
Inactivation of Alters Melanosome Shape But Has Only a Subtle Effect on Visible Pigmentation
PMEL is an amyloidogenic protein that appears to be exclusively expressed in pigment cells and forms intralumenal fibrils within early stage melanosomes upon which eumelanins deposit in later stages. PMEL is well conserved among vertebrates, and allelic variants in several species are associated with reduced levels of eumelanin in epidermal tissues. However, in most of these cases it is not clear whether the allelic variants reflect gain-of-function or loss-of-function, and no complete PMEL loss-of-function has been reported in a mammal. Here, we have created a mouse line in which the Pmel gene has been inactivated (Pmel−/−). These mice are fully viable, fertile, and display no obvious developmental defects. Melanosomes within Pmel−/− melanocytes are spherical in contrast to the oblong shape present in wild-type animals. This feature was documented in primary cultures of skin-derived melanocytes as well as in retinal pigment epithelium cells and in uveal melanocytes. Inactivation of Pmel has only a mild effect on the coat color phenotype in four different genetic backgrounds, with the clearest effect in mice also carrying the brown/Tyrp1 mutation. This phenotype, which is similar to that observed with the spontaneous silver mutation in mice, strongly suggests that other previously described alleles in vertebrates with more striking effects on pigmentation are dominant-negative mutations. Despite a mild effect on visible pigmentation, inactivation of Pmel led to a substantial reduction in eumelanin content in hair, which demonstrates that PMEL has a critical role for maintaining efficient epidermal pigmentation.
Published in the journal:
. PLoS Genet 7(9): e32767. doi:10.1371/journal.pgen.1002285
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002285
Summary
PMEL is an amyloidogenic protein that appears to be exclusively expressed in pigment cells and forms intralumenal fibrils within early stage melanosomes upon which eumelanins deposit in later stages. PMEL is well conserved among vertebrates, and allelic variants in several species are associated with reduced levels of eumelanin in epidermal tissues. However, in most of these cases it is not clear whether the allelic variants reflect gain-of-function or loss-of-function, and no complete PMEL loss-of-function has been reported in a mammal. Here, we have created a mouse line in which the Pmel gene has been inactivated (Pmel−/−). These mice are fully viable, fertile, and display no obvious developmental defects. Melanosomes within Pmel−/− melanocytes are spherical in contrast to the oblong shape present in wild-type animals. This feature was documented in primary cultures of skin-derived melanocytes as well as in retinal pigment epithelium cells and in uveal melanocytes. Inactivation of Pmel has only a mild effect on the coat color phenotype in four different genetic backgrounds, with the clearest effect in mice also carrying the brown/Tyrp1 mutation. This phenotype, which is similar to that observed with the spontaneous silver mutation in mice, strongly suggests that other previously described alleles in vertebrates with more striking effects on pigmentation are dominant-negative mutations. Despite a mild effect on visible pigmentation, inactivation of Pmel led to a substantial reduction in eumelanin content in hair, which demonstrates that PMEL has a critical role for maintaining efficient epidermal pigmentation.
Introduction
Vertebrates produce two types of pigment - red/yellow pheomelanins and black/brown eumelanins [1]. Premelanosome protein (PMEL also known as PMEL17, SILV or gp100) is an integral membrane protein exclusively expressed in pigment cells that synthesize primarily eumelanins [2]. In mouse, PMEL expression starts at E9.5 in the presumptive retinal pigment epithelium (RPE) and at E10.5 in neural crest-derived melanoblasts, suggesting a function in the early stages of melanosome biogenesis [3]. Several studies have shown that fragments derived from proteolytic maturation of PMEL form the fibrillar matrix within melanosomes upon which eumelanins are ultimately deposited [4]. A role for PMEL fibrils in melanosome maturation is suggested by their ability to template and accelerate polymerization of highly reactive eumelanin precursors [5]–[7]. The presence of PMEL has therefore been assumed to be critical for the normal production or stabilization of eumelanin but not pheomelanin. Fibrils formed by PMEL in vitro have biophysical hallmarks of amyloid such as those formed in Alzheimer's and Parkinson's diseases [6], providing a model for functional/non-pathological amyloid formation [8].
PMEL is well conserved among vertebrates and thus must play an important physiological role; the average amino acid sequence identity for PMEL among distantly related mammalian species (mouse-human) is in the range 75–80% (Table S1) and is as high as 50% between humans and zebrafish within certain subdomains [4]. Indeed, PMEL mutations resulting in hypopigmentation have been identified in a number of vertebrate species. The first reported mutation affecting PMEL function showed that the recessive silver allele in mouse (Pmelsi) is caused by a single nucleotide insertion that truncates the last 25 C-terminal residues of PMEL within the cytoplasmic domain [9], [10]. This results in intracellular transport defects that deplete PMEL from early stage melanosomes [11]. Based on this mutation, the gene encoding PMEL was named Silver but has recently been renamed Pmel to provide a consistent nomenclature across species; the human homolog is named PMEL accordingly. The silver allele dilutes black/brown eumelanin but has no observable effect on pheomelanin and causes hair graying over time on black backgrounds [12], [13]. Microscopic examination of hairs revealed that some have no pigment at all, others had a reduced number of scattered pigment granules, while some were white with sparsely pigmented areas [12]. The graying phenotype was more pronounced in silver mice with mutations at the Tyrosinase-related protein 1 locus (Tyrp1 or B), indicating a gene interaction between Pmel and Tyrp1 [13], [14]. Surprisingly the graying seemed to decrease with age in agouti mice, as opposed to the progressive graying on black backgrounds [12]. Relative to the elongated melanosomes in eumelanin-producing melanocytes from wild-type mice, melanosomes in immortalized melanocytes derived from silver mice are larger and round when analyzed by electron microscopy [11].
Mutations in Pmel homologs causing pigmentation phenotypes within a number of species have now been identified. In the domesticated chicken, a three amino acid insertion in the transmembrane (TM) region is associated with the Dominant white phenotype, inhibiting the production of all eumelanin in plumage, skin and uveal melanocytes in the choroid layer of the eye, whereas the RPE is not affected [15], [16]. Earlier studies showed that Dominant white was associated with irregularly shaped and assembled melanosomes [17] and pigment cell death [18]. In fact, Brumbaugh and Lee [19] suggested already 1975 on the basis of ultrastructural studies that Dominant white encodes a structural component involved in premelanosome formation which has now been confirmed by the identification of PMEL as the associated gene product. A five-amino acid deletion in the TM region and a point mutation downstream of the TM are associated with the Dun allele which has a similar effect on pigmentation as Dominant white [16]. The Smoky plumage color phenotype arose in a flock of Dominant white birds where pigmentation is partially restored due to a four amino acid deletion in the Polycystic Kidney Disease repeat domain [16], a lumenal region thought to be partially responsible for both PMEL localization [20] and fibril formation [21]. In horses, a missense mutation in the cytoplasmic region of PMEL is associated with dilution of black pigment in the mane and tail [22]. The zebrafish mutant fading vision (fdv) exhibits hypopigmentation in RPE and body melanocytes and a defect in vision, and is caused by a premature stop codon [23]. The Merle coat color pattern in dogs, characterized by patches of diluted pigment, is associated with the insertion of a short interspersed element (SINE) in intron 10 and the affected merle dogs exhibit auditory and ophthalmologic abnormalities resembling human patients with Waardenburg syndrome [24]. In cattle, a coat color dilution phenotype is associated with a missense mutation G22R in the signal peptide of PMEL, but conclusive evidence that this is the causative mutation has not yet been reported [25]. Importantly, in none of these cases is it clear that the mutation is a complete loss of PMEL function.
Mutations in genes important for coat pigmentation in laboratory mice are easily detected in mutagenesis screens and to date, 169 genes affecting pigmentation have been identified [26] (URL: http://www.espcr.org/micemut). In some of these genes, hundreds of mutations have been reported; for example, 104 alleles have been identified in the Tyrosinase (Tyr) gene, most of them causing albinism [27]. It is therefore somewhat surprising that only one Pmel mutation has been identified in mice as PMEL evidently has an important role in the melanocyte. This indicates that either (i) PMEL has other yet unknown functions and that inactivating mutations are embryonically lethal, or (ii) that complete loss-of-function mutations at the Pmel locus have no or only mild visible effects on pigmentation that are not readily detected in mouse mutagenesis screens.
To address the role of a non-functional PMEL in vivo, we have generated a knockout mouse line. In this study, we report that mice homozygous for the inactivated Pmel allele on black backgrounds have spherical melanosomes and display a weak dilution of eumelanin, similar to the one present in Pmelsi mice. The data show that inactivation of PMEL does not dramatically disrupt pigmentation in vivo, and that mutations in Pmel homologs that render a loss of pigmentation therefore possess dominant-negative activity. The latter is demonstrated in an accompanying paper showing that the Dominant white allele in chicken, as well as the Silver allele in horses, encode pathogenic forms of the PMEL amyloid [28].
Results
Generation and validation of Pmel knockout mice
To generate a knockout construct, we prepared a pFlrt3-vector that included a fragment spanning exon 2 to exon 3 of the Pmel gene flanked by loxP sites and a neomycin selection cassette flanked by FRT sites (Figure 1A). The target construct was introduced into R1 ES-cells by electroporation and gene targeting was confirmed by long range PCR (data not shown) and Southern blot analysis (Figure 1B), in which the targeted gene generates a novel 7.7 kb band. Positive ES-cell clones were injected into blastocysts, which subsequently were implanted into female foster mice. Chimeric male offspring were bred to C57BL/6J mice and their offspring carrying the targeted Pmel allele were subsequently bred to PGK-Cre transgenic mice (C57BL/6J), in which Cre is driven by the ubiquitously expressed PGK promoter [29], to generate Pmel+/− offspring. The Pmel+/− mice were further bred to C57BL/6J for two subsequent generations, and Pmel+/− offspring were then intercrossed to generate homozygous null offspring. The loss of Pmel mRNA expression was confirmed by a qPCR test, using cDNA synthesized from skin RNA, that showed about two-fold reduction of the Pmel transcript in skin from Pmel+/− mice relative to Pmel+/+ mice as expected, and a 279-fold reduction of the transcript level in Pmel−/− mice compared to Pmel+/+ littermates (Figure 1C); the latter likely reflects the transcript detection limit by this assay, suggesting that Pmel−/− mice lack Pmel expression.

Primary cultures of skin-derived melanocytes were established from neonatal wild-type C57BL/6 and Pmel−/− mice. Immunoblotting analyses with a panel of three PMEL antibodies showed that PMEL protein was readily detected in whole cell lysates of wild-type melanocytes (mel Pmel+/+) but absent from lysates of Pmel−/− melanocytes (mel Pmel−/−) (Figure 1D, upper panels). Lysates from transfected HeLa cells expressing human PMEL (HeLa hPMEL) or untransfected human fibroblasts (Fibrobl) were used as positive and negative controls, respectively. By contrast, two melanocyte-specific proteins, tyrosinase (TYR) and tyrosinase-related protein 1 (TYRP1), and a house-keeping protein, γ-tubulin, were expressed at similar levels in wild-type and Pmel−/− melanocytes (Figure 1D, lower panels). Thus we conclude that our construct effectively inactivated the expression of PMEL in skin-derived melanocytes, but did not affect the expression of other melanogenic proteins.
Inactivation of Pmel does not disrupt pigment synthesis but alters the shape of melanosomes
To assess the requirement for PMEL in pigmentation and melanosome distribution, primary cultures of skin-derived melanocytes from wild-type C57BL/6 and Pmel−/− mice were analyzed by bright-field microscopy (Figure 2A). No obvious alteration in the degree of pigmentation could be observed in Pmel−/− primary melanocytes compared to the wild-type by this analysis. To better appreciate the effect of the loss of PMEL on melanosome ultrastructure, primary melanocytes from Pmel+/+ and Pmel−/− mice were analyzed by electron microscopy. This analysis demonstrated that the loss of PMEL causes an altered shape of melanosomes (Figure 2B). Melanosomes in wild-type mice are rod-shaped; thus, in thin sections in which the rod-shaped structures are sectioned in random planes, the melanosomes appeared as a mixture of ellipsoids (transverse sections) and spheres (cross-section; Figure 2B). By contrast, melanosomes in Pmel−/− mice were always spherical, suggesting that they were no longer rod-shaped (Figure 2B). To quantify this effect, we measured the diameters of melanosomes along the long (length) and short (width) axes (Figure 2C). There was a weak correlation between the length and width of melanosomes in wild-type homozygotes (r = 0.52), consistent with their ellipsoidal shape, whereas in the knockout mice, the width and length of melanosomes were strongly correlated (r = 0.93), consistent with a spherical shape. Together, these data show that PMEL is not required for pigment production per se, but is necessary for the normal ellipsoidal shape of melanosomes.

The electron microscopy analysis revealed a number of additional features of melanosome architecture that were altered in Pmel−/− melanocytes relative to wild-type cells. First, whereas fibrillar stage II and III melanosomes were readily identified within wild-type melanocytes, no fibrillar intermediates were detected in Pmel−/− melanocytes although endosomal organelles were clearly visible (Figure 2B, compare Figure 2Bb and Figure 2Bd). This was expected given the previously established role for PMEL as the structural foundation for the fibrils. Second, many of the melanosomes in Pmel−/− melanocytes were associated with dense, granular deposits of melanin rather than the smooth, fibrillar deposits observed in wild-type melanocytes (Figure 2Bd). Together, these data suggest that PMEL is also required for the normal polymerization of melanin, with potential adverse consequences for melanosome integrity.
PMEL is not required for melanosome maturation or segregation from late endosomes/lysosomes
PMEL fibrils in wild-type melanocytes begin to form in stage I melanosomes, which mature into stage II melanosomes by fibril assembly and into stage III/IV melanosomes following the delivery of membrane-bound melanosome cargoes and consequent melanogenesis [30], [31]. However, stage I melanosomes are also accessible to endocytic cargo and appear to be intermediates in the maturation of late endosomes and lysosomes, which are separate from later stage melanosomes [31]. We therefore investigated the possibility that PMEL is required for the normal development of melanosomes and the segregation of cargoes destined for late stage melanosomes from those destined for late endosomes/ lysosomes. Primary melanocytes from wild-type C57BL/6 and Pmel−/− mice were analyzed by immunofluorescence and bright field microscopy for markers of melanosomes and late endosomes/ lysosomes (Figure 3). As expected, Pmel−/− melanocytes lacked labeling for the PMEL-specific antibody HMB45 (Figure 3Aa, d) but were robustly labeled for TYRP1 (Figure 3Ab, e). Importantly, labeling for TYRP1 predominantly surrounded pigment granules in both wild-type and Pmel−/− melanocytes (Figure 3Ac, f and insets). Furthermore, both TYRP1 and pigment granules showed only minimal overlap with LAMP2, a marker specific for late endosomes/ lysosomes (Figure 3B; note insets in panels c, f). These data demonstrate that PMEL is not required for either melanosome maturation or for the segregation of melanosomes from late endocytic compartments.

PMEL expression and function in skin, eye, and inner ear
We investigated whether the inactivation of PMEL expression caused changes in morphology or pigmentation in skin, eye, and inner ear. Skin from Pmel+/+ mice had pigmented cells in hair follicles that were immunoreactive for PMEL using the Pep13h antiserum (Figure 4Aa, b). The Pep13h antiserum detects the cytoplasmic domain only found on immature PMEL forms in early secretory organelles and the immunoreactivity thus reflects the site of PMEL synthesis [32]. Only background Pep13h-immunoreactivity was seen on pigmented cells in hair follicles in skin from the Pmel−/− mice (Figure 4Ac, d). The melanocytes in the knockout mice were intact and had pigmentation, thus PMEL is not required for pigmentation of melanocytes in situ.

Comparison of the choroid and outer layers of the retina in 3 day postnatal and 18-months old Pmel+/+ and Pmel−/− mice, showed that the choroidal melanocytes and RPE cells were Pep13h-immunoreactive in wild-type but not the knockout mice (Figure 4B). Weak Pep13h-staining in the RPE of Pmel−/− mice was also seen in the secondary antibody-controls in both wild-type and knockout retinas, and thus represented background immunoreactivity likely due to the well-established non-specific binding to Bruch's membrane. Western blot analysis of retina from wild-type and knockout mice using the Pep13h-antiserum confirmed the absence of PMEL in the eyes from the Pmel−/− mice (Figure S1).
To test whether melanosome shape was altered in eye pigment cells as in skin melanocytes, we analyzed thin sections of the retina by electron microscopy. Whereas melanosomes in the uveal melanocytes of the choroid layer and particularly in the RPE cells of wild-type mice were oblong, the melanosomes in both cell types in the Pmel−/− mice were spherical (Figure 5), in perfect agreement with the results obtained by electron microscopy of primary skin-derived melanocytes. Moreover, as in Pmel−/− skin melanocytes, many of the melanosomes in Pmel−/− RPE showed irregular melanin aggregates and poorly preserved membranes (Figure 5D). This demonstrates that PMEL is required for normal development of the melanosomes in all three cell types (skin melanocytes, choroid melanocytes, and RPE cells).

The striking changes in the structure of the RPE melanosomes suggested that the Pmel−/− mice might have alterations in retinal integrity that might impair their vision. To test this hypothesis, we performed electroretinography (ERG) on two 18-month old Pmel−/− mice and two age-matched controls. The results showed that amplitudes, implicit times, and waveforms for both dark-adapted and light-adapted responses were similar between Pmel−/− and Pmel+/+ mice. Hence, the results showed that despite the changes in melanosome architecture, loss of PMEL expression did not cause a severe impairment of retinal function detectable with full-field ERG.
To test whether loss of PMEL expression affects the morphology of the inner ear, thin sections of cochlear samples dissected from the inner ears of two Pmel−/− mice and one wild-type mouse were analyzed by electron microscopy (Figure S2). All cells, including the intermediate cells harboring the pigment were represented and there was no obvious difference in overall morphology between the two genotypes. Furthermore, the knockout mice had normal Preyer reflexes, a rough physiological indication of “normal” hearing.
Loss of PMEL affects coat and skin pigmentation in a qualitatively subtle but quantitatively substantial manner
The Pmel−/− mice are fully viable, fertile, and display no obvious developmental defects. Furthermore, Pmel−/− mice on a C57BL/6 background – which have the genotype Asipa/a at the locus encoding Agouti-signaling protein and thus make only eumelanin – did not display striking hypopigmentation. We therefore tested the effect of the Pmel- mutation on coat color dilution in different genetic backgrounds by intercrossing our C57BL/6 Pmel−/− line with BALB/c mice for three generations. In addition to segregating the Pmel locus, this intercross segregated three additional coat color loci: agouti-signaling protein (Asip), the classical Agouti (A) locus; tyrosinase-related protein 1 (Tyrp1), the classical Brown (B) locus; and Tyrosinase (Tyr), the classical Albino (C) locus. The genotypes at these four loci in the parental lines of the intercross were Pmel−/−, Asipa/a, Tyrp1B/B, TyrC/C for the C57BL/6 line and Pmel+/+, AsipA/A, Tyrp1b/b, Tyrc/c for Balb/c (lower case letters represent the recessive allele at each locus). The Pmel knockout showed a subtle effect on black (Asipa/a, Tyrp1B/B; Figure 6a), brown (Asipa/a, Tyrp1b/b; Figure 6b), agouti (AsipA/A, Tyrp1B/B; Figure 6c), and brown agouti (AsipA/A, Tyrp1b/b; Figure 6d) backgrounds. On a black background, a weak silvering effect in the coat color was seen, and the pigmentation on the paws and tail was reduced (Figure 6e, f), suggesting perhaps a more pronounced effect on pigmentation in interfollicular epidermis and dermis. The most pronounced effect of the Pmel knockout was observed on the brown background, where the brown pigmentation was markedly diluted. Brown agouti, Pmel−/− mice had an apparent dilution of the dark hairs, whilst the yellow pigmentation seemed unaffected. A more subtle effect of the Pmel−/− genotype was noted in agouti mice, but their paler tails distinguished the knockouts from the wild-type littermates. Together, these data confirm a previously described genetic interaction between the Pmel silver (Pmelsi/si) allele with the brown (Tyrp1) locus [13], [14]. However, a difference was that the previous studies indicated that silver showed a more pronounced effect in Tyrp1 heterozygotes whereas we only observed an altered pigmentation in Tyrp1b/b homozygotes. The results of this cross-breeding experiment show that PMEL only modestly influences visible pigmentation in hair and skin.

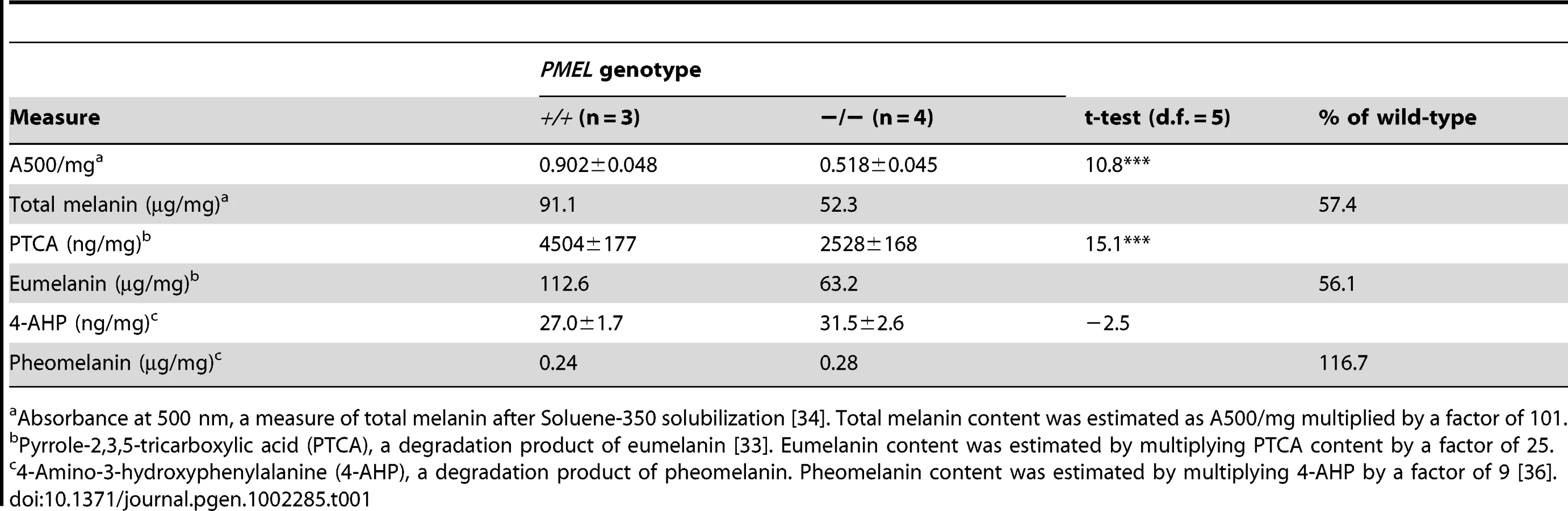
The effect of inactivating Pmel on pigment production was also assessed quantitatively by a chemical analysis using previously described methods [33]–[36]. A comparison of pigment content in hair from wild-type and Pmel−/− mice on a C57BL/6 background demonstrated that loss of PMEL expression causes a substantial reduction (−44%) of eumelanin content (Table 1); no significant effect on pheomelanin content was noted. This result was further corroborated by analyzing melanin content in the eight animals depicted in Figure 6. In each pairwise comparison on the same genetic background, the knockout mouse showed a lower amount of eumelanin in hair, ranging from 35–69% of the level observed in wild-type hair (Table 2). The most pronounced reduction was observed in animals on a Tyrp1b/b background, consistent with the visual appearance (Figure 6). No consistent difference in pheomelanin-content was noted among Pmel genotypes; by comparison, homozygosity for the non-agouti allele at the Asip locus caused a drastic reduction in pheomelanin content (Table 2).

The allele frequency distribution at the human PMEL locus is consistent with purifying selection
Given the reduced hair and skin pigmentation in Europeans it is conceivable that PMEL loss-of-function mutations have been tolerated or even under positive selection in this population. To address this question we exploited the low coverage resequencing data for 60 European, 59 African, and 60 Asian individuals that have recently been released by the 1000 Genomes Project [37]. The result clearly indicated that this is not the case and the data are consistent with purifying selection acting on the human PMEL locus (Table 3). In the European population only synonymous substitutions were found. In total, seven SNPs causing missense mutations were found, most of them in Africa, but the minor allele frequency at all loci were below 15%. Furthermore, in all three populations synonymous SNPs showed the highest degree of genetic diversity, a typical feature of a locus under purifying selection.

Discussion
The present study provides a substantial advance in understanding the role of PMEL and of the PMEL-derived amyloid fibrillar melanosome matrix in pigment cell biology. The PMEL protein has generally been thought to be important for pigmentation since mutations in PMEL orthologs in several species are associated with substantial pigment dilution, in some cases attributable to poor melanocyte survival. This study demonstrates that PMEL is essential for the normal development of the rod-shaped melanosomes in eumelanin-producing melanocytes in hair, skin, and eye, but not for the general survival of pigment cells in several tissues. Thus, while the amyloid fibrils formed by proteolytic fragments of PMEL play a critical role in generating the characteristic rod-shape of melanosomes, the Pmel knockout mutation has a relatively mild effect on visible pigmentation. A characteristic difference between melanosomes in eumelanin - versus pheomelanin-producing melanocytes is the shape of the melanosomes, rod-shaped versus spherical. The present study demonstrates that the shape of the melanosomes is not critical for which type of pigment is produced. Moreover, despite a dramatic effect on melanosome shape and melanin deposition in the RPE and choroid, visual function in Pmel−/− mice is not substantially impaired. Nonetheless, PMEL is well conserved throughout vertebrate evolution, and we show the presence of purifying selection at the human PMEL locus. Given that our results support pigment cell-specific expression of PMEL, this suggests that PMEL performs an essential function in pigment cells.
This study provides a potential explanation why PMEL function is well conserved among vertebrates. Despite only a subtle effect of Pmel inactivation on visible pigmentation, we show a 40–50% reduction in eumelanin content in hair from Pmel knockout mice relative to controls. This demonstrates a substantially reduced efficiency in pigmentation, implying that PMEL likely has an essential function in vertebrates exposed to damaging UV light. This function might reflect a role for PMEL in making pigment production more effective by evenly distributing melanin polymerization throughout melanosomes. Such an enhancement of pigment efficiency might be most critical in the RPE, in which pigment formation is temporally limited to a brief pre - and post-natal period and in which persistent interactions with phagolysosomes might expose poorly polymerized melanins to degradative enzymes; indeed, a number of mutations in melanosome biogenesis appear to more severely affect pigmentation in the eye than in the skin [38]. Another possibility is that the polymerization of eumelanins on PMEL fibrils facilitates melanin transfer to keratinocytes, such that transfer is reduced in Pmel−/− skin.
The phenotype of the Pmel knockout mice is similar to the silvering phenotype described for the mouse silver allele that arose spontaneously [12]–[14] and that is caused by a premature stop codon truncating the last 25 amino acid residues of PMEL [9], [10]. This allele dilutes eumelanin pigmentation but has no clear effect on pheomelanin pigmentation, consistent with the results in the present study. Furthermore, a previous analysis of melanin content showed that homozygosity for the silver allele caused a 20% reduction in eumelanin content on a recessive black (Asipa/a) background and 40% reduction on a brown (Tyrp1b/b) background [39], in good agreement with the present results for Pmel−/− mice (Table 1 and Table 2). The more pronounced effect of both Pmelsi/si and Pmel−/− on eumelanin production on a brown background might reflect a combined effect of TYRP1 on tyrosinase stability [40], [41], facilitating melanin formation, and of PMEL on melanin polymerization/stability. However, a clear difference between Pmelsi/si and Pmel−/− is that the silver allele shows a more pronounced dilution of pigmentation on a heterozygous brown background (Tyrp1b/+) than on the homozygous background (Tyrp1b/b) [12]–[14], whereas we only observed a phenotypic effect in the homozygous brown background. This difference may be explained by the fact that the knockout is a null allele, while silver encodes a truncated form of PMEL that lacks the 25 C-terminal amino acids from the cytoplasmic domain. The silver PMEL variant is depleted but not absent from melanosomes [11], and thus might confer partial function that in some way is more deleterious in the presence of limiting amounts of functional Tyrp1.
Consistent with the influence of PMEL primarily on eumelanin accumulation shown here, the Dominant white allele in chickens [16] and the Silver allele in horses [22] both show a dominant inheritance and inhibit the production of eumelanin but have no visible effect on pheomelanin. Surprisingly, the Dc allele in cattle associated with a G22R missense mutation in PMEL is associated with a dilution of both pheomelanin and eumelanin, but further work is required to demonstrate the causative nature of the G22R mutation [25].
Our results suggest that the alleles described in other species with a more drastic effect on pigmentation, such as Silver in horses [22], Merle in dogs [24], and Dominant white in chicken [16] represent dominant negative mutations. The Dominant white and Smoky alleles in chickens provide a good illustration [16]. Dominant white inhibits the expression of black eumelanin and is caused by an insertion of three amino acid residues in the transmembrane domain. Smoky arose in a line homozygous for Dominant white and restored pigmentation. The causative mutation for Smoky is a deletion of four highly conserved residues in the PKD domain of PMEL. Thus, a plausible interpretation of these data, in relation to the phenotypic effects of the Pmel knockout mice, is that Dominant white is a dominant negative mutation while the Smoky mutation is a loss-of-function that inhibits the effect of Dominant white. Interestingly, we have recently demonstrated that the Smoky mutation indeed generates an essentially inactive form of PMEL, countering a dominant gain-of-function by the Dominant white mutation [28].
The present study now provides a reasonable explanation why only a single Pmel mutant has been described in mice compared with the fairly high number of mutations at most other pigmentation loci – that a Pmel loss-of-function has only a subtle effect on visible pigmentation. By contrast, dominant PMEL mutations in domestic animals are rather common; the present study explains why dominant-negative mutations have been selected in domestic animals without adverse pleiotropic effects in other tissues, since we confirm that PMEL is a melanocyte-specific protein. In this context, Merle dogs are unusual in having pleiotropic defects. The phenotypic effects associated with Merle in dogs is assumed to be caused by a retrotransposon insertion at the boundary of intron 10/exon 11 of PMEL [24]. In the heterozygous condition, Merle causes patches of diluted pigmentation and the effect is restricted to the inhibition of eumelanin pigmentation consistent with the effects of Pmel alleles in mice, chickens, and horses. Merle homozygotes show very pale pigmentation associated with hearing loss and visually defective microphtalmic eyes, and is considered sublethal due to multiple abnormalities of the skeletal, cardiac, and reproductive system [24], [42]. Given our results that loss of PMEL expression has only modest phenotypic effects, and that PMEL appears to be a melanocyte-specific protein, the broad phenotype observed in Merle dogs appears to be inconsistent with a loss-of-function mutation that affects only PMEL expression. There are several possible explanations for this enigma. Firstly, there are not yet any published data showing that Merle is associated with any altered expression of PMEL at the transcript or protein level or that expression of a wild-type form of PMEL can rescue the Merle phenotype. Thus, although it appears likely that Merle is indeed a PMEL mutant because the effect on pigmentation is restricted to eumelanin, as expected for a PMEL mutant, it cannot be formally excluded that the non-pigment effects of Merle reflect an additional mutation. However, the high rate of germ-cell reversions [42] is consistent with a single mutation caused by an insertion of a transposable element. Secondly, the severe effects on hearing and vision in Merle homozygotes compared with the apparently normal phenotype observed in Pmel knockout mice might be explained if Merle was the most severe dominant-negative mutation at the Pmel locus detected so far. Thus it might result in extensive melanocyte death in skin, eye, the inner ear, and even in the heart in which melanocyte loss is associated with abnormalities [43]. A third possible explanation for the pleiotropic effects on many different tissues in Merle dogs is potential ectopic expression of PMEL induced by the transposon insertion, and consequent toxic effects of amyloid formation outside the melanosome. Finally, since PMEL is located in a gene dense region (six genes within 100 kb) it is possible that the Merle mutation alters the regulation of more than one gene in the region. Thus, some of the phenotypic effects of Merle may be associated with PMEL function whereas others may be caused by altered expression of other closely linked genes.
An important justification for the development of knockout mice models is to suggest possible pathological effects of corresponding mutations in humans. The clear indication that purifying selection acts at the human PMEL locus suggests that loss-of-function mutations will cause reduced fitness. No human PMEL mutation associated with any phenotypic effects or disorders has yet been reported. Based on the results of the present study and previously described spontaneous mutations we can make the following predictions. Dominant-negative PMEL mutations, like Dominant white in chicken and Silver in horses, are expected to be associated with red hair and fair skin due to the expected reduction of dark eumelanin. However, PMEL variants do not appear to be a common cause of red hair and fair skin in humans [44]. Loss-of-function mutations must exist in the human population and no observable phenotypic effects are expected in heterozygotes. Loss-of-function homozygotes are expected on the basis of the phenotype of Pmel−/− mice to be fully viable but with a reduced content of eumelanin pigmentation in skin and hair. Thus, they are expected to be more sensitive to damaging UV-light and have a higher risk to develop various forms of skin cancer. PMEL has not been associated with an increased risk of skin cancer in genome-wide association studies (GWAS) in humans [45], but GWAS have a low power to identify rare genetic variants as expected for PMEL loss-of-function mutations.
Our observation of an altered shape of the melanosomes both in the RPE cells and in the uveal melanocytes of the eye suggests that PMEL−/− humans may also show eye disorders, perhaps in particular a higher incidence of age-related disorders such as macular degeneration. The ERGs did not reveal a drastic effect on retinal function, but since the sample size was small we cannot exclude a subtle effect. Furthermore, visual impairment does not always manifest in abnormal full-field ERGs. In this context it is worth noting that the fading vision mutant at the PMEL locus in zebrafish, in which a premature stop codon leads to production of a truncated protein, causes a severe developmental defect of the RPE and a consequent vision defect [23]. Furthermore, many Silver horses show an eye disorder named Mutiple Congenital Ocular Anomalities (MCOA) that resembles congenital aniridia in humans [46], [47]. Thus, the fact that three previously described PMEL mutations - fading vision in zebrafish, Silver in horses and Merle in dogs – are associated with different eye disorders and that the melanosomes in pigment cells in the eye of Pmel−/− mice have an altered shape clearly calls for further studies to explore if changes in PMEL function may lead to impaired vision in humans. The mouse model described here constitutes an excellent resource for such studies.
Materials and Methods
Gene targeting and construct design
The strategy for building the targeting construct using the pFlrt3-vector is shown in Figure 1A. A PCR-based strategy was employed by using a proofreading enzyme (KOD Hot Start DNA Polymerase, Novagen, USA). A pFlrt3-vector harboring two loxP sites, two FRT sites, and a neomycin cassette was used. A 5.0-kb upstream targeting arm starting in intron 1 (forward primer with Cfr42I tail and reverse primer with NotI tail, Table S2) and a 2.4-kb downstream targeting arm spanning from intron 3 to the beginning of intron 6 (forward primer with Bsp119I tail and reverse primer with XhoI tail, Table S2) were amplified. The 5.0-kb fragment was cloned into the Cfr42I and NotI sites while the 2.4-kb fragment was cloned into the BstBI and XhoI sites in the pFlrt3-vector. A 589 bp fragment spanning exon 2, intron 2, and exon 3 (forward primer with BglII tail and reverse primer also with BglII tail, Table S2) was cloned into the BglII site flanked by two loxP-sites.
According to the Ensembl genome assembly available at the time of the construct design, a knockout of the 589 bp fragment would cause a frame-shift between exon 1 and exon 4 leading to a premature stop codon and a non-functional PMEL protein. However, during the process of generating our transgenic mice, the Ensembl genome assembly was updated and the deletion of the 589 bp fragment turned out to cause an in-frame skipping of exon 2 and exon 3. Fortunately, the RNA was later shown to be degraded and the PMEL protein absent in the knockout mice (see Figure 1C and 1D).
The final target construct was linearized by XhoI and used for homologous recombination in R1 ES cells [48]. 408 clones were obtained and screened for homologous recombination by long range PCR. In total, 30 clones were revealed as positive for a targeting event. These were confirmed by Southern blot to be positive, using a probe targeting upstream of the 5.0-kb fragment (Sp5F and Sp5R, Table S2). Genomic DNA was extracted and digested with the restriction enzyme KpnI and separated by 0.7% agarose gel electrophoresis. Five clones were injected into blastocysts taken from a cross between C57BL/6NCrl females and B6D2F1/Crl males. The blastocysts were subsequently implanted into CD1 females. Two chimeric mice (crossed with C57BL/6J mice) were successfully used for germ line transmission. The F1 agouti offspring were confirmed to be heterozygous at the modified allele by PCR. F1 males were subsequently crossed with C57BL/6J PGK-Cre females in order to generate animals heterozygous for the null allele. After four generations of backcrossing into a C57BL/6J background, heterozygous animals were intercrossed to get homozygous null animals.
Genotyping of mutant mice
PCR amplification of genomic DNA extracted from tail biopsies was performed to genotype mutant mice. Tail biopsies were incubated 45 min in 96°C in lysis buffer (250 mM NaOH and 2 mM EDTA) and the reaction was inhibited by the addition of 400 mM TRIS HCl pH 8.0. The offspring of the chimeric mice were genotyped for germline transmission using two primers amplifying the wild-type and the targeted allele (primer A in intron 1 and primer B in the loxP-flanked fragment, Table S2). Primers A and B generated a 236 bp band and a 392 bp band from the wild-type and the targeted allele, respectively (Figure 1A). The offspring of the PGK-Cre mice were genotyped to confirm the absence of the loxP-flanked fragment using three primers amplifying the lox and flox alleles (primers A, B, and D in the pFlrt3-vector flanked by the loxP and FRT sites, Table S2). Primers A and B amplified the floxed allele generating a 392 bp product, whilst no band was produced from the knocked allele. Primers A and D amplifying the floxed allele produced a 762 bp band and a 319 bp band was produced from the knocked allele.
RNA isolation and qPCR analysis
RNA was isolated from skin tissue samples with the RNeasy mini kit (Qiagen). The RNA samples were subjected to reverse transcription using the cDNA high capacity kit (Applied Biosystems). mRNA transcripts for the two alleles were measured by quantitative PCR analysis using TaqMan Gene Expression master mix (Applied Biosystems) on a 7900HT Fast RT-PCR System (Applied Biosystems). Data were analyzed with a threshold set in the linear range of amplification, based on a standard curve of serial 10-fold dilutions for each primer set. The Pmel data were normalized using two endogenous housekeeping genes (GAPDH and 18S rRNA) and plotted as fold change.
Tissue preparation and immunohistochemistry
Whole eyes and skin were dissected and fixed in 4% PFA for 60 min, washed 10 min in PBS and cryoprotected in 30% sucrose for 3 and 12 h, respectively, before being frozen in OCT (Sakura). Tissues were cryosectioned (10 µm sections for eyes and 12 µm sections for skin) and collected on Superfrost Plus glasses (Menzel-Gläser). For immunohistochemistry, the sections were rehydrated in PBS for 15 min and then blocked in PBS containing 1% fetal calf serum, 0.02% thimerosal and 0.1% Triton X-100 for 30 min. Primary and secondary antibodies were diluted in this solution. Samples were incubated with primary antibodies overnight at 4°C, and with secondary antibodies for 2 h at room temperature. Samples were analyzed using a Zeiss Axioplan2 microscope, equipped with Axiovision software. Images were formatted, resized, enhanced and arranged for publication using Axiovision, and Adobe Photoshop.
Antibodies
The following mouse monoclonal antibodies were used: HMB45 to PMEL (Lab Vision, Fremont, CA); TA99/Mel5 to TYRP1 (American Type Culture Collection, Manassas, VA; for immunofluorescence microscopy); and GTU-88 to γ-tubulin (Sigma-Aldrich, St. Louis, MO). Polyclonal rabbit antibodies included: H-90 to TYRP1 (Santa Cruz Biotechnologies, Santa Cruz, CA; for immunoblotting); Pep7 [49] to tyrosinase, αhPep13h [50] to the C-terminus of human PMEL, and αmPmel-N [11] to the N-terminus of mouse PMEL. Secondary antibodies included FITC labelled anti-rabbit immunoglobulin (Ig) antibodies (Vector laboratories) for immunofluorescence microscopy of eye tissues, or highly cross-absorbed goat or sheep antibodies to mouse Ig, rat Ig, or isotype-specific antibodies to mouse γ1 and γ2a heavy chains (Jackson Immunoresearch) conjugated to Alexafluor 488 or Alexafluor 594 (InVitroGen).
Cell culture
Primary cultures of skin-derived melanocytes were generated as described [51]. Briefly, skins from neonatal wild-type C57BL/6 or Pmel−/− mice were digested with trypsin to separate the epidermis from the dermis, and the epidermis (containing the melanocytes) was further digested to a single cell suspension with trypsin and seeded onto a feeder layer of XB2 keratinocytes. Cells were cultured in RPMI 1640 (InVitrogen, Carlsbad, CA) supplemented with 10% FBS, 200 nM TPA, and 2 nM cholera toxin in a humidified incubator with 10% carbon dioxide. For analyses, cells were cultured without feeders.
Immunoblotting—primary melanocytes
Cells were harvested with 5 mM EDTA in PBS, lysed with 0.5% SDS, 1% 2ME, and boiled to obtain whole cell lysates. Lysates corresponding to equivalent numbers of cells were fractionated by SDS-PAGE using Tris-glycine gels and then transferred to PVDF membranes (Millipore) for immunoblotting analysis as described [21], [50]. Protein bands were detected with alkaline phosphatase conjugated secondary antibodies, enhanced chemifluorescence, and phosphorimaging analysis using a Storm 860 fluorescence imaging system and ImageQUANT software (GE Biosciences).
Immunoblotting—sclera
Eyes were collected from wild-type and Pmel−/− mice. The pigmented sclera was isolated by dissection and homogenized using a Tissue Tearor in lysis buffer (20mM Tris, 0.1% Triton, 0.1% SDS, 50mM NaCl and 2.5mM EDTA, 1nM Na3VO4 and 1X protease inhibitor cocktail (Roche)). 25µg of total protein lysates were denatured in sample buffer and reducing agent. The samples were separated on a bistris 8–12% gel (Invitrogen). Proteins were transferred to a hybond-C extra membrane (Amersham Biosciences). Unspecific binding to the membrane was blocked using 5% skimmed milk in TBS 0.1% Tween. Primary αPep13h or actin (Santa Cruz Biotechnology) antibody was added in blocking buffer in a 1∶1000 dilution O/N. The membranes were washed in TBS 0.1% Tween and HRP conjugated anti rabbit (GE Healthcare) or anti goat secondary (DAKO) antibodies in a 1∶4000 dilution. The membranes were subsequently washed in TBS 0.1% Tween and developed using ECL plus (GE Healthcare).
Immunofluorescence and bright-field microscopy
Primary melanocytes were grown on matrigel-coated coverslips, fixed with 2% formaldehyde, labeled with primary and fluorochrome-conjugated secondary antibodies as described [21], [50], and analyzed on a DM IRBE microscope (Leica Microsystems, Wetzlar, Germany) equipped with an Orca digital camera (Hamamatsu, Bridgewater, NJ). Images were captured and manipulated using OpenLab software (Improvision, Lexington, MA) as described [52], and processed using Adobe Photoshop software.
Electron microscopy
Primary melanocyte cultures were fixed in a mixture of 4% paraformaldehyde and 2% glutaraldehyde in cacodylate buffer pH 7.4, post fixed with 2% Osmium tetroxide harvested by scraping, dehydrated in ethanol, and embedded in epon resin. Ultrathin sections were contrasted with 2% uranyl acetate, analyzed by transmission electron microscopy on a Philips CM120 electron microscope (FEI, Eindoven, The Netherlands), and digital acquisitions were made with a numeric camera Keen View (Soft Imaging System, Munster, Germany). Individual melanosomes within electron micrographs of epon-embedded sections were identified and the maximum width and height of each melanosome was measured using OpenLab software. At least 200 melanosomes were counted for each cell type. Statistical analyses were done using Microsoft Excel linear regression analysis to determine the correlation coefficient between melanosome width and length.
Electron microscopy analysis of stria vascularis of the inner ear cochlea was done as follows. The mice were deeply anaesthetized with pentobarbital and transcardially perfused with 0.9% saline followed by fixative containing 2.5% glutaraldehyde and 0.5% paraformaldehyde in a phosphate buffer pH 7.2. After the animals were decapitated, their temporal bones were removed and their cochleas were dissected out. Local perfusion was carried out with the same fixative and the cochleas were left in the solution over night at +4oC. The cochleae were decalcified in 0.1 M EDTA for approximately one week until the bone was soft. Rinsing with phosphate buffer was performed several times followed by post fixation in 1% osmium tetroxide. The tissues were dehydrated and embedded in Agar 100 (Agar 100 Resin kit, Agar Scientific Limited). To orient the tissue, 1 µm thick sections were made on an ultratome (LKB Cryo Nova), stained with toluidine blue and investigated in a light microscope. The stria vascularis at the lower apical turn of the cochlea were chosen and ultra-thin sections were cut in this area, mounted on formvar coated copper grids, stained with uranyl acetate and lead citrate to be examined with a transmission electron microscope (JEOL 1230).
In the perfused animals the entire eyes were extirpated and transported to the morphology laboratory in the same fixative as the previous perfusion. The anterior segment of the eyes from each animal was discarded leaving posterior eyecups used for the light-and electron microscopic studies. Eyecups were incubated with gentle agitation in the same fixative for at least 2 h at room temperature. The full length of each eyecup was then gross sectioned with two vertical sections through the optic nerve head and 2 mm temporally. Samples were post-fixed in 1% osmium tetroxide and embedded in epoxy resin. They were washed with 0.17M sodium cacodylate, pH 7.4, followed by secondary fixation in 1% osmium tetroxide. Subsequently, the samples were dehydrated via sequential incubation in increasing concentrations of acetone and embedded in epoxy resin. Sections of the embedded samples were cut for both LM and EM examinations. For LM, 1 µm thick sections were mounted on glass slides and were stained with Toluidine blue. For EM, sections were mounted on copper grids and were stained with uranyl acetate and lead citrate. Light microscopy was performed using a Zeiss Axiophot microscope in order to choose areas of interest for the EM studies. The electron microscopy was performed using a JEOL 1200 EX transmission electron microscope.
Electroretinography (ERG)
Two knockout mice (one male, one female, aged 23 months) and two age-matched mice were used for the experiment. The experiments were approved by the regional Ethical Committee and carried out following the guidelines of the ARVO guidelines for animal experimentation.
The ERGs were recorded under general anaesthesia induced by intraperitoneal injection of 3 mg/kg acepromazine (Plegicil vet 10 mg/ml, Pharmaxim Sweden AB, Alcon, Sweden), 100 mg/kg ketamine (Ketaminol vet 50 mg/ml, Intervet AB, Sollentuna, Sweden) and 20 mg/kg xylazine (Narcoxyl vet 20 mg/ml, Intervet AB, Sollentuna, Sweden).
The mice were placed in sternal recumbency. The ERGs were recorded using corneal electrodes (Goldring, 3 mm, Roland Consult, Brandenburg, Germany) with isotonic eye drops containing 0.2% hyaluronic acid (ZilkEye, Evolan Pharma AB, Danderyd, Sweden) as coupling agent. Subcutaneous platinum-iridium needle electrodes served as ground and reference electrodes (Technomed Subdermal EEG Needle Electrodes, Cephalon A/S, Nørresundby, Denmark).
Light stimulation was generated by a xenon strobe (Grass PS33+, Astro-Med Inc, West Warwick, RI, USA) and a diffuser on the inner surface of a Ganzfeld dome spread light. Neutral density filters (Kodak Wratten no. 96, Kodak Rochester, NY, USA) were used to decrease the stimulus intensity. Light intensity was measured by a light meter (IL-1700, International Lights, Peabody, MA, USA) at level with the tested eye in the Ganzfeld. Responses were amplified and filtered by a bandpass filter (0.1–1,000 Hz) and stored using a Powerlab system (Powerlab, SP8, ADInstruments (Europe) LTD, Chalgrove, UK).
The mice were dark-adapted overnight and anesthetized and prepared for ERG under dim red light. A heating pad was used to maintain the body temperature throughout the ERG. Dark-adapted responses were recorded over a 4.5 log unit range up to 3.0 cd/m2/s, starting with the dimmest stimulus. Four responses presented at a frequency of 0.05 Hz were averaged to each stimulus intensity. Cone responses were recorded after light adaptation in the Ganzfeld with a steady, white background light (25 cd/m2) for 10 min. Averaged cone transient responses were obtained in response to 3.0 cd/m2/s flashes presented at 1.1 Hz and finally a light adapted, 30 Hz, cone flicker response was recorded. Amplitudes and implicit times were determined for each response and measured according to convention.
Three-generation intercross
Two Pmel−/− males (backcrossed four generations to C57BL/6 background) were intercrossed with ten BALB/C females to generate F1 offspring heterozygous at the A (Agouti), B (Tyrp1), C (Tyrosinase) and Pmel loci. Subsequently, ten F1 males and 20 F1 females were intercrossed to generate 147 F2 offspring, with segregating genotypes at the four loci. SNP markers were developed for the Asip (rs27342000), Tyrp1 (rs32544046) and Tyr (rs31392322) loci (Table S2). The C57BL/6 (Pmel−/−) and BALB/C mice were fixed for different alleles at these SNP positions. The F2 offspring were genotyped at the four loci using pyrosequencing (Biotage, Uppsala, Sweden). Oligonucleotides are listed in Table S1. The coat and skin color of the mice in the F2 generation were ocularly examined at four weeks of age.
Assays of eumelanin, pheomelanin, and total melanin
Dorsal hairs of four-week-old mice were plucked and processed for chemical analysis of eumelanin to detect the specific degradation product, pyrrole-2,3,5-tricarboxylic acid (PTCA) upon alkaline hydrogen peroxide oxidation [33], and of pheomelanin to detect the specific degradation product, 4-amino-3-hydroxyphenylalanine (4-AHP) upon hydroiodic acid hydrolysis [36], as reported previously. Content of total melanin was determined as absorbance at 500 nm (A500) after solubilizing 1 mg hair in 900 µL Soluene-350 plus 100 µL water as previously described [34].
Ethical statement
The appropriate local Swedish ethical committees have approved all experiments involving live animals (permissions C85/10 and C154/7).
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. ItoSWakamatsuK 2008 Chemistry of mixed melanogenesis--pivotal roles of dopaquinone. Photochem Photobiol 84 582 592
2. FurumuraMSakaiCPotterfSBVieiraWDBarshGS 1998 Characterization of genes modulated during pheomelanogenesis using differential display. Proc Natl Acad Sci U S A 95 7374 7378
3. BaxterLLPavanWJ 2003 Pmel17 expression is Mitf-dependent and reveals cranial melanoblast migration during murine development. Gene Expr Patterns 3 703 707
4. TheosACTruschelSTRaposoGMarksMS 2005 The Silver locus product Pmel17/gp100/Silv/ME20: controversial in name and in function. Pigment Cell Res 18 322 336
5. ChakrabortyAKPlattJTKimKKKwonBSBennettDC 1996 Polymerization of 5,6-dihydroxyindole-2-carboxylic acid to melanin by the pmel 17/silver locus protein. Eur J Biochem 236 180 188
6. FowlerDMKoulovAVAlory-JostCMarksMSBalchWE 2006 Functional amyloid formation within mammalian tissue. PLoS Biol 4 e6 doi:10.1371/journal.pbio.0040006
7. LeeZHHouLMoellmannGKuklinskaEAntolK 1996 Characterization and subcellular localization of human Pmel 17/silver, a 110-kDa (pre)melanosomal membrane protein associated with 5,6,-dihydroxyindole-2-carboxylic acid (DHICA) converting activity. J Invest Dermatol 106 605 610
8. WattBRaposoGMarksSM 2010 Pmel17: An amyloid determinant of organelle structure. RigacciSBucciantiniM Functional Amyloid Aggregation Kerala Research Signpost
9. Martinez-EsparzaMJimenez-CervantesCSolanoFLozanoJAGarcia-BorronJC 2000 Regulation of the murine silver locus product (gp87) by the hypopigmenting cytokines TGF-beta1 and TNF-alpha. Pigment Cell Res 13 120 126
10. SolanoFMartinez-EsparzaMJimenez-CervantesCHillSPLozanoJA 2000 New insights on the structure of the mouse silver locus and on the function of the silver protein. Pigment Cell Res 13 Suppl 8 118 124
11. TheosACBersonJFTheosSCHermanKEHarperDC 2006 Dual loss of ER export and endocytic signals with altered melanosome morphology in the silver mutation of Pmel17. Mol Biol Cell 17 3598 3612
12. DunnLCThigpenLW 1930 The silver mouse: a recessive color variation. Journal of Heredity 21 495 498
13. LamoreuxMLDelmasVLarueLBennetCD 2010 The Colors of Mice: A Model Genetic Network: Wiley-Blackwell
14. SilversWK 1979 The Coat Colors of Mice. A Model for Mammalian Gene Action and Interaction New York, NY Springer-Verlag 379
15. KarlssonACKerjeSHallbookFJensenP 2009 The Dominant white mutation in the PMEL17 gene does not cause visual impairment in chickens. Vet Ophthalmol 12 292 298
16. KerjeSSharmaPGunnarssonUKimHBagchiS 2004 The Dominant white, Dun and Smoky color variants in chicken are associated with insertion/deletion polymorphisms in the PMEL17 gene. Genetics 168 1507 1518
17. BrumbaughJA 1971 The ultrastructural effects of the I and S loci upon black-red melanin differentiation in the fowl. Develop Biol 24 394 412
18. JimbowKSzaboGFitzpatrickTB 1974 Ultrastructural investigation of autophagocytosis of melanosomes and programmed death of melanocytes in White Leghorn feathers: a study of morphogenetic events leading to hypomelanosis. Dev Biol 36 8 23
19. BrumbaughJALeeKW 1975 The gene action and function of two dopa oxidase positive melanocyte mutants of the fowl. Genetics 81 333 347
20. TheosACTruschelSTTenzaDHurbainIHarperDC 2006 A lumenal domain-dependent pathway for sorting to intralumenal vesicles of multivesicular endosomes involved in organelle morphogenesis. Dev Cell 10 343 354
21. WattBvan NielGFowlerDMHurbainILukKC 2009 N-terminal domains elicit formation of functional Pmel17 amyloid fibrils. J Biol Chem 284 35543 35555
22. BrunbergEAnderssonLCothranGSandbergKMikkoS 2006 A missense mutation in PMEL17 is associated with the Silver coat color in the horse. BMC Genet 7 46
23. SchonthalerHBLampertJMvon LintigJSchwarzHGeislerR 2005 A mutation in the silver gene leads to defects in melanosome biogenesis and alterations in the visual system in the zebrafish mutant fading vision. Dev Biol 284 421 436
24. ClarkLAWahlJMReesCAMurphyKE 2006 Retrotransposon insertion in SILV is responsible for merle patterning of the domestic dog. Proc Natl Acad Sci U S A
25. KuhnCWeikardR 2007 An investigation into the genetic background of coat colour dilution in a Charolais x German Holstein F2 resource population. Anim Genet 38 109 113
26. MontoliuLOettingWSBennettDC 2010 European Society for Pigment Cell Research. World Wide Web (URL: http://www.espcr.org/micemut)
27. BultCJEppigJTKadinJARichardsonJEBlakeJA 2008 The Mouse Genome Database (MGD): mouse biology and model systems. Nucleic Acids Res 36 D724 728
28. WattBTenzaDLemmonMAKerjeSRaposoGAnderssonLMarksSM 2011 Mutations that alter the PMEL transmembrane domain confer toxicity to functional PMEL amyloid. PLoS Genet 7 e1002286 doi:10.1371/journal.pgen.1002286
29. LallemandYLuriaVHaffner-KrauszRLonaiP 1998 Maternally expressed PGK-Cre transgene as a tool for early and uniform activation of the Cre site-specific recombinase. Transgenic Res 7 105 112
30. HurbainIGeertsWJBoudierTMarcoSVerkleijAJ 2008 Electron tomography of early melanosomes: implications for melanogenesis and the generation of fibrillar amyloid sheets. Proc Natl Acad Sci U S A 105 19726 19731
31. RaposoGTenzaDMurphyDMBersonJFMarksMS 2001 Distinct protein sorting and localization to premelanosomes, melanosomes, and lysosomes in pigmented melanocytic cells. J Cell Biol 152 809 824
32. HarperDCTheosACHermanKETenzaDRaposoG 2008 Premelanosome amyloid-like fibrils are composed of only golgi-processed forms of Pmel17 that have been proteolytically processed in endosomes. J Biol Chem 283 2307 2322
33. ItoSNakanishiYValenzuelaRKBrilliantMHKolbeL 2011 Usefulness of alkaline hydrogen peroxide oxidation to analyze eumelanin and pheomelanin in various tissue samples: application to chemical analysis of human hair melanins. Pigment Cell Melanoma Res In press
34. OzekiHItoSWakamatsuKThodyAJ 1996 Spectrophotometric characterization of eumelanin and pheomelanin in hair. Pigment Cell Res 9 265 270
35. WakamatsuKItoS 2002 Advanced chemical methods in melanin determination. Pigment Cell Res 15 174 183
36. WakamatsuKItoSReesJL 2002 The usefulness of 4-amino-3-hydroxyphenylalanine as a specific marker of pheomelanin. Pigment Cell Res 15 225 232
37. DurbinRMAbecasisGRAltshulerDLAutonABrooksLD 2010 A map of human genome variation from population-scale sequencing. Nature 467 1061 1073
38. FutterCE 2006 The molecular regulation of organelle transport in mammalian retinal pigment epithelial cells. Pigment Cell Res 19 104 111
39. LamoreuxMLWakamatsuKItoS 2001 Interaction of major coat color gene functions in mice as studied by chemical analysis of eumelanin and pheomelanin. Pigment Cell Res 14 23 31
40. KobayashiTImokawaGBennettDCHearingVJ 1998 Tyrosinase stabilization by Tyrp1 (the brown locus protein). J Biol Chem 273 31801 31805
41. MangaPBoissyREPifko-HirstSZhouBKOrlowSJ 2001 Mislocalization of melanosomal proteins in melanocytes from mice with oculocutaneous albinism type 2. Exp Eye Res 72 695 710
42. SponenbergDPRothschildMF 2001 Genetics of coat colour and hair texture. RuvinskyASampsonJ The Genetics of the Dog New York, NY CABI Publishing 61 685
43. LevinMDLuMMPetrenkoNBHawkinsBJGuptaTH 2009 Melanocyte-like cells in the heart and pulmonary veins contribute to atrial arrhythmia triggers. J Clin Invest 119 3420 3436
44. SulemPGudbjartssonDFStaceySNHelgasonARafnarT 2007 Genetic determinants of hair, eye and skin pigmentation in Europeans. Nat Genet 39 1443 1452
45. BishopDTDemenaisFIlesMMHarlandMTaylorJC 2009 Genome-wide association study identifies three loci associated with melanoma risk. Nat Genet 41 920 925
46. AnderssonLSJurasRRamseyDTEason-ButlerJEwartS 2008 Equine Multiple Congenital Ocular Anomalies maps to a 4.9 megabase interval on horse chromosome 6. BMC Genet 9 88
47. RamseyDTEwartSLRenderJACookCSLatimerCA 1999 Congenital ocular abnormalities of Rocky Mountain Horses. Vet Ophthalmol 2 47 59
48. NagyARossantJNagyRAbramow-NewerlyWRoderJC 1993 Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc Natl Acad Sci U S A 90 8424 8428
49. JimenezMTsukamotoKHearingVJ 1991 Tyrosinases from two different loci are expressed by normal and by transformed melanocytes. J Biol Chem 266 1147 1156
50. BersonJFHarperDCTenzaDRaposoGMarksMS 2001 Pmel17 initiates premelanosome morphogenesis within multivesicular bodies. Mol Biol Cell 12 3451 3464
51. SviderskayaEVBennettDCHoLBailinTLeeST 1997 Complementation of hypopigmentation in p-mutant (pink-eyed dilution) mouse melanocytes by normal human P cDNA, and defective complementation by OCA2 mutant sequences. J Invest Dermatol 108 30 34
52. SettySRTenzaDTruschelSTChouESviderskayaEV 2007 BLOC-1 is required for cargo-specific sorting from vacuolar early endosomes toward lysosome-related organelles. Mol Biol Cell 18 768 780
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 9
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Retrotransposon-Induced Heterochromatin Spreading in the Mouse Revealed by Insertional Polymorphisms
- The Evolutionarily Conserved Longevity Determinants HCF-1 and SIR-2.1/SIRT1 Collaborate to Regulate DAF-16/FOXO
- Genome-Wide Analysis of Heteroduplex DNA in Mismatch Repair–Deficient Yeast Cells Reveals Novel Properties of Meiotic Recombination Pathways
- Association of eGFR-Related Loci Identified by GWAS with Incident CKD and ESRD
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy