin mRNA (Figure 5A). Next, we analyzed carefully the pre-mRNA sequence of NUMB. In the regions surrounding the 3′ splice site of intron 12, we found two QKI binding sites (AUUAAC and CUAAU), which are similar to the previously defined QKI binding consensus ACUAAY (Y = C or U) [44]–[46]. We made minigene constructs containing exons 11, 12, and 13 together with two shortened introns flanking exon 12. The second site or both sites were mutated to generate mut1 and mut2 constructs (Figure 5B). We then assayed the QKI binding affinities of the wildtype and mutant RNAs containing the last 56 nt of intron 12 and the first 25 nt of exon 12 sequences by a gel shift assay. The binding affinity of wildtype RNA to QKI is comparable to that of a control RNA, which carries a bipartite QKI consensus sequence determined previously using the SELEX method [45]. Compared with wildtype RNA, mut1 RNA exhibited reduced QKI-5 binding, while mut2 RNA lost QKI-5 binding completely (Figure 5C). This result indicates that both sites bind QKI-5. To determine whether QKI-5 inhibits exon 12 splicing through recognizing these two elements, we transfected NUMB minigene constructs together with a vector or a QKI-5 expression construct into HEK 293 cells. Overexpression of QKI-5 significantly inhibited exon 12 inclusion of the wildtype minigene (Figure 5D, lanes1 and 2). Mutation of the second site within exon 12 (mut1) increased exon 12 inclusion, indicating that the second site acts a silencing element for exon 12 inclusion. Ectopically expressed QKI-5 was still able to repress exon 12 inclusion, although the repression effect was less than that on the wildtype minigene (Figure 5D, lanes 3 and 4). This is not surprising, because QKI still binds but less efficiently to mut1 RNA compared to the wildtype RNA (Figure 5C). Mutation of the two sites resulted in a decrease of exon 12 inclusion (Figure 5D, lane 5). This is probably due to reduced binding of SF1 to mut2 pre-mRNA (Figure 5E, lanes 7–9). Interestingly, overexpression of QKI-5 had little effect on exon 12 inclusion of mut2 minigene (Figure 5D, lane 6). These data indicate that QKI inhibits NUMB exon 12 inclusion by recognizing two QKI binding sequences flanking the 3′ splice site of intron 12.

Since the first binding site (AUUAAC) is localized upstream of the polypyrimidine tract of intron 12 and since it is similar to the SF1 consensus ACUNAC (N = any nucleotides) sequence [53], we tested the SF1 binding activity of the wildtype and mutant RNAs by a gel shift assay. SF1 bound efficiently to the wildtype and mut1 RNAs, but the A to G mutation in mut2 RNA led to the reduction of SF1 binding (Figure 5E). We have shown that both SF1 and QKI bind to the first site (AUUAAC) upstream of the 3′ splice site (Figure 5C and E). Thus, our results suggest that QKI may repress exon 12 inclusion by competing with SF1 binding. We tested this idea by performing UV crosslink competition assay. QKI-5 inhibited SF1 binding to the wildtype RNA, but to a lesser extent for the mut1 RNA (Figure 5F, lanes 2–4 and 6–8). QKI-5 did not compete the weak SF1 binding to the mut2 RNA (Figure 5F, lanes 10–12). These data indicate that the two QKI binding sites may collaborate to enhance binding of one another, which explains why QKI competes less effectively with SF1 on the mut1 RNA. In addition, we show that QKI-5 did not affect U2AF65 binding to the wildtype and mutant RNAs (Figure 5G). Together, our results indicate that QKI-5 recognizes two binding sites located upstream of and within exon 12, and represses the inclusion of NUMB exon 12 by competing with SF1.
QKI-5 can inhibit splicing by selectively competing with SF1 for binding to the branchpoint
To gain further mechanistic insights into QKI-mediated splicing repression, we tested directly whether QKI inhibits splicing by competing with SF1 for targeting the branchpoint in an in vitro splicing system. We mutated the branchpoint sequence (ACUUAU, where the branchpoint adenosine is underlined) in MINX reporter pre-mRNA to the QKI binding consensus ACUAAU [54] (Figure 6A, mut1). Since it has been shown previously that QKI binds to the ACUAAY consensus, which is often accompanied by a so-called half site UAAY in close proximity [45], we also introduced a UAAU or UAAC site upstream or downstream of the ACUAAU motif (Figure 6A, mut2 and mut3). In the gel shift analysis, the wildtype MINX RNA bound poorly to the purified QKI-5. Mut2 and mut3 RNAs bound QKI more efficiently than mut1 at a low concentration of QKI-5, and all three RNAs formed larger RNA-protein complexes at a high concentration of QKI-5 (Figure 6B). These results indicate that the A4 in the consensus motif ACUAAY is crucial for QKI binding.

We next tested whether these mutations affect MINX splicing in HeLa nuclear extract with or without supplemented QKI-5 (Figure 6C and D). In HeLa nuclear extract alone, the splicing activity of mut1, mut2 and mut3 is comparable to that of wildtype (Figure 6C). This is likely due to the low abundance of QKI in HeLa nuclear extract. Addition of purified QKI-5 to the HeLa nuclear extract did not affect the splicing of wildtype MINX pre-mRNA, but inhibited the splicing of mut1, mut2 and mut3 pre-mRNAs (Figure 6D). Moreover, the result shows that QKI-5 inhibits splicing before the first step of splicing reaction, since production of the first exon was reduced after QKI-5 addition to the HeLa nuclear extract.
To determine whether QKI-5 affects the splicing of mutant pre-mRNAs by competing SF1 binding to branchpoint sequence, we conducted UV crosslink experiment. SF1 did not recognize a pre-mRNA in which the branchpoint sequence is destroyed (mutBP, Figure 6A), while SF1 interacted with the wildtype, mut1, mut2, and mut3 pre-mRNAs (Figure 6E). Notably, QKI-5 significantly inhibited SF1 binding to mut1, mut2, and mut3 pre-mRNAs, but had nearly no effect on SF1 binding to wildtype pre-mRNA (Figure 6E). Since the recognition of branchpoints by SF1 is essential for A complex formation, we constructed pre-mRNA substrates containing the 3′ half of MINX intron and the second exon and analyzed A complex assembly on these substrates. Addition of QKI-5 to the HeLa nuclear extract inhibited A complex assembly on the mutant pre-mRNAs, but had no effect on the wildtype pre-mRNA (Figure 6F). Thus, we conclude that QKI-5 can inhibit splicing by selectively competing with SF1 for binding to the branchpoint sequence.
Discussion
Emerging evidence indicates that splicing program is frequently deregulated during tumorigenesis, and cancer cells favor to produce protein isoforms that can promote growth and survival. However, how aberrant splicing takes place and how it contributes to tumor development are not well understood. In this study, we identified the RNA-binding protein QKI-5 as a new critical regulator of alternative splicing in NSCLC and as a potential marker for prognosis. Furthermore, we show that QKI-5 regulates the alternative splicing of NUMB, thereby inhibiting cancer cell proliferation and preventing the activation of the Notch signaling pathway. Our findings suggest a novel tumor suppression pathway and expand our understanding of splicing regulation in tumorigenesis.
Inappropriate activation of Notch signaling has been linked to a wide range of cancers including glioblastoma, colon, pancreatic, breast, and lung tumors [55], [56]. However, the molecular mechanisms of aberrant Notch activation in cancers are not well understood. In breast and lung cancers, there is frequently loss of NUMB-mediated suppression of Notch signaling due to ubiquitination-induced degradation of all NUMB protein isoforms [57], [58]. The authors of these two studies also noticed that when substantial levels of NUMB were detected in some NSCLC patient specimens, the Notch pathway was still highly activated. This implicates that additional mechanisms may lead to the dysregulation of NUMB-mediated repression of Notch signaling. Increased inclusion of NUMB exon 12 has been reported in several types of cancer including lung, ovarian, and breast cancers [12], [19], [20]. A previous study suggests that differential expression of NUMB isoforms has different effects on the Notch signaling [20]. However, how NUMB alternative splicing is regulated in cancer had remained unclear. Here, we show that alternative splicing of NUMB is regulated by QKI-5, and that this regulation is critically important for the control of cell proliferation (Figure 4E and F). Furthermore, our data clearly indicate that the two NUMB isoforms p72 and p66 have the opposite effects on the activation of the Notch pathway (Figure 4I). It is possible that NUMB isoform carrying exon 12, which encodes additional 48 aa in the protein-protein interaction domain PRR, may recruit other factors to stimulate NICD activation, or it may behave as a dominant negative form to antagonize the function of the NUMB isoform with shorter PRR domain. In summary, our results suggest a new QKI/NUMB/Notch pathway in the control of cell proliferation (Figure 7). Taken together that the alternative splicing of NUMB exon 12 is developmentally regulated [59]–[62] and that QKI is essential for embryonic development, it suggests that the QKI/NUMB/Notch pathway identified in this study may play a role during development.
In this work, we also show that QKI can either positively or negatively regulate exon inclusion (Figure 3C). To gain mechanistic insights into QKI-mediated splicing regulation, we generated an RNA map of QKI based on its known binding consensus sequence ACUAAY and our RNA-Seq data. This RNA map shows that QKI often activates exon inclusion when its binding sites are located in the downstream intron regions of the regulated exons, and represses exon inclusion when its binding sites are located within regulated exons or in the upstream intron regions (Figure S4). This indicates that QKI regulates alternative splicing in a position-dependent manner. The position-dependent effect of splicing regulators has emerged as a key mechanism in splicing regulation. QKI shares the same positional principles with several other splicing regulators including Nova, Fox1/2, and MBNL [63]–[66]. Recently, Hall et al showed that QKI depletion in myoblasts induces widespread alternative splicing changes and exhibits similar aforementioned positional effects on splicing regulation [32]. However, so far the mechanisms underlying the position-dependent splicing regulation remain to be investigated. A recent large-scale analysis of splicing lariats reveals that most human branchpoints are located between 18 and 35 nt upstream of the 3′ splice site [67]. In the QKI RNA map we generated, we noted that a peak of ACUAAY motif, which represents the potential branchpoint, is located within 40 nt upstream of the two 3′ splice sites in the randomly selected control pre-mRNAs (Figure S4). Remarkably, ACUAAY motif is significantly enriched within 40 nt upstream of QKI-repressed exons. This observation suggests that QKI binding sites overlap with the branchpoints upstream of QKI-repressed exons. During the initial step of spliceosome assembly, the branchpoint is first recognized by SF1. Both SF1 and QKI contain the KH and QUA2 domains which contribute to the sequence-specific RNA recognition. The optimal binding site of human SF1 (ACUNAC, where the branchpoint adenosine is underlined) is very similar to the QKI binding consensus (ACUAAY). Using minigene and in vivo splicing assay, we showed that QKI-5 inhibits the inclusion of NUMB exon 12 by competing with SF1 (Figure 5). In addition, although the RNA recognition motifs of SF1 and QKI are similar, we found that they display differential binding affinities to RNA elements containing similar sequences. Using in vitro reporter assay, we showed that QKI-5 can inhibit splicing by selectively competing with a core splicing factor SF1 for binding to the branchpoint (Figure 6). These findings suggest that QKI plays a vital role in fine-tuning the activity of SF1.
This study and those of others strongly suggest that QKI functions as a tumor suppressor [40], [68]. However, the functional targets of QKI in cancers are largely unknown. Our genome-wide analysis of QKI targets by RNA-Seq established QKI-5 as a master regulator of alternative splicing in lung cancer cells (Figure 3B). Down-regulation of QKI in NSCLC causes a number of cancer-associated splicing events (Figure 3D and Table S3). Beside NUMB, we also found other critical targets of QKI that have been previously implicated in tumorigenesis. For example, BIN1, a nucleocytoplasmic adaptor protein, functions as a tumor suppressor that directly binds and inhibits c-Myc. As shown in Figure S5, QKI-5 in BEAS2B cells induced the exclusion of BIN1 exon12A, which has been shown to abolish its binding to c-Myc and lose its anti-oncogenic activity in melanoma cells [69]. Since down-regulation of QKI is significantly associated with poor prognosis at early cancer stages (Figure 1F), it indicates that QKI and its targets identified in this study may serve as earlier markers for lung cancer. We also show that increasing QKI-5 levels in lung cancer cells suppresses cell proliferation and transformation both in vitro and in vivo, implicating that QKI is a potential drug target for the cancer treatment. In summary, our study demonstrates that QKI is a critical regulator of aberrant splicing in lung cancer and that the Notch signaling regulator, NUMB, is a key target of QKI in the control of cell proliferation.
Materials and Methods
Oligonucleotides
The sequences of all the oligonucleotides used in this study are listed in Table S4.
Plasmid construction
Plasmid construction is described in Protocol S1.
Ethics statement
All paired lung normal and tumor tissues were collected from informed, consenting patients in Fudan University Shanghai Cancer Center (Shanghai, China) from October 2007 to February 2012 with approval from the Institute Research Ethics Committee. All mice were treated according to the protocols approved by the Institutional Animal Care and Use Committee of the Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences.
Cell culture
The human bronchial epithelial BEAS2B cells were grown in Dulbecco's Modified Eagle's medium supplemented with 10% fetal bovine serum. The human lung cancer A549, H1373, H520 and H358 cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum.
Real-time quantitative reverse-transcription polymerase chain reaction (qRT-PCR)
Total RNA was extracted from patient tissues and cultured cells using Trizol (Invitrogen, USA) and reverse transcribed into first-strand cDNA from random hexamers using MMLV reverse transcriptase (Promega, USA). PCR was then performed using SYBR Green PCR Master Mix (Applied Biosystems, USA) on a 7500 Fast Real-Time PCR system according to manufacturer's instruction (Applied Biosystems, USA).
Lentivirus production and infection
To generate recombinant lentivirus, HEK 293T cells were transfected with lentiviral expression constructs together with two helper plasmids pVSVG and delta-R-8.2 using a calcium phosphate method. A549 and H520 cells were infected by recombinant lentiviruses and screened for stable expression of QKI-5 and NUMB in the presence of puromycin according to manufacturer's instruction (System Biosciences, USA).
Western blotting
To extract the protein from patient samples, normal and tumor tissues were homogenized in RIPA buffer containing 50 mM Tris-Cl pH 7.4, 150 mM NaCl, 1% NP-40, 1 mM EDTA-free protease inhibitor cocktail (Roche, Germany) plus 1.5 mM phenylmethylsulfonyl fluoride (PMSF). Lysates were collected following the removal of insoluble material from tissue extracts by centrifugation at 14,000 rpm for 20 min at 4°C. Protein lysates were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by gel transfer to a nitrocellulose membrane (BioRad, USA). The membranes were incubated first with the primary antibodies, and then with secondary antibodies coupled to horseradish peroxidase (HRP). Band signals were detected with an enhanced chemiluminescence (ECL) system (Thermo Scientific) and visualized by image analyzer (Fujifilm, Japan). The primary antibodies used for this study are anti-GAPDH (Kang Cheng Bio-tech, China), anti-QKI (Sigma, USA), anti-γ-tubulin (Sigma, USA), anti-FLAG (Sigma, USA), anti-NUMB (Cell Signaling, USA), anti-NICD (Val1744, Cell Signaling, USA), and anti-HA (Roche, Germany). The HRP-conjugated secondary antibodies anti-mouse IgG and anti-rabbit IgG were purchased from Promega.
MTT cell proliferation assay
Cells were seeded at a density of 2000/well in 24-well culture plates. After 24, 48, 72, 96, and 120 h of incubation, cells were treated with 3-(4, 5-methylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) with a final concentration of 5 µg/ul for 4 h. The resulting formazan was solubilized with dimethylsulfoxide (DMSO), and the absorption was measured at 570 nm using a spectrophotometer (Thermo Scientific, USA). The cell viability was expressed as the optical density at 570 nm.
Anchorage-independent soft agar colony assay
Cells were plated at a density of 10,000/well in a top layer of RPMI-1640 medium containing 10% FBS and 0.4% low melting point agar (Invitrogen, USA) which is over a bottom layer of RPMI-1640 medium containing 10% FBS and 1% low melting point agar. Each cell line was plated in triplicates. After 4 weeks, cells were stained with 0.5 ml crystal violet overnight. The colonies were counted from 3 random fields under an inverted microscope.
Tumorigenicity assay
Six-week-old male nude mice were injected subcutaneously with 1×106 cells of A549 stable cell lines. Tumor growth was monitored by caliper measurement once a week for at least 6 weeks. The tumor volume (cm3) was calculated using the following formula: 0.5× (L×W2), where L is tumor length and W is tumor width. Six weeks after inoculation, the mice were sacrificed by CO2 asphyxiation.
Activation of Notch1
C2C12 cells were transfected with a vector and two NUMB cDNA constructs using Lipofectamine 2000 reagent (Invitrogen, USA). 48 h after transfection, C2C12 cells were washed with PBS twice, and incubated for 15 min with pre-warmed PBS containing 5 mM EDTA. Treated cells were then washed with PBS and chased for 30 min in regular culture medium.
Protein purification
E. coli BL21 DE3 cells transformed with expression plasmids encoding GST-tagged SF1 (aa 1–361), GST-tagged U2AF65, and His-tagged QKI-5 were induced with 0.3 mM isopropyl beta-D-1-thiogalactopyranoside (IPTG) for 5 h at 37°C. Purification of GST - and His-tagged proteins was performed using glutathione-Sepharose 4B or Ni-NTA according to manufacturer's instructions (GE Healthcare, USA; QIAGEN, Germany).
In vitro splicing
In vitro splicing was performed as previously described [70]. Briefly, a 25 µl of splicing reaction was setup by incubating 10 ng of 32P-labeled RNA substrates with HeLa nuclear extract (Cil Biotech, Belgium) at 30°C.
RNA immunoprecipitation (RIP)
BEAS2B cells were harvested and lysed in lysis buffer (50 mM Tris-HCl, pH 7.4, 100 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, 1/100 protease inhibitor cocktail from Roche). After centrifuging at 10,000 g for 10 min, an aliquot (10%) of supernatant was removed and served as input. The remaining supernatant was immunoprecipitated with either a mouse IgG or a QKI antibody immobilized on Protein G Sepharose. The bound RNAs were washed extensively and isolated using TRIzol (Invitrogen, USA) and reverse transcribed into first-strand cDNA using random hexamers and MMLV reverse transcriptase (Promega, USA). PCRs were then performed using primers T7-NUMB-for/rev for detecting NUMB pre-mRNA and ACTB-for/rev for detecting β-actin mRNA. The resulting DNA fragments were separated on a 2% agarose gel and visualized by ethidium bromide staining.
Gel shift assay
DNA templates for in vitro transcription were obtained by PCR using primer sets T7-MINX-For/MINX-2, T7-NUMB-For/-Rev, and T7-NUMB-For/-mut-Rev. 10 ng of 32P-labeled RNAs were incubated with different molar excess of recombinant His-tagged QKI-5 and GST-tagged SF1 (aa 1–361) protein under standard splicing conditions at 30°C for 20 min. An aliquot of 10 µl was removed and incubated with 2 µl of tRNA (1 mg/ml) for 5 min. RNA-protein complexes were fractionated by a 5% native polyacrylamide gel and visualized with a phosphoimager (Fujifilm, Japan).
UV crosslinking
20 ng of 32P-labeled RNAs were incubated with recombinant His-tagged QKI-5 in the absence or presence of GST-tagged SF1 (aa 1–361) and U2AF65 under standard splicing conditions at 30°C for 20 min. UV crosslinking was done on ice for 20 min with 254-nm UV light. Unprotected RNAs were removed by RNase A at 37°C for 20 min. Crosslinked species were analyzed on a 12.5% SDS polyacrylamide gel and visualized with a phosphoimager (Fujifilm, Japan).
In vivo splicing
The day before transfection, 2×105 BEAS2B cells were seeded in 35 mm culture dishes. Minigene plasmids were transfected using Lipofectamine 2000. 24 h after transfection, total RNAs were isolated using Trizol Reagent. The first-strand cDNAs were reverse - transcribed using BGH-rev as primer and MMLV reverse-transcriptase following the manufacturer's instruction. PCR was performed using target gene-specific primers.
Spliceosome assembly
To prepare RNA substrates for A complex assembly, DNA templates for in vitro transcription were obtained by PCR using primer set T7-MINX-For/MINX-2. 10 ng of 32P-labeled RNA substrate was incubated in HeLa cell nuclear extract supplemented with different amount of recombinant His-tagged QKI-5 protein under standard splicing condition for 15 min. 5 µl of the reaction was removed and mixed with 1 µl of heparin (5 mg/ml) and 1 µl of glycerol (87%) to stop the reaction. The samples were separated by a 1.5% low melting agarose gel. After drying the gel, signals were visualized with a phosphoimager (Fujifilm, Japan).
RNA-Seq and data analysis
Total RNAs isolated from BEAS2B cells expressing a control shRNA or a QKI shRNA were subjected to single-end RNA-Seq using Illumina GAII platform according to the manufacturer's instruction. We performed RNA-Seq twice with read lengths 50 nt and 75 nt, individually.
Data analysis was carried out according to a previous publication [71]. Briefly, we first obtained the annotations of human protein coding genes from Ensembl database and generated splicing-junctions by joining every exon with all possible downstream exons within the same gene. To force an alignment overhang of at least 6 nt from one side of the junction to the other, the exon sequences on either side of the junction were 44 and 69 nt for 50 and 75 nt reads, respectively. Secondly, the RNA-Seq reads were aligned to the human genome (hg19) and the splicing-junctions by Bowtie allowing for up to three mismatches. The ambiguously aligned reads were excluded from further analysis. The junctions that were covered by at least two independent reads were reported. Thirdly, we searched for the alignments that gave the evidence of seven types of alternative splicing events: cassette exon (SE), retained intron (IR), alternative 5′ splicing site (A5SS), alternative 3′ splicing site (A3SS), mutually exclusive exon (MXE), alternative first exon (AFE), and alternative last exon (ALE). To identify the significantly changed alternative splicing events, a Fisher's exact test was performed for each event using a 2*2 contingency table consisting of the read counts from either the inclusion or exclusion isoforms in the control and treated sample. The RNA-Seq data are available at NCBI's GEO database under the accession number GSE55215.
Supporting Information
Zdroje
1. SiegelR, NaishadhamD, JemalA (2012) Cancer statistics, 2012. CA Cancer J Clin 62 : 10–29.
2. YouldenDR, CrambSM, BaadePD (2008) The international epidemiology of lung cancer: geographical distribution and secular trends. J Thorac Oncol 3 : 819–831.
3. NilsenTW, GraveleyBR (2010) Expansion of the eukaryotic proteome by alternative splicing. Nature 463 : 457–463.
4. CooperTA, WanL, DreyfussG (2009) RNA and disease. Cell 136 : 777–793.
5. VenablesJP (2006) Unbalanced alternative splicing and its significance in cancer. Bioessays 28 : 378–386.
6. DavidCJ, ManleyJL (2010) Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev 24 : 2343–2364.
7. BlackDL (2003) Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem 72 : 291–336.
8. MatlinAJ, ClarkF, SmithCW (2005) Understanding alternative splicing: towards a cellular code. Nat Rev Mol Cell Biol 6 : 386–398.
9. ChenM, ManleyJL (2009) Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat Rev Mol Cell Biol 10 : 741–754.
10. KarniR, de StanchinaE, LoweSW, SinhaR, MuD, et al. (2007) The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol 14 : 185–193.
11. AnczukówO, RosenbergAZ, AkermanM, DasS, ZhanL, et al. (2012) The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat Struct Mol Biol 19 : 220–228.
12. VenablesJP, KlinckR, KohC, Gervais-BirdJ, BramardA, et al. (2009) Cancer-associated regulation of alternative splicing. Nat Struct Mol Biol 16 : 670–676.
13. LefaveCV, SquatritoM, VorlovaS, RoccoGL, BrennanCW, et al. (2011) Splicing factor hnRNPH drives an oncogenic splicing switch in gliomas. EMBO J 30 : 4084–4097.
14. DavidCJ, ChenM, AssanahM, CanollP, ManleyJL (2010) HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 463 : 364–368.
15. GrossoAR, MartinsS, Carmo-FonsecaM (2008) The emerging role of splicing factors in cancer. EMBO Rep 9 : 1087–1093.
16. PioR, MontuengaLM (2009) Alternative splicing in lung cancer. J Thorac Oncol 4 : 674–678.
17. XiL, FeberA, GuptaV, WuM, BergemannAD, et al. (2008) Whole genome exon arrays identify differential expression of alternatively spliced, cancer-related genes in lung cancer. Nucleic Acids Res 36 : 6535–6547.
18. PioR, BlancoD, PajaresMJ, AibarE, DuranyO, et al. (2010) Development of a novel splice array platform and its application in the identification of alternative splice variants in lung cancer. BMC Genomics 11 : 352.
19. LangerW, SohlerF, LederG, BeckmannG, SeidelH, et al. (2010) Exon array analysis using re-defined probe sets results in reliable identification of alternatively spliced genes in non-small cell lung cancer. BMC Genomics 11 : 676.
20. Misquitta-AliCM, ChengE, O'HanlonD, LiuN, McGladeCJ, et al. (2011) Global profiling and molecular characterization of alternative splicing events misregulated in lung cancer. Mol Cell Biol 31 : 138–150.
21. SeoJS, JuYS, LeeWC, ShinJY, LeeJK, et al. (2012) The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Res 22 : 2109–2119.
22. SuLJ, ChangCW, WuYC, ChenKC, LinCJ, et al. (2007) Selection of DDX5 as a novel internal control for Q-RT-PCR from microarray data using a block bootstrap re-sampling scheme. BMC Genomics 8 : 140.
23. LandiMT, DrachevaT, RotunnoM, FigueroaJD, LiuH, et al. (2008) Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PLoS One 3: e1651.
24. WrageM, RuosaariS, EijkPP, KaifiJT, HollménJ, et al. (2009) Genomic profiles associated with early micrometastasis in lung cancer: relevance of 4q deletion. Clin Cancer Res 15 : 1566–1574.
25. FengY, BankstonA (2010) The star family member QKI and cell signaling. Adv Exp Med Biol 693 : 25–36.
26. WuJ, ZhouL, TonissenK, TeeR, ArtztK (1999) The quaking I-5 protein (QKI-5) has a novel nuclear localization signal and shuttles between the nucleus and the cytoplasm. J Biol Chem 274 : 29202–29210.
27. HardyRJ, LoushinCL, FriedrichVLJr, ChenQ, EbersoleTA, et al. (1996) Neural cell type-specific expression of QKI proteins is altered in quakingviable mutant mice. J Neurosci 16 : 7941–7949.
28. EbersoleTA, ChenQ, JusticeMJ, ArtztK (1996) The quaking gene product necessary in embryogenesis and myelination combines features of RNA binding and signal transduction proteins. Nat Genet 12 : 260–265.
29. KondoT, FurutaT, MitsunagaK, EbersoleTA, ShichiriM, et al. (1999) Genomic organization and expression analysis of the mouse qkI locus. Mamm Genome 10 : 662–669.
30. WuJI, ReedRB, GrabowskiPJ, ArtztK (2002) Function of quaking in myelination: regulation of alternative splicing. Proc Natl Acad Sci USA 99 : 4233–4238.
31. NovikovL, ParkJW, ChenH, KlermanH, JallohAS, et al. (2011) QKI-mediated alternative splicing of the histone variant MacroH2A1 regulates cancer cell proliferation. Mol Cell Biol 31 : 4244–4255.
32. HallMP, NagelRJ, FaggWS, ShiueL, ClineMS, et al. (2013) Quaking and PTB control overlapping splicing regulatory networks during muscle cell differentiation. RNA 19 : 627–638.
33. van der VeerEP, de BruinRG, KraaijeveldAO, de VriesMR, BotI, et al. (2013) Quaking, an RNA-binding protein, is a critical regulator of vascular smooth muscle cell phenotype. Circ Res 113 : 1065–1075.
34. LarocqueD, PilotteJ, ChenT, CloutierF, MassieB, et al. (2002) Nuclear retention of MBP mRNAs in the quaking viable mice. Neuron 36 : 815–829.
35. LiZ, ZhangY, LiD, FengY (2000) Destabilization and mislocalization of myelin basic protein mRNAs in quaking dysmyelination lacking the QKI RNA-binding proteins. J Neurosci 20 : 4944–4953.
36. LarocqueD, GalarneauA, LiuHN, ScottM, AlmazanG, et al. (2005) Protection of p27(Kip1) mRNA by quaking RNA binding proteins promotes oligodendrocyte differentiation. Nat Neurosci 8 : 27–33.
37. ZhaoL, KuL, ChenY, XiaM, LoPrestiP, et al. (2006) QKI binds MAP1B mRNA and enhances MAP1B expression during oligodendrocyte development. Mol Biol Cell 17 : 4179–4186.
38. DoukhanineE, GavinoC, HainesJD, AlmazanG, RichardS (2010) The QKI-6 RNA binding protein regulates actin-interacting protein-1 mRNA stability during oligodendrocyte differentiation. Mol Biol Cell 21 : 3029–3040.
39. ZearfossNR, ClingmanCC, FarleyBM, McCoigLM, RyderSP (2011) Quaking regulates Hnrnpa1 expression through its 3′ UTR in oligodendrocyte precursor cells. PLoS Genet 7: e1001269.
40. ChenAJ, PaikJH, ZhangH, ShuklaSA, MortensenR, et al. (2012) STAR RNA-binding protein Quaking suppresses cancer via stabilization of specific miRNA. Genes Dev 26 : 1459–1472.
41. SaccomannoL, LoushinC, JanE, PunkayE, ArtztK, et al. (1999) The STAR protein QKI-6 is a translational repressor. Proc Natl Acad Sci U S A 96 : 12605–12610.
42. ZhaoL, MandlerMD, YiH, FengY (2010) Quaking I controls a unique cytoplasmic pathway that regulates alternative splicing of myelin-associated glycoprotein. Proc Natl Acad Sci USA 107 : 19061–19066.
43. WangY, VogelG, YuZ, RichardS (2013) The QKI-5 and QKI-6 RNA binding proteins regulate the expression of microRNA 7 in glial cells. Mol Cell Biol 33 : 1233–1243.
44. RyderSP, WilliamsonJR (2004) Specificity of the STAR/GSG domain protein Qk1: implications for the regulation of myelination. RNA 10 : 1449–1458.
45. GalarneauA, RichardS (2005) Target RNA motif and target mRNAs of the Quaking STAR protein. Nat Struct Mol Biol 12 : 691–698.
46. HafnerM, LandthalerM, BurgerL, KhorshidM, HausserJ, et al. (2010) Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141 : 129–141.
47. GyőrffyB, SurowiakP, BudcziesJ, LánczkyA (2013) Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One 8: e82241.
48. GuoM, JanLY, JanYN (2007) Control of daughter cell fates during asymmetric division: interaction of Numb and Notch. Neuron 17 : 27–41.
49. McGillMA, McGladeCJ (2003) Mammalian numb proteins promote Notch1 receptor ubiquitination and degradation of the Notch1 intracellular domain. J Biol Chem 278 : 23196–23203.
50. McGillMA, DhoSE, WeinmasterG, McGladeCJ (2009) Numb regulates post-endocytic trafficking and degradation of Notch1. J Biol Chem 284 : 26427–26438.
51. DhoSE, FrenchMB, WoodsSA, McGladeCJ (1999) Characterization of four mammalian numb protein isoforms. Identification of cytoplasmic and membrane-associated variants of the phosphotyrosine binding domain. J Biol Chem 274 : 33097–33104.
52. RandMD, GrimmLM, Artavanis-TsakonasS, PatriubV, BlacklowSC, et al. (2000) Calcium depletion dissociates and activates heterodimeric notch receptors. Mol Cell Biol 20 : 1825–1835.
53. CorioniM, AntihN, TanackovicG, ZavolanM, KrämerA (2011) Analysis of in situ pre-mRNA targets of human splicing factor SF1 reveals a function in alternative splicing. Nucleic Acids Res 39 : 1868–1879.
54. GaurRK, ValcárcelJ, GreenMR (1995) Sequential recognition of the pre-mRNA branch point by U2AF65 and a novel spliceosome-associated 28-kDa protein. RNA 1 : 407–417.
55. RoyM, PearWS, AsterJC (2007) The multifaceted role of Notch in cancer. Curr Opin Genet Dev 17 : 52–59.
56. RanganathanP, WeaverKL, CapobiancoAJ (2011) Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer 11 : 338–351.
57. PeceS, SerresiM, SantoliniE, CapraM, HullemanE, et al. (2004) Loss of negative regulation by Numb over Notch is relevant to human breast carcinogenesis. J Cell Biol 167 : 215–221.
58. WesthoffB, ColalucaIN, D'ArioG, DonzelliM, TosoniD, et al. (2009) Alterations of the Notch pathway in lung cancer. Proc Natl Acad Sci USA 106 : 22293–22298.
59. VerdiJM, BashirullahA, GoldhawkDE, KubuCJ, JamaliM, et al. (1999) Distinct human NUMB isoforms regulate differentiation vs. proliferation in the neuronal lineage. Proc Natl Acad Sci USA 96 : 10472–10476.
60. ToriyaM, TokunagaA, SawamotoK, NakaoK, OkanoH (2006) Distinct functions of human numb isoforms revealed by misexpression in the neural stem cell lineage in the Drosophila larval brain. Dev Neurosci 28 : 142–155.
61. Bani-YaghoubM, KubuCJ, CowlingR, RochiraJ, NikopoulosGN, et al. (2007) A switch in numb isoforms is a critical step in cortical development. Dev Dyn 236 : 696–705.
62. KimKK, NamJ, MukouyamaYS, KawamotoS (2013) Rbfox3-regulated alternative splicing of Numb promotes neuronal differentiation during development. J Cell Biol 200 : 443–458.
63. UleJ, StefaniG, MeleA, RuggiuM, WangX, et al. (2006) An RNA map predicting Nova-dependent splicing regulation. Nature 444 : 580–586.
64. YeoGW, CoufalNG, LiangTY, PengGE, FuXD, et al. (2009) An RNA code for the FOX2 splicing regulator revealed by mapping RNA-protein interactions in stem cells. Nat Struct Mol Biol 16 : 130–137.
65. ZhangC, ZhangZ, CastleJ, SunS, JohnsonJ, et al. (2008) Defining the regulatory network of the tissue-specific splicing factors Fox-1 and Fox-2. Genes Dev 22 : 2550–2563.
66. DuH, ClineMS, OsborneRJ, TuttleDL, ClarkTA, et al. (2010) Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat Struct Mol Biol 17 : 187–193.
67. TaggartAJ, DeSimoneAM, ShihJS, FillouxME, FairbrotherWG (2012) Large-scale mapping of branchpoints in human pre-mRNA transcripts in vivo. Nat Struct Mol Biol 19 : 719–721.
68. YangG, FuH, ZhangJ, LuX, YuF, et al. (2010) RNA-binding protein quaking, a critical regulator of colon epithelial differentiation and a suppressor of colon cancer. Gastroenterology 138 : 231–240.
69. GeK, DuHadawayJ, DuW, HerlynM, RodeckU, et al. (1999) Mechanism for elimination of a tumor suppressor: aberrant splicing of a brain-specific exon causes loss of function of Bin1 in melanoma. Proc Natl Acad Sci USA 96 : 9689–9694.
70. WeiWJ, MuSR, HeinerM, FuX, CaoLJ, et al. (2012) YB-1 binds to CAUC motifs and stimulates exon inclusion by enhancing the recruitment of U2AF to weak polypyrimidine tracts. Nucleic Acids Res 40 : 8622–8636.
71. BrooksAN, YangL, DuffMO, HansenKD, ParkJW, et al. (2011) Conservation of an RNA regulatory map between Drosophila and mammals. Genome Res 21 : 193–202.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 4
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- The Sequence-Specific Transcription Factor c-Jun Targets Cockayne Syndrome Protein B to Regulate Transcription and Chromatin Structure
- The Mechanism of Gene Targeting in Human Somatic Cells
- Genetic Predisposition to In Situ and Invasive Lobular Carcinoma of the Breast
- Widespread Use of Non-productive Alternative Splice Sites in
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy