
Nondisjunction of a Single Chromosome Leads to Breakage and Activation of DNA Damage Checkpoint in G2
The resolution of chromosomes during anaphase is a key step in mitosis. Failure to disjoin chromatids compromises the fidelity of chromosome inheritance and generates aneuploidy and chromosome rearrangements, conditions linked to cancer development. Inactivation of topoisomerase II, condensin, or separase leads to gross chromosome nondisjunction. However, the fate of cells when one or a few chromosomes fail to separate has not been determined. Here, we describe a genetic system to induce mitotic progression in the presence of nondisjunction in yeast chromosome XII right arm (cXIIr), which allows the characterisation of the cellular fate of the progeny. Surprisingly, we find that the execution of karyokinesis and cytokinesis is timely and produces severing of cXIIr on or near the repetitive ribosomal gene array. Consequently, one end of the broken chromatid finishes up in each of the new daughter cells, generating a novel type of one-ended double-strand break. Importantly, both daughter cells enter a new cycle and the damage is not detected until the next G2, when cells arrest in a Rad9-dependent manner. Cytologically, we observed the accumulation of damage foci containing RPA/Rad52 proteins but failed to detect Mre11, indicating that cells attempt to repair both chromosome arms through a MRX-independent recombinational pathway. Finally, we analysed several surviving colonies arising after just one cell cycle with cXIIr nondisjunction. We found that aberrant forms of the chromosome were recovered, especially when RAD52 was deleted. Our results demonstrate that, in yeast cells, the Rad9-DNA damage checkpoint plays an important role responding to compromised genome integrity caused by mitotic nondisjunction.
Published in the journal:
. PLoS Genet 8(2): e32767. doi:10.1371/journal.pgen.1002509
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002509
Summary
The resolution of chromosomes during anaphase is a key step in mitosis. Failure to disjoin chromatids compromises the fidelity of chromosome inheritance and generates aneuploidy and chromosome rearrangements, conditions linked to cancer development. Inactivation of topoisomerase II, condensin, or separase leads to gross chromosome nondisjunction. However, the fate of cells when one or a few chromosomes fail to separate has not been determined. Here, we describe a genetic system to induce mitotic progression in the presence of nondisjunction in yeast chromosome XII right arm (cXIIr), which allows the characterisation of the cellular fate of the progeny. Surprisingly, we find that the execution of karyokinesis and cytokinesis is timely and produces severing of cXIIr on or near the repetitive ribosomal gene array. Consequently, one end of the broken chromatid finishes up in each of the new daughter cells, generating a novel type of one-ended double-strand break. Importantly, both daughter cells enter a new cycle and the damage is not detected until the next G2, when cells arrest in a Rad9-dependent manner. Cytologically, we observed the accumulation of damage foci containing RPA/Rad52 proteins but failed to detect Mre11, indicating that cells attempt to repair both chromosome arms through a MRX-independent recombinational pathway. Finally, we analysed several surviving colonies arising after just one cell cycle with cXIIr nondisjunction. We found that aberrant forms of the chromosome were recovered, especially when RAD52 was deleted. Our results demonstrate that, in yeast cells, the Rad9-DNA damage checkpoint plays an important role responding to compromised genome integrity caused by mitotic nondisjunction.
Introduction
Chromosomes lagging or bridging during anaphase are believed to be one of the main sporadic causes of cytokinesis failure, which leads to tetraploid cells with multicentrosomes, a hallmark of early tumourigenesis [1], [2]. Conversely, if these anaphase bridges break apart, chromosomes could enter the so-called breakage-fusion-bridge cycle [3]–[5], which has been related to oncogene amplification and intratumour heterogeneity [6]–[8]. Carcinogens such as cigarette smoke, dysfunction of key cancer genes, bacterial toxins, and, paradoxically, many antitumour chemotherapeutic treatments (e.g. topoisomerase inhibitors) are known to cause anaphase bridges [9]–[12].
Chromosomes bridge in anaphase because they have either more than one centromere or problems in resolving the sister chromatids. Most of our knowledge on the biology of sister chromatid resolution comes from studies in yeast. In Saccharomyces cerevisiae, as in the rest of eukaryotes, sister chromatids are kept together after replication by both the cohesin complex and DNA-DNA topological entanglement arising from DNA metabolism (i.e., catenations) [13]. During anaphase onset, cohesion is lost through the regulated cleavage of cohesin by separase [14], and catenations are removed by the combined actions of condensin [15] and type 2 topoisomerase (Top2) [16]. Yeast mutants for any of these players show knotted nuclear masses in anaphase with trailing distal chromosome regions which cannot be resolved in otherwise bipolarly attached centromeres [15]–[20]. Despite these anaphase problems, all these mutants often perform cytokinesis, leading to a “cut” phenotype characterized by aneuploid daughter cells carrying broken chromosomes [14], [17], [21]. Not surprisingly, many daughter cells are not able to enter a new cell cycle after cytokinesis [14]–[16]. This has precluded use of those mutants as tools to follow up the short-term consequences in the progeny of anaphase bridges formed by unresolved sister chromatids.
The last genomic region to get resolved in yeast is the ribosomal DNA array (rDNA) [19], [22]–[24]. Importantly, resolution at this locus depends on a third player, besides condensin and Top2: the late mitotic phosphatase Cdc14 [22]–[27]. This is because Cdc14 inactivates transcription by RNA polymerase I in late anaphase, which allows the loading of condensin to the rDNA, its condensation and further resolution with the help of Top2 [22], [23], [27]–[31]. Other findings also suggest that these Cdc14 actions could serve to finish up replication within this locus [32], [33]. When Cdc14 is inactivated by means of thermosensitive conditional alleles such as cdc14-1, the anaphase segregation problem is much milder than that observed for the other aforementioned mutants. Indeed, cdc14-1 cells get arrested in telophase with the bulk of the nuclear masses segregated yet the rDNA bridging between mother and daughter cells [23], [24].
In a previous report we demonstrated that re-activation of the thermosensitive protein Cdc14-1 restores its cell cycle functions and is enough to exit mitosis [28]. Nevertheless, a portion of cells do this in spite of failing, in the end, to segregate the rDNA. Because little is known about the behaviour and fate of cells that commit to a new cell cycle once they have failed to resolve sister chromatids, we decided to address these questions taking advantage of this cdc14-1 re-activation phenotype. Herein, we show that cdc14-1 release leads to severing of the rDNA anaphase bridge and a new Rad9-dependent G2/M arrest. We followed the DNA damage response (DDR) in these cells and observed that they elicit a Rad52 long-lasting response that is independent of Mre11. We further discuss how our system provides a model for the study of DNA double strand breaks (DSB) where the ends finish up in different compartments (i.e., “one-ended”).
Results
Release from a cdc14-1 telophase block leads to a pre-anaphase arrest in the following cell cycle
Since the pioneering works by Hartwell and collaborators on yeast cell cycle control, it is known that conditional mutants for two essential genes, CDC14 and CDC15, give a telophase block with mostly binucleated dumbbell cells [34]. Nevertheless, we also now know that at least some cdc14 mutants have problems in the resolution and segregation of chromosomes during anaphase [22]–[24], [33]. As for the rDNA-bearing chromosome XII right arm (cXIIr), the telophase block elicited by the cdc14-1 allele prevents sister chromatid resolution, and therefore segregation, of regions that extend from somewhere within the large rDNA locus to the end of that chromosome arm [23]. Consequently, cXIIr forms an anaphase bridge between the connected daughter nuclei (Figure 1 for a scheme, “a” phenotype). In a previous work, we surprisingly found that, after reactivating the thermosensitive Cdc14-1 protein, cells were able to resume the cell cycle in spite of often failing to complete the resolution and segregation of such distal regions [28]. In general, around 50% of the cells coming out of a cdc14-1 block do not change their missegregation pattern, whereas the other 50% fully complete segregation of cXIIr (Figure 1, “a” & “b” phenotypes respectively).

We began our study by closely monitoring the cell cycle that follows cdc14-1 release in a strain where the cXIIr telomere is labelled (tetO:1061). We further included a side-by-side isogenic cdc15-2 strain as a control, after confirming that the cXIIr is fully segregated in its telophase block (Figure S1). In the same experiment we monitored: (i) the budding pattern after the release (Figure 2A); (ii) the morphological changes of the nuclei and the overall resolution and segregation of the cXIIr telomere (Figure 2B and 2C); (iii) the changes in DNA content by flow cytometry (i.e., bulk replication) (Figure 2D); and (iv) chromosome behaviour in a pulsed-field gel electrophoresis (PFGE) (i.e., individual chromosome replication and integrity) (Figure 2E). For the first two, we looked under the microscope and counted individual cells. The telophase release led to rebudding of the initial dumbbell mitotic cell for both mutants (Figure 2A). Virtually all cells were able to resume the cell cycle in a synchronous way as indicated by the drop of the dumbbell category (in red) to values below 10%. Around 120 minutes after the release, most cells had rebudded again. Since most daughter cells remain together for a while after the release, this rebudding gave “threesomes” (i.e., single-rebudded or three cell bodies) and “foursomes” (i.e., double-rebudded or four cell bodies). Foursomes (in blue) remained the most abundant category from 120 minutes onwards (∼90% in the cdc14-1 release, and ∼50% in the cdc15-2 release). The lesser amount of foursomes in the cdc15-2 release occurred because daughter cells from this mutant were eventually able to separate from each other, whereas daughters for cdc14-1 remained tightly together even long after becoming foursomes (see below). When we followed the release for longer periods we noticed that the budding pattern of the cdc15-2 release became complex and tended to be oscillatory. By contrast, cdc14-1 was much simpler and many cells stalled as the foursome category throughout (data not shown and Figure S2). A critical difference between the observed threesomes and foursomes for cdc15-2 and cdc14-1 became evident when we looked at the nuclei by DAPI. Thus, cdc15-2 foursomes had 4 nuclei (i.e., both daughters have entered and completed another nuclear division round) in ∼70% of the cases at minute 180. By contrast, the cdc14-1 release had less than 10% of the foursomes in this situation at that time (Figure 2B and Figure S2). When we looked at the segregation pattern of the cXIIr telomere (tetO:1061) in these strains, we found that cdc15-2 always segregated it faithfully in the two cell division that took place (94.0±0.1% [mean ± SEM, n = 3] of foursomes with four nuclei had one tetO in each nucleus) (Figure 2C for a representative micrograph). In the case of cdc14-1, 63.6±1.7% (mean ± SEM, n = 3) of foursomes with 2 nuclei had already missegregated cXIIr as expected [28], and no more cell divisions proceeded in that period (Figure 2C).

We repeated this block-and-release experiment in different yeast strains and backgrounds and found that the main conclusion was conserved (i.e., long-lasting arrest of many cdc14-1 cells as foursomes in a pre-anaphase stage). However, we observed slight differences in terms of synchrony after the release, time of rebudding, and number of daughter cells able to separate from each other. For instance, in the W303 background, cdc15-2 cells got released earlier and the synchrony was much better throughout (Figure S2).
We further explored the spindle apparatus (spindle itself using Tub1-GFP and spindle pole bodies with Tub4-CFP) in the cdc14-1 foursomes and observed that each of the two nuclei contained duplicated spindle pole bodies and a metaphase-like spindle (Figure S3). This shows that the single nucleus observed for each daughter cell in the cdc14-1 foursome represents a genuine pre-anaphase arrest and not a highly tangled anaphase.
Daughter cells complete DNA replication after a cdc14-1 release
The fact that cdc14-1 cells stalled as binucleated foursomes after the telophase release indicated that cells got arrested somewhere between S phase (whose beginning coincides with the rebudding event) and anaphase. We next narrowed the window of this arrest to G2/M by demonstrating that cells completed DNA replication after the cdc14-1 release. This was possible because, at the time we took samples for microscopy in the above-mentioned experiment, we also took samples for following DNA replication in the cell population by flow cytometry and PFGE (Figure 2D and 2E).
When we performed flow cytometry analysis, we observed a duplication of the DNA amount in cells coming from a cdc14-1 release (Figure 2D). Since these cells ended up as foursomes, replication could be clearly assessed by simply observing how cells transited from a 2 N to a 4 N peak. In the case of cdc15-2, the assessment was a little more difficult since the release gave rise to a complex mixture of single cells (both unbudded and budded) and rebudded cells still connected through the cell wall (threesomes and foursomes). However, the three major peaks for DNA content visible during this release accounted well for the observed amounts of each cell type (Figure 2D, left panel), and indicate that these cells also replicated their DNA. An important conclusion we reached from these data is that replication started and finished at the same time for both mutants, at least for the bulk of their DNA.
When we performed PFGE for those samples, we further confirmed that chromosome replication is mostly completed for all chromosomes after the cdc14-1 release. We ascertained this using the fact that yeast chromosomes cannot enter a PFGE while being replicated [35]. Thus, we observed that a new replication round for all chromosomes started at around minute 90 in both mutants and that most chromosomes re-entered the gel ∼60 minutes later (Figure 2E, upper panel; and S4). This individual chromosome replication behaviour fits well with the bulk replication seen by flow cytometry in Figure 2D.
Chromosome XII integrity is compromised after a cdc14-1 release
Although chromosome XII also started replication after the cdc14-1 release, the recovery of the whole band was incomplete. In fact, we observed just by ethidium bromide staining that chromosome XII became fainter than any other chromosome after the cdc14-1 release (Figure 2E, upper panels; and S4). This did not happen during the cdc15-2 release. Importantly, when we performed a southern blot with a probe against the rDNA we could see that other shorter bands appeared (Figure 2E, lower panels, double - and triple-asterisks). These new bands were visible after the new round of replication was completed, but they were also visible if we prevented replication after the cdc14-1 release by blocking daughter cells in G1. Again, this G1 block also led to a 50% drop of chromosome XII band intensity in the ethidium bromide staining; and this drop was specific to the cdc14-1 release (Figure S4). Besides, a smear above the band for the entire chromosome was also seen during the cdc14-1 release, especially after chromosome replication (Figure 2E, lower panels, single-asterisk). Although we do not know what this smear might be, we speculate that it could account for chromosome XII with replication or recombination intermediates. Interestingly, the cell population in this cdc14-1 strain may have up to three rDNA sizes (Figure 2E, lower panel, cXII arrow); which would indicate that the rDNA array is more unstable in this mutant.
The new arrest is long-lasting for those cells that were unable to resolve and segregate the chromosome XII right arm
Because the tetO:1061 can still be segregated in ∼50% of the cells coming from a cdc14-1 release [28], we next decided to specifically assess whether these cells eventually bypass the arrest as foursomes. For that purpose we filmed any re-budding beyond that point and compared it to the previous cXIIr segregation outcome. We followed up 22 dumbbell cdc14-1 cells as they transited out of the telophase arrest on minimal medium agarose patches. Twelve out of these 22 cells ended up missegregating the tetO (54.5%). As expected, all daughter cells rebudded again, although it took around an hour longer than when we performed the release in liquid cultures (half-life of the dumbbell phenotype was ∼135 minutes for agarose patches versus ∼75 minutes for cells in culture). Importantly, no foursomes that originally missegregated the cXIIr had rebudded a third time by 6 hours after the telophase release (n = 12); whereas 80% of foursomes had done so when cXIIr segregation had been correct (n = 10). This difference is statistically very significant (p<0.001, Fisher's exact test on the 2×2 contingency table).
Cells complete karyokinesis and cytokinesis after the cdc14-1 release, even in those cells that missegregated the chromosome XII right arm
At the telophase block, cdc14-1 strains have daughter-to-be cells still connected through the bud neck as cytokinesis has not yet been completed [36]. A key question to understand the observed G2/M block is to address the fate of the cXIIr anaphase bridge after the release; importantly, whether or not cdc14-1 cells complete cytokinesis and hence sever the bridge. We addressed this question two ways.
First we looked at karyokinesis microscopically (i.e., nuclear fission) in a strain where the distal part of the rDNA is tagged (tetO:487) and the nuclear TetR-YFP is overexpressed. In this strain we can see both the cXIIr bridge and the nucleoplasm. When Z stacks of microscope pictures were taken at the cdc14-1 block, the tetO:487 was seldom segregated and a clear nucleoplasm bridge was visible across the bud neck (Figure 3A, 0′ picture; Figure S5, hollow pointers). The nucleoplasm bridge (soluble TetR-YFP, do not mistake nucleoplasm bridge for anaphase bridge) was also observed for all cells blocked with the cdc15-2 allele. This suggests that karyokinesis has not yet taken place in both telophase blocks. Noticeably, the nucleoplasm bridge had bulges in the cdc14-1 block (Figure S5, filled pointers), yet was a thin and straight line in the cdc15-2 block. We tested whether this bulge accommodates the unresolved rDNA and distal regions of the cXIIr by using another yeast strain that also carries the nucleolar marker Net1 fused to CFP. We found that the nucleolus colocalized with the bulge in more than 95% of the cells (Figure S5B). After the cdc14-1 telophase release, the nucleoplasm bridge eventually disappeared in dumbbell cells and was never visible in daughter cells that had already rebudded (Figure 3A, dumbbells [D] and foursomes [F] at minute 90). A similar behaviour was seen for cdc15-2 release (data not shown). Importantly, no cdc14-1 foursomes had a nucleoplasm bridge, even if they had previously missegregated cXIIr. A conclusive proof that karyokinesis took place before the daughter cells became foursomes was obtained when cells were arrested in G1 right after the cdc14-1 release. Thus, the nucleoplasm bridge was never visible after the release in cells treated with alpha-factor, irrespective of the cXIIr segregation status (Figure 3A, photo α-F at minute 90).

Although rDNA-missegregating cells ended up performing karyokinesis, it was somehow striking we did not observe a delay in the cell cycle during the cdc14-1 release. Such delay is expected since a checkpoint has been described to sense the presence of anaphase bridges in yeast (i.e., NoCut checkpoint) [37], [38]. We took advantage of having the aforementioned strain to look at the nucleoplasm and the cXIIr bridges simultaneously and check whether the maintenance of the cXIIr bridge after the release correlated with a delay in the karyokinesis (Figure 3B). We performed a time course of the cdc14-1 release and followed daughter cells (as dumbbells) throughout the new G1. We observed that the nucleoplasm bridge (again do not mistake for the cXIIr bridge) took around 20 minutes longer to be severed in those cells that finally failed to segregate the cXIIr (Figure 3B, half-life for the nucleoplasm bridge was ∼30 minutes for cells with segregated cXIIr versus ∼50 minutes for missegregated cXIIr). We believe that this 20 minute delay in karyokinesis may account for the NoCut checkpoint. In any case, the time of disappearance of the nucleoplasm bridge (i.e., karyokinesis) was short and the NoCut checkpoint did not preclude cells with the cXIIr bridge from finally completing karyokinesis.
In order to confirm that the fate of the cXIIr bridge is to be severed after the cdc14-1 release, we also looked at cytokinesis indirectly. We employed an assay based on the fact that formaldehyde-fixed cells that have not completed cytokinesis are resistant to separation by cell wall digesting enzymes (i.e., zymolyase) [39]. At the cdc14-1 block, when most cells were dumbbells, zymolyase treatment was not able to separate the daughters, although the bud neck that connects them became very thin (Figure 3C, left-corner photos). By contrast, foursomes seen two hours after the release could be split in two (Figure 3C, main lower photo). The drop of foursomes after zymolyase treatment was high (from ∼70% to ∼3%). In another set of experiments, we also counted cell number after zymolyase treatment under different chemical conditions to inhibit either cytokinesis or S-phase. To inhibit cytokinesis, we added the F-actin inhibitor Latrunculin A (LatA) [40]. Since its action against cytokinesis is optimal if cells are incubated before they reach telophase but after they have budded, we employed an initial arrest in G2/M [40]. Then we let them transit from the G2/M arrest to the telophase arrest. To inhibit the new S-phase, we blocked cells in G1 with alpha-factor, added at the telophase release. As expected, overall cell number doubled two hours after the release relative to the telophase arrest in a culture without LatA (Figure 3D). Importantly, this separation could be partially prevented by incubating the cdc14-1 cells with LatA (cdc14-1 release with versus without LatA gave a p = 0.036, Student's T test). Moreover, we also demonstrated that cytokinesis occurred before the daughters entered the new S-phase. Thus, alpha-factor prevented dumbbells from becoming foursomes after the release, but it did not circumvent cell separation after zymolyase treatment (Figure 3D). Again, this separation was prevented when LatA was added during the G2/M to telophase transition (cdc14-1 release to alpha-factor with versus without LatA gave a p = 0.028, Student's T test).
On the whole, we can conclude from this set of experiments about chromosome XII integrity (Figure 2E) and karyo/cytokinesis (Figure 3) that cells physically separate from each other irrespective of the presence of the cXIIr bridge. The logical consequence of this should be the generation of at least a DSB near or within the rDNA.
The new G2/M arrest that follows the cdc14-1 release is dependent on Rad9
The observed karyokinesis, cytokinesis and cXIIr breakage, followed by the arrest in G2/M in the new cell cycle, likely implies that a DSB-mediated DNA damage checkpoint is activated after the cdc14-1 release. A critical component of this checkpoint is Rad9. Mutants for this protein allow cells with DSBs to enter a new segregation round [41]. Hence, we decided to check whether our observed pre-anaphase arrest could be overcome by deleting RAD9. We did this in our cdc14-1 TUB1-GFP strain to follow spindle morphology as well as nuclear division after the release (Figure 4A). In contrast to the single mutant cdc14-1 TUB1-GFP, the double mutant cdc14-1 rad9Δ TUB1-GFP could enter anaphase by 3 hours after the release, becoming foursomes with more than two nuclei masses (i.e., at least one of the daughter cells entered a new anaphase) (Figure 4A, upper panels). Accordingly, when we looked at spindle morphology using Tub1-GFP, we observed a transition from metaphase-like spindles to other patterns in the cdc14-1 rad9Δ double mutant (mainly G1-like Tub1 dot signals) (Figure 4A, lower panels).
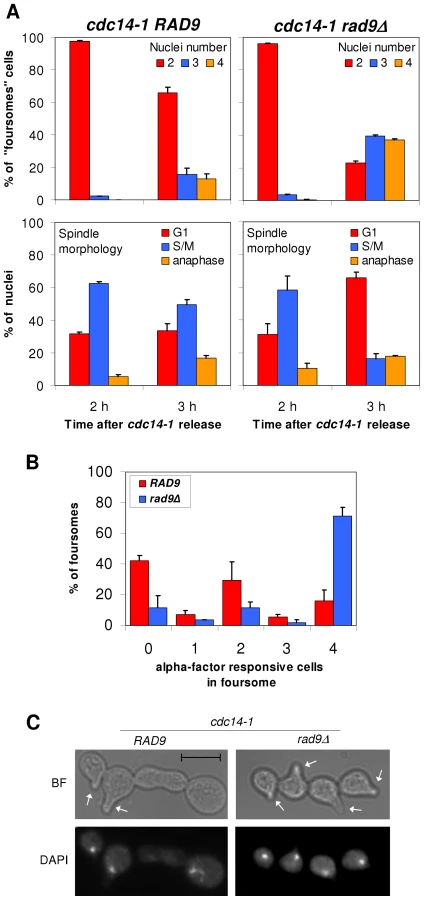
Besides this, we tested the responsiveness of foursomes to the G1-specific pheromone alpha-factor. We reasoned that if cells were able to progress beyond the G2/M arrest, they would become responsive to the pheromone and change their morphology accordingly (i.e., acquire the shmoo phenotype). Thus, we treated cdc14-1 cells with the pheromone, not at the time of the release as in other experiments above, but after they became foursomes (2 hours after the release). Then, we left them in alpha-factor for another 3 hours. We first noticed that some rad9Δ backgrounds, like the one that carries the TUB1-GFP, were able to split the foursomes after this 5 hours incubation time. Therefore, we used one of the W303 backgrounds that kept the foursome category in these conditions. Importantly, all cells in most cdc14-1 rad9Δ foursomes were responsive to the pheromone (Figure 4B). Also, these foursomes had four segregated nuclei (Figure 4C). Interestingly, cells in the cdc14-1 RAD9 foursome distributed in three peaks: one with no responsive cells, one with all four cells responsive, and a third subgroup with just two cells responding to the pheromone (Figure 4B). Within this subgroup, the two responsive cells were always a partner and each has a nucleus (Figure 4C). In order to determine where these two cells come from, we repeated this assay with the cdc14-1 strain that carries the labelled cXIIr telomere. We found that in 92% of foursomes (n = 39) both responsive cells had a tetO, whereas there was no tetO in either of the two non-responsive cells. This means that: (i) this subgroup came from cdc14-1 cells where missegregation occurred in the first place, and (ii) the cell that retained the intact chromosome XII plus the broken cXIIr was able to eventually pass the G2/M arrest.
Therefore, we concluded that cells coming from a cdc14-1 release activated the Rad9 checkpoint to prevent daughter cells from entering anaphase, and that this G2/M arrest persists for a long time in the daughter cell that lost an intact copy of chromosome XII (i.e., DC1).
Missegregation of chromosome XII right arm leads to accumulation of Rad52 foci after a cdc14-1 release
The Rad9-dependent cell cycle arrest means that daughter cells sense the DSB(s). Therefore, they must accordingly trigger a DNA damage response (DDR). At this point, we started looking at proteins that cytologically mark this DDR by appearance in nuclear foci. Rad52 is a key mediator in the DDR that comprises the preferred homologous recombination (HR) pathway for repair [42]. This pathway is central in the DDRs that occur throughout S-phase and well into mitosis [43]. We reasoned that, because daughter cells reached and completed S-phase on schedule after a cdc14-1 release (Figure 2) and then get arrested in G2/M in a Rad9-dependent manner (Figure 2 and Figure 4), Rad52 should be involved in the DDR. Importantly, Rad52-YFP forms widely studied nuclear foci after induced DNA damage [43]. Thus, we looked at Rad52 foci in our telophase block-and-release experiments. We indeed observed foci after a cdc14-1 release for a subset of cells (Figure 5). Rad52 foci number and intensity were clearly superior in the cdc14-1 release relative to a side-by-side experiment with the cdc15-2 strain (Figure 5A). Importantly, there was no difference between the strains when growing asynchronously at 25°C (only ∼5% of budded cells had foci). Foci were observed at the telophase block for neither cdc14-1 nor cdc15-2, further indicating that DNA damage has not yet taken place at this stage. Foci started around 90 minutes after the release and always after rebudding (Figure 5B for a typical time-lapse movie). Furthermore, Rad52 foci were rather dynamic at the beginning of the new S-phase and, within the subgroup that, at some point, had Rad52-YFP foci, tended to end up as either just one major focus in the foursomes (in one of its two nuclei) or 2 foci, one located in each nucleus (Figure 5A and 5B).

Remarkably, the percentage of foursomes acquiring at least one long-lasting single Rad52-YFP focus during the release was ∼50% (Figure 5A). We noticed that this percentage of cells was equivalent to that of rDNA/cXIIr missegregation [28]. We therefore hypothesised that cells with Rad52 foci may represent those that had failed in rDNA segregation. We addressed this important question in two ways. First we made use of a second mutation that worsens rDNA segregation after a cdc14-1 release (i.e., deletion of FOB1 gene) [28] (Figure S6). Second, we double labelled two cdc14-1 strains (one in the S288C background and the other one in W303) with a tag for the rDNA and a tag for the Rad52 protein. For the S288C background, we employed our strain with the tetO:487 and added a RAD52-RedStar2 allele (Figure S7). As for W303, we employed a previously described strain that bears both Rad52-YFP and a tag inserted within the rDNA (tetOs/TetR-mRFP system) [44] and that we made cdc14-1 (Figure 5C). By using the cdc14-1 fob1Δ double mutant, we could correlate worsening of the rDNA segregation with a higher frequency of foursomes carrying at least one bright Rad52 focus (Figure S6; p<0.0001, Pearson's chi-square test). On the other hand, double labelling of Rad52 and the rDNA further and strongly confirmed that Rad52 foci are more frequent in cells that missegregated the rDNA (Figure 5C and Figure S7; p<0.0001, Pearson's chi-square test). Moreover, these strains allowed us to determine that the first and strongest Rad52 focus appeared in the daughter cell that does not carry the tetOs (i.e., cell DC1). Thus, 75% of these foci were located in that cell versus only 8% of Rad52 single foci seen in the daughter cell that carries the tetOs (the remaining 17% of foursomes had one Rad52 focus in each daughter cell). We believe that this is an important result since the genetic material that each daughter carries in the anaphase bridge is different as stated above (Figure 1, “a” phenotype). Hence, “DC1” cell (the one without the tetOs) bears just one broken copy of the resolved part of chromosome XII (from left telomere to somewhere within the rDNA), whereas “DC2” cell carries one entire sister chromatid plus the unresolved part of the other one (from somewhere within the rDNA to the right telomere). The fact that Rad52 foci are stronger and long-lasting in DC1 might indicate that this cell struggles to repair the DSB, while DC2 might end up repairing its broken end. This is in agreement with what we observed when deleted RAD9 (Figure 4B). Finally, it is worth mentioning that, for those Rad52 foci visible in the tetO-carrying nuclei, the fluorescent dots were almost always in close proximity (Figure 5D). However, these Rad52 foci did not localize within the nucleolus when we used a nucleolar marker (Figure S8). This is not surprising though as broken rDNA sequences are transported out of the nucleolus towards nuclear Rad52 factories [44].
Overall, these observations fit well with the prediction of a DDR occurring preferentially in those daughter cells that failed in rDNA segregation during the preceding division.
Release from the cdc14-1 block also leads to Rfa1 foci, yet only after cells reach the new S-phase
We expected the DSB to occur shortly after the release into G1, when karyo - and cytokinesis took place (Figure 3) and chromosome XII appeared partly broken (Figure 2E). We were intrigued by the fact that cells did not, however, delay G1 (Figure 2). A possible explanation for this anomaly would be that the DSB is clean (i.e., with little associated single-stranded DNA [ssDNA] at the edges). This is the type of DSB generated by inducible endonucleases like HO as opposed to DSBs obtained after ionizing irradiation, which are rich in ssDNA (i.e., ragged ends) [45], [46]. It has been shown that clean ends are poorly resected in G1, forming little ssDNA, whereas ragged DSBs can already bind ssDNA-binding proteins such as the RPA complex. The formation of ssDNA and the binding of the RPA complex to it are key steps in checkpoint activation [47], [48]. Because of this, we also included the RPA complex as a reporter to study the DDR that follows the cdc14-1 release. YFP-tagged Rfa1 (one of the complex subunits) also forms foci under the presence of DSBs [49]. Crucially, Rfa1 can form foci in G1 provided that the DSB takes place at this stage and, as just mentioned, the break is ragged [46], [49], [50]. Thus, we observed Rfa1 foci for a subset of cells coming from a cdc14-1 release (Figure 6A and 6B). Rfa1 foci were as dynamic as those of Rad52 and also tended to end up as a major focus, one per nucleus in the foursome at the most (Figure 6C). Moreover, it was noticeable that Rfa1-YFP eventually gave very intense foci (Figure 6B). Nevertheless, all foci began to appear around 60′–90′ after the release (Figure 6A), and always after rebudding (Figure 6C). Accordingly, foci were not observed in cdc14-1 cells transiting from telophase to a G1 arrest with alpha-factor (Figure 6B). This likely means that the new type of DSB generated by the severing of the cXIIr bridge has little associated ssDNA (i.e., the DSB is clean) and therefore it is not recognized by the RPA complex in G1.

The Rad52 response to the severing of the chromosome XII right arm anaphase bridge is independent of Mre11
In the canonical model for DSB recognition and repair by HR, RPA and Rad52 are downstream players to the MRX complex (Mre11-Rad50-Xrs2) [49]. This complex is supposed to recognize each DSB end, bring them together and help in the first stages of end processing to allow template searching for HR. The expected “one-ended” nature of the DSB, a consequence of the anaphase bridge severing (i.e., the two ends cannot be brought together), prompted us to further study this important component in the DSB signalling and repair. We made use of Mre11-YFP as a reporter of the DSB-specific MRX complex. Unlike Rad52 and Rfa1, Mre11 foci have been observed in all cell cycle stages (including G1) and for all types of “two-ended” DSBs generated [46], [49]. We were not able to observe Mre11 foci for cdc14-1 throughout the release (less than 1% of cells at any one time point in 10 minutes intervals, data not shown). Nevertheless, Mre11 was fully functional in cdc14-1 cells growing at the permissive temperature since it forms foci when DSBs were chemically generated (Figure S9). We further ruled out any role of the MRX complex in the observed Rad52-dependent response after the release by looking at Rad52 foci in a cdc14-1 mre11Δ double mutant. Indeed, we saw Rad52 foci at a number and intensity comparable to that of a cdc14-1 MRE11 strain in foursomes taken 2 and 4 hours after the telophase release (Figure 7A and 7B). Because mre11Δ gave some background of Rad52 foci at the cdc14-1 arrest (Figure 7A, time 0′), we filmed cells during the release and observed that about 75% of cells with Rad52 foci at the arrest never entered a new cell cycle (data not shown). Therefore, almost all foci measured in the foursomes likely came from cells without foci at the previous arrest. We thus concluded that Rad52 foci in cdc14-1 foursomes were independent of Mre11.

The cdc14-1 anaphase bridge comprises few chromosome arms aside from the chromosome XII right arm
Although the cXIIr is a hotspot for missegregation in cdc14 mutants, it seems not to be the only genomic region affected. Thus, at least one telomere of a chromosome other than XII appeared missegregated at the telophase block in previous works that made used of another thermosensitive allele (i.e., cdc14-3) [22], [33]. Therefore, we decided to address whether telomeres other than cXIIr were also missegregated during the cdc14-1 release. We looked at four different telomeres located in two chromosomes (V & XIV) [51]. Chromosome V has the right telomere labelled with the lacO/LacI-CFP system, whereas its left telomere (V-L) is labelled with the tetO/TetR-YFP system. On the other hand, chromosome XIV has the right telomere (XIV-R) labelled with the tetO/TetR-YFP system and its left telomere labelled with the lacO/LacI-CFP system. It is important to note that telomere V-L was the one used in the above-mentioned cdc14-3 studies. When we carried out the cdc14-1 telophase block at 37°C we often failed to detect the CFP signal, so we focused on missegregation after the release (CFP signal recovered after the temperature drop). Since the cdc15-2 release gave only few binucleated foursomes (Figure 2 and Figure S2), and in order to avoid a possible bias, we compared the cdc14-1 release to the cdc15-2 telophase block. To preserve the CFP signal, we arrested cdc15-2 cells at 34°C (at least for the YFP-labelled telomeres there was no difference between 34°C and 37°C in terms of segregation, data not shown). We observed that missegregation in cdc14-1 binucleated foursomes was low for all four telomeres and comparable to that observed at the cdc15-2 block (Table 1). From these data, and from the pattern of chromosome integrity shown in Figure S4, we can conclude that in a cdc14-1 release many chromosomes are expected to be fully segregated. Thus, the anaphase bridge severed after the cdc14-1 release must be relatively enriched with cXIIr fragments.

Aberrant forms of chromosome XII can be recovered from daughter cells that survived the cdc14-1 release
In a previous paper, we demonstrated that less than 1% of daughter cells can survive passage through multiple mitoses (>25) without Cdc14 (regulated overexpression of the cyclin-dependent kinase inhibitor Sic1 through GAL-SIC1 was employed to overcome Cdc14 roles in the Mitotic Exit Network) [28]. In that work, we found that all survivors had dramatically shortened the rDNA locus (although other chromosome rearrangements were not obvious from the PFGE analysis). We also showed that the small survival capability depended on Rad52 (i.e., HR is needed to repair the DNA damage and survive). This prompted us to study whether our cdc14-1 block-and-release approach, where only one cell cycle is compromised, leads to similar results. Because Rad52 also seems to play an important role after the transient Cdc14 inactivation (Figure 5, Figures S6 and S7), we also tested whether RAD52 was essential in this system by including a double mutant cdc14-1 rad52Δ. Thus, we performed the block-and-release experiment for both cdc14-1 and cdc14-1 rad52Δ and plated the foursomes to obtain isolated colonies after 3–5 days (Figure 8A). We also plated cells right before the block-and-release experiment, while growing asynchronously at 25°C. At the time of plating, we counted cells with a haemocytometer to determine overall viability. Surprisingly, we did not see a great loss of viability after the transient Cdc14 inactivation (Figure 8A and table underneath). As for the single cdc14-1 mutant, this loss was around 25% at the most, whereas double mutant cdc14-1 rad52Δ showed no drop in viability at all. This demonstrates that at least one of the daughter cells of the foursome often survives and gives raise to a colony. Taking into account that 50% of foursomes missegregated the cXIIr (equivalent values of missegregation were seen during a cdc14-1 rad52Δ release: 48%), the observed percentage of viable cells might indicate that 50% and 100% of the daughter cells that carry an intact cXII (i.e., DC2) must survive in cdc14-1 and cdc14-1 rad52Δ respectively. Other results we showed above already pointed towards this possibility. For instance, DC2 was often able to pass the G2/M block after a while (Figure 4B). Besides, Rad52 foci seem to eventually disappear in that cell. All these data indicate that DC2 might sometimes repair the damage and carry on dividing until it forms a colony. Importantly, we did notice that around one third of those colonies grew much more slowly in cdc14-1 (Figure 8A, “s” colonies). These slow-growing colonies were also observed when cdc14-1 cells where plated while normally growing at the permissive temperature. However, there was a three-fold increase in their number when plated after the transient Cdc14 inactivation (Figure 8A and table underneath). Strikingly, deletion of RAD52 prevented these very slow-growing colonies from appearing, although most colonies grew ∼30% more slowly after the cdc14-1 release (Figure 8A and table underneath).

The different effects of both the block-and-release experiment and the presence of Rad52 on the colony size of survivors prompted us to analyse the state of chromosome XII in the different outcomes (Figure 8B). Thus, we grew several colonies at the permissive temperature and carried out PFGE. We found that chromosome XII was shorter in the cdc14-1 rad52Δ strain we used (Figure 8B) than in its parental cdc14-1 RAD52 strain. Interestingly, chromosome XII was highly unstable in the cdc14-1 rad52Δ survivors, whereas it remained more constant in cdc14-1 RAD52, even in the slow-growing survivors (Figure 8B). One of the cdc14-1 RAD52 survivors (#b4) could have duplicated chromosome XII as suggested by the presence of two rDNA-containing bands.
From this set of experiments we conclude that many foursomes where cXIIr missegregation occurred can still carry on dividing for many generations (DC2 likely seeds these survivors). In addition to this, chromosome XII rearrangements and a reduced fitness are frequent outcomes of transient inactivation of Cdc14 for one cell cycle.
Discussion
The cdc14-1 release experiment as a model to study severing of anaphase bridges comprising unresolved sister chromatids
The major manifestation of entering anaphase without completing sister chromatid resolution is the appearance of anaphase bridges. Herein, we have introduced a new model to study the short-term consequences of these bridges based on the primary phenotype observed for the cdc14-1 mutant of Saccharomyces cerevisiae [23], [24], [28]. From a technical point of view this model presents several key advantages that facilitate cell biology studies on anaphase bridges: (i) non-resolution specificity for few genomic regions (e.g., cXIIr, see below); (ii) cell mixtures of segregated and missegregated cXIIr in the same population and experiment [28] (Figure 2); (iii) synchrony of the cells exiting mitosis (Figure 2); (iv) capability to monitor and cross-compare both daughter cells as they remain together after a cdc14-1 release (Figure 2 and Figure 3); and (v) availability of a proper parallel control that mostly behaves like cdc14-1 but does segregate the cXIIr (i.e., cdc15-2 conditional allele) [22] (Figure 2, Figures S1 and S4).
The cdc14-1 anaphase bridge and its fate in comparison to what is observed in separase, condensin, and top2 mutants
Because Cdc14 controls condensin and Top2 in anaphase and directs their activities to the rDNA [22], [27], [28], [30], [31], the overall expectation of our system is that the cdc14-1 anaphase bridge is like those of condensin and top2 mutants, however mainly restricted to a single chromosome arm (i.e., cXIIr). Therefore, it is interesting to compare our results to those previously reported for condensin and top2 mutants. We also include here mutants for cohesin removal due to their similarities. All these mutants form anaphase bridges comprised of trailing and distally unresolved sister chromatids as we depict in Figure 1 and Figure 9 (just for chromosome XII in those figures [i.e., cdc14-1], more chromosome arms are like cXIIr for these other mutants). This pattern of non-resolution likely arises from the spindle forces being able to slide cohesin and catenations away from bipolarly attached centromeres. Importantly, these mutants differ in the extent of non-resolution along the chromosome arms and the number of affected chromosomes, cohesin-removal mutants having the strongest phenotype and cdc14-1 the mildest [14], [19], [20], [23]. Accordingly, a common outcome in cells where cohesin or catenation removal have been impaired is the appearance of an anaphase where the nuclear mass cannot be split in two. For instance, condensin or top2 conditional mutants show rod-like nuclei in anaphase [15], [16]. The same outcome is seen in mutants where cohesin cleavage is inhibited in anaphase (e.g., separase mutants or non-cleavable forms of cohesin) [14]. Importantly, this unresolved nucleus does not abort cytokinesis, which eventually takes place leading to a “cut” phenotype in all cases. This phenotype is characterized by aneuploid daughter cells carrying broken chromosomes [14], [16], [32]. Another common feature of those daughter cells is that many are unable to resume the cell cycle, likely because of the massive chromosome breakage observed. The results we present in this work indicate that the anaphase bridge in cdc14-1 and its fate is somewhat different. First, the cdc14-1 block does not lead to a rod-like nucleus in anaphase, rather it is able to split the two DNA masses, which end up in each daughter cell [23]–[25], [34]. Likely, this is the consequence of most chromosome arms being able to segregate at the block. It is important to point out that we have assessed four telomeres of two other chromosomes (V and XIV) and found little missegregation in cdc14-1 foursomes relative to a cdc15-2 block (Table 1). Moreover, the drop of band intensity in the PFGE was only seen for chromosome XII in the cdc14-1 release (Figure 2E and Figure S4). Taking into account that, even in top2 and condensin mutants, small and medium-sized chromosomes segregate despite the rod-like nuclear phenotype [19], [21], we believe that the anaphase bridge in cdc14-1 mutants must comprise few chromosome arms; and that those severed by cytokinesis after the cdc14-1 release are fewer than for the other mutants. This in turn would explain why both daughter cells reach G2/M (Figure 2 and Figure S2). If more than four chromosome arms were severed, we would expect a G1 delay [45], which we did not observe.

On the other hand, in all mutants but cdc14-1, cytokinesis is followed by cell separation. This makes difficult to follow up and cross-compare both daughter cells, a key advantage we show for cdc14-1. It is unclear why cdc14-1 daughter cells are unable to separate from each other. However, this phenotype can be also seen when overcoming the cdc14-1 block by overexpressing Sic1 [28], [52]; and it was then shown that cytokinesis was completed [52]. Our data also suggest that cytokinesis is completed after cdc14-1 re-activation (Figure 3). Thus, a possible role of Cdc14 in cell septation could be responsible for this phenotype (Figure 2). A role that could actually be extended to the mitotic exit network as cdc15-2 also has a partial defect in cell separation.
Finally, the anaphase bridges formed in cohesin and some top2 mutants have been employed to define a checkpoint that delays cytokinesis (i.e., NoCut checkpoint) [37]. The actual length of such delay has been difficult to measure. In our case, the cdc14-1 anaphase bridge gave a short delay of about 20 minutes when comparing karyokinesis relative to the maintenance of the cXIIr bridge after the release (Figure 3B), and likely accounts for the NoCut checkpoint. Nevertheless, this delay was difficult to see by other means. For instance, it was observed neither relative to cdc15-2 (at least in the S288C background) nor as a biphasic drop of dumbbell cells in cdc14-1 (i.e., cells able to segregate the cXIIr versus those with the cXIIr bridge) (Figure 2A and Figure S2). Besides, the dynamics of the spindle disassembly after the release were quick for dumbbell cells (within the first hour, Figure S3A, left panel). It may be possible that a greater number of chromosomes in the anaphase bridge obtained by other mutants may trigger a stronger checkpoint signal.
The cdc14-1 anaphase bridge and its fate in comparison to bridges of a different physical nature
As stated above, the cdc14-1 anaphase bridge is supposed to be similar to condensin and top2 mutants, yet restricted to cXIIr. Notably, there are other situations where we can predict anaphase bridges of a different nature. For instance, anaphase bridges formed by partly replicated chromatids which nevertheless enter anaphase. For instance, sic1 and smc5/6 mutants behave this way [53], [54]. As with the difference between top2 and cdc14-1, sic1 and smc5/6 also differ in the actual number of chromosomes in the bridge, cXIIr being enriched in mutants for the Smc5/6 complex [44], [53], [54]. Despite the cytological similarities of the anaphase bridges between top2 and sic1, and between cdc14-1 and smc5/6, the bridge appeared broken in anaphase before completing cytokinesis in sic1 and smc5/6 mutants and a DDR can be also observed in that cell cycle stage [53], [54]. These findings highlight a key difference between the anaphase bridges formed by tangled sister chromatids and those where replication is incomplete: breakage before cytokinesis occurs in the latter, perhaps due to more fragile DNA in the unreplicated material. Finally, it is interesting to point out that other cdc14 mutants might enter anaphase with unreplicated DNA as well [33]. However, the behaviour of our cdc14-1 bridge is much closer to top2 and condensin than to what is observed in sic1 and smc5/6. This is, the chromosome can enter a PFGE in the cdc14-1 block (Figure 2E) and no DDR is observed at the block (Figure 5A and Figure 6A).
Another distinct anaphase bridge is that accomplished by the use of conditional dicentric chromosomes [55]–[57]. Like our cdc14-1 model, this approach has multiple technical advantages such as: (i) an anaphase bridge formed by a single chromosome; and (ii) cells with and without the bridge in the same population and experiment (∼50% chance of having two centromeres within a single sister chromatid attached to opposing SPBs). Nevertheless, the physical nature of the bridge is rather different. In the dicentric model, the bridge is formed by the sister chromatids being in an anti-parallel conformation. Moreover, sisters are supposed to be completely resolved from each other. In cdc14-1, the bridge is often formed by just one sister, the other one being out of the cytokinetic plane (Figure 1 and Figure 9) [23]. Thus, the expected DSBs and the broken genetic material in daughter cells are different when a dicentric chromosome is used. In relation to this system, it is interesting to note that the conditional dicentric chromosome triggers a Rad9-dependent mid-anaphase checkpoint (characterized by short spindles) that we did not see (data not shown) [24], [56]. Regardless, this and other dicentric models, like top2/condensin/cdc14-1 mutants, do not seem to break the anaphase bridge until cytokinesis takes place [57], [58].
On the DNA damage generated after severing the cdc14-1 chromosome XII right arm anaphase bridge
A key conclusion of this work is that at least one DSB near or within the rDNA is produced after the release from the cdc14-1 block (Figure 2, Figure 3, Figure 4). Unlike DSBs generated by endonucleases, radiation or any other means within a single nucleus [49], DSBs generated during anaphase bridge severing cause the ends of the broken DNA molecule(s) to migrate to opposing compartments which cannot be brought together anymore (i.e., nuclei of daughter cells). In the first scenario, the two ends of the DSB can be physically tied again and repaired by either non-homologous end joining (NHEJ) or HR. However, in the second case, the severing of the DNA molecule during nuclear division leads to a DSB where only one end can be found in each daughter nucleus (i.e., a “one-ended” DSB). Importantly, the state of each daughter cell is actually different with regard to chromosome XII dose (see Figure 1 and Figure 9 for schemes). While one cell (i.e., “DC2”) would have an entire chromosome XII plus the broken distal region of the right arm of the same chromosome (from the DSB to the telomere), the other one (i.e., “DC1”) would retain a fragment of a single chromosome XII (from the left telomere to the DSB, including its centromere). It is difficult to envisage how each DSB end might be repaired. For instance, break-induced replication, de novo telomere addition, chromosome translocation and/or elimination of the broken sisters might well be possible. Confounding matters, if the DSB takes place within the rDNA, which may happen often according to our data, cells can find a template for HR in another copy of the array. This latter situation can lead to an uncertain outcome (e.g., extrachromosomal circles?). Whichever way daughter cells face the problem, our results provide several interesting observations: i) the DSB(s) does not trigger a strong DDR in the new G1 (Figure 2 and Figure 6); ii) the MRX complex (i.e., Mre11) has no role in DSB(s) processing (Figure 7 and Figure S9); iii) the Rad9 checkpoint protein, the RPA complex and Rad52 are part of the mechanism to deal with these DSBs as soon as both daughter cells reach S-phase (Figure 4, Figure 5, Figure 6, Figure S6, and Figure S7); iv) the activity of these key proteins is long lasting and cumulative, especially in the daughter cell that only carries a fragmented cXIIr copy (i.e., DC1) (Figure 4, Figure 5, Figure 6); and v) DC2 often survives and might get rid of the broken distal fragment of cXIIr in order to do so (without using it as a template for HR, see below and Figure 8).
In relation to the absence of both a G1 arrest and Rfa1 foci in the new G1, our results indicate that the DSB generated after cdc14-1 release is similar to that generated by endonucleases (i.e., a “clean” DSB) as opposed to those generated by ionizing radiation (i.e., “ragged” DSBs) [45], [46], [49]. Also, it indicates that the number of DSBs should be below four or five (i.e., few chromosome arms are part of the anaphase bridge) [45]. Besides this, it is interesting that the processing of these DSBs is independent of Mre11 (Figure 7). Perhaps the MRX complex is not needed because the one-ended nature of each DSB means that there is no need to join both broken ends. Perhaps other molecular players are required in this context. In any case, others have previously reported that cells deficient in Mre11 and other MRX components can still generate a strong DDR and repair DSB through HR [59]. An alternative explanation for the Rfa1/Rad52 foci would be related to a sort of de novo damage generated as a consequence of DNA replication through an unrepaired or faultily-repaired chromosome XII. Future works are also needed in this direction.
As for the long-term consequences of this type of one-ended DSB, it is interesting that we observed that the DC2 cell can eventually recover in many cases (Figure 4 and Figure 8). Most surprising was the fact that viability got better when RAD52 was deleted. We interpret this as indicating that checkpoint adaptation followed by loss of the acentric fragment might be the main pathway that allows DC2 to progress. Accordingly, surviving cdc14-1 rad52Δ foursomes grew ∼30% more slowly (they actually took around one extra day to be visible on plates) relative to the same cells plated before the block-and-release experiment (Figure 8A). Indeed, the presence of Rad52 might compromise the chances of cdc14-1 DC2 cells surviving with a good fitness (Figure 8A). Contrary to expectations, though, PFGE of survivors showed that chromosome XII was more unstable in cdc14-1 rad52Δ. Also, the slow-growing colonies of cdc14-1 RAD52 did not show visible abnormal chromosome patterns. Despite our having checked only a few surviving colonies, it is interesting that none show evidence of chromosome XII rearrangements that involve translocations, although one might have duplicated the chromosome. Thus, we concluded that the DC2 cell very often survives and that it might repair the broken cXIIr in two ways; one which is dependent on Rad52 (e.g., through break induced replication, a likely event at least in the survivor with two chromosome XIIs), and a second Rad52-independent manner that somehow makes more likely changes in chromosome XII size.
As far as we know, this is the first time that an analysis of the DNA damage generated by cytokinetic severing of a single chromosome is conducted in yeast. A recent paper has just described the DDR after cytokinesis severs lagging chromosomes in human cells [60]. Many conclusions from that paper agree with those we observed in our work, although the system is clearly distinct (i.e., more than one chromosome is affected, both sister chromatids are severed, etc). In these human cells, DSBs arise after cytokinesis and are often repaired by NHEJ in G1, leading to aberrant chromosomes. The difference in the mechanism of repair in our yeast system is nevertheless expected, since yeast basically rely on HR acting through S and G2 rather than NHEJ in G1 [42]. Another key difference between both systems is the ploidy of the dividing cells. Human cells are diploids and may repair broken sisters using homologous chromosomes as templates. Our yeast strains were all haploids. It would be interesting to study whether cdc14-1 diploids also missegregate cXIIr and whether the DDR is different from what we describe here for haploids. Future work will be carried out to this aim.
Conclusion
In this study we have assessed the fate of cells that have an anaphase bridge formed by the right arm of chromosome XII (Figure 9 for a model and summary). We show how cells can go through a new G1, although they sever the bridge as they complete cytokinesis, and reach G2/M where they get arrested in a Rad9-dependent manner. We also show that the expected DNA damage response comprised RPA and Rad52, but is independent of Mre11. All these data shed light on how one-ended DSBs generated by a “cut” phenotype may be processed in eukaryotic cells. This work provides the first systematic study of the cell responses to a previous failure in sister chromatid resolution.
Materials and Methods
Yeast strains, growth, and experimental conditions
All yeast strains used in this work are listed in Table 2. Strains with the tetOs along chromosome XII right arm and those with tags for chromosome XIV telomeres were S288C background. Those with Rad52-YFP, Rfa1-YFP, Mre11-YFP, GFP-Tub1 and Tub4-CFP tags, and those with tags for chromosome V telomeres were W303. C-terminal tagging with GFP variants, gene deletions and allele replacements were performed using PCR methods [61], [62]. All strains were grown overnight at 25°C in YPD media. For telophase block-and-release experiments, asynchronous cultures were first adjusted to OD660/ml = 0.2, incubated at 37°C for 3 h in air orbital incubators and then shifted back to 25°C. To arrest cells in G1 in the cell cycle that follows the telophase release, cells were treated with alpha-factor (50 ng/ml) for 2 hours after the 25°C shift (all tested strains were bar1Δ). Flow cytometry analysis was carried out as described [54] in a BD FACScalibur machine, adjusting the peaks for 1 N and 2 N with an asynchronous culture at 25°C before reading the samples. PFGE to see all yeast chromosomes was performed using a CHEF DR-III system (Bio-Rad) in a 0.8% agarose gel in 0.5× TBE buffer and run at 12°C for 20 h at 6 V/cm with an initial switching time of 80 seconds, a final of 150 seconds, and an angle of 120°. PFGE to assess the size of chromosome XII was performed at 3 V/cm for 68 h with 300 and 900 seconds of initial and final switching time respectively. Ethidium bromide was used to visualize the chromosome bands in the gel. Band quantifications were performed with ImageJ software (NIH). Chromosome XII band(s) was identified by Southern blot using a Digoxigenin-labelled probe (Roche) against the NTS2 region within the rDNA.

Fluorescence microscopy
Fluorescent proteins and chromosome tags were analysed by wide-field fluorescence microscopy. Series of z-focal plane images (10–20 planes, 0.15–0.3 µm depth) were collected on a Leica DMI6000, using a 63×/1.30 immersion objective and an ultrasensitive DFC 350 digital camera, and processed with the AF6000 software (Leica). Scale bars in micrographs depict 5 µm. For nuclear morphology studies, DNA was stained using DAPI at 4 µg/ml final concentration after short cell treatment with 1% Triton X-100. Time-lapse movies were filmed without Triton/DAPI treatment on minimal medium agarose patches. Imaging was done at room temperature. Nucleoplasm pictures using nuclear-tagged TetR-YFP was also done without Triton/DAPI treatment. Rad52 foci recognition was performed either manually or using the CellProfiler software [63]. For the latter, whole images were normalized following the procedure: most intense focus in the first photo taken was set to 1, least intense pixel of the background was set to 0. A lower threshold of 0.1 was set for foci recognition.
Cytokinesis assays
Cytokinesis was monitored as previously described [39] with minor modifications. Briefly, aliquots of cells were fixed directly in the growth media by the addition of formaldehyde to 5% final concentration. After incubation at 25°C for 1 h with gentle rocking, fixed cells were washed twice with PBS and then once with 1 M sorbitol in 50 mM KPO4, pH 7.5. Cells were incubated with 0.2 mg/ml zymolyase 20T (Zymo Research) in the above sorbitol buffer containing 2 mM DTT for 20 minutes at 37°C. After zymolyase treatment, cell numbers were counted on a haemocytometer.
Special chemical treatments in these assays (i.e., nocodazole, alpha-factor and latrunculin A) were performed as follows: The arrest in G2/M was carried with 15 µg/ml of nocodazole at 25°C for 2.5 hours. Latrunculin A (100 µM) was added right at the G2/M release and alpha-factor (50 ng/ml) was added right at the telophase release. Release from the G2/M arrest was accomplished by washing away the nocodazole. Incubation for 1.5 hours at 37°C was used to block cells in telophase after a G2/M release. For the telophase release, cultures were shifted back to 25°C. Samples for this cytokinesis assay were taken at the telophase block and two hours after the telophase release. We used a cdc14-1 strain of the W303 background in these assays because it gave better synchrony, especially during the double block-and-release experiments (first at G2/M and then at telophase).
Statistics
Error bars in graphs represent the standard error of the mean (SEM) unless stated otherwise. The number of experiments is indicated in the corresponding figure legend or table. Statistic inference for cross-comparison of categorical variables distributions were performed by the Fisher's exact test when a 2×2 contingency table could be built (e.g., segregation vs. missegregation). For other categorical variables with more than two possible outcomes (e.g., number of Rad52 foci), the Pearson's chi-square test was employed. Individual comparisons between means of independent experiments were performed by the Student's T test. All tests were two-tailed.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. FujiwaraTBandiMNittaMIvanovaEVBronsonRT 2005 Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 437 1043 7
2. ShiQKingRW 2005 Chromosome nondisjunction yields tetraploid rather than aneuploid cells in human cell lines. Nature 437 1038 42
3. BajerA 1964 Cine-Micrographic Studies On Dicentric Chromosomes. Chromosoma 15 630 51
4. McClintockB 1939 The Behavior in Successive Nuclear Divisions of a Chromosome Broken at Meiosis. Proc Natl Acad Sci USA 25 405 16
5. McClintockB 1941 The Stability of Broken Ends of Chromosomes in Zea Mays. Genetics 26 234 82
6. GisselssonDPetterssonLHöglundMHeidenbladMGorunovaL 2000 Chromosomal breakage-fusion-bridge events cause genetic intratumor heterogeneity. Proc Natl Acad Sci USA 97 5357 62
7. ShimizuNShingakiKKaneko-SasaguriYHashizumeTKandaT 2005 When, where and how the bridge breaks: anaphase bridge breakage plays a crucial role in gene amplification and HSR generation. Exp Cell Res 302 233 43
8. VukovicBBeheshtiBParkPLimGBayaniJ 2007 Correlating breakage-fusion-bridge events with the overall chromosomal instability and in vitro karyotype evolution in prostate cancer. Cytogenet Genome Res 116 1 11
9. FrenchJDDunnJSmartCEManningNBrownMA 2006 Disruption of BRCA1 function results in telomere lengthening and increased anaphase bridge formation in immortalized cell lines. Genes Chromosomes Cancer 45 277 89
10. Cuevas-RamosGPetitCRMarcqIBouryMOswaldE 2010 Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc Natl Acad Sci USA 107 11537 42
11. Giménez-AbiánJFClarkeDJGiménez-MartínGWeingartnerMGiménez-AbiánMI 2002 DNA catenations that link sister chromatids until the onset of anaphase are maintained by a checkpoint mechanism. Eur J Cell Biol 81 9 16
12. LuoLZWernerKMGollinSMSaundersWS 2004 Cigarette smoke induces anaphase bridges and genomic imbalances in normal cells. Mutat Res 554 375 85
13. Díaz-MartínezLaGiménez-AbiánJFClarkeDJ 2008 Chromosome cohesion - rings, knots, orcs and fellowship. J Cell Sci 121 2107 14
14. UhlmannFLottspeichFNasmythK 1999 Sister-chromatid separation at anaphase onset is promoted by cleavage of the cohesin subunit Scc1. Nature 400 37 42
15. StrunnikovAVHoganEKoshlandD 1995 SMC2, a Saccharomyces cerevisiae gene essential for chromosome segregation and condensation, defines a subgroup within the SMC family. Genes Dev 9 587 599
16. HolmCGotoTWangJCBotsteinD 1985 DNA topoisomerase II is required at the time of mitosis in yeast. Cell 41 553 63
17. HolmCStearnsTBotsteinD 1989 DNA topoisomerase II must act at mitosis to prevent nondisjunction and chromosome breakage. Mol Cell Biol 9 159 68
18. DiNardoSVoelkelKSternglanzR 1984 DNA topoisomerase II mutant of Saccharomyces cerevisiae: topoisomerase II is required for segregation of daughter molecules at the termination of DNA replication. Proc Natl Acad Sci USA 81 2616 20
19. FreemanLAragon-AlcaideLStrunnikovA 2000 The condensin complex governs chromosome condensation and mitotic transmission of rDNA. J Cell Biol 149 811 24
20. BhallaNBigginsSMurrayAW 2002 Mutation of YCS4, a budding yeast condensin subunit, affects mitotic and nonmitotic chromosome behavior. Mol Biol Cell 13 632 45
21. SpellRMHolmC 1994 Nature and distribution of chromosomal intertwinings in Saccharomyces cerevisiae. Mol Cell Biol 14 1465 76
22. D'AmoursDStegmeierFAmonA 2004 Cdc14 and condensin control the dissolution of cohesin-independent chromosome linkages at repeated DNA. Cell 117 455 69
23. MachínFTorres-RosellJJarmuzAAragónL 2005 Spindle-independent condensation-mediated segregation of yeast ribosomal DNA in late anaphase. J Cell Biol 168 209 19
24. Torres-RosellJMachínFJarmuzAAragónL 2004 Nucleolar segregation lags behind the rest of the genome and requires Cdc14p activation by the FEAR network. Cell cycle 3 496 502
25. GranotDSnyderM 1991 Segregation of the nucleolus during mitosis in budding and fission yeast. Cell Motil Cytoskeleton 20 47 54
26. StegmeierFAmonA 2004 Closing mitosis: the functions of the Cdc14 phosphatase and its regulation. Annu Rev Genet 38 203 32
27. SullivanMHiguchiTKatisVLUhlmannF 2004 Cdc14 phosphatase induces rDNA condensation and resolves cohesin-independent cohesion during budding yeast anaphase. Cell 117 471 82
28. MachínFTorres-RosellJPiccoliGDeCarballoJAChaRS 2006 Transcription of ribosomal genes can cause nondisjunction. J Cell Biol 173 893 903
29. GuacciVHoganEKoshlandD 1994 Chromosome condensation and sister chromatid pairing in budding yeast. J Cell Biol 125 517 30
30. Clemente-BlancoAMayán-SantosMSchneiderDAMachínFJarmuzA 2009 Cdc14 inhibits transcription by RNA polymerase I during anaphase. Nature 458 219 22
31. WangB-DYong-GonzalezVStrunnikovAV 2004 Cdc14p/FEAR pathway controls segregation of nucleolus in S. cerevisiae by facilitating condensin targeting to rDNA chromatin in anaphase. Cell cycle 3 960 7
32. BaxterJDiffleyJFX 2008 Topoisomerase II inactivation prevents the completion of DNA replication in budding yeast. Mol Cell 30 790 802
33. DulevSRentyCdeMehtaRMinkovISchwobE 2009 Essential global role of CDC14 in DNA synthesis revealed by chromosome underreplication unrecognized by checkpoints in cdc14 mutants. Proc Natl Acad Sci USA 106 14466 71
34. CulottiJHartwellLH 1971 Genetic control of the cell division cycle in yeast. 3. Seven genes controlling nuclear division. Exp Cell Res 67 389 401
35. HennessyKMLeeAChenEBotsteinD 1991 A group of interacting yeast DNA replication genes. Genes Dev 5 958 969
36. BurkeDJChurchD 1991 Protein synthesis requirements for nuclear division, cytokinesis, and cell separation in Saccharomyces cerevisiae. Mol Cell Biol 11 3691 8
37. MendozaMNordenCDurrerKRauterHUhlmannF 2009 A mechanism for chromosome segregation sensing by the NoCut checkpoint. Nat Cell Biol 11 477 83
38. NordenCMendozaMDobbelaereJKotwaliwaleCVBigginsS 2006 The NoCut pathway links completion of cytokinesis to spindle midzone function to prevent chromosome breakage. Cell 125 85 98
39. HartwellLH 1971 Genetic control of the cell division cycle in yeast. IV. Genes controlling bud emergence and cytokinesis. Exp Cell Res 69 265 76
40. TollidayNVerPlankLLiR 2002 Rho1 directs formin-mediated actin ring assembly during budding yeast cytokinesis. Curr Biol 12 1864 70
41. WeinertTAHartwellLH 1988 The RAD9 gene controls the cell cycle response to DNA damage in Saccharomyces cerevisiae. Science 241 317 22
42. PâquesFHaberJE 1999 Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev 63 349 404
43. LisbyMRothsteinRMortensenUH 2001 Rad52 forms DNA repair and recombination centers during S phase. Proc Natl Acad Sci USA 98 8276
44. Torres-RosellJSunjevaricIPiccoliGDeSacherMEckert-BouletN 2007 The Smc5-Smc6 complex and SUMO modification of Rad52 regulates recombinational repair at the ribosomal gene locus. Nat Cell Biol 9 923 31 doi:10.1038/ncb1619
45. ZierhutCDiffleyJFX 2008 Break dosage, cell cycle stage and DNA replication influence DNA double strand break response. EMBO J 27 1875 85
46. BarlowJHLisbyMRothsteinR 2008 Differential regulation of the cellular response to DNA double-strand breaks in G1. Mol Cell 30 73 85
47. GarvikBCarsonMHartwellL 1995 Single-stranded DNA arising at telomeres in cdc13 mutants may constitute a specific signal for the RAD9 checkpoint. Mol Cell Biol 15 6128 38
48. ZouLElledgeSJ 2003 Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300 1542 8
49. LisbyMBarlowJHBurgessRCRothsteinR 2004 Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell 118 699 713
50. GeraldJNFBenjaminJMKronSJ 2002 Robust G1 checkpoint arrest in budding yeast: dependence on DNA damage signaling and repair. J Cell Sci 115 1749 57
51. BystrickyKLarocheTHouweGvanBlaszczykMGasserSM 2005 Chromosome looping in yeast: telomere pairing and coordinated movement reflect anchoring efficiency and territorial organization. J Cell Biol 168 375 87
52. FitzpatrickPJToynJHMillarJBJohnstonLH 1998 DNA replication is completed in Saccharomyces cerevisiae cells that lack functional Cdc14, a dual-specificity protein phosphatase. Mol Gen Genet 258 437 41
53. LengronneASchwobE 2002 The yeast CDK inhibitor Sic1 prevents genomic instability by promoting replication origin licensing in late G(1). Mol Cell 9 1067 78
54. Torres-RosellJMachínFFarmerSJarmuzAEydmannT 2005 SMC5 and SMC6 genes are required for the segregation of repetitive chromosome regions. Nat Cell Biol 7 412 9
55. HillABloomK 1989 Acquisition and processing of a conditional dicentric chromosome in Saccharomyces cerevisiae. Mol Cell Biol 9 1368 70
56. YangSSYehESalmonEDBloomK 1997 Identification of a mid-anaphase checkpoint in budding yeast. J Cell Biol 136 345 54
57. ThrowerDABloomK 2001 Dicentric chromosome stretching during anaphase reveals roles of Sir2/Ku in chromatin compaction in budding yeast. Mol Biol Cell 12 2800 12
58. HaberJEThorburnPC 1984 Healing of broken linear dicentric chromosomes in yeast. Genetics 106 207 26
59. HaberJE 1998 The many interfaces of Mre11. Cell 95 583 6
60. JanssenAvan der BurgMSzuhaiKKopsGJMedemaRH 2011 Chromosome Segregation Errors as a Cause of DNA Damage and Structural Chromosome Aberrations. Science 333 1895 1898
61. KnopMSiegersKPereiraGZachariaeWWinsorB 1999 Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast 15 963 72
62. JankeCMagieraMMRathfelderNTaxisCReberS 2004 A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21 947 62
63. CarpenterAEJonesTRLamprechtMRClarkeCKangIH 2006 CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7: R100. Available: http://genomebiology.com/content/7/10/R100
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 2
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Gene Expression and Stress Response Mediated by the Epigenetic Regulation of a Transposable Element Small RNA
- Contrasting Properties of Gene-Specific Regulatory, Coding, and Copy Number Mutations in : Frequency, Effects, and Dominance
- Homeobox Genes Critically Regulate Embryo Implantation by Controlling Paracrine Signaling between Uterine Stroma and Epithelium
- Nondisjunction of a Single Chromosome Leads to Breakage and Activation of DNA Damage Checkpoint in G2
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy