
A Synthetic Community Approach Reveals Plant Genotypes Affecting the Phyllosphere Microbiota
The leaves of plants are inhabited by a diverse community of microorganisms. These leaf inhabitants influence their hosts with respect to growth and resistance to abiotic and biotic stresses. Recent studies revealed that the bacterial communities associated with leaves undergo selection, resulting in conserved microbial communities. However, the factors that are involved in structuring of bacterial communities are not well understood. In order to uncover host genetic factors that determine the community composition and to exclude confounding environmental effects, we inoculated Arabidopsis thaliana with a synthetic bacterial community under controlled conditions We screened a panel of Arabidopsis mutants defective in various traits for alterations in community structure and abundance and were able to show that cuticle synthesis and ethylene perception affect the bacterial community. In addition, we identified plant ecotypes with drastic differences in the community composition. Our system can thus be used to identify additional host genes and to broaden insights into plant microbe interactions, potentially providing a basis for applied plant protection through the identification of traits that enhance growth of plant probiotic bacteria.
Published in the journal:
. PLoS Genet 10(4): e32767. doi:10.1371/journal.pgen.1004283
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004283
Summary
The leaves of plants are inhabited by a diverse community of microorganisms. These leaf inhabitants influence their hosts with respect to growth and resistance to abiotic and biotic stresses. Recent studies revealed that the bacterial communities associated with leaves undergo selection, resulting in conserved microbial communities. However, the factors that are involved in structuring of bacterial communities are not well understood. In order to uncover host genetic factors that determine the community composition and to exclude confounding environmental effects, we inoculated Arabidopsis thaliana with a synthetic bacterial community under controlled conditions We screened a panel of Arabidopsis mutants defective in various traits for alterations in community structure and abundance and were able to show that cuticle synthesis and ethylene perception affect the bacterial community. In addition, we identified plant ecotypes with drastic differences in the community composition. Our system can thus be used to identify additional host genes and to broaden insights into plant microbe interactions, potentially providing a basis for applied plant protection through the identification of traits that enhance growth of plant probiotic bacteria.
Introduction
The aerial parts of the plants, which are dominated by leaves, represent one of the largest terrestrial habitats for microorganisms [1]–[3]. This habitat, called the phyllosphere, is occupied by a diverse community of bacteria and fungi, which is important for plant health and growth [1]–[3]. Microorganisms in the phyllosphere can promote plant growth through the production of hormones. They can also be involved in plant protection, which is due to direct interactions of microorganisms through the production of antibiotic compounds and competition for resources [3]. Additionally, microorganisms may protect plants against pathogens by inducing systemic resistance [4], [5]. Commensals belonging to the genus Sphingomonas and their closely related species might represent part of the core phyllosphere community that protect plants against pathogens [6]. In addition, certain Pseudomonas strains have been shown to be plant protective agents [7].
Given the functional importance of the phyllosphere community on plant traits, it is relevant to understand the processes that are responsible for determining the composition of this community. This pertains to the fundamental question in community ecology of what principles underlie the assembly of strains into communities. A large body of theoretical and empirical work addresses this question (for a recent review see [8]), and has implied a vast number of different processes that play a role in community assembly [9] A recent synthesis groups this diversity of processes in just four classes – selection, drift, speciation and dispersal [10]. While this synthesis has not been specifically developed for microbial communities, it is well suited as a conceptual framework to describe and analyze the assembly of microbial communities [11]; furthermore, the suitability of the phyllosphere to test ecological concepts has been pointed out [12].
Here, we focus on selective factors that shape the assembly of the phyllosphere community, that is, on factors that have a consistent and reproducible effect on the composition of the microbial community on plant leaves. Previous studies have established that the bacterial phyllosphere communities are dominated by few phyla: Proteobacteria, Actinobacteria and Bacteroidetes [1], [13]. It is assumed that different factors contribute to the shaping of bacterial communities in the phyllosphere, including environmental cues, microbial interactions, the plant genotypes and phenotypes [1], and environmental factors such as temperature, water availability [14], [15], and geographic location (for example, [16]). The effects of plant factors on community composition have been demonstrated for leaf age [17], plant species [18], and cultivars [19]–[21]. Moreover, total population size is also affected by the plant species [22]. Several quantitative trait loci (QTL) have been identified as associated with bacterial diversity in corn [23] or with disease suppression in tomato [24]. However, no direct effects of specific genes on the composition of the phyllosphere community could be established in these studies. Using the model plant Arabidopsis thaliana, one study identified jasmonic acid synthesis as a factor driving epiphytic diversity in the phyllosphere [25], whereas another study did not reveal any effect for trichomes on bacterial diversity [26].
There are a number of plant factors that could potentially have selective effects on phyllosphere microbial communities. A first potentially important factor is the hosts' innate immune system. Plants recognize bacteria at two levels of their immune system: the first level is pattern-triggered immunity (PTI), whereby plant receptors recognize microbial-associated molecular patterns (MAMPS), for example, flagellin [27]; and the second level is effector-triggered immunity (ETI), where intracellular plant receptors recognize microbial effectors, which are virulence factors transferred by pathogens into the host cytoplasm to dampen PTI [28]. It is not known how plants discriminate between pathogenic and commensal or beneficial microorganisms and whether plant receptors recognize these non-pathogenic phyllosphere bacteria and trigger plant immune signaling networks downstream of PTI or ETI activation, with potential effects on community structure. The habitat is scarce in nutrients [1] so other potential traits that may influence the presence of plant-associated microorganisms include, for example, mutants in sugar transporters [29] or amino acid transporter [30]. Similarly, it is not known whether mutants defective in secondary metabolites used for defense, such as camalexin, glucosinolates [31], and flavonoids [32], affect their associated microbial populations. More specifically, mutants in pectin synthesis are hypothesized to affect the abundance of methylotrophic bacteria because methanol, as a by-product of pectin synthesis, is an important factor for bacterial growth under competitive conditions [1], [33].
In general, identifying host genetic factors using field experiments is challenging because of the confounding influence of the external environment as well as the diversity of natural microbial communities. To reduce environmental complexity, gnotobiotic model systems with well-defined communities represent an alternative approach. The advantages of such controlled systems are that they allow for reproducible experimentation and the use of molecular fingerprinting methods to characterize the defined community. In mice, bacterial synthetic communities have been successfully used to study how diet impacts the microbiota [34], [35]. To our knowledge, synthetic bacterial communities have not yet been used to identify plant host genotypes that shape the associated microbiota.
Using a synthetic community approach, we aimed to identify plant genetic factors that influence community composition and/or the bacterial abundance of the leaf-associated community of A. thaliana. A set of 55 plant mutants was screened for such phenotypic effects, resulting in the identification of three mutants with significant community alterations. In addition, of the nine natural accessions tested, four were found to modify community composition and abundance, indicating that natural variation can be used for future experiments with the synthetic community to identify novel host genes affecting phyllosphere microbiota.
Results
Establishment of a core synthetic community of the A. thaliana Col-0 phyllosphere
Knowledge of the overall composition of the microbiota of the A. thaliana phyllosphere [1], [13], [36] provides invaluable information for formulating a core microbiota based on cultivated model strains. A laboratory strain collection was used to establish a bacterial synthetic community, which allowed for the reproducible colonization of the phyllosphere in a gnotobiotic system. First, 20 strains were tested as individual inoculates. To be included in the synthetic community, strains were chosen that met two criteria: i) they were able to colonize the phyllosphere (higher than 107 CFU/g leaf fresh weight upon single inoculation, see Table 1), and ii) they did not induce disease symptoms nor cause a reduction in growth. In addition, the strains needed to represent the most abundant phylogenetic groups detected in the phyllosphere. Because Alphaproteobacteria is the most abundant sub-phylum in the phyllosphere of A. thaliana [1], four species were selected to represent this phylogenetic group: Sphingomonas phyllosphaerae and Sphingomonas sp. Fr1, which both have a plant-protective effect on A. thaliana [6], and Methylobacterium radiotolerans and M. extorquens PA1, which are efficient colonizers of the phyllosphere [37] but do not show a plant-protective effect [6]. In addition, two representatives of the Actinobacteria and one Betaproteobacteria were chosen for these abundant phyla of the phyllosphere (Table 1). Although five different strains of Gammaproteobacteria were tested as single isolates (Table S1), none could be included in the community because those strains either reduced plant growth or induced a strong disease phenotype under the experimental conditions. Mixing these strains with the rest of the community did not mask the disease phenotype.

To monitor changes in community composition in a high-throughput manner, automated ribosomal intergenic spacer analysis (ARISA) was used. Briefly, the 16S–23S rRNA intergenic spacer region was amplified by PCR using fluorescence-tagged universal primers. The PCR products were separated using a capillary sequencer. Each species in the community could be distinguished from the others based on its unique ARISA profile. Because some species were characterized by multiple peaks due to multiple 16S rRNA gene copies and variable length of the intergenic spacer regions, one representative peak was chosen for each species (Table 1). In addition, the peak area was normalized by the 16S rRNA gene copy number so that the abundance of each species was roughly proportional to the peak area in a semi-quantitative approach [38], [39] (for a validation experiment in which the DNA of one species was diluted against a mixture of DNA background see Figure S1).
A time course experiment was performed where the seven-member synthetic community (Table 1) was assessed immediately after spray inoculation of wild-type Col-0 plants and once a week for four weeks thereafter. Community composition was compared using the Bray-Curtis dissimilarity index (the more different two communities are, the closer to 1 their index is). Figure S2A shows that community comparisons of the inoculum to leaves sampled immediately after spraying were indistinguishable from community comparisons of leaf samples with each other (P>0.05). After one week, community comparisons of plant samples to inoculation solution was significantly greater relative to community comparisons made within plant samples (P = 0.0048). Paralleled determination of the population sizes by leaf washings revealed that the number of bacteria per plant was 1.18*103 immediately after spraying and increased steadily through time (Figure S2B). Based on the time-course experiment, we decided for the remainder of the study to harvest the leaves two weeks post-inoculation to allow growth of and competition between bacteria.
Using ARISA to analyze the community associated with wild-type Col-0 samples from ten independent biological experiments, we found that the synthetic community colonizes the phyllosphere in a reproducible manner (Figure S3). The average relative fluorescence intensity after colonization ranged from 3% for S. phyllosphaerae to 40% for Rhodococcus (Table 1).
A real-time qPCR method was developed to estimate the bacterial abundance in the phyllosphere. First, we confirmed that the PCR primers amplify the 16S rRNA gene in a linear fashion in the absence (Figure S4A) and in the presence of plant DNA (Figure S4B). The relative abundance of the 16S rRNA gene was calculated by normalizing with a plant gene and is proportional to the amount of bacterial DNA (Figure S4C).
Screening Arabidopsis genotypes
To identify plant host genetic factors that influence community composition, a priori candidate genes were selected from six different classes: cuticle and trichome, cell wall and pectin synthesis, secondary metabolism, sugar and amino acid transporters, defense signaling and pattern-triggered immunity (see Table S2 for a complete list of mutants). In addition, a small panel of natural accessions was screened. For each plant genotype, community composition and the 16S rRNA gene copy number were assessed (Figure 1).

A total of 55 A. thaliana mutants and accessions were tested in 10 independent experiments, each including Col-0 as a control and Landsberg erecta (Ler) as well as Wassilewskija (WS) when needed, dependent on the genetic background of each mutant. Results are shown in Figure S5 for individual screens and Figure S6 for all plant genotypes as a clustering analysis based on the Bray-Curtis dissimilarity index. We observed that the communities from some of the selected mutants clustered together and separate from Col-0. To compare independent experiments, community comparisons of each genotype to wild-type samples were calculated with the Bray-Curtis Dissimilarity index (Figure S7). Col-0 was used as the wild-type, except for mutants with a different background. In order to exclude genotypes where samples showed high variability from each other, community comparisons were also made within plant samples. Figure 2A shows the ratio of comparisons between each genotype and the corresponding wild-type over the comparison within each genotype. For each of these experiments, the 16S rRNA gene copy number was determined by qPCR (Figure 2B).
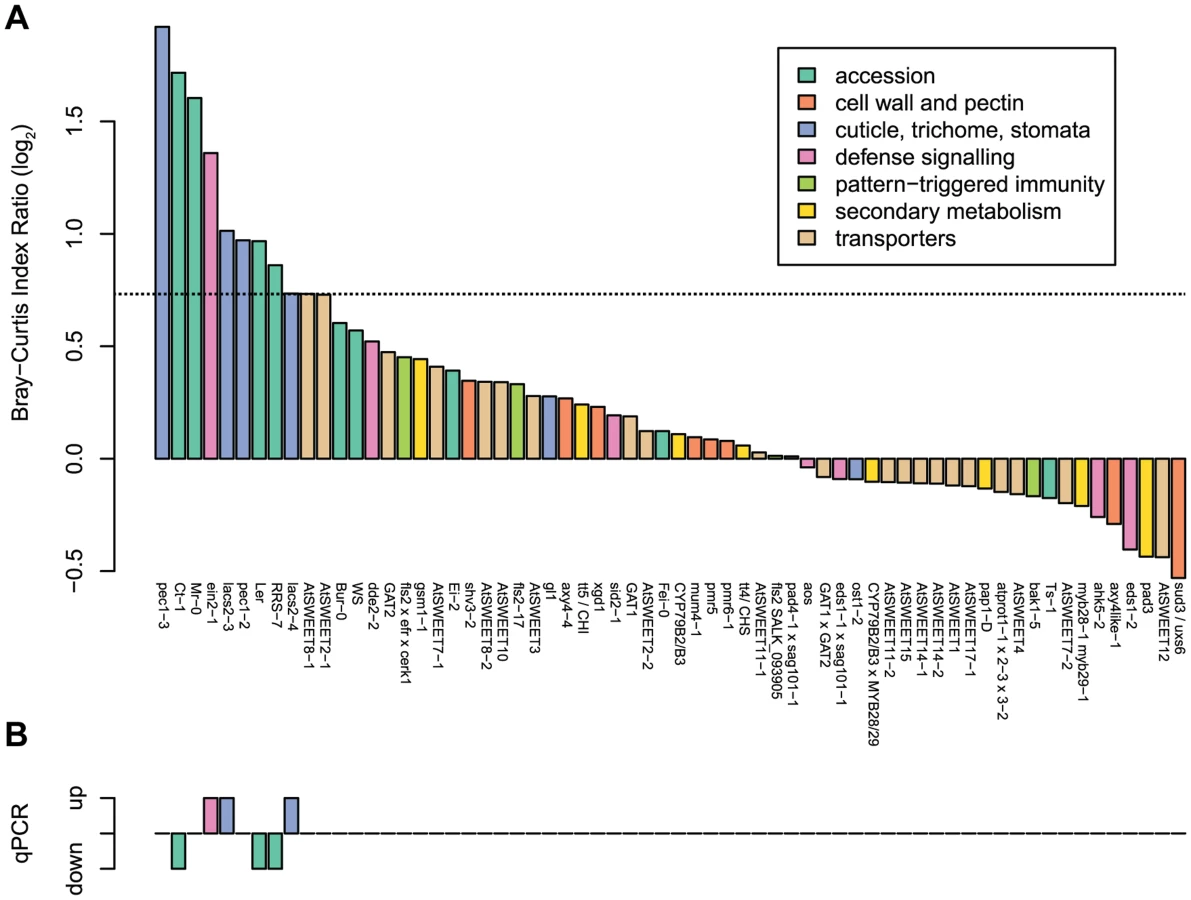
From the initial screen, the ten plant genotypes showing the highest dissimilarity to the wild-type (Figure 2A) and the six ecotypes with higher or lower bacterial abundances (Figure 2B) were selected for validation experiments using ARISA and qPCR. In addition, colony forming units were determined to verify altered community abundances and compositions using a cultivation-dependent method. From these sets of genotypes, 3 mutants (lacs2, pec1, and ein2) and 4 accessions (Mr-0, Ler, RRS-7, and Ct-1) with an altered community composition and/or overall abundance could be verified (see below). In contrast, results with the ‘sweet’ mutants could not be confirmed in validation experiments, and no genotype effect for community composition nor for bacterial abundance was observed (Figure S8).
Cuticle synthesis is an important factor for community composition and abundance
A different community composition of lacs2 and pec1 samples was confirmed with independent replicate experiments using two independent alleles each (Figure 3A). Multivariate analysis of variance confirmed a significant effect of these genotypes across replicate experiments (Table 2 and Table S3). LACS2 encodes for long-chain acyl-coenzyme A synthetase 2, an enzyme involved in cutin biosynthesis. The mutant plants are characterized by the absence of the cuticular membrane and a reduction of cuticular polyesters, which normally compose the wild-type cuticle [40]. The pec1 mutant has an intermediate phenotype between Col-0 and lacs2 and carries a mutation in an ATP-binding cassette transporter involved in the export of cuticle precursors [41]. Statistical tests indicated that there was a genotype effect for the relative fluorescence intensity (RFI) of Rhodococcus, Sphingomonas sp. Fr1, S. phyllosphaerae and Variovorax. Compared to the wild-type, both mutants harbored more Variovorax and less Rhodococcus, whereas pec1-3 had less Sphingomonas sp. Fr1. Using the qPCR method to estimate relative 16S rRNA gene copy numbers, we found that lacs2 harbored a higher bacterial abundance compared to the wild-type (Figure 2B). Multivariate analysis of variance indicated that there is a genotype effect for 16S rRNA gene copy numbers of both lacs2 and pec1 (Table 2 and Table S4) which was further confirmed by the fact that both lacs2 alleles carried a higher bacterial abundance compared to wild-type plants (Figure 4).



The results of the ARISA and qPCR analysis were partially confirmed using bacterial enumeration (Figure S9). For example, Variovorax cells were more abundant on the lacs2 and pec1 mutants, in-line the ARISA results. Contrary to the ARISA results, Sphingomonas were more abundant on both the pec1 and lacs2 mutants, suggesting that the relative abundance (ARISA results) does not necessarily reflect the absolute abundance (bacterial enumeration). This discrepancy might be due to differences in the protocols. ARISA profiles were determined using DNA extracted from the whole plants (both epiphytic and endophytic communities), whereas the protocol for leaf enumeration possibly extracts more epiphytic than endophytic bacteria.
Ethylene signaling is a factor involved in community composition
Independent replicate experiments confirmed a shift in the synthetic bacterial community colonizing the ein2 mutant plants compared to the wild-type plants (Figure 3B). The adonis test validated a significant effect of genotype across replicate experiments (Table 2 and Table S3). There was a plant genotype effect for the RFI of Variovorax, which was higher on the ein2 mutant. The RFI of other bacterial species were not affected by plant genotype. The ein2 mutant is ethylene insensitive [42] and carries a mutation in EIN2 [43], which plays a central role in the ethylene response, an important hormone for response to the environment and plant defense [44]. On the contrary, when we tested other mutants in the plant defense signaling pathways, we found that the jasmonate mutant aos and the salicylic acid mutant sid2 harbored a similar community composition compared to the wild-type plants (Figure 3B). In the initial screen, bacterial abundance, as measured by relative 16S rRNA gene copy number, was found to be higher on the ein2 mutant (Figure 2B). ANOVA confirmed a significant effect for this genotype (Table 2 and Table S4); however, this effect was weak and not significant for a single experiment (Figure S10). The ARISA results were confirmed using bacterial enumeration (Figure S9). Variovorax cells were more abundant on the ein2 mutant, whereas the abundance of other bacterial species was not affected by this mutation.
Different Arabidopsis accessions harbor different communities
The four Arabidopsis accessions identified in the first round of screening with a different community composition, Mr-0, Ler, Ct-1, and RRS-7, were confirmed in independent replicate experiments (Figure 5A). Multivariate analysis of variance confirmed a significant effect of each genotype on community composition (Table 2 and Table S3). Statistical tests revealed that there was a genotype effect for the RFI of Arthrobacter (lower in Mr-0), M. extorquens PA1 (higher in RRS-7), M. radiotolerans (higher in Ler and RRs-7), and Sphingomonas sp. Fr1 (lower in Ct-1, Ler and RRS-7. In addition, bacterial abundance was also different on the natural accessions as indicated by quantitative qPCR of the 16S rRNA gene (Table 2 and Table S4). The 16S rRNA gene copy numbers were higher on Mr-0 and lower on Ler, Ct-1, and RRS-7 (Figure 5B).

Discussion
In this study, we established a synthetic community approach to examine the effect of host genotype on the bacterial community composition and total abundance on plant leaves. We demonstrate that a model microbiota developed in a reproducible manner in the phyllosphere, allowing for the monitoring of perturbations dependent on the host's genotype. This system has several advantages, including its relatively short time scale and small space requirements, facilitating the independent biological repetition of experiments. Although in the future, more complex communities might be tested to link plant genotype to bacterial abundance, the low complexity community applied in this study has already allowed for the identification of three A. thaliana mutants with effects on community composition and/or total abundance. The plant mutants with the strongest effect on the associated bacteria were lacs2 and pec1, which are characterized by a more permeable cuticle compared to wild-type plants [40], [41], [45]. The cuticle is present on the outside surface of epidermal cells and is composed of the aliphatic polyester cutin and waxes [46]. In addition to its function as a diffusion barrier that diminishes water loss and as a protection against abiotic stresses, such as UV radiation [47], the cuticle also serves as a key interface for plant-microbe interactions. First, the cuticle represents the initial interaction surface with microorganisms colonizing the phyllosphere, therefore, features of the cuticle affect adhesion and, thus, microbial immigration [48]. Second, the cuticle controls transpiration, thus reducing water availability, which is a limiting factor for the growth of phyllosphere bacteria [15]. Third, the cuticle is involved in the transport of polar solutes and lipophilic organic compounds, thus reducing nutrient availability [47]. Furthermore, the question of whether components of the cuticle themselves can be used as substrate by bacteria is open. Evidence for the importance of the cuticle for the phyllosphere community comes from both sides of the study of plant-bacteria interactions. In terms of bacteria, epiphytic bacteria have been shown to alter the leaf surface permeability of isolated intact cuticles [49]. In terms of the plants, epicuticular wax synthesis has been shown to impact the colonization of single bacteria inoculates using maize mutant plants [50], and to impact the phyllosphere community composition, as shown very recently in Arabidopsis [51]. Moreover, epidermal thickness was correlated with the bacterial population size colonizing eight Mediterranean plant species [52].
In this study, we present evidence that cuticle permeability affects not only bacterial abundance but also community composition. We observed that lacs2 and pec1 mutants harbored more Variovorax. The average RFI of Variovorax in Col-0 was 16%; however, for the cuticle mutants, the average RFI ranged from 27% (lacs2-4) up to 54% (pec1-3). One explanation is that an enhanced permeability of the cuticle leads to an increase in nutrient availability, which favors the growth of the Betaproteobacterium Variovorax, the name of which notably refers to its ability to consume many different substrates. In contrast, the RFI of Rhodococcus decreased for both cuticle mutants, ranging from 23% (pec1-3) to 30% (lacs2-4) compared to 40% in Col-0. One possible explanation is that Rhodococcus feeds on cuticle components, which are less abundant in the mutants. In contrast, the abundance of both Methylobacterium strains was not different on the cuticle mutants, which might indicate that methanol availability is not affected by either mutation.
We found that the lacs2 mutant carried a higher bacterial abundance compared to both the wild-type and pec1 mutant (Figure 4). The pec1 mutant was shown to have an intermediate phenotype to lacs2 in terms of cuticular permeability, as measured using toluidine blue staining, sensitivity to herbicides and water loss [41]. Moreover, analysis of the cuticle ultrastructure revealed that pec1 retains a thick layer of electron-dense material, representing insoluble lipid-derived polymers, that is missing in lacs2 [40]. Analyses of the leaf polyester monomers demonstrated that the monomer composition of the lacs2 mutant was reduced by 20–25% compared to wild-type amounts [40], whereas only minor changes were observed for pec1 [41]. Interestingly, both lacs2 and pec1 mutants are more resistant to the fungal pathogen Botrytis cinerea, with the increased resistance proposed to be due to the induction of antifungal compounds by elicitors diffusing through the cuticle [40]. In contrast, lacs2 is more susceptible to avirulent Pseudomonas syringae, with the increased susceptibility hypothesized to be due to enhanced tissue collapse upon infiltration of the pathogen [53]. However, tissue collapse likely does not play a role in our study because the synthetic community was not syringe-infiltrated but rather sprayed onto the leaves. Therefore, we hypothesize that the higher bacterial abundance phenotype measured on lacs2 was due to the increased leaching of nutrients from this mutant compared to the wild-type and pec1 mutant.
The ethylene-insensitive mutant ein2 harbored a different community composition. In particular, Variovorax was more abundant in the phyllosphere of the mutant compared to the wild-type plants. Ethylene is a plant hormone with multiple roles in development, such as seed germination, fruit ripening, and root hair formation [54], [55]. In addition, ethylene modulates plant resistance to pathogens, which is dependent on the type of attacker. Generally, ethylene is found to reduce the appearance of diseases caused by necrotrophic and hemibiotrophic pathogens and to increase disease symptoms caused by other types of pathogens [56]. Interestingly, several pathogenic bacteria and fungi interfere with the plant defense-signaling pathway by producing ethylene [57], [58], which, in this case, can be considered a virulence factor. In contrast, some plant growth-promoting bacteria colonizing roots can degrade the compound 1-aminocyclopropane-1-carboxylic acid (ACC), the precursor of ethylene, using the enzyme ACC deaminase, thereby increasing root length [59]. In this study, we found the phyllosphere of the ein2 mutant to show quantitative differences in the community composition. In the A. thaliana rhizosphere, the ein2 mutant was found to harbor a lower bacterial abundance and was not associated with any changes in bacterial community composition [60]. In contrast, in the tobacco rhizosphere, Long et al. found a lower bacterial diversity and a different community in ethylene-insensitive transgenic plants compared to wild-type plants [61]. The distinct functions of ethylene in roots and leaves might affect the associated communities differently, for example, ethylene is involved in the formation of root hairs, which are hypothesized to serve as an entry point for the bacterial colonization of roots. Wild-type tobacco plants have also been demonstrated to have more root hairs compared to ethylene-insensitive plants and have been found to be associated with a different bacterial community [61].
Natural accessions of A. thaliana are a source of genetic diversity that can be harnessed to identify novel genes underlying phenotypic variations, which can then be used for quantitative trail locus (QTL) analyses of recombinant inbred lines (RIL) [62]. Furthermore, the recent development of cheaper SNP arrays and sequencing technology has enabled genome wide associations (GWA) in A. thaliana [63]. Natural accessions thus provide a valuable resource to begin identifying the intricate relationship of plants and associated microorganisms. Recently, using 16S rRNA gene amplicon sequencing of root samples and analyzing eight Arabidopsis accessions, Lundberg et al. [64] identified a small subset of 12 operational taxonomic units (OTU) out of 778 that showed host genotype dependent quantitative differences. In another study only one OTU of the root endophyte community showed significantly different quantitative enrichment when analyzing two Arabidopsis accessions [65]. Field and sample types (rhizosphere versus bulk soil) were found to be more important than the plant genotype for bacterial root community composition [65], [66].
Notably, here using a synthetic community approach applied to the phyllosphere of Arabidopsis we found that 4 out of the 9 accessions tested harbor a different community composition compared to Col-0, indicating that natural accessions offer significant potential for the discovery of new genes affecting community composition and/or abundance. In addition, several of the tested accessions harbor different Methylobacterium and Arthrobacter abundances that were not affected by the lacs2, pec1 and ein2 mutants. Interestingly, we found accessions with both lower (Ct-1, Ler, and RRS-7) and higher (Mr-0) bacterial abundances compared to the Col-0 accession. Future experiments with the model synthetic community and methods developed in this study will represent a valuable approach to map and identify novel genes affecting community composition and bacterial abundance.
Materials and Methods
Bacterial strains
The isolates and type strains used for the synthetic community are listed in Table 1. Variovorax sp., Arthrobacter sp. and Rhodococcus sp. were isolated from wild plants growing at different sites located near Madrid, Spain and described in [16]. The 16S rRNA genes of these three strains were sequenced for verification. The 16S rRNA gene copy number was determined by Southern blot.
Plant growth conditions
A. thaliana plants were cultivated on half-strength MS nutrient medium including vitamins and 0.55% plant agar (both from Duchefa, Haarlem, Netherlands) and supplemented with 1% sucrose. Seventy milliliters of medium was poured into microboxes outfitted with a XXL filter (Combiness, Nazareth, Belgium). To avoid leaves touching the medium, a sterile Lumox Film 25 (Sarstedt, Nümbrecht, Germany) with 6 holes (diameter, 4 mm) was placed on the agar surface. A. thaliana seeds were surface sterilized using a standard protocol [6] and stratified for 3 days (at 4°C) before being placed at the holes. Plants were grown under short-day conditions (a 9-h photoperiod) in a standard growth chamber, as previously described [6].
Plant inoculation
Both Methylobacterium strains were grown on mineral salt medium [67] supplemented with 0.5% succinate as a carbon source. All other strains were grown on nutrient broth (NB) without additional NaCl (Sigma-Aldrich, St. Louis, MO, USA). The two Sphingomonas strains were grown in liquid cultures, whereas the other strains were grown on solid media. Strains were grown at 28°C for three days (both Methylobacterium strains), two days (Rhodococcus sp., Variovorax sp., Arthrobacter sp. and S. phyllosphaerae), or one day (Sphingomonas sp. Fr1). Before inoculation, cells from the liquid cultures were washed once and resuspended in 10 mM MgCl2 solution. For cultures grown on solid medium, a loop of material was resuspended in 10 mM MgCl2 solution. The optical density at 600 nm (OD600) of each solution was adjusted to 0.2. The synthetic community was obtained by mixing the seven strains at 1∶1∶1∶1∶1∶1∶1 OD600. This solution was then diluted to an OD600 of 0.02. Plants were inoculated by spraying 200 µl of bacterial suspension with an airbrush paint gun [68].
Harvesting
For the time course experiment, samples were harvested immediately after spraying (four DNA pools, each with ten plants), and 1, 2, 3, and 4 weeks after inoculation (for each time point, four DNA pools, each with five plants). For the screening of plant genotypes, plants were harvested two weeks after inoculation (five plants from five different microboxes were pooled for one DNA extraction). In total, between 3 and 6 DNA pools per genotype were sampled depending on the experiment. Plants were taken out of the microboxes, and the roots and cotyledons were removed using flame-sterilized scalpels and forceps. In addition, for the validation experiments, the population size colonizing individual plants was determined using a dilution series (see below for the protocol).
DNA extraction
DNA was extracted from the plant tissues using the NucleoSpin Plant II kit (Macherey-Nagel, Düren, Germany). Plant samples were lyophilized, and one metal bead was added to each sample in a 2 ml - centrifuge tube before chilling in liquid nitrogen. Samples were homogenized for 2 min at 25 Hz using a Retsch TissueLyser (Retsch, Haan, Germany). The SDS-based lysis buffer PL2 was added to the homogenized samples, and the standard protocol according to the manifacturer's instructions was used thereafter.
ARISA
Primer 1492F (reverse complement of 1492R, [69]), and 23Sr [70] were used to amplify the 16S–23S rRNA intergenic space region. These primers were tested with DNA from plants grown axenically; no product was detected, indicating that they do not amplify mitochondrial or chloroplast DNA. Primer 1492F was labeled at the 5′ end with fluorescein. The reaction volume was 25 µl and contained 1-fold Phusion HF reaction buffer, 200 µM dNTP, 250 nM of each primer, 3% DMSO, 0.4 units of Phusion polymerase and approximately 20 ng DNA. The PCR program consisted of an initial denaturation step of 4 min at 94°C, followed by 35 cycles of denaturation at 94°C for 30 sec, annealing at 60°C for 30 sec, elongation at 72°C for 1 min followed by a final elongation step at 72°C for 7 min. PCR products were verified on agarose gels before preparing for ARISA. PCR products (2 µl, diluted 10 - and 20-fold) were mixed with 8 µl of HiDi formamide (Applied Biosystems) and 0.2 µl MapMarker 1000-ROX (BioVentures, Murfreesboro, USA). After denaturation at 95°C for five minutes, the samples were analyzed using a 3130 ABI capillary sequencer. Genemapper version 3.7 (Applied Biosystems) was used for the data analysis. Sizing tables were exported for analysis with R.
qPCR
The reaction volumes were 20 µl and contained 1-fold FastStart Universal SYBR green Master (Roche Applied Science), UltraPure DNase/RNase-free water (Life Technologies), 600 nM primer mix (16S rRNA primers) or 300 nM (plant gene primers), and approximately 5 ng DNA. The 16S rRNA gene was amplified using primers 799F [71] and 904R [72], see Table S5 for the primer sequences. Although these primers were designed to exclude organelle DNA, a product is amplified with DNA from plants grown axenically (deltaCt = 6–9 between inoculated Col-0 plants and axenically grown plants). This PCR product was cloned and sequenced and found to include sequences from organelles (chloroplast and mitochondria) confirming that the plants were axenic. Because we compared the samples with each other, we assumed that the small amount of plant DNA amplified by those primers did not affect our estimations. The primers ExpF1 and ExpF2 (Table S5) amplify the plant gene AT4G33380, a reference gene used for transcript normalization [73], which was used to normalize the 16S rRNA gene copy number. Controls, including no template, were included for each run. PCR assays were run in duplicate on a Rotorgene 3000 (Corbett Life Science, Qiagen). The PCR program consisted of a touchdown program with an initial denaturation step of 10 min at 95°C, followed by 35 cycles of 15 sec of denaturation at 95°C, 25 sec for the annealing step with the temperature decreasing from 65°C to 55°C (2 degrees per cycle), and 45 sec of elongation at 72°C followed by a melting curve analysis.: The raw data were exported directly from Corbett Research Software version 1.7 and imported into LinRegPCR version 12.8 [74] to determine cycle number to threshold (Ct) and efficiency (E). The 16S rRNA gene copy number was normalized to the plant gene AT4G33380 and calculated as follows: 16S rRNA/plant gene = Eplant gene Ct plant gene/E16SCt 16S, where Ct is the mean of the 2 duplicate reactions and E is the mean for all reactions with a particular primer pair for each run. To test for linearity of the qPCR method, a two-fold dilution series of Variovorax DNA was prepared (starting concentration 1 ng/µl DNA). qPCR was run following the standard protocol with 5 µl of bacterial DNA in the absence (Figure S4A) or presence of plant DNA (5 ng per reaction) (Figure S4B).
Enumeration of phyllosphere bacteria
Cell numbers were determined on randomly selected plants from several microboxes using a previously described protocol [6]. Briefly, leaves were washed in 100 mM phosphate buffer (pH 7) containing 0.2% Silwett by shaking for 15 minutes on a Retsch TissueLyser and sonicating for 5 minutes in a water bath. This protocol has been demonstrated to release both the epiphytic and endophytic Pseudomonas syringae associated with leaves [75]. Ten-fold dilution series were plated on different media. Naturally rifampicin-resistant Variovorax cell numbers were determined on King's B plates containing rifampicin (50 µg/ml). Sphingomonas cell numbers were determined on NB plates containing streptomycin (20 µg/ml). Methylobacterium extorquens PA1 could be distinguished from other members of the community because of its pink colony color on minimal media supplemented with 0.5% methanol. Total bacterial cell counts were determined by counting cell numbers on minimal media supplemented with 0.5% succinate.
Statistical analysis
The R statistical environment was used for all the statistical analyses and plotting (R Development Core Team; http://www.R-project.org). Relative fluorescent intensity (RFI) was calculated by dividing individual peak area by the total peak area for each sample using the R binning script written by Ramette [38]. Parameters for the script were: a range from 500 bp to 1000 bp, a minimum RFI cutoff of 0.2%, a window size of 5 bp and shift of 1 bp. One peak was chosen to represent each species of the community (Table 1). Furthermore, RFI was normalized by the 16S rRNA gene copy number to take into account variations in copy numbers among strains. Abundance tables were analyzed using the package vegan [76]. The function vegdist with default parameters (binary = FALSE) was used to calculate the Bray-Curtis index. This function calculates the Bray-Curtis index based on proportions of different types in a sample, in contrast to the binary version of the vegdist function, which only takes into account the presence and absence of different types. Hclust was used for hierarchical clustering with the average method. The Wilcox rank sum test was used to contrast Bray-Curtis indices of comparisons between plants samples and the inoculum and within plant samples (Figure S7). Multivariate analysis of variance was conducted with the vegan functions adonis [77] to assess the effect of genotype and experiment on community composition (Table 2 and Table S3). To test for the effect of genotype on the RFI of each bacterial population, a generalized linear model was used with a quasibinomial distribution to correct for overdispersion. The Dunnet's test was used to test significance in the comparison to the appropriate wild-type. Multivariate analysis of variance (ANOVA) was used to test the effect of genotype and experiment on 16S rRNA gene copy numbers (Table 2 and Table S4). After normalizing to the wild-type, the numbers were log-transformed. A qqplot indicated that the standardized residuals were normally distributed; furthermore, this was confirmed by the Shapiro-Wilk test. The Student's t-Test was used to test whether each mutant differed significantly from the wild-type. Similarly, ANOVA was used to test the effect of genotype on CFU/g FW, for which the data were log10 transformed (Figure S9). The P values were adjusted for multiple testing using the Bonferroni correction.
Nucleotide sequence accession numbers
The full-length 16S rRNA gene sequences of the Variovorax sp., Arthrobacter sp. and Rhodococcus sp. strains used in this study have been deposited in the European Nucleotide Archive under accession numbers HG737356, HG737357, HG737358, respectively.
Supporting Information
Zdroje
1. VorholtJA (2012) Microbial life in the phyllosphere. Nature Rev Microbiol 10 : 828–840.
2. BerendsenRL, PieterseCM, BakkerPA (2012) The rhizosphere microbiome and plant health. Trends Plant Sci 17 : 478–486.
3. BergG (2009) Plant-microbe interactions promoting plant growth and health: perspectives for controlled use of microorganisms in agriculture. Appl Microbiol Biotechnol 84 : 11–18.
4. ConrathU, BeckersGJ, FlorsV, Garcia-AgustinP, JakabG, et al. (2006) Priming: getting ready for battle. Mol Plant Microbe Interact 19 : 1062–1071.
5. PieterseCM, Van der DoesD, ZamioudisC, Leon-ReyesA, Van WeesSC (2012) Hormonal modulation of plant immunity. Annu Rev Cell Dev Biol 28 : 489–521.
6. InnerebnerG, KniefC, VorholtJA (2011) Protection of Arabidopsis thaliana against leaf-pathogenic Pseudomonas syringae by Sphingomonas strains in a controlled model system. Appl Environ Microbiol 77 : 3202–3210.
7. CabrefigaJ, BonaterraA, MontesinosE (2007) Mechanisms of antagonism of Pseudomonas fluorescens EPS62e against Erwinia amylovora, the causal agent of fire blight. Intern Microbiol 10 : 123–132.
8. WeiherE, FreundD, BuntonT, StefanskiA, LeeT, et al. (2011) Advances, challenges and a developing synthesis of ecological community assembly theory. Philos Trans R Soc Lond B Biol Sci 366 : 2403–2413.
9. LawtonJH (1999) Are there general laws in ecology? Oikos 84 : 177–192.
10. VellendM (2010) Conceptual synthesis in community ecology. Quart Rev Biol 85 : 183–206.
11. NemergutDR, SchmidtSK, FukamiT, O'NeillSP, BilinskiTM, et al. (2013) Patterns and processes of microbial community assembly. Microbiol Mol Biol Rev 77 : 342–356.
12. MeyerKM, LeveauJHJ (2012) Microbiology of the phyllosphere: a playground for testing ecological concepts. Oecologia 168 : 621–629.
13. DelmotteN, KniefC, ChaffronS, InnerebnerG, RoschitzkiB, et al. (2009) Community proteogenomics reveals insights into the physiology of phyllosphere bacteria. Proc Natl Acad Sci U S A 106 : 16428–16433.
14. LindowSE, BrandlMT (2003) Microbiology of the phyllosphere. Appl Environ Microbiol 69 : 1875–1883.
15. BeattieGA (2011) Water relations in the interaction of foliar bacterial pathogens with plants. Annu Rev Phytopathol 49 : 533–555.
16. KniefC, RametteA, FrancesL, Alonso-BlancoC, VorholtJA (2010) Site and plant species are important determinants of the Methylobacterium community composition in the plant phyllosphere. ISME J 4 : 719–728.
17. ErcolaniGL (1991) Distribution of epiphytic bacteria on olive leaves and the influence of leaf age and sampling time. Microbial Ecol 21 : 35–48.
18. YangCH, CrowleyDE, BornemanJ, KeenNT (2001) Microbial phyllosphere populations are more complex than previously realized. Proc Natl Acad Sci U S A 98 : 3889–3894.
19. RascheF, TrondlR, NaglreiterC, ReichenauerT, SessitschA (2006) Chilling and cultivar type affect the diversity of bacterial endophytes colonizing sweet pepper (Capsicum anuum L.). Can J Microbiol 52 : 1036–1045.
20. HunterPJ, HandP, PinkD, WhippsJM, BendingGD (2010) Both leaf properties and microbe-microbe interactions influence within-species variation in bacterial population diversity and structure in the Lettuce (Lactuca species) phyllosphere. Appl Environ Microbiol 76 : 8117–8125.
21. RastogiG, SbodioA, TechJJ, SuslowTV, CoakerGL, et al. (2012) Leaf microbiota in an agroecosystem: spatiotemporal variation in bacterial community composition on field-grown lettuce. ISME J 6 : 1812–1822.
22. KinkelLL, WilsonM, LindowSE (2000) Plant species and plant incubation conditions influence variability in epiphytic bacterial population size. Microb Ecol 39 : 1–11.
23. Balint-KurtiP, SimmonsSJ, BlumJE, BallareCL, StapletonAE (2010) Maize leaf epiphytic bacteria diversity patterns are genetically correlated with resistance to fungal pathogen infection. Mol Plant Microbe Interact 23 : 473–484.
24. SmithKP, HandelsmanJ, GoodmanRM (1999) Genetic basis in plants for interactions with disease-suppressive bacteria. Proc Natl Acad Sci U S A 96 : 4786–4790.
25. KniskernJM, TrawMB, BergelsonJ (2007) Salicylic acid and jasmonic acid signaling defense pathways reduce natural bacterial diversity on Arabidopsis thaliana. Mol Plant Microbe Interact 20 : 1512–1522.
26. ReisbergEE, HildebrandtU, RiedererM, HentschelU (2012) Phyllosphere bacterial communities of trichome-bearing and trichomeless Arabidopsis thaliana leaves. Ant Van Leeuwenhoek 101 : 551–560.
27. Gomez-GomezL, BollerT (2000) FLS2: an LRR receptor-like kinase involved in the perception of the bacterial elicitor flagellin in Arabidopsis. Mol Cell 5 : 1003–1011.
28. JonesJD, DanglJL (2006) The plant immune system. Nature 444 : 323–329.
29. ChenLQ, HouBH, LalondeS, TakanagaH, HartungML, et al. (2010) Sugar transporters for intercellular exchange and nutrition of pathogens. Nature 468 : 527–532.
30. GrallathS, WeimarT, MeyerA, GumyC, Suter-GrotemeyerM, et al. (2005) The AtProT family. Compatible solute transporters with similar substrate specificity but differential expression patterns. Plant Physiol 137 : 117–126.
31. BuerCS, MudayGK (2005) The transparent testa4 mutation prevents flavonoid synthesis and alters auxin transport and the response of Arabidopsis roots to gravity and light. Plant Cell 17 : 2614–2614.
32. BuerCS, IminN, DjordjevicMA (2010) Flavonoids: new roles for old molecules. J Integr Plant Biol 52 : 98–111.
33. SyA, TimmersAC, KniefC, VorholtJA (2005) Methylotrophic metabolism is advantageous for Methylobacterium extorquens during colonization of Medicago truncatula under competitive conditions. Appl Environ Microbiol 71 : 7245–7252.
34. FaithJJ, McNultyNP, ReyFE, GordonJI (2011) Predicting a human gut microbiota's response to diet in gnotobiotic mice. Science 333 : 101–104.
35. McNultyNP, YatsunenkoT, HsiaoA, FaithJJ, MueggeBD, et al. (2011) The impact of a consortium of fermented milk strains on the gut microbiome of gnotobiotic mice and monozygotic twins. Sci Transl Med 3 : 106ra106.
36. BodenhausenN, HortonMW, BergelsonJ (2013) Bacterial communities associated with the leaves and the roots of Arabidopsis thaliana. Plos One 8: e56329.
37. KniefC, FrancesL, VorholtJA (2010) Competitiveness of diverse Methylobacterium strains in the phyllosphere of Arabidopsis thaliana and identification of representative models, including M. extorquens PA1. Microb Ecol 60 : 440–452.
38. RametteA (2009) Quantitative community fingerprinting methods for estimating the abundance of operational taxonomic units in natural microbial communities. Appl Environ Microbiol 75 : 2495–2505.
39. KembelSW, WuM, EisenJA, GreenJL (2012) Incorporating 16S gene copy number information improves estimates of microbial diversity and abundance. PLoS Comput Biol 8: e1002743.
40. BessireM, ChassotC, JacquatAC, HumphryM, BorelS, et al. (2007) A permeable cuticle in Arabidopsis leads to a strong resistance to Botrytis cinerea. EMBO J 26 : 2158–2168.
41. BessireM, BorelS, FabreG, CarracaL, EfremovaN, et al. (2011) A member of the PLEIOTROPIC DRUG RESISTANCE family of ATP binding cassette transporters is required for the formation of a functional cuticle in Arabidopsis. Plant Cell 23 : 1958–1970.
42. AlonsoJM, HirayamaT, RomanG, NourizadehS, EckerJR (1999) EIN2, a bifunctional transducer of ethylene and stress responses in Arabidopsis. Science 284 : 2148–2152.
43. GuzmanP, EckerJR (1990) Exploiting the triple response of Arabidopsis to identify ethylene-related mutants. Plant Cell 2 : 513–523.
44. ZhaoQ, GuoHW (2011) Paradigms and paradox in the ethylene signaling pathway and interaction network. Mol Plant 4 : 626–634.
45. SchnurrJ, ShockeyJ, BrowseJ (2004) The acyl-CoA synthetase encoded by LACS2 is essential for normal cuticle development in Arabidopsis. Plant Cell 16 : 629–642.
46. Jeffree CE (2006) Biology of the Plant Cuticle. In: Riederer M, Muller C, editors. Annu Plant Rev. Oxford: Blackwell Publishing.
47. Riederer M (2006) Introduction: biology of the plant cuticle. In: Riederer M, Muller C, editors. Annual Plant Reviews Volume 23: Biology of the Plant Cuticle. Oxford: Blackwell Publishing.
48. Beattie GA (2002) Leaf surface waxes and the process of leaf colonization by microorganisms. In: Lindow SE, Hecht-Poinar EI, Elliott VJ, editors. Phyllosphere microbiology. St. Paul, Minnesota: The American Phytopathological Society. pp. 3–26.
49. SchreiberL, KrimmU, KnollD, SayedM, AulingG, et al. (2005) Plant-microbe interactions: identification of epiphytic bacteria and their ability to alter leaf surface permeability. New Phytologist 166 : 589–594.
50. MarcellLM, BeattieGA (2002) Effect of leaf surface waxes on leaf colonization by Pantoea agglomerans and Clavibacter michiganensis. Mol Plant Microbe Interact 15 : 1236–1244.
51. ReisbergEE, HildebrandtU, RiedererM, HentschelU (2013) Distinct phyllosphere bacterial communities on Arabidopsis wax mutant leaves. Plos One 8: e78613.
52. YadavRKP, KaramanoliK, VokouD (2005) Bacterial colonization of the phyllosphere of Mediterranean perennial species as influenced by leaf structural and chemical features. Microb Ecol 50 : 185–196.
53. TangDZ, SimonichMT, InnesRW (2007) Mutations in LACS2, a long-chain acyl-coenzyme a synthetase, enhance susceptibility to avirulent Pseudomonas syringae but confer resistance to Botrytis cinerea in Arabidopsis. Plant Physiology 144 : 1093–1103.
54. BleeckerAB, KendeH (2000) Ethylene: A gaseous signal molecule in plants. Annu Rev Cell Dev Biol 16 : 1–18.
55. MudayGK, RahmanA, BinderBM (2012) Auxin and ethylene: collaborators or competitors? Trends Plant Sci 17 : 181–195.
56. van LoonLC, GeraatsBPJ, LinthorstHJM (2006) Ethylene as a modulator of disease resistance in plants. Trends Plant Sci 11 : 184–191.
57. FukudaH, OgawaT, TanaseS (1993) Ethylene production by microorganisms. Adv Microbial Physiol 35 : 275–306.
58. VolkschB, WeingartH (1998) Toxin production by pathovars of Pseudomonas syringae and their antagonistic activities against epiphytic microorganisms. J Basic Microbiol 38 : 135–145.
59. SaleemM, ArshadM, HussainS, BhattiAS (2007) Perspective of plant growth promoting rhizobacteria (PGPR) containing ACC deaminase in stress agriculture. J Indust Microbiol Biotechnol 34 : 635–648.
60. DoornbosRF, GeraatsBPJ, KuramaeEE, Van LoonLC, BakkerPAHM (2011) Effects of jasmonic acid, ethylene, and salicylic acid signaling on the rhizosphere bacterial community of Arabidopsis thaliana. Mol Plant Microbe Interact 24 : 395–407.
61. LongHH, SonntagDG, SchmidtDD, BaldwinIT (2010) The structure of the culturable root bacterial endophyte community of Nicotiana attenuata is organized by soil composition and host plant ethylene production and perception. New Phytologist 185 : 554–567.
62. KoornneefM, Alonso-BlancoC, VreugdenhilD (2004) Naturally occurring genetic variation in Arabidopsis thaliana. Annu Rev Plant Biol 55 : 141–172.
63. AtwellS, HuangYS, VilhjalmssonBJ, WillemsG, HortonM, et al. (2010) Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature 465 : 627–631.
64. LundbergDS, LebeisSL, ParedesSH, YourstoneS, GehringJ, et al. (2012) Defining the core Arabidopsis thaliana root microbiome. Nature 488 : 86–90.
65. BulgarelliD, RottM, SchlaeppiK, Ver Loren van ThemaatE, AhmadinejadN, et al. (2012) Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 488 : 91–95.
66. PeifferJA, SporA, KorenO, JinZ, TringeSG, et al. (2013) Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc Natl Acad Sci U S A 110 : 6548–6553.
67. PeyraudR, KieferP, ChristenP, MassouS, PortaisJC, et al. (2009) Demonstration of the ethylmalonyl-CoA pathway by using 13C metabolomics. Proc Natl Acad Sci U S A 106 : 4846–4851.
68. Remus-EmsermannMN, TeconR, KowalchukGA, LeveauJH (2012) Variation in local carrying capacity and the individual fate of bacterial colonizers in the phyllosphere. ISME J 6 : 756–765.
69. Lane DS (1990) 16S and 23S rRNA sequencing. In: Stackebrandt E, Goodfellow M, editors. Nucleic acid techniques in bacterial systematics. New York: John Wiley and Sons. pp. 115–148.
70. BornemanJ, TriplettEW (1997) Molecular microbial diversity in soils from eastern Amazonia: Evidence for unusual microorganisms and microbial population shifts associated with deforestation. Appl Environ Microbiol 63 : 2647–2653.
71. CheliusMK, TriplettEW (2001) The diversity of archaea and bacteria in association with the roots of Zea mays L. Microb Ecol 41 : 252–263.
72. HodkinsonBP, LutzoniF (2009) A microbiotic survey of lichen-associated bacteria reveals a new lineage from the Rhizobiales. Symbiosis 49 : 163–180.
73. CzechowskiT, StittM, AltmannT, UdvardiMK, ScheibleWR (2005) Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol 139 : 5–17.
74. RuijterJM, RamakersC, HoogaarsWM, KarlenY, BakkerO, et al. (2009) Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res 37: e45.
75. TorneroP, DanglJL (2001) A high-throughput method for quantifying growth of phytopathogenic bacteria in Arabidopsis thaliana. Plant J 28 : 475–481.
76. Oksanen J, Blanchet FG, Kindt R, Legendre P, O'Hara RB, et al. (2011) Vegan: Community Ecology Package. R package version 1.17–10.
77. AndersonMJ (2001) A new method for non-parametric multivariate analysis of variance. Austral Ecol 26 : 32–46.
78. ItoH, IizukaH (1971) Taxonomic studies on a radio-resistant Pseudomonas. XII. Studies on microorganisms of cereal grain. Agricultural Biol Chem 35 : 1566–1571.
79. RivasR, AbrilA, TrujilloME, VelazquezE (2004) Sphingomonas phyllosphaerae sp nov., from the phyllosphere of Acacia caven in Argentina. Int J System Evol Microbiol 54 : 2147–2150.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 4
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- The Sequence-Specific Transcription Factor c-Jun Targets Cockayne Syndrome Protein B to Regulate Transcription and Chromatin Structure
- The Mechanism of Gene Targeting in Human Somatic Cells
- Genetic Predisposition to In Situ and Invasive Lobular Carcinoma of the Breast
- Widespread Use of Non-productive Alternative Splice Sites in
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy