
Reduction of NADPH-Oxidase Activity Ameliorates the Cardiovascular Phenotype in a Mouse Model of Williams-Beuren Syndrome
A hallmark feature of Williams-Beuren Syndrome (WBS) is a generalized arteriopathy due to elastin deficiency, presenting as stenoses of medium and large arteries and leading to hypertension and other cardiovascular complications. Deletion of a functional NCF1 gene copy has been shown to protect a proportion of WBS patients against hypertension, likely through reduced NADPH-oxidase (NOX)–mediated oxidative stress. DD mice, carrying a 0.67 Mb heterozygous deletion including the Eln gene, presented with a generalized arteriopathy, hypertension, and cardiac hypertrophy, associated with elevated angiotensin II (angII), oxidative stress parameters, and Ncf1 expression. Genetic (by crossing with Ncf1 mutant) and/or pharmacological (with ang II type 1 receptor blocker, losartan, or NOX inhibitor apocynin) reduction of NOX activity controlled hormonal and biochemical parameters in DD mice, resulting in normalized blood pressure and improved cardiovascular histology. We provide strong evidence for implication of the redox system in the pathophysiology of the cardiovascular disease in a mouse model of WBS. The phenotype of these mice can be ameliorated by either genetic or pharmacological intervention reducing NOX activity, likely through reduced angII–mediated oxidative stress. Therefore, anti-NOX therapy merits evaluation to prevent the potentially serious cardiovascular complications of WBS, as well as in other cardiovascular disorders mediated by similar pathogenic mechanism.
Published in the journal:
. PLoS Genet 8(2): e32767. doi:10.1371/journal.pgen.1002458
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002458
Summary
A hallmark feature of Williams-Beuren Syndrome (WBS) is a generalized arteriopathy due to elastin deficiency, presenting as stenoses of medium and large arteries and leading to hypertension and other cardiovascular complications. Deletion of a functional NCF1 gene copy has been shown to protect a proportion of WBS patients against hypertension, likely through reduced NADPH-oxidase (NOX)–mediated oxidative stress. DD mice, carrying a 0.67 Mb heterozygous deletion including the Eln gene, presented with a generalized arteriopathy, hypertension, and cardiac hypertrophy, associated with elevated angiotensin II (angII), oxidative stress parameters, and Ncf1 expression. Genetic (by crossing with Ncf1 mutant) and/or pharmacological (with ang II type 1 receptor blocker, losartan, or NOX inhibitor apocynin) reduction of NOX activity controlled hormonal and biochemical parameters in DD mice, resulting in normalized blood pressure and improved cardiovascular histology. We provide strong evidence for implication of the redox system in the pathophysiology of the cardiovascular disease in a mouse model of WBS. The phenotype of these mice can be ameliorated by either genetic or pharmacological intervention reducing NOX activity, likely through reduced angII–mediated oxidative stress. Therefore, anti-NOX therapy merits evaluation to prevent the potentially serious cardiovascular complications of WBS, as well as in other cardiovascular disorders mediated by similar pathogenic mechanism.
Introduction
Williams-Beuren syndrome (WBS [MIM 194050]) is a developmental disorder with multisystemic manifestations and a prevalence of ∼1/10,000 newborns, caused by a segmental aneusomy of 1.55–1.83 Mb at chromosomal band 7q11.23, which includes ELN (coding for elastin [MIM 130160]) and 25–27 additional genes [1], [2]. The recurrent WBS deletion common to most patients is mediated by nonallelic homologous recombination between regional segmental duplications that flank the WBS critical region [3]. In addition to distinctive craniofacial characteristics and mild mental retardation with social disinhibition and hyperacusis, a hallmark feature of WBS is a generalized arteriopathy presenting as narrowing of the large elastic arteries [4]. Histological characterization of arterial vessel walls of WBS patients showed increased number and disorganized lamellar structures, fragmented elastic fibers, and hypertrophy of smooth muscle cells [5]. This large arterial vessel remodeling which is a consequence of abnormalities in vascular development, is thought to be responsible for the cardiovascular disease manifested in 84% of WBS patients [4], [6]. Identical vascular features, most prominently supravalvular aortic stenosis, are also found in patients with heterozygous deletions or disruptions of the ELN gene, implicating elastin haploinsufficiency in this phenotype [5], [7]. The arteriopathy is the main cause of serious morbidity in WBS, including systemic hypertension and possible complications such as stroke, cardiac ischemia, and sudden death [8], [9].
Animal models provide further evidence for elastin deficiency as the main cause of cardiovascular disease in WBS, underscoring the prominent role of the elastic matrix in the morphogenesis and homeostasis of the vessel wall [10]. Heterozygous knockout mice with only one copy of the Eln gene reproduce many of the alterations observed in the WBS vascular phenotype [11], [12]. Hypertension is a consistent feature of Eln+/− mice, associated with elevated plasma renin activity (PRA) and angiotensin II (angII) levels, that can be blocked by the administration of angII type 1 receptor (AT1R) antagonists [13]. In addition to direct effects on the vasculature, many of the cellular actions of angII are mediated by the activation of the NADPH-oxidase (NOX), thus stimulating the formation of reactive oxygen species (ROS). Evidence is accumulating that increased oxidative stress has a relevant pathophysiological role in cardiovascular disease, including hypertension, atherosclerosis, and heart failure [14].
In WBS, the dosage of the NCF1 gene, encoding the p47phox subunit of NOX, is a strong modifier of the risk of hypertension. Hypertension was significantly less prevalent in patients whose deletion included NCF1, indicating that hemizygosity for NCF1 was a protective factor against hypertension in WBS. Decreased p47phox protein, superoxide anion production, and protein nitrosylation levels, were all observed in cell lines from patients hemizygous at NCF1 [15]. Reduced angII-mediated oxidative stress in the vasculature was the proposed mechanism behind this protective effect. Indeed, studies performed in Ncf1 knockout mice have revealed that p47phox is one of the major effectors of angII action. The administration of angII did not lead to increased superoxide production or blood pressure elevation in homozygous knockout animals, as it did in wild-type mice [16].
The aim of the present study was to evaluate whether oxidative stress significantly contributes to the cardiovascular phenotype of a mouse model for WBS, and whether reduction of NOX activity by genetic modification and/or by pharmacological inhibition might have a potential benefit in the rescue of this phenotype. By using non-invasive blood pressure measurements, histological, biochemical and molecular analyses, we have documented a negative correlation between NOX activity and the cardiovascular phenotype in a mouse model of WBS, as well as prevention of many of the manifestations by using anti-NOX therapies.
Results
Cardiovascular phenotype of DD mice related to elevated angII and oxidative stress
Previously reported mice bearing a heterozygous deletion of half of the orthologous region of the WBS locus (0.67 Mb, from Limk1 to Trim50, including Eln), called DD, were used as a model for the WBS cardiovascular phenotype [11], [17].
We confirmed the elevated systolic, diastolic and mean blood pressures of 16-weeks old DD mice, ∼40% higher than their wild-type littermates on average, without increased heart rate (Table 1 and Table S1). Hypertension was already present at 8 weeks of age and persisted throughout life (Table S2) without reducing life-expectancy, since these animals have been kept alive for more than 2 years with no instances of early death or unexpected morbidity [11]. As previously reported [17], body weight was significantly reduced for DD mice at all ages when compared to wild-type (P<0.001) (Table 1).

Post-mortem evaluation at 16 and 32 weeks revealed significantly larger hearts in DD mice, measured as the heart wet-weight relative to the body weight (P<0.001). The cardiac hypertrophy was associated with increased cardiomyocyte size both in left and right ventricles (P<0.001) (Table 1, Tables S3 and S4). Vascular histology and morphologic examination provided insight into the structure of the aorta. DD mice showed fragmented, disorganized and jagged elastin sheets when compared to wild-type vessels in sections of the ascending aortic wall stained with elastic VVG, as previously reported [11]. We also observed a significantly increased arterial wall thickness (P<0.001), with small changes in the number of lamellar units (Table 1).
The expression of three genes encoding components of the angII biosynthetic pathway, angII precursor (angiotensinogen, Agt), renin (Ren) and angII converting enzyme (Ace), was increased 2 to 5 fold by qRT-PCR on mRNA of several tissues (heart, aorta, lung and kidney) (Figure S1). Accordingly, DD mice showed significantly elevated angII peptide plasma levels (P<0.001) (Table 1). Plasma renin activity levels were, however, highly variable even within groups, thus preventing inter-group comparisons.
The level of oxidative stress in ascending aortas was determined using two experimental avenues, quantifying the levels of superoxide anion and protein nitrosylation (Table 1). These assays demonstrated higher levels of oxidative stress in DD mice when compared to those in wild-type littermates. These results confirm the relationship between hypertension, elevated angII and increased oxidative stress in these mice.
Ncf1 gene expression as a modulator of the cardiovascular phenotype
NCF1 gene dosage had been shown to modify the risk of hypertension in WBS patients [15]. Interestingly, while DD mice were consistently hypertensive, PD mice (heterozygous 0.45 Mb deletion, from Gtf2i to Limk1) were normotensive and mean blood pressure in D/P mice (harboring both deletions in trans) was only slightly increased by ∼10% [11]. Although the Ncf1 gene is located outside the PD deletion (Figure 1A), we investigated whether the expression levels of Ncf1 could be affected in these mice, by qRT-PCR in three different tissues and using Eln (hemizygously deleted in DD and D/P and not deleted in PD) as control. DD mice showed a ∼3 fold increase of Ncf1 mRNA, while the expression was reduced in PD animals, and elevated but only ∼2 fold in D/P mice, correlating with the blood pressure (Figure 1B). The low basal Ncf1 expression in PD and the relatively lower (compared to DD) in D/P strongly suggest that there may be a cis regulator element controlling Ncf1 expression in the PD deletion. It also indicates that Ncf1 is a strong modifier for the cardiovascular phenotype secondary to elastin deficiency.

We then investigated the expression of other genes related to the NOX system. On average, transcript levels of all genes but Cyba were significantly increased in DD animals with respect to wild-type. In contrast, they were not significantly different in PD mice, and D/P mice showed elevated expression of Nox2 along with the ∼2-fold increase of Ncf1 levels (Figure 1C and Figure S2). The elevated transcriptional NOX levels observed in DD mice could be the basis for the excessive ROS and protein nitrosylation documented in the aortic wall.
The cardiovascular phenotype of DD mice is partially rescued upon reduction of Ncf1 gene dosage
We then crossed mice homozygous for a spontaneous loss of function mutation in Ncf1 [18] with DD mice, in order to generate double heterozygotes in trans (DD/Ncf1−), resembling the genotype of WBS patients with lower risk of hypertension and deletions that include the NCF1 locus. At 16 weeks of age, DD/Ncf1 − animals had normal blood pressure similar to wild-type littermates (Figure 2A). AngII plasma levels were reduced with respect to DD mice (P = 0.036), although still elevated compared to wild-type values (P = 0.002) (Figure 2B), and were accompanied by a significant reduction of mRNA expression of the angII biosynthetic pathway genes (Figure 2C).
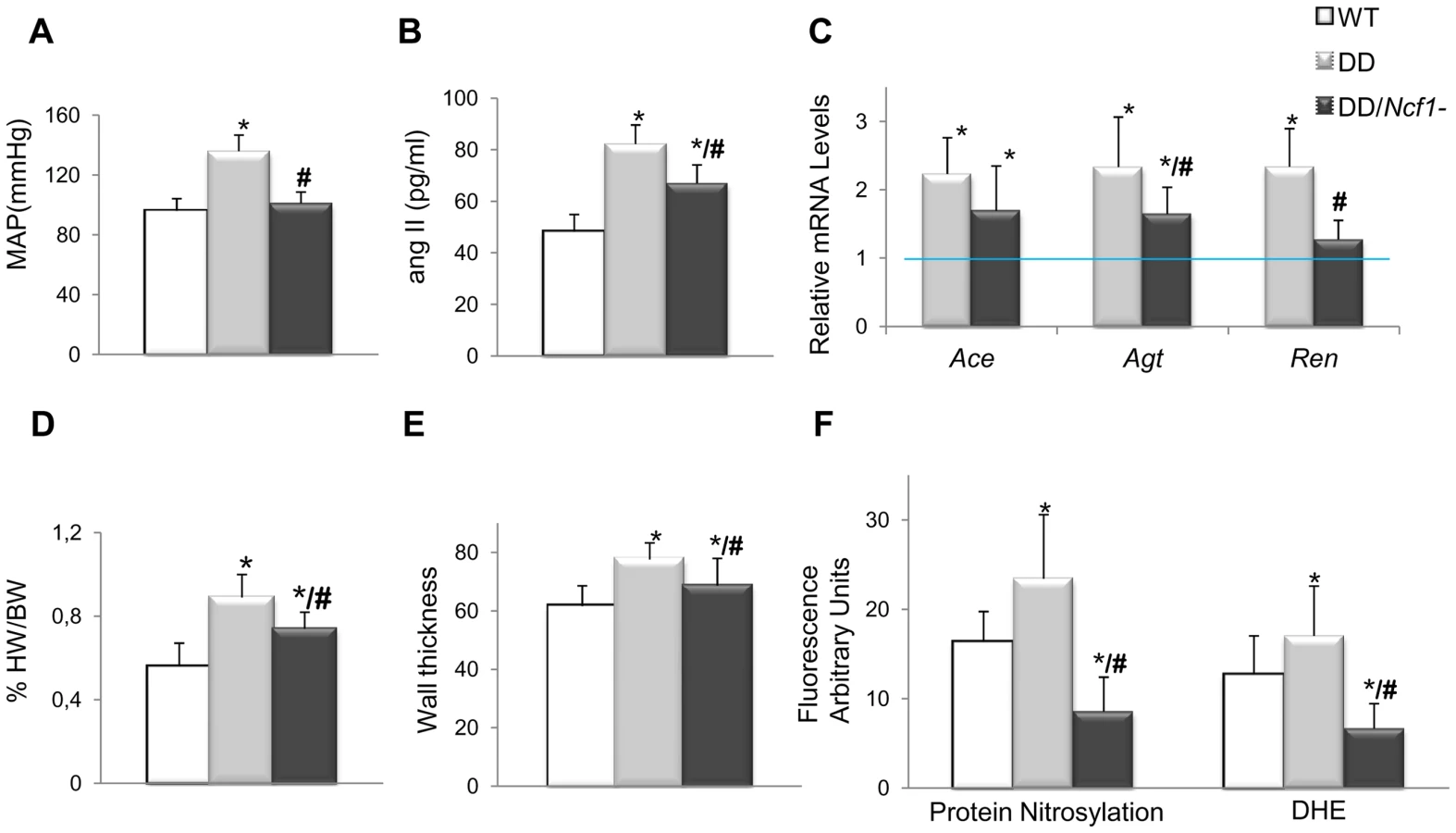
The hearts of DD/Ncf1− mice were 20% smaller than those of DD mice (P = 0.039), although they still were slightly larger than those of wild-type animals (P = 0.046) (Figure 2D and Table S3). Heart size reduction in DD/Ncf1− mice was accompanied with a decrease in the size of the cardiomyocytes of the left and right ventricles (P = 0.025 and 0.039 respectively). We also observed a reduction of the aortic wall thickness (P = 0.007), with a slight improvement in the organization of the elastin sheets (Figure 2E).
Expression of NOX-related genes was down regulated in DD/Ncf1− as compared to DD animals, reaching values similar to wild-type littermates. Consistently with these data, DD/Ncf1− mice showed significantly reduced levels of protein nitrosylation (P<0.001) and superoxide anion (P<0.001) in their ascending aortas compared to DD mice (Figure 2F).
Pharmacological rescue of the cardiovascular phenotype of DD mice
The evidence for genetic complementation prompted us to investigate whether the treatment of DD mice with either losartan (an AT1R antagonist) or apocynin (a NOX inhibitor) could rescue the abnormal cardiovascular parameters. Both pre - and postnatal-onset treatments with losartan or apocynin corrected the elevated blood pressure levels seen in 16 week-old DD mice (Figure 3A). Blood pressure control was associated with a significant reduction of angII plasma levels in all treated with respect to untreated DD mice, although the levels still remained higher than those of wild-type mice (Figure 3B). Both drugs acted synergistically with the genetic reduction of Ncf1 gene dosage, as shown by the below normal values of angII in treated DD/Ncf1 − mice (Table S5). The therapeutic effect was evident at the gene transcription level, since a significant reduction of transcripts encoding three angII biosynthetic pathway proteins was observed both in DD (Figure 3C) and DD/Ncf1 − mice (Table S6). A reduction in ROS production was also noted in the ascending aortas of treated DD mice (Figure 4A and Table S7), as well as down regulation of several oxidative stress genes, including Ncf1, Ncf2, Nox2 and Nox4 (Figure 4B and Table S8).


Either apocynin or losartan therapy also completely prevented the cardiac hypertrophy of DD mice. Treated DD animals displayed heart weights and cardiomyocyte sizes similar to those of wild-type counterparts, significantly smaller than those of the untreated DD group (Figure 5A and 5B). A mild improvement of the arterial wall thickness was also evident in all animals treated with both medications, along with reduced elastic fiber fragmentation during histological observation (Figure 5C and 5D). All evaluated parameters of cardiovascular phenotypic rescue (blood pressure, heart size, vascular morphology) persisted at 32 weeks of age in treated mice.

Secondary effects of pharmacological treatments
We found a high proportion of fetal deaths (∼32%) associated with the prenatal administration of losartan (Table S9). No difference in the expected Mendelian proportions was observed in the offspring of heterozygous crosses (DD x Ncf1+/−). We also observed premature postnatal deaths in ∼15% of the treated mice with prenatal-onset losartan (Table S9), mostly due to renal failure before the age of sacrifice, with similar frequencies among genotypes. Our data are in agreement with previous reports contraindicating losartan in pregnancy due to its potential teratogenicity [19], [20]. No specific genotype was associated with increased susceptibility to losartan toxicity. On the other hand, no instances of prenatal death or early postnatal complications were observed in apocynin treated animals, and no other complications were observed in any of the groups treated with postnatal onset.
Note: The full dataset of clinical, morphological, biochemical and molecular parameters at the different time-points, including the effects of treatment on wild-type and DD/Ncf1 − animals, is provided as supplementary information (Tables S1, S2, S3, S4, S5, S6, S7, S8, S9).
Discussion
The majority of patients with WBS (84%) manifest cardiovascular problems throughout their lives, particularly an arteriopathy consisting of stenosis of medium and large size arteries that can be present at birth [4]. Hypertension is found in 40%–70% of patients, even during childhood, and there is a significant risk of other cardiovascular complications, such as stroke, cardiac ischemia, and sudden death [4], [6], [8], [21], [22], [23], [24], [25]. Surgical treatment of focal vascular lesions is required in ∼20% of cases and frequently relies on vascular grafts or balloon dilatation angioplasty [4]. Although β-adrenergic blocker and calcium channel blocker drugs have been utilized, there is insufficient evidence to recommend a specific drug therapy for hypertension [8], [22], [26], [27]. Molecules that can either promote elastin biosynthesis or suppress vascular smooth muscle cell proliferation and migration, such as minoxidil [28], glucocorticoids [29], and retinoids [30] have been proposed as possible approaches to the treatment of cardiovascular disease, but none of them have shown to be clinically effective yet. Therefore, additional insight into the pathophysiology is needed to define better-targeted therapies as alternatives to current protocols to prevent the common complications of WBS arteriopathy.
A recently developed mouse model with elastin deficiency (DD) has provided further insight into the cardiovascular disease of WBS [17]. DD mice develop morphological changes in the aortic wall (thickening with disorganized elastin fibers) leading to chronic hypertension and cardiac hypertrophy. As in the case of Eln+/− mice, hypertension of DD mice is related to elevated angII plasma levels, and we have also shown over-expression of several NOX-related genes (Ncf1, Ncf2, Nox2/Cybb and Nox4) and significantly increased oxidative stress in these mice. AngII is an important physiological regulator of blood pressure and cardiac function, with hypertensive, growth, and remodeling effects mediated through AT1R. AngII acting through AT1R is also known to stimulate NOX generating ROS in a variety of cells [31]. Chronic infusion of angII in rats increases vascular NOX-derived ROS preceded by a prominent expression of the p47phox subunit of NOX in the vasculature and kidney [32]. Although some of the ROS serve as signaling molecules in the cells, excessive production is damaging and has been implicated in the progression of many disease processes.
In WBS patients, deletions are almost identical in size and mediated by non allelic homologous recombination, but the deletion breakpoints determine whether a functional copy of the NCF1 gene is included or not in the deleted interval [33]. Patients with ELN deletion and only one functional NCF1 allele have a 4-fold decreased risk of hypertension compared with those with more than one copy of NCF1 [15]. Interestingly, mean blood pressure in adult D/P mice, combining DD and PD deletions, was only slightly increased by ∼10%, suggesting a modifying effect of gene(s) within or near the PD region on blood pressure in these mice [11]. In addition, the presence of the PD deletion somehow decreased Ncf1 expression, being likely the main modifier for the non-significant blood pressure increase in D/P mice. Similarly, by genetic crossing, we have demonstrated that the loss of a functional copy of Ncf1 in DD mice completely restored oxidative stress and plasma angII levels preventing development of serious cardiovascular anomalies. These data reinforced the idea that pharmacological NOX inhibition could be efficient in the treatment of DD mice. Genetic ablation or pharmacological inhibition of Nox4 has proven to have a remarkable neuroprotective role in a mouse model for cerebral ischemia [34]. Antioxidants could also decrease blood pressure in several models of hypertension with proven implication of angII and the redox system, acting to scavenge the ROS produced by NOX, but their clinical effectiveness is limited [35].
The AT1R blocker losartan is known to lower blood pressure and rescue vascular wall alterations in other connective tissue defects by inhibiting the TGF-β signaling [14], [36]. Losartan inhibits the growth and remodeling effects of angII but also the NOX generated ROS, all mediated through AT1R.
On the other hand, apocynin is a naturally occurring methoxy-substituted catechol, experimentally used as a more specific inhibitor of NOX with anti-inflammatory activity demonstrated in a variety of cell and animal models. In resting cells, p47phox is folded in on itself through intramolecular interactions between the autoinhibitory region and the bis-SH3 and PX domains. These interactions are destabilized by phosphorylated serine residues within the autoinhibitory region, allowing p47phox to adopt an activated open conformation. Apocynin is thought to inhibit NADPH-oxidase assembly by preventing phosphorylation of the autoinhibitory region of p47phox, along with some scavenger activity of hydrogen peroxide [37]. Despite the controversy about the specific mode of action to decrease NOX activity, apocynin has been successfully used in a mouse model to treat hypertension and faster arterial thrombosis [38].
We have evaluated the efficacy and safety of both drugs, losartan and apocynin, in our mouse model of WBS cardiovascular pathology using previously titrated dosages with prenatal and postnatal onset of the therapies [36], [38]. Both treatments were highly effective in the prevention of the development of cardiovascular anomalies in DD mice. Similar effects were manifested by reducing the consequences of NOX activity, with almost complete control of hormonal and biochemical parameters in plasma and tissues, and resulting in normalized blood pressure and improved cardiovascular histology. There was an improvement in aortic wall thickness and architecture without complete reversion of the developmental anomalies secondary to elastin deficiency. Apocynin was as efficient as losartan in prenatal onset, with excellent tolerance and without secondary effects. However, a high proportion of fetal and premature postnatal deaths were associated with the prenatal administration of losartan, supporting its contraindication in pregnancy [19], [20]. Both drugs had significant beneficial effects after postnatal onset of the intervention, with excellent tolerance and no secondary effects.
In conclusion, both, losartan and apocynin, have significant efficacy in the treatment of the cardiovascular phenotype of a mouse model for WBS. Losartan is already approved for human use, while apocynin has been used by inhaler in some clinical trials [39]. The validation of apocynin for human use and the development of additional specific inhibitors of NOX are of great interest, given their potential therapeutic utility in some forms of cardiovascular disease [40]. We believe that these drugs merit evaluation as potential therapeutic agents to prevent the serious cardiovascular problems in human patients with WBS.
Methods
The study has been performed in accordance with the ARRIVE guidelines, reporting of in vivo experiments (http://www.nc3rs.org/ARRIVE).
Ethics statement
Animal procedures were conducted in strict accordance with the guidelines of the European Communities Directive 86/609/ EEC regulating animal research and were approved by the local Committee of Ethical Animal Experimentation (CEEA-PRBB).
Animal models
Previously reported mice bearing a heterozygous deletion of half of the orthologous region of the WBS locus on chromosomal band 5G1 (0.67 Mb from Limk1 to Trim50, including Eln), called DD for distal deletion, were used as a model for the WBS cardiovascular phenotype [11], [17]. Mice with the proximal half-deletion of the orthologous WBS locus (0.45 Mb from Limk1 to Gtf1i), called PD, and the double mutants in trans (with homozygous Limk1 deletion), D/P [17] were also used for some studies. Heterozygous DD animals were crossed with mice bearing a homozygous loss of function mutation of the Ncf1 gene (B6 (Cg)-Ncf1m1J)[18] to obtain double mutants in the first generation (DD/Ncf1−), harbouring then a mutant allele (DD deletion and Ncf1 mutation) in each chromosome (Figure 1A). All mice were bred on a majority C57BL/6J background (97%). Tail clipping was performed within 4 weeks of birth to determine the genotype of each mouse using PCR and appropriate primers (See primer sequences in Table S10).
Animal treatments
Fifteen different groups of mice (7–15 littermate animals per group, 5 groups per genotype: wild-type, DD or DD/Ncf1−), were used in this study for a total of n = 208. The 5 groups per genotype corresponded to untreated animals (NT), treated with losartan (Coozar, MSD) with prenatal (LP) or postnatal onset (LN), and treated with apocynin (Sigma) with prenatal (AP) or postnatal onset (AN). As previously described, drugs were administered in the drinking water with final concentrations of 0.002 g/day for losartan [36] and 2.5×10−4 g/day for apocynin [41]. In the groups of prenatal initiation, pregnant females started treatment at 14.5 dpc and therapy was continued throughout lactation. Postnatal treatments started at 7 weeks of age. In both cases, mice continued on oral therapy until 16 or 32 weeks of age, when they were sacrificed. Drinking water with drugs were refreshed every 3 days and protected from light by wrapping the drinking water container with aluminum foil. We recorded drinking volumes for untreated and treated mice in order to avoid any interference in the drinking water because of drugs supplement (Table S11).
Blood pressure measurements
Systolic, mean, and diastolic blood pressure were measured in conscious male mice on three separate occasions by using a tail cuff system (Non-Invasive Blood Pressure System, PanLab), while holding the mice in a black box on a heated stage. In order to improve measurement consistency, multiple sessions were performed to train each mouse. At least 12 readings (4 per session) were made for each mouse (n = 7–15 per group).
Histopathology
Animals were sacrificed at two time points (16 or 32-week-old). Immediately following sacrifice, all the organs in the thoracic cage (thymus, lung, heart and aorta) were removed in block and fixed in 10% buffered formalin at 4°C for 16 hours. Hearts and aorta were dissected, washed, and weighed (wet weight). Hearts and vessels were processed for paraffin embedding. Wall thickness and lamellar units were analyzed using 5 µm cross-sections of the ascending aorta (transected immediately below the level of the brachiocephalic artery) stained with Verhoeff-van Gieson (VVG) to visualize elastic lamina. Wall thickness at 10 different representative locations was measured and averaged by an observer blinded to genotype and treatment arm for each mouse. The number of medial lamellar units (MLUs) at 4 sites was assessed and averaged by 2 separate blinded observers. These axial cross-sections were imaged with an Olympus BXS1 microscope with epifluorescence and phase-contrast optics equipped with the Olympus DP71 camera, and images were captured with the CellB Digital Imaging system software. MLUs counting and wall thickness were quantified using Adobe Photoshop CS (Adobe Systems).
Quantification of mRNA
RNA was extracted from the visceral organs of the thorax by using TRIZOL reagent (Invitrogen) according to the manufacturer's instructions, followed by a second spin columns (Qiagen) purification. To avoid possible contamination of gDNA, all samples were analyzed before conversion to cDNA using standard PCR. In addition, primers were designed in different exons to avoid undesired amplification. cDNA was prepared from 1 µg total RNA using random hexamers and SuperScript II RNase H - reverse transcriptase (Invitrogen). The expression of genes involved in the angII biosynthetic pathway (Agt, Ren and Ace) and NOX-related oxidative stress (Ncf1, Ncf2, Nox2/Cybb, Nox4, Cyba and Rac2) were evaluated by quantitative real-time PCR (qRT-PCR). After diluting the cDNA (from 1∶10 to 1∶100, depending on the tissue), 5 µl were used as template for qRT-PCR using an ABI5700 thermocycler (Applied Biosystems) with the FastStart DNA Master SYBR Green Kit (Roche) and gene specific primers. Characteristics of primers are given in Table S10. Amplification of the Rps28 transcript served as RNA control for relative quantification. Each sample and the corresponding negative controls for each pair of primers were analyzed in triplicate at least in two independent experiments. Threshold cycle values were set manually and analyzed using the comparative method [42].
Measurements of plasma angII levels
Blood was collected from the mouse heart into EDTA tubes immediately after sacrifice. Plasma was collected after centrifugation at 1,500 g for 10 minutes and stored at −80°C until use. AngII levels were determined with the Renin Fluorometric Assay Kit Sensolyte 520 following the manufacturer's instructions.
Superoxide anion and nitrotyrosine detection
Formalin-fixed, paraffin-embedded transverse sections (5 µm in thickness) were mounted on polylysine-coated glass slides. After blockade with 5% bovine serum albumin plus 0.1% Triton X-100 in phosphate-buffered saline overnight at 4°C, the sections were incubated for 90 min at 37°C with the fluorescent probe DHE (Calbiochem, Darmstadt, Germany). In the presence of O2-, DHE is oxidized to ethidium, which intercalates with DNA, and yields bright red fluorescence. After washing with PBS plus 0.1% Triton X-100, sections were mounted and visualized by fluorescence microscopy (Olympus BX51, Japan). DHE fluorescence intensity was analyzed with NIH ImageJ software (v1.43, April 2010;U.S. National Institutes of Health, Bethesda, MD) as previously described [43]. The fluorescence intensity is proportional to the amount of superoxide anion. Thereafter, sections were incubated with 40,6-diamindino-2-fenilindol (DAPI) (300 nM) for 5 min at 37°C, reactive with fluorescent blue, marking the interlayer between DNA base pairs of cell nuclei. DAPI staining of cell nuclei helps detect true DHE staining (present in the nucleus) versus nonspecific staining. The specificity of the immunostaining was evaluated by the omission of the dye (negative controls). For the quantification of fluorescence, we also subtracted the background present in the negative control, in an attempt to eliminate any autofluorescence. All comparisons were made on cuts prepared with the same experimental conditions and the same day.
The distribution of 3-nitrotyrosine residues, as an indirect marker of peroxynitrite (ONOO-) production, was evaluated by indirect immunofluorescence. In brief, arterial sections were blocked for 2 h at 37°C and incubated overnight at 4°C with a polyclonal anti-nitrotyrosine antibody (dilution 1∶100; Chemicon International, Temecula, CA, USA).
Statistics
All data are presented as means ± SD. Statistical analysis was performed using ANOVA with a post hoc Bonferroni comparison between multiple groups. In specific cases of two-group comparisons we performed t-test. Values of p<0.05 were considered significant.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. PeoplesRFrankeYWangYKPerez-JuradoLPapernaT 2000 A physical map, including a BAC/PAC clone contig, of the Williams-Beuren syndrome–deletion region at 7q11.23. Am J Hum Genet 66 47 68
2. PoberBR 2010 Williams-Beuren syndrome. N Engl J Med 362 239 252
3. BayesMMaganoLFRiveraNFloresRPerez JuradoLA 2003 Mutational mechanisms of Williams-Beuren syndrome deletions. Am J Hum Genet 73 131 151
4. PoberBRJohnsonMUrbanZ 2008 Mechanisms and treatment of cardiovascular disease in Williams-Beuren syndrome. J Clin Invest 118 1606 1615
5. O'ConnorWNDavisJBJrGeisslerRCottrillCMNoonanJA 1985 Supravalvular aortic stenosis. Clinical and pathologic observations in six patients. Arch Pathol Lab Med 109 179 185
6. ReinAJPremingerTJPerrySBLockJESandersSP 1993 Generalized arteriopathy in Williams syndrome: an intravascular ultrasound study. J Am Coll Cardiol 21 1727 1730
7. TassabehjiMMetcalfeKDonnaiDHurstJReardonW 1997 Elastin: genomic structure and point mutations in patients with supravalvular aortic stenosis. Hum Mol Genet 6 1029 1036
8. BroderKReinhardtEAhernJLiftonRTamborlaneW 1999 Elevated ambulatory blood pressure in 20 subjects with Williams syndrome. Am J Med Genet 83 356 360
9. WesselAGravenhorstVBuchhornRGoschAPartschCJ 2004 Risk of sudden death in the Williams-Beuren syndrome. Am J Med Genet A 127A 234 237
10. DietzHCMechamRP 2000 Mouse models of genetic diseases resulting from mutations in elastic fiber proteins. Matrix Biol 19 481 488
11. GoergenCJLiHHFranckeUTaylorCA 2011 Induced chromosome deletion in a Williams-Beuren syndrome mouse model causes cardiovascular abnormalities. J Vasc Res 48 119 129
12. LiDYFauryGTaylorDGDavisECBoyleWA 1998 Novel arterial pathology in mice and humans hemizygous for elastin. J Clin Invest 102 1783 1787
13. FauryGPezetMKnutsenRHBoyleWAHeximerSP 2003 Developmental adaptation of the mouse cardiovascular system to elastin haploinsufficiency. J Clin Invest 112 1419 1428
14. LeeMYGriendlingKK 2008 Redox signaling, vascular function, and hypertension. Antioxid Redox Signal 10 1045 1059
15. Del CampoMAntonellAMaganoLFMunozFJFloresR 2006 Hemizygosity at the NCF1 gene in patients with Williams-Beuren syndrome decreases their risk of hypertension. Am J Hum Genet 78 533 542
16. LandmesserUSpiekermannSDikalovSTatgeHWilkeR 2002 Vascular oxidative stress and endothelial dysfunction in patients with chronic heart failure: role of xanthine-oxidase and extracellular superoxide dismutase. Circulation 106 3073 3078
17. LiHHRoyMKuscuogluUSpencerCMHalmB 2009 Induced chromosome deletions cause hypersociability and other features of Williams-Beuren syndrome in mice. EMBO Mol Med 1 50 65
18. HultqvistMOlofssonPHolmbergJBackstromBTTordssonJ 2004 Enhanced autoimmunity, arthritis, and encephalomyelitis in mice with a reduced oxidative burst due to a mutation in the Ncf1 gene. Proc Natl Acad Sci U S A 101 12646 12651
19. AlwanSPolifkaJEFriedmanJM 2005 Angiotensin II receptor antagonist treatment during pregnancy. Birth Defects Res A Clin Mol Teratol 73 123 130
20. QuanA 2006 Fetopathy associated with exposure to angiotensin converting enzyme inhibitors and angiotensin receptor antagonists. Early Hum Dev 82 23 28
21. BirdLMBillmanGFLacroRVSpicerRLJariwalaLK 1996 Sudden death in Williams syndrome: report of ten cases. J Pediatr 129 926 931
22. EronenMPeippoMHiippalaARaatikkaMArvioM 2002 Cardiovascular manifestations in 75 patients with Williams syndrome. J Med Genet 39 554 558
23. GiordanoUTurchettaAGiannottiADigilioMCVirgiliiF 2001 Exercise testing and 24-hour ambulatory blood pressure monitoring in children with Williams syndrome. Pediatr Cardiol 22 509 511
24. RoseCWesselAPankauRPartschCJBurschJ 2001 Anomalies of the abdominal aorta in Williams-Beuren syndrome–another cause of arterial hypertension. Eur J Pediatr 160 655 658
25. WollackJBKaiferMLaMonteMPRothmanM 1996 Stroke in Williams syndrome. Stroke 27 143 146
26. CherniskeEMCarpenterTOKlaimanCYoungEBregmanJ 2004 Multisystem study of 20 older adults with Williams syndrome. Am J Med Genet A 131 255 264
27. WesselAMotzRPankauRBurschJH 1997 [Arterial hypertension and blood pressure profile in patients with Williams-Beuren syndrome]. Z Kardiol 86 251 257
28. TsoporisJKeeleyFWLeeRMLeenenFH 1998 Arterial vasodilation and vascular connective tissue changes in spontaneously hypertensive rats. J Cardiovasc Pharmacol 31 960 962
29. PierceRAMariencheckWISandefurSCrouchECParksWC 1995 Glucocorticoids upregulate tropoelastin expression during late stages of fetal lung development. Am J Physiol 268 L491 500
30. McGowanSEDoroMMJacksonSK 1997 Endogenous retinoids increase perinatal elastin gene expression in rat lung fibroblasts and fetal explants. Am J Physiol 273 L410 416
31. SowersJR 2002 Hypertension, angiotensin II, and oxidative stress. N Engl J Med 346 1999 2001
32. ChabrashviliTTojoAOnozatoMLKitiyakaraCQuinnMT 2002 Expression and cellular localization of classic NADPH oxidase subunits in the spontaneously hypertensive rat kidney. Hypertension 39 269 274
33. CuscoICorominasRBayesMFloresRRivera-BruguesN 2008 Copy number variation at the 7q11.23 segmental duplications is a susceptibility factor for the Williams-Beuren syndrome deletion. Genome Res 18 683 694
34. KleinschnitzCGrundHWinglerKArmitageMEJonesE 2010 Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biol 8 e1000479 doi:10.1371/journal.pbio.1000479
35. ParaviciniTMTouyzRM 2006 Redox signaling in hypertension. Cardiovasc Res 71 247 258
36. HabashiJPJudgeDPHolmTMCohnRDLoeysBL 2006 Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 312 117 121
37. DrummondGRSelemidisSGriendlingKKSobeyCG 2011 Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov 10 453 471
38. AdamsGNLaruschGAStavrouEZhouYNiemanMT 2011 Murine prolylcarboxypeptidase depletion induces vascular dysfunction with hypertension and faster arterial thrombosis. Blood
39. StefanskaJSokolowskaMSarniakAWlodarczykADoniecZ 2010 Apocynin decreases hydrogen peroxide and nitrate concentrations in exhaled breath in healthy subjects. Pulm Pharmacol Ther 23 48 54
40. WindSBeuerleinKEuckerTMullerHScheurerP 2010 Comparative pharmacology of chemically distinct NADPH oxidase inhibitors. Br J Pharmacol 161 885 898
41. TangXNCairnsBCairnsNYenariMA 2008 Apocynin improves outcome in experimental stroke with a narrow dose range. Neuroscience 154 556 562
42. LivakKJSchmittgenTD 2001 Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods 25 402 408
43. LauYEGalliganJJKreulenDLFinkGD 2006 Activation of ETB receptors increases superoxide levels in sympathetic ganglia in vivo. Am J Physiol Regul Integr Comp Physiol 290 R90 95
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 2
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Gene Expression and Stress Response Mediated by the Epigenetic Regulation of a Transposable Element Small RNA
- Contrasting Properties of Gene-Specific Regulatory, Coding, and Copy Number Mutations in : Frequency, Effects, and Dominance
- Homeobox Genes Critically Regulate Embryo Implantation by Controlling Paracrine Signaling between Uterine Stroma and Epithelium
- Nondisjunction of a Single Chromosome Leads to Breakage and Activation of DNA Damage Checkpoint in G2
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy