
Separation of Recombination and SOS Response in RecA Suggests LexA Interaction Sites
RecA plays a key role in homologous recombination, the induction of the DNA damage response through LexA cleavage and the activity of error-prone polymerase in Escherichia coli. RecA interacts with multiple partners to achieve this pleiotropic role, but the structural location and sequence determinants involved in these multiple interactions remain mostly unknown. Here, in a first application to prokaryotes, Evolutionary Trace (ET) analysis identifies clusters of evolutionarily important surface amino acids involved in RecA functions. Some of these clusters match the known ATP binding, DNA binding, and RecA-RecA homo-dimerization sites, but others are novel. Mutation analysis at these sites disrupted either recombination or LexA cleavage. This highlights distinct functional sites specific for recombination and DNA damage response induction. Finally, our analysis reveals a composite site for LexA binding and cleavage, which is formed only on the active RecA filament. These new sites can provide new drug targets to modulate one or more RecA functions, with the potential to address the problem of evolution of antibiotic resistance at its root.
Published in the journal:
. PLoS Genet 7(9): e32767. doi:10.1371/journal.pgen.1002244
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002244
Summary
RecA plays a key role in homologous recombination, the induction of the DNA damage response through LexA cleavage and the activity of error-prone polymerase in Escherichia coli. RecA interacts with multiple partners to achieve this pleiotropic role, but the structural location and sequence determinants involved in these multiple interactions remain mostly unknown. Here, in a first application to prokaryotes, Evolutionary Trace (ET) analysis identifies clusters of evolutionarily important surface amino acids involved in RecA functions. Some of these clusters match the known ATP binding, DNA binding, and RecA-RecA homo-dimerization sites, but others are novel. Mutation analysis at these sites disrupted either recombination or LexA cleavage. This highlights distinct functional sites specific for recombination and DNA damage response induction. Finally, our analysis reveals a composite site for LexA binding and cleavage, which is formed only on the active RecA filament. These new sites can provide new drug targets to modulate one or more RecA functions, with the potential to address the problem of evolution of antibiotic resistance at its root.
Introduction
Genetic material is under constant environmental assault. The bacterial recombinase protein RecA is pivotal to DNA repair [1]–[4] and to orchestrate the bacterial DNA damage response (SOS response) against natural, or drug-induced, genotoxic conditions. It is part of an ancient and evolutionarily widespread protein family and, except for a few endosymbionts [5], homologs carry out related functions in archaea [6] and eukaryotes [7], and in some cases mutants are linked to human cancers [8], [9].
To perform its many roles, RecA interacts with multiple partners in E. coli [3]. It normally exists in an inactive conformation without bound DNA [10], [11]. Upon DNA damage, an essential first step is the RecA polymerization around a single stranded DNA (ssDNA) in an ATP-dependent fashion [12]–[14]. In this active filament form, it can direct homologous recombination [15], bind to DinI [16], [17] and RecX [18]–[20] to control filament growth [21], [22], and bind the RecFOR complex to repair ssDNA breaks [23]–[25]. RecA is also a co-protease that promotes cleavage of the transcriptional repressor LexA [26] to trigger the expression of over 40 SOS response genes [27]. It also promotes cleavage of UmuD [28]–[31] to become a constituent of the error-prone DNA polymerase V (pol V) [32], [33], in addition to direct interaction with pol V for its activity [33]. Alternately it also interacts with another Y family of DNA polymerase, DinB (pol IV), to directly modulate its mutagenic potential in the translesion DNA synthesis [34]–[36]. It also promotes cleavage of the phage repressor, λCI, triggering induction of the lytic cycle [37], [38]. Every one of these interactions is a potential target to design drugs or mutants that dissect the molecular basis of RecA-dependent genomic repair and stability.
There are many crystallographic structures of RecA, or homologs, but most do not include bound DNA, and so are thought to represent the inactive conformation [39]–[50]. More recently, the crystal structure of E. coli RecA bound to DNA in the active conformation was solved (hereafter PDB:3cmx) [51]. It showed the ATP binding site, the DNA binding site and RecA-RecA interfaces in a likely active form (Figure 1A). Still, the interaction sites for other partners (such as DinI, RecX, RecFOR, LexA, UmuD, UmuD2C, DinB and λCI, as mentioned above) remain unknown. Separately, several mutational studies sought to identify residue determinants of diverse RecA functions, but without yet producing a fully coherent view [52].

To investigate the biological roles of known structural sites and to discover other RecA functional sites, we turned to the Evolutionary Trace (ET). This phylogenomic method [53]–[55] ranks a protein's residues by relative evolutionary importance. A structural map of the top-ranked residues then reveals clusters that indicate active sites and binding sites on the protein surface and that efficiently guide site-directed mutations that block, separate, or rewire functions in eukaryotic proteins [56]–[68]. ET analysis revealed many clusters of top-ranked residues on the E. coli RecA surface, which were targeted for mutagenesis followed by functional analysis. This extended and confirmed the biological role of the interfaces revealed in the inactive and active filament structure [51] and, critically, revealed new sites in other regions where mutations separated recombinase activity from co-protease activity for LexA cleavage. Two structurally distant amino acids (G108 and G22) are linked to the RecA-LexA interaction, and their location on RecA subunits i and i+6 apart in the helical active filament, across the groove, suggests a constraint on a low-resolution, illustrative model of the LexA-RecA interaction.
Results
Evolutionary Trace (ET) analysis identified clusters of residues in RecA
In order to identify novel, biologically relevant functional sites in the E. coli RecA protein, ET analysis was performed on 201 RecA homologs of bacterial origin. Each residue sequence position was ranked by ET based on how well its variations among homologs correlated with phylogenetic divergences (Figures S1 and S2) [56], [69]. Residue positions ranked in the top 40th percentile rank (thereafter ET40) were mapped onto the monomer of the RecA crystal structure, in the active form [51] (Figure 1B, shaded red and maroon). ET40 residues formed statistically significant clusters, with a z-score of 1.9, and suggested a number of functional surfaces, including as expected known sites such as the RecA-ATP interface, RecA-DNA interface and the two RecA-RecA interfaces (Figure 1A and contoured with a thick line in Figure 1B).
One area of interest includes a cluster of ET40 residues that borders the RecA-RecA interface in the inactive structure (residues highlighted in cyan in Figure 1B) but within the RecA-RecA, RecA-DNA interfaces in the active structure (Figure 1A and 1B). It includes residues E123, E154, L126, G212, G165 and A168. The structural data and the ET rank of these residues suggest they may be functionally important for oligomerization, although no experimental evidence has indicated such a role. It is likewise for residues S172, R176 and Q173, which are within the RecA-RecA structural interface common to both active and inactive structures (residue positions shown in Figure 1B). All of these residues were therefore grouped together as the extended RecA-RecA/DNA interface patch and chosen for mutational and functional analysis, described below.
Besides this interface patch, other ET40 residues formed various other clusters elsewhere on the RecA structure. These were named, arbitrarily, ET site-1 (D224, R226 and K245), ET site-2 (G288, Q300 and N304), ET site-3 (G87, K88 and G108) and ET site-4 (G22, K23 and G24) (residue positions shown in Figure 1C). Since each of these sites suggest a new putative structural interface without any known function, site-specific mutagenesis was performed to probe their function. For all site-directed mutagenesis, amino acids were individually mutated to alanine unless the alanine substitution already existed in a member of the ET sequence dataset. For such exceptions, tyrosine, tryptophan or glycine residues were used depending on their absence from the substitution profile of the targeted position. All mutations were constructed on a low-copy plasmid-borne recA gene and transformed into a ΔrecA E. coli strain [70]. The mutant RecA strains were tested for their UV sensitivity to assess the global impact on RecA function. Representative mutant strains from each ET clusters were also tested for their sensitivity to mitomycin C to demonstrate that the survival phenotypes of these mutants were not specific to UV induced DNA damage (Figure S3). Then, to pinpoint the molecular basis of UV sensitivity, both a P1 recombination assay and a LexA western blot assay were performed to probe the recombinase activity and the induction of LexA cleavage of each RecA variant in vivo, respectively.
Finally, to validate our ET analysis on RecA, several poorly-ranked ET residues located on the RecA surface (in the worst quartile of importance) were analyzed through site-directed mutagenesis and functional analysis as described above. Such bottom-ranked residues near the known RecA interfaces included T89, N181, N186 and V238, and others that were away from any known RecA interfaces included K294 and N312 (Figure 1B, shown in blue letters). As expected, mutation of these residues displayed no UV sensitivity (Figure 2A), had relatively intact recombination efficiencies that ranged from 56 to 72% compared to wild-type RecA strain as judged by P1 recombination assay (Figure 2B) and were all fully capable of inducing LexA cleavage leading to upregulation of RecA protein (Figure 2C).
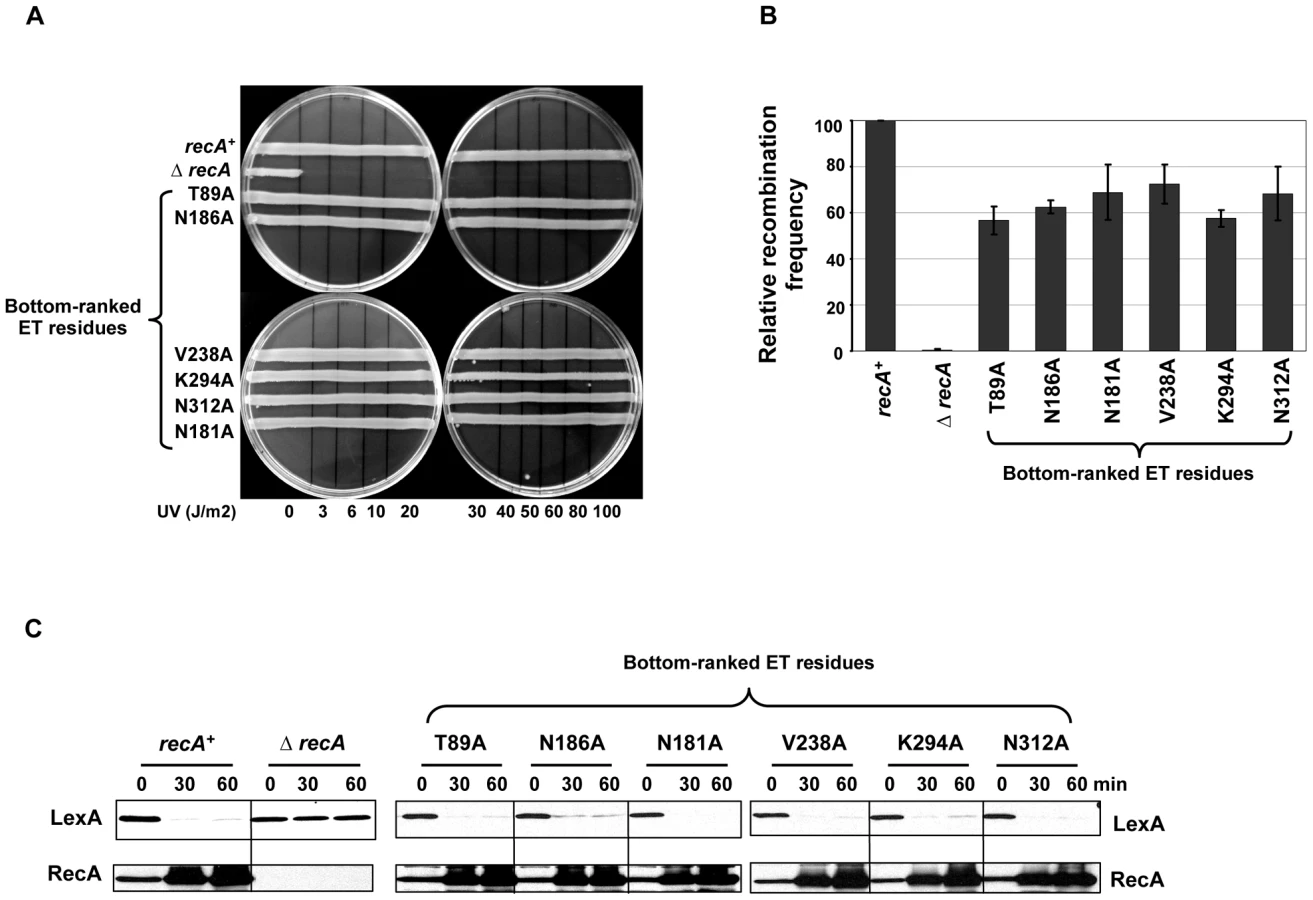
Functional validation of the extended interface patch in RecA active filament
In order to test the functional role of the extended interface patch residues (Figure 1B), site-directed mutagenesis was performed. As expected from interference with RecA-RecA or RecA-DNA interactions, either of which would disrupt the basic ability of RecA to form active nucleoprotein filament, these RecA mutant strains within the interface patch were strongly sensitive to very low doses of UV damage (Figure 3A) similar to the empty vector in a ΔrecA background (Figure 2A) with the exception of the Q173A substitution. As a positive control, the E. coli strain with wild-type RecA overcame UV damage (Figure 2A).

To characterize the recombination efficiency, a P1 transduction assay was performed. All variants, except for Q173A, are disrupted for recombination similarly to the ΔrecA strain (Figure 3B). Likewise, these variants, significantly hindered RecA's ability to promote LexA cleavage upon DNA damage and subsequently failed to up-regulate RecA (Figure 3C). The observation that Q173A mutation showed no effect on RecA activity could be attributed to the lesser importance of this residue, which has the worst rank of the ET40 residues in this patch (30th percentile-rank).
Taken together, these mutations confirmed that top-ranked ET40 residues in this extended interface patch impair both the recombination and co-protease activities of RecA, and thus are crucial for UV survival. This is consistent with the structural data on the active RecA filament [51]. These residues are directly involved in RecA-RecA and RecA-DNA interaction, and their mutations are thus likely to interfere with the basic assembly or working of the nucleoprotein RecA-DNA filament.
ET site-1 may be involved in RecA–RecA interaction
Two of three ET site-1 RecA variants (R226A and D224A) are UV-sensitive (Figure 1C and Figure 4A). These mutations strongly disrupt recombinase activity of RecA (Figure 4B) and LexA self-cleavage (Figure 4C), similarly to the extended interface patch variants. Assuming that RecA folding is not affected, and given the structural contiguity to the RecA-RecA interface-1 (see Figure 1A and 1C) one reason may be some involvement in RecA-RecA interaction and filament formation. Another possibility is that ET site-1 could play a role in binding to proteins such as RecX, DinI and RecFOR that modulate RecA function.

ET site-2 specifically affects the recombination function of RecA
Mutational analysis of ET site-2 residues (Figure 1C) showed separation of RecA function. First, two of the three mutant strains have abnormal UV sensitivity (Figure 5A). The N304D variant was the most sensitive, followed by Q300A. The G288Y variant displayed no UV sensitivity. Next, all three mutant strains had reduced recombination efficiency (Figure 5B), with the N304D variant being as deficient as the ΔrecA strain. Finally, LexA cleavage upon DNA damage was intact (Figure 5C), suggesting that the RecA folding and active filament formation required for SOS induction were unaffected. Thus, overall, all these ET site-2 mutations have more severe impact on the recombinase activity than on the SOS response. The N304D mutant, which is completely defective for recombinase activity, displays the clearest example of separation of function. Therefore, these data suggest that the ET site-2 is essential for the recombinase function of RecA. One explanation is that this site may bind to the dsDNA or to other partners essential for recombination events.

Residues in ET site-3 and site-4 specifically affect LexA cleavage
Mutagenesis of ET site-3 (G87, K88 and G108) and ET site-4 (G22, K23 and G24) (Figure 1C) also produced partial separation of function. In contrast to mutations affecting ET site-2 residues, these variants displayed no UV sensitivity (Figure 6A), except for the G22Y variant, which only becomes UV sensitive at higher doses. All variants displayed lower recombination efficiency compared to wild-type RecA but none as complete as the ΔrecA strain. The efficiency was down approximately to 9, 19 and 25% for G87Y, K88Y and G108Y in ET site-3 and to 5, 11 and 22% for G22Y, K23Y and G24Y, respectively, in ET site-4 (Figure 6B). Among these residues, the G108Y (in ET site-3), G22Y and K23Y (in ET site-4) showed strongly reduced LexA proteolysis upon DNA damage (Figure 6C, highlighted in red). Strikingly, these three mutant strains exhibited 3.5 to 4.6-fold upregulation of RecA levels, consistent with SOS induction, even in the absence of LexA cleavage after DNA damage. These variants show some similarity to a well-known recA mutant, recA430 (corresponding to G204S) [71]–[73], which is only slightly affected for recombination but deficient for LexA cleavage. In our assays, this variant showed an increase in UV-sensitivity (Figure 6A, lowermost panel), with relatively intact recombination activity, and no ability to induce LexA cleavage. However, this variant did not up-regulate RecA, unlike the G108Y, G22Y and K23Y mutant strains (Figure 6C). Thus RecA upregulation without LexA cleavage upon DNA damage is unique to our three mutant strains. Finally we asked whether this defect in co-protease activity was specific to LexA by testing another substrate of RecA, UmuD, which is also activated upon LexA cleavage. In this case, the active RecA filament mediates UmuD autoproteolysis yielding UmuD'. Since UmuD cleavage induction is a later SOS response than that of LexA, it was analyzed at later time points. Upon DNA damage, we observed an upregulation of UmuD levels in G108Y mutant strain and also a slight upregulation in G22Y and K23Y mutants respectively (data not shown). To analyze UmuD cleavage, we used a lexA (def) E. coli strain which is constitutive for UmuD expression. Formation of UmuD', the cleavage product of UmuD was visible in G108Y and G22Y mutant strains indicative of self-proteolysis of UmuD induced by RecA (Figure 6D). However in the G24Y mutant, that was shown to cause a slow LexA cleavage, there was a robust upregulation of UmuD like wild-type RecA (data not shown). In addition, the recA430 mutant strain, in our assay, could not cleave UmuD as well (Figure 6D). The upregulation of UmuD and its subsequent cleavage to UmuD' in G108Y, G22Y and to some extent in K23Y, strengthens the possibility that these variants alter most likely RecA-LexA interaction rather than affecting the overall co-protease activity of RecA.

Role of G108 and G22 residues in initiating LexA cleavage
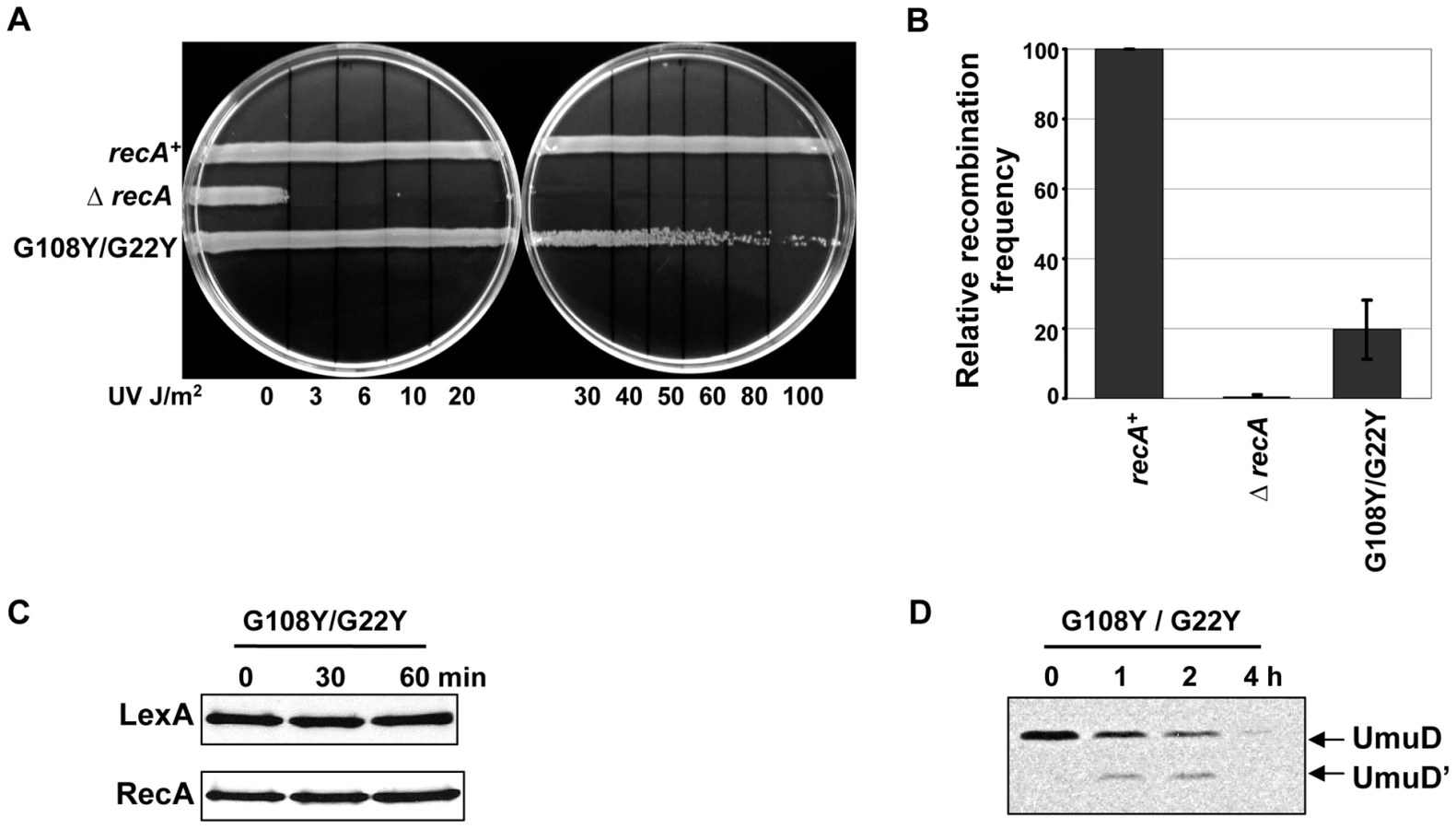
The unexpected upregulation of RecA without LexA cleavage after DNA damage could suggest that LexA is sequestered by active RecA filaments, leading to SOS induction. Specifically, mutation of just one of the two residues, G108 or G22, could leave the ET-site with the other residue intact and able to bind LexA to RecA, effectively titrating it away from transcriptional repression irrespective of cleavage.
To test the possibility that LexA cleavage induction might require binding RecA at G108 and G22 residues at the same time, we made the double mutant G108Y/G22Y. We reasoned that with both ET-sites 3 and 4 mutated, LexA could not bind to RecA anymore, allowing it to repress the SOS response. In our assays, the double mutant (G108Y/G22Y) was weakly sensitive to UV (Figure 7A) comparable to the recA430 mutant (G204S) (Figure 6A). The recombination efficiency of the double mutant was intermediate between G108Y and G22Y individual mutants (20% as that of wild-type RecA) (Figure 7B). The mutant also did not induce LexA cleavage (Figure 7C), but could still cleave UmuD to UmuD' although, less efficiently (Figure 7D). Importantly, RecA upregulation was much reduced compared to the individual mutants. This supports our hypothesis that a joint disruption at ET-sites 3 and 4, via double mutations at residues G108 and G22, impairs LexA binding and prevents its sequestration to RecA with subsequent upregulation of SOS genes. The similar impact of individual mutations at residues G108 and G22 and the synergy of their coupled mutations suggest that they may play joint roles in both LexA binding and subsequent cleavage.
Discussion
This work identifies new surface exposed domains of RecA critical for its recombinase and LexA cleavage functions. The discovery of these residues with the Evolutionary Trace (ET) shows that this computational strategy, based on phylogenetically correlated sequence variations, applies equally well here in prokaryotes as previously in eukaryotes, and that it can efficiently identify key functional residues that evaded detection by several past studies on this highly mutagenized protein [52]. Finally, this work validates the functional role of recent crystallographic evidence for RecA-RecA and RecA-DNA interfaces [51], and suggests a model for the RecA filament-LexA interaction.
Overall RecA function
To confirm past observations at RecA functional sites and also to validate the ET strategy, we targeted for site-directed mutagenesis the top-ranked Evolutionary Trace residues at the interface defined in the active RecA-ssDNA filament structure [51]. This structure has a ∼12 Å shift of the RecA-RecA interface upon ssDNA binding compared to the inactive structure [39], and it now includes additional residues important for filament formation (G165, S172, R176 and G212 and E123, A168). Consistent with both the inactive and active structures, top-ranked ET residues significantly overlapped the RecA-RecA and RecA-DNA dimer interfaces (Figure 1A and 1B) and their mutations prohibit both recombinase and LexA cleavage activities (Figure 3). This is in line with previous mutations of neighboring residues with similar defects in recombinase or co-protease activities [74]–[84], and it establishes a functional role for the residues in the extended RecA-RecA interface observed structurally in the active RecA filament.
A second set of top-ranked ET residues, ET site-1 (D224, R226, K245), was found in the cleft region of RecA and adds details on the determinants of overall RecA function. This cleft is located in between two adjacent RecA monomers and was previously proposed to bind repressors [39], [79], [80], [85]–[87] and dsDNA [88], [89], possibly through several of the positively charged side chains [86], [89]. The ET site-1 overlaps this region and mutations of D224 and R226 eradicated both recombinase activity and SOS functions of RecA. This suggests that, as above, this site also takes part in forming a functionally active filament, possibly as a functional extension of the neighboring extended interface patch. The inactive RecA filament structure (PDB:1u99) [39] shows ET site-1 next to the RecA homodimerization site. In addition, the active filament structure (PDB:3cmt) [51] shows both R226 and D224 residues binding to the previously disordered DNA binding loop 2 (L2). In fact, the binding partner of R226 in L2 is Glu207, which is an absolutely conserved residue among 64 RecA enzymes [90], [91] and does not tolerate any amino acid substitution without some loss of function, as seen from saturation mutagenesis [74]. Thus, the severe impact of R226 and D224 mutations on both recombination and SOS induction is consistent with ET site-1 contributing to the formation of the active filament and, indirectly, to DNA binding.
Recombination function
Another set of top-ranked ET residues, ET site-2 (N304, Q300 and G288), is located in the RecA C-terminal domain (CTD). The CTD region of RecA has been previously implicated in recombinase function [79], [92]–[94], acting as a secondary DNA (dsDNA) binding pocket on the outer surface of the filament. Mutations of all three ET site-2 residues impair recombinase activity but not LexA cleavage (Figure 5B and 5C). These residues are in the edge of the filament groove, and might provide binding stability to dsDNA for its efficient uptake into the filament. Of note, mutation N304D showed a striking separation of function with a complete destruction of recombinase activity similar to ΔrecA strain. Such marked defect in recombinase function was previously reported by a point mutation involving Gly301 to Asp in the CTD [95], [96], suggesting the intolerance of negatively charged amino acid side chains in the RecA CTD in dsDNA binding during the recombination process. Alternatively, these residues might modulate interaction between RecA and DinI [97], RecX [22] or RecFOR proteins.
RecA–LexA interaction
The induction of the SOS response by RecA-mediated cleavage of LexA has been extensively studied both in vivo and in vitro, yet the sites involved in the interaction of these proteins remain unclear. Our ET analysis reveals two new sites with some potential to be determinants of the RecA-LexA interaction. Mutation of these sites preserves recombination but in majority, inhibits LexA cleavage. Paradoxically, levels of the LexA-repressed proteins, RecA (Figure 6C) and UmuD (data not shown) were up-regulated upon DNA damage; indicating that there was SOS induction, independent of LexA cleavage. These mutants could promote UmuD cleavage; indicating that this defect in co-protease function was highly specific to LexA (Figure 6D). To our knowledge, the activation of the SOS response by UV, independent of LexA cleavage, has not been previously observed. Electron micrograph [20], [98], [99] and mutational studies [71], [72], [79], [80], [82], [83], [85]–[87] point to the binding of LexA, cI and UmuD structural homologs deep within the RecA filament's helical groove. However, structural elements on the edge of the helical groove including the dynamic N-terminal helix/strand (1–30) and C-terminal domain (270–333), have also been found to contribute to cleavage of LexA, cI and UmuD [79], [99]. Consistent with these findings, the residues that we find to be highly specific to LexA hydrolysis lie between the N-terminal α-helix A and β-strand 0 (G22 and K23) or adjacent to the CTD (G108). We propose that LexA binds across the RecA filament's groove through direct contacts at both of the two distant ET-sites 3 and 4, and that these sites cooperate to enable LexA proteolysis (see Figure 8A). Then, as observed, the disruption of either one could permit binding but not cleavage of LexA, leading to SOS induction without efficient LexA degradation. Previous mutational studies [74], [79], [82], [83] implicating residues facing the helical groove in repressor cleavage functions (Figure 8A, shown in magenta) are consistent with this model. Moreover, this model predicts that the simultaneous disruption of both LexA binding sites 3 and 4 would prevent LexA binding and sequestration. Indeed, the G108Y/G22Y double-mutant does not up-regulate expression of RecA (Figure 7C) and UmuD (data not shown). An alternative possibility would be that each point-mutation changes the conformation of the active RecA filament to prevent LexA cleavage. However, such an allosteric effect would have to be subtle since both the recombination function of RecA and its co-protease activity towards UmuD are still present in both the point-mutant and the double-mutant (Figure 7B and 7D). Nevertheless, the less efficient co-protease activity of the double mutant towards UmuD also suggests that the binding sites for UmuD might be shared among these residues or their neighbors, so that the double mutation either directly disrupts the efficiency of UmuD binding and/or cleavage or indirectly affects the protein fold for UmuD binding. This is in agreement with previous electron micrograph and mutational studies suggesting the possibility of repressors sharing similar binding sites on the RecA filament [79], [87], [98].

Consistent with our model of LexA binding to a composite site, a geometric docking analysis of LexA dimer binding to the RecA filament identifies, among many other possible solutions, one in which the LexA dimer binds to ET site-3 and ET site-4 from RecA units at positions i and i+6, or one helical turn apart, across the filament groove (Figure 8B). This illustrates how, by wedging itself into the groove, the DNA binding domain of LexA may bind the core of the RecA filament and at the same time allow the catalytic C-terminal domain of LexA to span the helical filament's edge. The model could be further addressed by direct assays measuring LexA binding and proteolysis in these mutant proteins in vitro. In the future, these RecA mutants may become a useful tool for trapping the RecA-LexA interaction towards efforts to obtain a co-crystal structure. Overall our results suggest that a cooperative binding at RecA residues G108 and G22 is essential for triggering LexA proteolysis.
In conclusion, ET identified new functional sites and efficiently guided their mutational validation in RecA. These sites form important new targets for future biochemical studies of RecA function, and may prove useful for creating separation of function mutants that will help dissect the network of interactions responsible for DNA damage repair. The emergence of bacterial resistance to antibiotics is mediated in part by the SOS response and it has been proposed that blocking the SOS pathway may prevent the evolution of bacteria in contact with these antibiotics [100], [101]. The new RecA sites identified in this work may become useful for the design of new drugs preventing the evolution of bacteria to antibiotics.
Materials and Methods
Materials
The low copy plasmid pGE591 containing wild-type recA [81] was a kind gift from Dr. George Weinstock, Washington University in St. Louis, MO, and the E. coli strain SMR6765 [70] lacking functional RecA was provided by Dr. Susan Rosenberg, Baylor College of Medicine, TX. E. coli strain CH458 (lacZYA::gfp-cat) was used as donor strain for P1 phage lysate preparation. Rabbit anti-UmuDD' polyclonal antiserum [102] was generously provided by Dr. Roger Woodgate (Laboratory of Genomic Integrity, NIH, MD).
Bacterial strains and plasmids
The genotypes and sources of the E. coli strains and plasmids used in this study are listed in Table S1. Strains made in this study were constructed by classical P1 transduction [103].
Sequence and structure analysis
The Evolutionary Trace analysis [104] used a sequence alignment consisting of 201 RecA protein sequences, nearly all bacterial, that have LexA or LexA homolog (Table S2). The primary source of the alignment was the HSSP database and it was retrieved using the Evolutionary Trace Report Maker Server [105]. Each sequence was BLASTed against the NCBI non-redundant protein sequences (nr) database and the sequences with at most 20 gaps or additions relative to the RecA sequence of E. coli were aligned using MUSCLE [106]. This dedicated alignment spanned greater evolutionary distances than the one provided automatically by the ET viewer software [107] (ET servers and viewing tools are available for public use at http://mammoth.bcm.tmc.edu/). The ET phylogenetic tree and multiple sequence alignment of RecA sequences in text and image formats are also available at http://mammoth.bcm.tmc.edu/AdikesavanEtAl/Sup. The interfaces of RecA with ATP, DNA and other monomers were defined as the amino acids that are closer than 5 Å from the ligand in atom to atom distances, excluding hydrogens. The figures of RecA monomer and filament structures were generated by PyMOL (The PyMOL Molecular Graphics System, Version 1.3, Schrödinger, LLC) using the PDB structure: 3cmw. The RecA filament was extended by repeated duplications and space alignment of the terminal monomers. 34045 rigid-body protein-protein docking models of a LexA dimer bound to the RecA filament with good molecular shape complementarity were created with the program PatchDock [108].
Site-directed mutagenesis of E. coli RecA protein
The wild-type RecA plasmid (pGE591-recA-WT) was used as template for site-directed mutagenesis of RecA protein using QuikChange II XL site-directed mutagenesis kit (Stratagene) as per manufacturer's protocol. The plasmids containing mutations in the recA gene obtained by site-directed mutagenesis were transformed in E. coli SMR6765 (ΔrecA) strain. All the recA mutant plasmids were sequence verified. The mutant RecA proteins expressed from these strains were also checked for their stability by western analysis using anti-RecA antibody.
UV survival assay
The semi-quantitative measurement of UV sensitivities of wild-type RecA and RecA mutants was done as described previously [79]. E. coli SMR6765 strains expressing either wild-type RecA or RecA mutants were grown overnight in Luria-Bertani (LB) medium containing selective antibiotic (kanamycin 25 µg/mL). The next day, subcultures were made and grown further till the OD600 reached 0.5. The bacterial cultures were streaked onto sterile LB/Kanamycin plates using sterile Q-tips. The plates were exposed to increasing doses (J/m2) of UV light using a UV Stratalinker, and incubated at 37°C for a further period of 16 hours protected from light. Different levels of UV survival between wild-type RecA and RecA mutant strains were analyzed. The assay was repeated at least three times independently and the representative results are shown.
In vivo LexA cleavage analysis by Western blot
Western blot analysis of in vivo LexA cleavage was carried out as described previously [79], [109] with minor modifications. E. coli SMR6765 strains carrying either wild-type RecA or RecA mutants were grown overnight and the next day, subcultures made and grown at 37°C till the OD600 reached 0.5. The DNA damaging agent, nalidixic acid (Sigma) was added to each culture at 100 µg/mL final concentration. The cultures were grown further at 37°C and 1 mL of culture from each strain was aliquoted at 0, 30 and 60 minutes. The culture aliquots were washed once in cold PBS and were stored at −80°C until further processing. Subsequently, the pellets were lysed using BugBuster Master Mix (Novagen) and the total lysate made as per the manufacturer's protocol. Total proteins in the lysates were estimated using the Micro BCA Protein Assay Kit (Thermo Scientific). The RecA protein levels were normalized to bacterial growth by using equal amount (50 µg) of total protein lysate collected at different time points for resolving in SDS-PAGE. The resolved bands were blotted to nitrocellulose membranes and probed with anti-LexA (1∶7000, ABR bioreagents) and anti-RecA (1∶15000, MBL International) antibodies. Goat anti-rabbit IgG-HRP (Chemicon International) was used as the secondary antibody at 1∶7000 dilutions. Chemiluminescence detection was done using Amersham ECL western blotting kit and autoradiographed as per manufacturer's protocol. All the experiments were repeated at least three times for each RecA mutants and the representative results are shown.
P1 transduction assay
The recombination efficiency of the E. coli strains carrying wild-type RecA and RecA mutant proteins were assayed by P1 transduction as described [110]. The assay measures the efficiency of the wild-type RecA or its variants, to recombine the selectable genetic marker (gfp-cat gene) into their chromosome, using P1 phage mediated transduction. P1 lysate was prepared by growing the donor bacterial strain (CH458≡MG1655 lacZYA::gfp-cat) overnight in LB medium with chloramphenicol antibiotic. The overnight culture was diluted 1∶4 in fresh LB+ 5 mM CaCl2 and 0.2% glucose and allowed to stand for 30 min at room temperature. Then wild-type P1 phage lysate was added to the diluted overnight culture, incubated with shaking @ 37°C for 20 min followed by plating them on LB plates with 5 mM CaCl2 and 0.2% glucose. Next day after overnight incubation of the plates, the top layer of lysed cells were scrapped-off into sterile centrifuge tubes, and ∼300 µl of chloroform added to the lysate, vortexed and allowed to stand for 30 min at room temperature with intermittent vortexing followed by centrifugation @ 10000 rpm for 10 min to collect the supernatant P1 lysate. The P1 phage lysate was subsequently titred against E. coli strain SMR6765 containing wild-type RecA on pGE591 plasmid. The viable cell numbers for wild-type RecA and RecA mutant strains was also assayed, so that approximately 1 phage for every 100 viable cells was used in the P1 transduction assay. During the assay, the recipient bacterial strains (wild-type RecA and the RecA-mutant strains) were grown overnight and subcultured the next day till the OD600 reached 0.5. P1 lysate was added to the cultures in such a way that the ratio of phage to viable cell count was ∼1∶100, vortexed, and incubated with shaking @ 37°C for 18 min followed by centrifugation for 2 min at 7000 rpm to pellet the cells. The cells were resuspended in LB medium with 100 mM sodium citrate and plated on LB-citrate plates with chloramphenicol, incubated overnight at 37°C. Next day, the number of transductant colonies in each strain was counted. The transduction or recombination efficiency of the wild-type RecA and mutant RecA strains were calculated by the number of transductants relative to the phage titer. The assay was repeated at least 3 times for all the wild-type RecA or RecA mutant strains and the mean standard error values for recombination efficiency were used for graphical representation.
Analysis of UmuDD' proteins by Western blot
The cleavage of UmuD protein to UmuD' upon DNA damage were shown individually in E. coli strains with plasmid-borne wild-type RecA or empty vector or RecA mutants (G108Y, G22Y, K23Y and G24Y) by western blot [111]. The E. coli strains (OL53) used in this assay were lexA (def) to enable constitutive UmuD expression. UmuD cleavage to UmuD' was assayed similar to LexA cleavage analysis except that after DNA damage induction, the aliquots were collected at 0, 1, 2 and 4 hours (since UmuD induction is a late process in the SOS response). The culture aliquots were processed similarly as mentioned above for LexA cleavage analysis. 200 µg of total protein from lysates were resolved in SDS-PAGE and immunoblotting was done with anti-UmuDD' antisera (1∶2000). The analyses were repeated at least 3 times independently for each wild-type RecA or mutant strains and the representative data were shown.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. RaddingCM 1981 Recombination activities of E. coli recA protein. Cell 25 3 4
2. LusettiSLCoxMM 2002 The bacterial RecA protein and the recombinational DNA repair of stalled replication forks. Annu Rev Biochem 71 71 100
3. CoxMM 2007 Regulation of bacterial RecA protein function. Crit Rev Biochem Mol Biol 42 41 63
4. CoxMM 2007 Motoring along with the bacterial RecA protein. Nat Rev Mol Cell Biol 8 127 138
5. TamasIKlassonLCanbackBNaslundAKErikssonAS 2002 50 million years of genomic stasis in endosymbiotic bacteria. Science 296 2376 2379
6. SeitzEMBrockmanJPSandlerSJClarkAJKowalczykowskiSC 1998 RadA protein is an archaeal RecA protein homolog that catalyzes DNA strand exchange. Genes Dev 12 1248 1253
7. ShinoharaAOgawaHOgawaT 1992 Rad51 protein involved in repair and recombination in S. cerevisiae is a RecA-like protein. Cell 69 457 470
8. VamvakasSVockEHLutzWK 1997 On the role of DNA double-strand breaks in toxicity and carcinogenesis. Crit Rev Toxicol 27 155 174
9. KhannaKKJacksonSP 2001 DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet 27 247 254
10. FloryJTsangSSMuniyappaK 1984 Isolation and visualization of active presynaptic filaments of recA protein and single-stranded DNA. Proc Natl Acad Sci U S A 81 7026 7030
11. YuXJacobsSAWestSCOgawaTEgelmanEH 2001 Domain structure and dynamics in the helical filaments formed by RecA and Rad51 on DNA. Proc Natl Acad Sci U S A 98 8419 8424
12. KuzminovA 1999 Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda. Microbiol Mol Biol Rev 63 751 813 table of contents
13. KowalczykowskiSCDixonDAEgglestonAKLauderSDRehrauerWM 1994 Biochemistry of homologous recombination in Escherichia coli. Microbiol Rev 58 401 465
14. KowalczykowskiSC 2000 Initiation of genetic recombination and recombination-dependent replication. Trends Biochem Sci 25 156 165
15. CoxMM 1999 Recombinational DNA repair in bacteria and the RecA protein. Prog Nucleic Acid Res Mol Biol 63 311 366
16. YasudaTMorimatsuKHoriiTNagataTOhmoriH 1998 Inhibition of Escherichia coli RecA coprotease activities by DinI. Embo J 17 3207 3216
17. LusettiSLVoloshinONInmanRBCamerini-OteroRDCoxMM 2004 The DinI protein stabilizes RecA protein filaments. J Biol Chem 279 30037 30046
18. DreesJCLusettiSLCoxMM 2004 Inhibition of RecA protein by the Escherichia coli RecX protein: modulation by the RecA C terminus and filament functional state. J Biol Chem 279 52991 52997
19. StohlEABrockmanJPBurkleKLMorimatsuKKowalczykowskiSC 2003 Escherichia coli RecX inhibits RecA recombinase and coprotease activities in vitro and in vivo. J Biol Chem 278 2278 2285
20. VanLoockMSYuXYangSGalkinVEHuangH 2003 Complexes of RecA with LexA and RecX differentiate between active and inactive RecA nucleoprotein filaments. J Mol Biol 333 345 354
21. LusettiSLDreesJCStohlEASeifertHSCoxMM 2004 The DinI and RecX proteins are competing modulators of RecA function. J Biol Chem 279 55073 55079
22. RenzetteNGumlawNSandlerSJ 2007 DinI and RecX modulate RecA-DNA structures in Escherichia coli K-12. Mol Microbiol 63 103 115
23. MorimatsuKKowalczykowskiSC 2003 RecFOR proteins load RecA protein onto gapped DNA to accelerate DNA strand exchange: a universal step of recombinational repair. Mol Cell 11 1337 1347
24. ShanQBorkJMWebbBLInmanRBCoxMM 1997 RecA protein filaments: end-dependent dissociation from ssDNA and stabilization by RecO and RecR proteins. J Mol Biol 265 519 540
25. WangTCChangHYHungJL 1993 Cosuppression of recF, recR and recO mutations by mutant recA alleles in Escherichia coli cells. Mutat Res 294 157 166
26. LittleJW 1991 Mechanism of specific LexA cleavage: autodigestion and the role of RecA coprotease. Biochimie 73 411 421
27. Fernandez De HenestrosaAROgiTAoyagiSChafinDHayesJJ 2000 Identification of additional genes belonging to the LexA regulon in Escherichia coli. Mol Microbiol 35 1560 1572
28. BurckhardtSEWoodgateRScheuermannRHEcholsH 1988 UmuD mutagenesis protein of Escherichia coli: overproduction, purification, and cleavage by RecA. Proc Natl Acad Sci U S A 85 1811 1815
29. NohmiTBattistaJRDodsonLAWalkerGC 1988 RecA-mediated cleavage activates UmuD for mutagenesis: mechanistic relationship between transcriptional derepression and posttranslational activation. Proc Natl Acad Sci U S A 85 1816 1820
30. ShinagawaHIwasakiHKatoTNakataA 1988 RecA protein-dependent cleavage of UmuD protein and SOS mutagenesis. Proc Natl Acad Sci U S A 85 1806 1810
31. PhamPSeitzEMSavelievSShenXWoodgateR 2002 Two distinct modes of RecA action are required for DNA polymerase V-catalyzed translesion synthesis. Proc Natl Acad Sci U S A 99 11061 11066
32. TangMShenXFrankEGO'DonnellMWoodgateR 1999 UmuD'(2)C is an error-prone DNA polymerase, Escherichia coli pol V. Proc Natl Acad Sci U S A 96 8919 8924
33. JiangQKarataKWoodgateRCoxMMGoodmanMF 2009 The active form of DNA polymerase V is UmuD'(2)C-RecA-ATP. Nature 460 359 363
34. GodoyVGJaroszDFSimonSMAbyzovAIlyinV 2007 UmuD and RecA directly modulate the mutagenic potential of the Y family DNA polymerase DinB. Mol Cell 28 1058 1070
35. JaroszDFBeuningPJCohenSEWalkerGC 2007 Y-family DNA polymerases in Escherichia coli. Trends Microbiol 15 70 77
36. MaulRWSuttonMD 2005 Roles of the Escherichia coli RecA protein and the global SOS response in effecting DNA polymerase selection in vivo. J Bacteriol 187 7607 7618
37. LittleJW 1984 Autodigestion of lexA and phage lambda repressors. Proc Natl Acad Sci U S A 81 1375 1379
38. LittleJW 1993 LexA cleavage and other self-processing reactions. J Bacteriol 175 4943 4950
39. StoryRMWeberITSteitzTA 1992 The structure of the E. coli recA protein monomer and polymer. Nature 355 318 325
40. DattaSPrabuMMVazeMBGaneshNChandraNR 2000 Crystal structures of Mycobacterium tuberculosis RecA and its complex with ADP-AlF(4): implications for decreased ATPase activity and molecular aggregation. Nucleic Acids Res 28 4964 4973
41. DattaSKrishnaRGaneshNChandraNRMuniyappaK 2003 Crystal structures of Mycobacterium smegmatis RecA and its nucleotide complexes. J Bacteriol 185 4280 4284
42. DattaSGaneshNChandraNRMuniyappaKVijayanM 2003 Structural studies on MtRecA-nucleotide complexes: insights into DNA and nucleotide binding and the structural signature of NTP recognition. Proteins 50 474 485
43. ConwayABLynchTWZhangYFortinGSFungCW 2004 Crystal structure of a Rad51 filament. Nat Struct Mol Biol 11 791 796
44. RajanRBellCE 2004 Crystal structure of RecA from Deinococcus radiodurans: insights into the structural basis of extreme radioresistance. J Mol Biol 344 951 963
45. WuYHeYMoyaIAQianXLuoY 2004 Crystal structure of archaeal recombinase RADA: a snapshot of its extended conformation. Mol Cell 15 423 435
46. XingXBellCE 2004 Crystal structures of Escherichia coli RecA in a compressed helical filament. J Mol Biol 342 1471 1485
47. XingXBellCE 2004 Crystal structures of Escherichia coli RecA in complex with MgADP and MnAMP-PNP. Biochemistry 43 16142 16152
48. QianXWuYHeYLuoY 2005 Crystal structure of Methanococcus voltae RadA in complex with ADP: hydrolysis-induced conformational change. Biochemistry 44 13753 13761
49. WuYQianXHeYMoyaIALuoY 2005 Crystal structure of an ATPase-active form of Rad51 homolog from Methanococcus voltae. Insights into potassium dependence. J Biol Chem 280 722 728
50. KrishnaRManjunathGPKumarPSuroliaAChandraNR 2006 Crystallographic identification of an ordered C-terminal domain and a second nucleotide-binding site in RecA: new insights into allostery. Nucleic Acids Res 34 2186 2195
51. ChenZYangHPavletichNP 2008 Mechanism of homologous recombination from the RecA-ssDNA/dsDNA structures. Nature 453 489 484
52. McGrewDAKnightKL 2003 Molecular design and functional organization of the RecA protein. Crit Rev Biochem Mol Biol 38 385 432
53. LichtargeOBourneHRCohenFE 1996 An evolutionary trace method defines binding surfaces common to protein families. J Mol Biol 257 342 358
54. LuaRCLichtargeO PyETV: a PyMOL evolutionary trace viewer to analyze functional site predictions in protein complexes. Bioinformatics 26 2981 2982
55. WilkinsADLuaRErdinSWardRMLichtargeO Sequence and structure continuity of evolutionary importance improves protein functional site discovery and annotation. Protein Sci 19 1296 1311
56. LichtargeOYamamotoKRCohenFE 1997 Identification of functional surfaces of the zinc binding domains of intracellular receptors. J Mol Biol 274 325 337
57. SowaMEHeWWenselTGLichtargeO 2000 A regulator of G protein signaling interaction surface linked to effector specificity. Proc Natl Acad Sci U S A 97 1483 1488
58. SowaMEHeWSlepKCKercherMALichtargeO 2001 Prediction and confirmation of a site critical for effector regulation of RGS domain activity. Nat Struct Biol 8 234 237
59. MadabushiSGrossAKPhilippiAMengECWenselTG 2004 Evolutionary trace of G protein-coupled receptors reveals clusters of residues that determine global and class-specific functions. J Biol Chem 279 8126 8132
60. RajagopalanLPatelNMadabushiSGoddardJAAnjanV 2006 Essential helix interactions in the anion transporter domain of prestin revealed by evolutionary trace analysis. J Neurosci 26 12727 12734
61. RaviscioniMHeQSalicruEMSmithCLLichtargeO 2006 Evolutionary identification of a subtype specific functional site in the ligand binding domain of steroid receptors. Proteins 64 1046 1057
62. ShenoySKDrakeMTNelsonCDHoutzDAXiaoK 2006 beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J Biol Chem 281 1261 1273
63. Ribes-ZamoraAMihalekILichtargeOBertuchAA 2007 Distinct faces of the Ku heterodimer mediate DNA repair and telomeric functions. Nat Struct Mol Biol 14 301 307
64. RajagopalanLPereiraFALichtargeOBrownellWE 2009 Identification of functionally important residues/domains in membrane proteins using an evolutionary approach coupled with systematic mutational analysis. Methods Mol Biol 493 287 297
65. RodriguezGJYaoRLichtargeOWenselTG Evolution-guided discovery and recoding of allosteric pathway specificity determinants in psychoactive bioamine receptors. Proc Natl Acad Sci U S A 107 7787 7792
66. LichtargeOWilkinsA Evolution: a guide to perturb protein function and networks. Curr Opin Struct Biol 20 351 359
67. BaameurFMorganDHYaoHTranTMHammittRA Role for the regulator of G-protein signaling homology domain of G protein-coupled receptor kinases 5 and 6 in beta 2-adrenergic receptor and rhodopsin phosphorylation. Mol Pharmacol 77 405 415
68. KobayashiHOgawaKYaoRLichtargeOBouvierM 2009 Functional rescue of beta-adrenoceptor dimerization and trafficking by pharmacological chaperones. Traffic 10 1019 1033
69. YaoHKristensenDMMihalekISowaMEShawC 2003 A Sensitive, Accurate, and Scalable Method to Identify Functional Sites in Protein Structures. J Mol Bio In Press
70. PenningtonJMRosenbergSM 2007 Spontaneous DNA breakage in single living Escherichia coli cells. Nat Genet 39 797 802
71. MorandPBlancoMDevoretR 1977 Characterization of lexB mutations in Escherichia coli K-12. J Bacteriol 131 572 582
72. RobertsJWRobertsCW 1981 Two mutations that alter the regulatory activity of E. coli recA protein. Nature 290 422 424
73. KawashimaHHoriiTOgawaTOgawaH 1984 Functional domains of Escherichia coli recA protein deduced from the mutational sites in the gene. Mol Gen Genet 193 288 292
74. HortnagelKVoloshinONKinalHHMaNSchaffer-JudgeC 1999 Saturation mutagenesis of the E. coli RecA loop L2 homologous DNA pairing region reveals residues essential for recombination and recombinational repair. J Mol Biol 286 1097 1106
75. LarminatFCazauxCGermanierMDefaisM 1992 New mutations in and around the L2 disordered loop of the RecA protein modulate recombination and/or coprotease activity. J Bacteriol 174 6264 6269
76. KonolaJTLoganKMKnightKL 1994 Functional characterization of residues in the P-loop motif of the RecA protein ATP binding site. J Mol Biol 237 20 34
77. LoganKMKnightKL 1993 Mutagenesis of the P-loop motif in the ATP binding site of the RecA protein from Escherichia coli. J Mol Biol 232 1048 1059
78. NguyenTTMuenchKABryantFR 1993 Inactivation of the recA protein by mutation of histidine 97 or lysine 248 at the subunit interface. J Biol Chem 268 3107 3113
79. MustardJALittleJW 2000 Analysis of Escherichia coli RecA interactions with LexA, lambda CI, and UmuD by site-directed mutagenesis of recA. J Bacteriol 182 1659 1670
80. DutreixMMoreauPLBailoneAGalibertFBattistaJR 1989 New recA mutations that dissociate the various RecA protein activities in Escherichia coli provide evidence for an additional role for RecA protein in UV mutagenesis. J Bacteriol 171 2415 2423
81. WeisemannJMWeinstockGM 1988 Mutations at the cysteine codons of the recA gene of Escherichia coli. DNA 7 389 398
82. NastriHGKnightKL 1994 Identification of residues in the L1 region of the RecA protein which are important to recombination or coprotease activities. J Biol Chem 269 26311 26322
83. NastriHGGuzzoALangeCSWalkerGCKnightKL 1997 Mutational analysis of the RecA protein L1 region identifies this area as a probable part of the co-protease substrate binding site. Mol Microbiol 25 967 978
84. KelleyJAKnightKL 1997 Allosteric regulation of RecA protein function is mediated by Gln194. J Biol Chem 272 25778 25782
85. OgawaHOgawaT 1986 General recombination: functions and structure of RecA protein. Adv Biophys 21 135 148
86. KonolaJTNastriHGLoganKMKnightKL 1995 Mutations at Pro67 in the RecA protein P-loop motif differentially modify coprotease function and separate coprotease from recombination activities. J Biol Chem 270 8411 8419
87. KonolaJTGuzzoAGowJBWalkerGCKnightKL 1998 Differential cleavage of LexA and UmuD mediated by recA Pro67 mutants: implications for common LexA and UmuD binding sites on RecA. J Mol Biol 276 405 415
88. MazinAVKowalczykowskiSC 1998 The function of the secondary DNA-binding site of RecA protein during DNA strand exchange. Embo J 17 1161 1168
89. KurumizakaHIkawaSSaraiAShibataT 1999 The mutant RecA proteins, RecAR243Q and RecAK245N, exhibit defective DNA binding in homologous pairing. Arch Biochem Biophys 365 83 91
90. KarlinSBrocchieriL 1996 Evolutionary conservation of RecA genes in relation to protein structure and function. J Bacteriol 178 1881 1894
91. RocaAICoxMM 1997 RecA protein: structure, function, and role in recombinational DNA repair. Prog Nucleic Acid Res Mol Biol 56 129 223
92. AiharaHItoYKurumizakaHTeradaTYokoyamaS 1997 An interaction between a specified surface of the C-terminal domain of RecA protein and double-stranded DNA for homologous pairing. J Mol Biol 274 213 221
93. KurumizakaHAiharaHIkawaSKashimaTBazemoreLR 1996 A possible role of the C-terminal domain of the RecA protein. A gateway model for double-stranded DNA binding. J Biol Chem 271 33515 33524
94. LusettiSLWoodEAFlemingCDModicaMJKorthJ 2003 C-terminal deletions of the Escherichia coli RecA protein. Characterization of in vivo and in vitro effects. J Biol Chem 278 16372 16380
95. TessmanESPetersonPK 1985 Isolation of protease-proficient, recombinase-deficient recA mutants of Escherichia coli K-12. J Bacteriol 163 688 695
96. WangTCSmithKC 1986 Inviability of dam recA and dam recB cells of Escherichia coli is correlated with their inability to repair DNA double-strand breaks produced by mismatch repair. J Bacteriol 165 1023 1025
97. GalkinVEBrittRLBaneLBYuXCoxMM Two modes of binding of DinI to RecA filament provide a new insight into the regulation of SOS response by DinI protein. J Mol Biol 408 815 824
98. YuXEgelmanEH 1993 The LexA repressor binds within the deep helical groove of the activated RecA filament. J Mol Biol 231 29 40
99. GalkinVEYuXBielnickiJNdjonkaDBellCE 2009 Cleavage of bacteriophage lambda cI repressor involves the RecA C-terminal domain. J Mol Biol 385 779 787
100. CirzRTRomesbergFE 2007 Controlling mutation: intervening in evolution as a therapeutic strategy. Crit Rev Biochem Mol Biol 42 341 354
101. SmithPARomesbergFE 2007 Combating bacteria and drug resistance by inhibiting mechanisms of persistence and adaptation. Nat Chem Biol 3 549 556
102. WoodgateRRajagopalanMLuCEcholsH 1989 UmuC mutagenesis protein of Escherichia coli: purification and interaction with UmuD and UmuD'. Proc Natl Acad Sci U S A 86 7301 7305
103. MillerJH 1972 Experiments in molecular genetics Cold Spring Harbor, N.Y. Cold Spring Harbor Laboratoryxvi, 466
104. MihalekIResILichtargeO 2004 A family of evolution-entropy hybrid methods for ranking protein residues by importance. J Mol Biol 336 1265 1282
105. MihalekIResILichtargeO 2006 Evolutionary trace report_maker: a new type of service for comparative analysis of proteins. Bioinformatics 22 1656 1657
106. EdgarRC 2004 MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5 113
107. MorganDHKristensenDMMittelmanDLichtargeO 2006 ET viewer: an application for predicting and visualizing functional sites in protein structures. Bioinformatics 22 2049 2050
108. Schneidman-DuhovnyDInbarYNussinovRWolfsonHJ 2005 PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res 33 W363 367
109. LinLLLittleJW 1989 Autodigestion and RecA-dependent cleavage of Ind - mutant LexA proteins. J Mol Biol 210 439 452
110. GoldbergRBBenderRAStreicherSL 1974 Direct selection for P1-sensitive mutants of enteric bacteria. J Bacteriol 118 810 814
111. FrankEGGonzalezMEnnisDGLevineASWoodgateR 1996 In vivo stability of the Umu mutagenesis proteins: a major role for RecA. J Bacteriol 178 3550 3556
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 9
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Retrotransposon-Induced Heterochromatin Spreading in the Mouse Revealed by Insertional Polymorphisms
- The Evolutionarily Conserved Longevity Determinants HCF-1 and SIR-2.1/SIRT1 Collaborate to Regulate DAF-16/FOXO
- Genome-Wide Analysis of Heteroduplex DNA in Mismatch Repair–Deficient Yeast Cells Reveals Novel Properties of Meiotic Recombination Pathways
- Association of eGFR-Related Loci Identified by GWAS with Incident CKD and ESRD
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy