
A Noncoding Point Mutation of Causes Multiple Developmental Malformations and Obesity in Twirler Mice
Heterozygous Twirler (Tw) mice develop obesity and circling behavior associated with malformations of the inner ear, whereas homozygous Tw mice have cleft palate and die shortly after birth. Zeb1 is a zinc finger protein that contributes to mesenchymal cell fate by repression of genes whose expression defines epithelial cell identity. This developmental pathway is disrupted in inner ears of Tw/Tw mice. The purpose of our study was to comprehensively characterize the Twirler phenotype and to identify the causative mutation. The Tw/+ inner ear phenotype includes irregularities of the semicircular canals, abnormal utricular otoconia, a shortened cochlear duct, and hearing loss, whereas Tw/Tw ears are severely malformed with barely recognizable anatomy. Tw/+ mice have obesity associated with insulin-resistance and have lymphoid organ hypoplasia. We identified a noncoding nucleotide substitution, c.58+181G>A, in the first intron of the Tw allele of Zeb1 (Zeb1Tw). A knockin mouse model of c.58+181G>A recapitulated the Tw phenotype, whereas a wild-type knockin control did not, confirming the mutation as pathogenic. c.58+181G>A does not affect splicing but disrupts a predicted site for Myb protein binding, which we confirmed in vitro. In comparison, homozygosity for a targeted deletion of exon 1 of mouse Zeb1, Zeb1ΔEx1, is associated with a subtle abnormality of the lateral semicircular canal that is different than those in Tw mice. Expression analyses of E13.5 Twirler and Zeb1ΔEx1 ears confirm that Zeb1ΔEx1 is a null allele, whereas Zeb1Tw RNA is expressed at increased levels in comparison to wild-type Zeb1. We conclude that a noncoding point mutation of Zeb1 acts via a gain-of-function to disrupt regulation of Zeb1Tw expression, epithelial-mesenchymal cell fate or interactions, and structural development of the inner ear in Twirler mice. This is a novel mechanism underlying disorders of hearing or balance.
Published in the journal:
. PLoS Genet 7(9): e32767. doi:10.1371/journal.pgen.1002307
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002307
Summary
Heterozygous Twirler (Tw) mice develop obesity and circling behavior associated with malformations of the inner ear, whereas homozygous Tw mice have cleft palate and die shortly after birth. Zeb1 is a zinc finger protein that contributes to mesenchymal cell fate by repression of genes whose expression defines epithelial cell identity. This developmental pathway is disrupted in inner ears of Tw/Tw mice. The purpose of our study was to comprehensively characterize the Twirler phenotype and to identify the causative mutation. The Tw/+ inner ear phenotype includes irregularities of the semicircular canals, abnormal utricular otoconia, a shortened cochlear duct, and hearing loss, whereas Tw/Tw ears are severely malformed with barely recognizable anatomy. Tw/+ mice have obesity associated with insulin-resistance and have lymphoid organ hypoplasia. We identified a noncoding nucleotide substitution, c.58+181G>A, in the first intron of the Tw allele of Zeb1 (Zeb1Tw). A knockin mouse model of c.58+181G>A recapitulated the Tw phenotype, whereas a wild-type knockin control did not, confirming the mutation as pathogenic. c.58+181G>A does not affect splicing but disrupts a predicted site for Myb protein binding, which we confirmed in vitro. In comparison, homozygosity for a targeted deletion of exon 1 of mouse Zeb1, Zeb1ΔEx1, is associated with a subtle abnormality of the lateral semicircular canal that is different than those in Tw mice. Expression analyses of E13.5 Twirler and Zeb1ΔEx1 ears confirm that Zeb1ΔEx1 is a null allele, whereas Zeb1Tw RNA is expressed at increased levels in comparison to wild-type Zeb1. We conclude that a noncoding point mutation of Zeb1 acts via a gain-of-function to disrupt regulation of Zeb1Tw expression, epithelial-mesenchymal cell fate or interactions, and structural development of the inner ear in Twirler mice. This is a novel mechanism underlying disorders of hearing or balance.
Introduction
Twirler (Tw) spontaneously arose in a crossbred stock of mice segregating multiple recessive mutant alleles [1]. Heterozygous Tw mice develop obesity after three months of age, and exhibit stereotypic behavior that includes waltzing, spinning, and horizontal head-shaking [1]. This behavior is thought to result from malformed vestibular labyrinths that include hypomorphic or absent lateral semicircular canals, irregular contours of the anterior and posterior semicircular canals, and absent otoconia in the utricle and saccule [1]. In contrast, all homozygous Tw mice are born with cleft palate and die soon after birth [1].
Tw is located on proximal chromosome 18 but the causative mutation has not been identified [1], [2]. A transgene insertional mutant, Tg9257, exhibits a similar inner ear phenotype and is also located on proximal chromosome 18, raising the possibility that these phenotypes are allelic [3]. However, complementation testing is inconclusive [3]. Similarly, the Irxl1 gene, located within a broad critical map interval for Tw and expressed in developing palate, has also been ruled out as a candidate for Tw [4].
Zeb1 is also located on proximal chromosome 18 and encodes a transcription factor, Zeb1, that binds E-box-like elements to either repress [5], [6], or activate transcription [7]–[9]. Mice that are homozygous for a targeted deletion of exon 1 of Zeb1 (Zeb1ΔEx1) die soon after birth with cleft palate, limb defects and other skeletal abnormalities, and T-cell deficiency [10], whereas heterozygous Zeb1ΔEx1/+ mice are viable and adult females show increased adiposity [11]. This partial phenotypic overlap with Twirler does not include stereotypic vestibular behavior or inner ear malformations, although these were likely not examined in Zeb1ΔEx1 mice. Ectopic expression of Zeb1 in neoplastic epithelium has been implicated in the epithelial-to-mesenchymal transition (EMT) leading to local tumor invasion and metastasis [12]. In normally developing mesenchymal tissue, Zeb1 is thought to repress epithelial-specific genes such as E-cadherin and activate mesenchyme-specific genes such as collagen, smooth muscle actin and myosin [9]. Genome-wide expression profiling reveals a probable similar role for Zeb1 in the regulation of gene expression in developing mouse inner ear mesenchyme [13]. In humans, heterozygous mutations of ZEB1 cause posterior polymorphous corneal dystrophy, characterized by an epithelial transition and abnormal proliferation of corneal endothelium [14]. Zeb1ΔEx1/+ mice also show corneal abnormalities and further implicate Zeb1 in the suppression of an epithelial phenotype [15].
In the current study we show that Twirler is caused by a noncoding point substitution in the first intron of Zeb1. The mutation does not affect splicing, but does disrupt a consensus binding site sequence for Myb proteins [16]. The maintenance of inner ear mesenchyme - and epithelium-specific gene expression is disrupted in Twirler inner ears [13], demonstrating a novel mutation and developmental mechanism for the pathogenesis of hearing or balance disorders.
Results
Lymphoid phenotype of Twirler mice
Heterozygous Tw/+ adult mice had smaller spleens (38±2 mg vs. 68±7 mg, P<0.013) in comparison to wild type littermates. Tw/+ thymi were also smaller although the difference was not significant (13±2 mg vs. 31±6 mg, P<0.06). Tw/+ mice had lower counts of white blood cells (1×103/µl vs. 7.2×103/µl, P<0.0001), lymphocytes (0.5×103/µl vs. 5.9×103/µl, P<0.0004) and polymorphonuclear neutrophils (0.4×103/µl vs. 1.3×103/µl, P<0.04). No abnormalities were found in other adult Tw/+ tissues. Histopathological examination of P0 animals revealed no abnormalities in the thymus or spleen of wild type, Tw/+ or homozygous Tw/Tw mice. Tw/Tw mice had cleft palates.
Obesity and metabolic phenotype of Twirler mice
There was no significant difference in average body weight between Tw/+ and wild type littermates of either sex until 12 weeks of age (Figure 1A and 1B). Beginning at seven weeks of age, Tw/+ mice consumed approximately 15 to 20% more food than wild type littermates (Figure 1C and 1D). There was a significant increase in the percentage of body fat and slightly reduced lean body mass in Tw/+ mice of both sexes (Table 1), indicating that fat accounts for the increased body mass. Body weight-adjusted energy expenditure, estimated from oxygen consumption, revealed a reduced metabolic rate in Tw/+ mice that did not reach statistical significance (Table 1). Tw/+ mice had normal serum glucose levels but elevated levels of serum free fatty acids, triglycerides, insulin, leptin, corticosterone and adiponectin (Table 1). Insulin and glucose tolerance tests of 15-week-old females showed insulin resistance and slight glucose intolerance in Tw/+ mice (Figure 1E and 1F), consistent with data for other obese mice with hyperinsulinemia [17].


Inner ear phenotype of Twirler mice
We evaluated the morphology of mutant inner ears using the paint-filling technique (Figure 2). The Tw/+ inner ears had grossly intact semicircular canals and neurosensory cristae ampullaris, but the contours of the canals were irregular due to small bulges and projections (Figure 2B). The most anatomically consistent malformation was found at the non-ampullated end of the lateral canal where it normally narrows to join the vestibule in wild type ears (Figure 2D). In contrast, the non-ampullated ends of Tw/+ lateral canals were irregular or constricted (Figure 2E). Tw/Tw inner ears have more severe malformations that include absence of the lateral semicircular canal, truncation of the posterior semicircular canal, and shortening of the cochlear duct (Figure 2C, 2F and 2I).

The average length of Tw/+ cochlear ducts (Figure 2H) was 91% (±5%) that of wild type ears (P<0.00002; Figure 2G). Binaural average ABR thresholds were elevated for Tw/+ mice in comparison to wild type controls at one month of age (33±1.6 dBSPL vs. 55±5.3 dBSPL at 8 kHz, p<0.0006; 33±1.8 dBSPL vs. 46±4 dBSPL at 16 kHz, p<0.01; 29±1.9 dBSPL vs. 39±3.6 dBSPL at 32 kHz, p<0.023; Figure 2J). Tw/+ mice showed no significant change in ABR thresholds measured at three months of age in comparison to thresholds measured at one month of age (not shown).
Tw/+ utricles had giant otoconia that were transparent by light microscopic examination but visible by scanning electron microscopy (Figure 2N). In contrast, Tw/+ saccular otoconia appeared normal (Figure 2L).
Linkage mapping and positional cloning of Tw
We screened 1679 [(C57BL/6J-Tw/+ x CAST/Ei)F1-Tw/+ x C57BL/6J]N2 progeny for recombinations. Recombination locations were refined with additional markers to narrow the Tw interval to 814 kb between D18Nih6 and D18Nih42 (Figure 3A). This interval was five Mb proximal to the Tg9257 transgene insertion site [3]. The Tw interval contained three genes: Zeb1, Zeb1os (Zeb1 opposite strand transcript, annotated in MGI as predicted gene Gm10125) and Zfp438 (Figure 3A). Zeb1 encodes a transcription factor with two zinc finger motifs and one homeobox motif. Zeb1os is predicted to encode a long noncoding RNA of unknown function. It is located on the opposite strand of Zeb1 where the two overlapping genes share parts of their first introns. Finally, Zfp438 is predicted to encode a zinc finger protein whose biological function is unknown [18].

Zeb1 was a good candidate for the gene mutated in Tw based upon the phenotype associated with a targeted deletion allele, Zeb1ΔEx1. Homozygous Zeb1ΔEx1 mice are born with cleft palate, skeletal and thymus abnormalities, and die shortly after birth [10]. We observed that Zeb1ΔEx1/+ heterozygotes have inner ear morphology and hearing thresholds that are indistinguishable from those of wild type littermates, whereas Zeb1ΔEx1/ΔEx1 homozygotes have a subtle constriction of the midportion of the lateral semicircular canal that differs in location and severity from that observed in Tw/+ mice (Figure S1). This difference is probably not due to genetic background since both lines were congenic on a C57BL/6J background.
To determine if Tw and Zeb1ΔEx1 can complement to form a normal palate or inner ear, we crossed heterozygous Tw and heterozygous Zeb1ΔEx1 mice. We observed an approximate Mendelian ratio of genotypes: five +/+, five Tw/+, seven Zeb1ΔEx1/+ and eight Tw/Zeb1ΔEx1. All Tw/Zeb1ΔEx1 mice were born with normal palates and developed into adults with circling behavior typical of Tw/+ mice. The lateral semicircular canals resembled those of Tw/+ mice (Figure S1). These results suggest these mutations exert their effects via different genes or mechanisms. While the Zeb1 pathway may be altered in Twirler mice, it is unlikely to be due to a loss-of-function allele of Zeb1.
To identify the Tw mutation, we first used 5′-RACE and 3′-RACE to identify novel exons of Zeb1, Zeb1os and Zfp438. 5′-RACE revealed Zeb1 transcripts with each of five additional alternative first exons (designated 1b, 1c, 1d, 1e and 1f) between exon 1 (heretofore termed exon 1a) and exon 2 (Figure S2). We amplified and sequenced all novel and annotated exons of Zeb1, Zeb1os and Zfp438 from genomic DNA of Tw/Tw, Tw/+ and wild type mice. We also amplified and sequenced cDNA transcripts of these genes from embryonic mRNA. All major transcripts of these genes were amplified from mice with each genotype. We found no sequence differences in the cDNAs or genomic exons. Sequence analysis of the 192-bp region of overlap of Zeb1 and Zeb1os revealed a single nucleotide substitution (G>A) 181 bp downstream of Zeb1 exon 1 and 12 bp downstream of Zeb1os exon 1 in Tw (Figure 3B). We designated this Tw variant as c.58+181G>A, which was the only sequence variation we detected. The wild type variant c.58+181G was conserved among 13 normal control inbred mouse strains as well as other vertebrate species (Figure 3B). In silico analyses (NNsplice, GeneSplicer, Net2Gene) predict that c.58+181G>A does not affect splicing of the adjacent splice donor site for exon 1 of Zeb1os. Sequence analysis of Zeb1 and Zeb1os cDNA transcripts confirmed no effect of c.58+181G>A on splicing.
Electrophoretic mobility shift assay of Tw DNA
c.58+181G>A disrupts a predicted site for Myb protein binding (Figure 3B)[16]. To test if this change can alter the binding of a Myb protein, recombinant mouse C-Myb was expressed and purified for an electrophoretic mobility shift assay (EMSA) of its binding to oligonucleotide probes containing either c.58+181G or c.58+181A and the flanking genomic sequences. There was a shift of the mobility of the wild type DNA probe in the presence of C-Myb, while the Tw DNA probe mobility was unchanged (Figure 4A). The binding of C-Myb to wild type DNA was inhibited by both the wild type probe and a mim-1 control probe which has been shown to interact with C-Myb [19], but not by the Tw probe (Figure 4B). These data provide in vitro evidence that the Tw mutation can disrupt binding of a Myb protein (C-Myb) to the mutated first intronic sequence of Zeb1.

Quantitative RT-PCR analysis of Zeb1Tw and Zeb1ΔEx1 transcripts
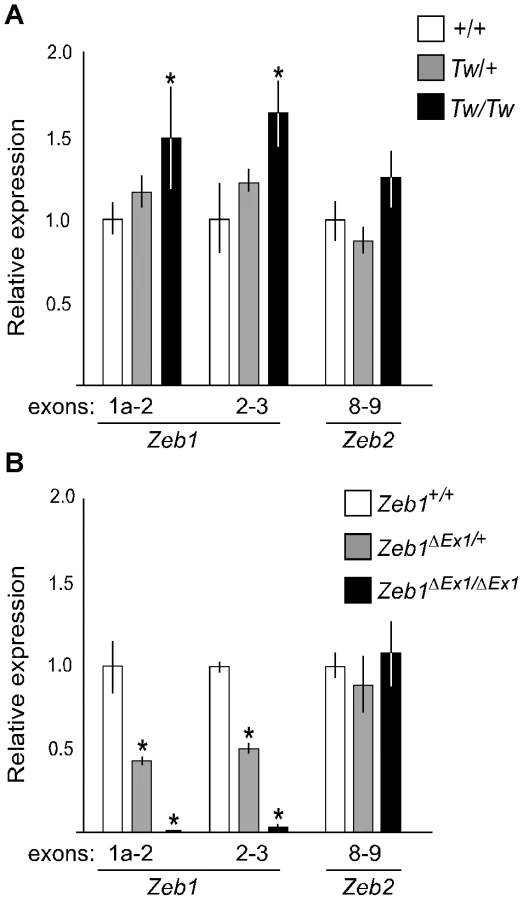
We analyzed mRNA expression levels of Zeb1, Zeb1os and Zfp438 from inner ears of Tw/Tw, Tw/+ or wild type mice at E13.5. We performed the same analysis with Zeb1ΔEx1 heterozygotes, homozygotes, and wild type littermates. We designed primer pairs to specifically amplify Zeb1 transcripts starting from each of exons 1a, 1b, 1c, 1d, 1e or 1f. One primer pair for constitutively spliced exons 2 and 3 was designed to amplify all Zeb1 transcripts. The levels of Zeb1 transcripts containing exon 1b, 1c, 1d, 1e, or 1f, as well as the Zeb1os and Zfp438 transcripts, were too low to be reliably quantified by RT-PCR. The levels of transcripts containing exons 1a and 2, as well as exons 2 and 3, were significantly increased from the Tw allele of Zeb1 (Zeb1Tw) in comparison to wild type Zeb1 (Figure 5A). In contrast, Zeb1ΔEx1 expressed no Zeb1 transcripts containing exons 1a and 2, and nearly non-detectable levels of any other Zeb1 transcripts containing other exons (Figure 5B). Transcripts levels for the closely related Zeb2 gene were unchanged among all three Zeb1 genotypes (Figure 5B). These results indicate that Zeb1ΔEx1 is a loss-of-function allele whereas Zeb1Tw is likely to act via gain-of-function.
A mouse knockin of the Zeb1Tw intron 1 sequence variant recapitulates the Twirler phenotype
To confirm the pathogenic effect of c.58+181G>A, we generated two knockin mouse lines: KIA segregates the Tw variant c.58+181A and KIG segregates the wild type variant c.58+181G (Figure 6). Compound heterozygous KIG/KIA mice consumed more food and grew heavier with increased adiposity in comparison to KIG/KIG control males and females (Figure 7A–7D, Table 2). The energy expenditure and circulating hormone levels in KIG/KIA mice recapitulated the Tw/+ phenotype (Table 2). The reduction in body weight-adjusted energy expenditure reached statistical significance in KIG/KIA female mice, whereas it did not in Tw/+ females (Table 1). Insulin and glucose tolerance tests showed insulin resistance and slight glucose intolerance in KIG/KIA mice (Figure 7E and 7F). Although KIG/KIA mice showed neither circling behavior nor constricted semicircular canals, the semicircular canals were irregular (Figure 8B) and the utricles contained giant otoconia (Figure 8N). Average ABR thresholds for KIG/KIA and KIG/KIG mice were not significantly different (Figure 8J). KIA/KIA and KIA/Tw inner ears displayed the same malformations as Tw/Tw ears (Figure 8C, 8F, 8I, and Figure 9B, 9D, 9F). KIG/KIA average spleen weight was decreased by 15% (P<0.05) but average thymus weight did not differ relative to KIG/KIG littermates (Table 2). We observed cleft palate with or without cleft lip in KIA/KIA and KIA/Tw mice with 50% and 90% penetrance, respectively (not shown). We did not observe cleft palate or cleft lip in KIG/KIA, KIG/KIG or KIG/Tw mice, indicating that the recapitulation of the Tw phenotype is specific.





The different phenotypic severity and penetrance of KIA in comparison to Tw could result from genetic background differences, since Tw arose on a different undefined stock. However, we serially backcrossed Tw to wild type C57BL/6J for over 30 generations, and KIA was generated from C57BL/6-derived Bruce4 ES cells and maintained on an isogenic C57BL/6J background. Therefore the differences in severity and penetrance could result from closely linked sequence variation, the residual loxP site in KIA, or a combination of these effects.
Zeb1 protein expression in Twirler inner ears
To determine if Zeb1 protein is expressed from the Tw allele, we stained inner ears of Tw/Tw mice with anti-Zeb1 antibodies (Figure 10A and 10B). We observed Zeb1 expression in non-epithelial (mesenchymal) cells surrounding Tw/Tw inner ears in which epithelial and mesenchymal tissue compartments could be microanatomically differentiated (Figure 10B). Other Tw/Tw inner ears had poorly preserved microarchitecture, precluding a differentiation of epithelium versus mesenchyme (Figure 10C). We conclude that Zeb1 protein is expressed in Tw/Tw ears, consistent with the result of real-time RT-PCR.

To determine if Zeb1 protein levels are altered by Tw, we performed a western blot analysis of inner-ear or whole-head protein extracts from E13.5 mice. We compared Tw/Tw, Tw/+ and wild type littermates, as well as KIG/KIG, KIG/KIA and KIA/KIA littermates. We were unable to detect Zeb1 in inner-ear protein extracts, but were able to reliably detect it in samples from whole heads. Total Zeb1 protein levels appeared to be slightly increased by Tw in comparison to wild type littermates (Figure 10D). This difference was not significant (ANOVA, P>0.05), possibly due to small numbers of animals and the degree of variation of Zeb1 band intensities within genotypes (Figure 10F). In contrast, Zeb1 protein levels in KIG/KIA and KIA/KIA mice were 2 - to 3-fold higher than in KIG/KIG littermates (Figure 10E). The variation within knockin genotype groups was smaller, resulting in differences between knockin genotype groups that were significant (P<0.05) (Figure 10G).
Discussion
This study demonstrates that the phenotype of Twirler is caused by a noncoding nucleotide substitution within a shared first intron of the Zeb1 and Zeb1os genes on mouse chromosome 18. This is a rare example of a Mendelian noncoding point mutation that does not affect a splice site or promoter. Our results demonstrate the potential for complex phenotypic effects of noncoding point variants, which are increasingly implicated in association studies of genetically complex traits. Our recombinant knockin mouse model and wild type knockin control for testing the pathogenic potential of the Tw mutation may be a useful paradigm to explore the effects of other noncoding variants of unknown pathogenic potential. The altered penetrance potentially associated with a residual loxP site in the Tw knockin line serves a cautionary note to include a wild type knockin control.
Although the initial study by Lyon [1] described abnormal development of the sensory neuroepithelium in the cristae ampullaris of some semicircular canals of Tw/+ mice, we have not observed the same alteration. Instead we observed a highly penetrant constriction of the non-ampullated end of the lateral semicircular canal that could impede or prevent the flow of endolymph and disrupt neurosensory detection of angular acceleration. A difference in strain background [1] may account for the different result. Moreover, Lyon reported that utricular otoconia were absent in Tw/+ ears whereas we observed giant utricular otoconia. This difference could also result from the strain background difference, loss of giant otoconia during the dissection process, or our use of scanning electron microscopy in addition to light microscopy. Nevertheless, either of the described utricular phenotypes could impair the detection of linear acceleration by Tw/+ utricles. We conclude that our observed semicircular canal and utricular anomalies underlie the vestibular behaviors of Tw/+ mice, although we cannot estimate their relative contributions to the observed vestibular functional phenotype. Correlating mouse vestibular structural or functional abnormalities with behavior is difficult due to a complex interrelationship between vestibular behavior and anxiety that is also dependent upon strain background [20].
The cause of hearing loss observed in some Tw/+ mice also remains obscure. Postmortem examination of middle ears did not reveal otitis media or developmental malformations of the external or middle ears that could account for the hearing loss. Although severe hearing loss has been observed in other mouse mutants with much shorter cochlear ducts [21], the severity of hearing loss in Tw/+ mice was highly variable but the degree of shortening of the cochlear duct was nearly constant. This lack of correlation leads us to conclude that associated physiologic defects or undetected structural anomalies underlie hearing loss in Tw/+ mice.
The Tw/+ phenotype includes hyperphagia with elevated levels of circulating corticosterone and adiponectin that are similar to those in a corticotropin-releasing factor (CRF) transgenic mouse model of Cushing syndrome [22], [23]. Other phenotypic similarities of that model to Tw/+ include increased body weight and adiposity, alopecia, atrophy of the thymus and spleen, and muscle wasting. This may suggest that Tw disrupts, at least in part, the hypothalamus-pituitary-adrenal axis. This is consistent with expression of Zeb1 in the pituitary gland [24]. However, Zeb1 protein is also expressed in adipose tissue and increases during adipogenesis in cell culture [11]. Moreover, Zeb1ΔEx1/+ mice develop obesity that is not associated with hyperphagia [11], unlike Tw/+ mice (Figure 1C and 1D). Therefore different mechanisms or tissues may underlie obesity phenotypes associated with Zeb1ΔEx1 and Zeb1Tw. The pathogenetic mechanism for one or both of these Zeb1 alleles may also underlie a locus for susceptibility to obesity on human chromosome 10p11 [25]-[28], which includes the human ZEB1 gene.
The results presented here and in Hertzano et al. [13], in combination with the body of published data on Zeb1 in cancer and normal development, show that Zeb1 is a master regulator of mesenchyme-specific gene expression in the developing mouse ear. Twirler is a novel example of a disorder of hearing or balance caused by a disruption of mesenchymal-epithelial identities or interactions. A similar lateral semicircular canal phenotype is seen in other hyperactive circling mice, including epistatic circler mice [29] and mice segregating a gene-trap allele of Chd7 [30]. Chd7 encodes a chromodomain protein required for the development of multipotent migratory neural crest cells [31], which includes an epithelial-to-mesenchymal transition. An auditory-vestibular phenotype approximating that of Twirler and Chd7 mutant mice is also observed in human patients with CHARGE syndrome and mutations of the human CHD7 gene [32], [33]. Semicircular canal formation is also known to require Bmp4 [34] and heterozygosity for a knockout allele of mouse Bmp4 primarily affects the lateral semicircular canal [35]. Bmp4 is a member of the transforming growth factor-β (TGF-β) super-family [36], providing another link to Zeb1 since Zeb1 and Zeb2 have been implicated in TGF-beta/BMP signaling [8].
Why do Twirler mice have a different inner ear phenotype than Zeb1ΔEx1 mice? Genetic background differences seem unlikely to account for this difference since Zeb1ΔEx1 and Twirler were both maintained on a congenic C57BL/6J background. It is possible that other Zeb1 transcripts could compensate for the loss of exon 1 in Zeb1ΔEx1 ears, but our quantitative RT-PCR and expression profiling results [13] render this hypothesis unlikely. Alternatively, the closely related Zeb2 gene may be able to compensate for the loss of Zeb1 expression in the inner ear, but not other affected tissues such as the palate or lymphoid system. However, our quantitative RT-PCR results revealed no compensatory change in Zeb2 transcript levels in the mutants. It is also possible that disruption of Zeb1os may contribute to the Tw phenotype. Ectopic expression of an analogous long noncoding antisense RNA in epithelial cells leads to altered Zeb2 RNA splicing, increased Zeb2 protein levels, and epithelial-to-mesenchymal transition [37]. However, Zeb1os RNA levels were too low for us to reliably detect and monitor by either qRT-PCR or Northern blot analyses to confidently address this possibility (data not shown). Finally, perhaps Twirler does not exert its pathogenic effect via Zeb1. This also seems unlikely since there is significant phenotypic overlap of Twirler with Zeb1ΔEx1, including abnormalities of the semicircular canals associated with both mutant alleles. Furthermore, the phenotypic effects of compound heterozygosity for Zeb1ΔEx1 and Tw are consistent with the conclusion that Zeb1ΔEx1 is an amorphic or hypomorphic allele whereas Twirler acts as a hypermorphic or neomorphic allele to misregulate Zeb1 expression.
Our electrophoretic mobility shift experiment (Figure 4) suggests that Tw exerts its pathogenic effect by disruption of binding of C-Myb or other Myb proteins to the first intron of Zeb1. There are also published observations supporting the general hypothesis that loss of Myb protein binding leads to de-repression of Zeb1Tw and inner ear malformations: First, C-Myb can function as either an activator or repressor of gene transcription [38] and is thought to function in regulation of epithelial-mesenchymal cell identity [39]. Second, a pathogenic effect of up-regulation of developmental transcription factors has been demonstrated for Pax6 in the eye [40] and Tbx1 in the inner ear [41]. In the inner ear, increased expression of Tbx1 can cause malformations that include incomplete coiling and reduced extension of the cochlear duct [41]. Furthermore, Tbx1 expression in the periotic mesenchyme is required for cochlear duct outgrowth [42], suggesting a potential link to the observed inner ear phenotype of Twirler. Taken together, these observations and our results support the hypothesis that Twirler disrupts inner ear development via mis-regulation of Zeb1.
The cell type-specific gene expression profiles of Twirler ears [13] suggest that a pathologic disruption of epithelial and mesenchymal cell identities underlies the inner ear malformations. This could arise from a loss of mesenchymal cell identity leading to mesenchymal-epithelial transition (MET), a loss of epithelial cell identity leading to epithelial-mesenchymal transition (EMT), or a combination of these mechanisms. Although the gene expression profiles [13] seem consistent with MET, it is difficult to conceive a simple MET pathway that does not invoke a loss-of-function mechanism in Tw mesenchyme. In contrast, EMT would involve a gain-of-function with ectopic expression of Zeb1 in Tw inner ear epithelium. Indeed, ectopic expression of Zeb proteins in other epithelial tissues has been shown to lead to EMT in other neoplastic and developmental processes [43]. Distinguishing among EMT and MET mechanisms may be difficult if they involve complex regulatory pathways mediated by Zeb1os, Zeb2, microRNAs or other genes.
In summary, we have identified the pathogenic mutation of Twirler as a noncoding point mutation that leads to over - or mis-expression of Zeb1, pathologic alterations of gene expression [13], cell fate and interactions in the developing inner ear. The ultimate result is a gross alteration of the structure and function of the vestibular and auditory organs. Disruption of epithelial-mesenchymal identity or interactions may be a shared pathogenetic mechanism underlying phenotypes that primarily affect development of the lateral semicircular canals, extension of the cochlear duct, or both.
Materials and Methods
Animals
Mice were maintained on a 12∶12-h light-dark cycle. All experiments and procedures were approved by the Animal Care and Use Committees of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institute of Neurological Disorders and Stroke and National Institute on Deafness and Other Communication Disorders. Twirler mice were a kind gift from Drs. Miriam Meisler and Siew-Ging Gong at the University of Michigan and were maintained on a C57BL/6J background by backcrossing heterozygous Tw males to C57BL/6J females for at least 30 generations. Zeb1ΔEx1 mice [10] were a generous gift from Dr. Douglas Darling and were serially backcrossed to C57BL/6J to maintain the line.
Generation of knockin mice
Bacterial artificial chromosome (BAC) clone RP23-135A18 containing mouse genomic DNA encoding exon 1 of Zeb1 was digested with PacI/SphI and SphI to yield 7.6-kb and 2.6-kb homology arms, respectively, for targeting constructs (Figure 6). Each targeting construct included loxP sites flanking a splice acceptor site and internal ribosomal entry site (IRES) (pGT1.8IresBgeo, provided by Austin Smith at University of Edinburgh) [44], E. coli lacZ, and a reverse-oriented pPGK-neomycin resistance cassette cloned into the pPNT plasmid [45] (Figure 6). The wild type (KIG) and Twirler (KIA) 7.6-kb PacI/SphI homology arms contained G and A at position c.58+181, respectively. Bruce4 embryonic stem (ES) cells [46] were electroporated with the KIG or KIA targeting constructs and grown in the presence of G418 and ganciclovir, using standard protocols at the University of Michigan Transgenic Animal Model Core [47]. G418-resistant ES clones were screened for homologous recombination by PCR and Southern blot analyses. At least three recombinant ES cell lines for each targeting construct were injected into C57BL/6 blastocysts. Chimeric males were mated with C57BL/6 females and offspring were analyzed by Southern blot and PCR analyses for germline transmission of KIG or KIA (Figure S3). Table S1 shows PCR primer pairs used to genotype KI alleles before and after neomycin cassette removal (Figure 6). Mice transmitting KIG or KIA in the germline were crossed to Cre recombinase-expressing mice (C57BL/6-TgN(Zp3-Cre)93Knw, Jackson laboratory, ME) to delete the IRES-lacZ-neomycin resistance cassette, leaving a single loxP site 606 bp downstream from Zeb1 exon 1.
Phenotype survey
A comprehensive gross anatomical, histological, and serological analysis of three 15-week-old Tw/+ and three wild type littermate males was performed as described [48]. Tissue sections from two Tw/Tw, two Tw/+ and two wild type mice at postnatal day 0 (P0) were analyzed.
Inner ear phenotype analyses
Heterozygous Tw males and females were mated. Pregnant females were identified by the presence of a vaginal plug and gestational stage was estimated by defining that morning as 0.5 days post-conception (dpc). Embryos at 14.5 dpc were harvested and processed for paint-filling as described [49]. The length of the cochlear duct was measured along its outer contour from a ventral view [50]. For scanning electron microscopy (SEM), whole-mounted inner ears were fixed in 2.5% glutaraldehyde in 0.1 M sodium cacodylate with 2 mM CaCl2 for 90 min. The organ of Corti, saccule, utricle, and crista ampullaris were dissected free in water and dehydrated with a serial dilution series of acetone. Samples were critical point-dried and sputter-coated followed by visualization with a field-emission scanning electron microscope (S-4800, Hitachi). Auditory brainstem response (ABR) thresholds were measured in response to click or pure-tone stimuli of 8, 16, or 32 kHz as described [51].
Obesity and metabolic phenotype analyses
Six Tw/+ male, six Tw/+ female, six wild type male and six wild type female mice were housed individually with regular mouse chow and water provided ad libitum. Body weights were measured weekly from 5 weeks of age. Weekly food intake was measured from weeks 6 through 22 to calculate average daily food intake. At 23 weeks of age, mice were transferred to the NIDDK Mouse Metabolism Core Laboratory for measurement of oxygen consumption, carbon dioxide production and motor activity as described [52]. Body composition was measured using Echo3-in-1 NMR analyzer (Echo Medical Systems, Houston, TX). Tail vein blood was used for serologic analyses. Fifteen-week-old female mice (eight Tw/+, six wild type) were tested for glucose and insulin tolerance as described [52]. All data are expressed as a mean ± SEM. Student's t-test was used to identify statistically significant differences between genotype groups.
Linkage backcross
Twirler males (C57BL/6J-Tw/+) were crossed with DBA/2J or Castaneus (CAST/Ei) females since Twirler females are poor caretakers of offspring. Male (C57BL/6J-Tw/+ x DBA/2J)F1-Tw/+ or (C57BL/6J-Tw/+ x CAST/Ei)F1-Tw/+ progeny were backcrossed with DBA/2J or C57BL/6J females, respectively, to generate 337 and 1679 N2 backcross progeny, respectively. Progeny were scored for circling behavior or obesity by visual inspection.
Recombination mapping
We genotyped short tandem repeat (STR) markers on 337 DBA/2J N2 backcross progeny to identify two STR markers (D18Mit65, D18Mit64, D18Mit19 and D18Umi1) flanking each side of Tw. These markers were genotyped in the 1679 CAST/Ei N2 backcross progeny to identify recombinations in the Tw region. The Tw map interval was defined by genotypes of additional markers in the recombinants. We genotyped MIT markers between D18Mit65 and D18Umi1, as well as 40 novel STR markers (denoted D18Nih1 through D18Nih44; PCR primer sequences listed in Table S1) located between D18Mit19 and D18Mit219.
Mutation analyses
Genomic DNA of Tw/Tw, Tw/+ and wild type mice were isolated for PCR amplification as described [53]. The primers were designed to amplify and sequence all of the annotated exons of the Zeb1, Zeb1os (MGI predicted gene Gm10125) and Zfp438 genes in the Tw critical interval. Additional novel exons were identified by 5′ and 3′ - RACE (5′ and 3′ rapid amplification of cDNA ends) of the Zeb1, Zeb1os and Zfp438 genes. This revealed multiple alternative first exons for Zeb1 that were also sequenced. Reverse transcription (RT)-PCR was performed to amplify and sequence full-length cDNA clones of the three genes using whole body mRNA collected from embryonic Tw/Tw, Tw/+ and wild type littermates.
PCR reaction conditions were modified to amplify and sequence the overlapping genomic region of Zeb1 and Zeb1os. Fifty-µl PCR reactions contained 50 to 100 ng of genomic DNA, 5 pmol each of forward and reverse primers, 200 mM each dNTP, 0.5 M betaine, 10% dimethyl sulfoxide (DMSO), 2.5 mM MgCl2, and 0.5 U of thermostable polymerase. Thermal cycling conditions were: 95°C for 1 min; 33 cycles of 20 s at 95°C, 20 s at 57°C, and 45 s at 72°C; and a final 2-min extension at 72°C. For sequencing, 50 µl PCR reaction products were purified with a QIAquick PCR purification kit (Qiagen, Hilden, Germany) and eluted with 30 µl elution buffer. Three µl of purified products were added to a 10-µl sequencing reaction containing 3.2 pmol primer, 0.25 µl Big Dye Terminator Ready Reaction mix (PE Biosystems), sequencing buffer and 10% DMSO. Cycling conditions were 96°C for 2 min and 33 cycles of 96°C for 10 s, 55°C for 10 s, and 60°C for 4 min. We also amplified and sequenced the overlapping genomic region of Zeb1 and Zeb1os from normal mouse control strains 129/J, AKR/J, BALB/cJ, C3H/HeJ, C57BL/6J, C58/J, CBA/J, CE/J, DBA/2J, P/J, RF/J, SEA/GnJ and SWR/J DNA.
Electrophoretic mobility shift assay
Double-stranded oligodeoxyribonucleotide probes were synthesized to encode genomic sequences containing c.58+181G (5′-TGCTGGACTGGACCGTTATGTCTTACCTGC and 5′-GCAGGTAAGACATAACGGTCCAGTCCAGCA), c.58+181A (5′-TGCTGGACTGGACCATTATGTCTTACCTGC and 5′-GCAGGTAAGACATAATGGTCCAGTCCAGCA), or a C-Myb binding site control from the mim-1 gene [19] (5′-GCTCTAAAAAACCGTTATAATGTACAGATATCTT and 5′-AAGATATCTGTACATTATAACGGTTTTTTAGAG). Probes were end-labeled with [γ-32P]ATP by T4 Polynucleotide Kinase (New England Biolabs). Mouse C-Myb cDNA was cloned in pET-41a(+) (Novagen), and the protein was expressed in E.coli strain BL21(DE3)pLys (Invitrogen) and purified with Ni-NTA columns (Qiagen). Twenty-μl reactions were performed with the EMSA Accessory Kit (Novagen). Unlabeled oligonucleotide competitors were added at 25 - or 50-fold molar excess. Binding reaction products were separated by 6% DNA retardation gel electrophoresis (Invitrogen) and visualized with a Typhoon Trio+ (GE Healthcare).
Quantitative RT-PCR analyses
Inner ears with adjacent mesenchyme were microdissected from E13.5 offspring of Tw/+ x Tw/+ matings. Total RNA was isolated from inner ears using PicoPure (Applied Biosystems, Foster City, CA). Total RNA from 10 to 14 ears of the same genotype was pooled and purified with the RNAeasy MinElute Cleanup kit (Qiagen). RNA integrity was measured with an Agilent 2100 Bioanalyzer (Applied Biosystems). One µg of total RNA was reverse-transcribed with oligo(dT) primers and SuperScriptIII (Invitrogen, Carlsbad, CA, USA). For TaqMan real-time PCR, PCR primers were designed to amplify Zeb1 exons 1a to 2, 1b to 2, 1c to 2, 1d to 2, 1e to 2, 1f to 2, and 2 to 3, Zeb1os exons 1 to 2, and Zfp438 exons 3 to 4 with ZEN double-quenched probes containing a 5′ FAM fluorophore, 3′ IBFQ quencher, and an internal ZEN quencher (IDT, Coralville, IA). Sequences for the primers and probes are listed in Table S1.
Comparative TaqMan assays were performed in triplicate on an ABI 7500 real-time PCR system (Applied Biosystems). PCR reactions were performed in a 50-µl volume containing 5 µl cDNA, 5 µl primer mix (IDT), and 25 µl of Universal PCR Master Mix (Applied Biosystems). Cycling conditions were 50°C for 2 min, 95°C for 10 min, followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. Relative expression was normalized as the percentage of β-actin expression, and calculated using the comparative threshold cycle method of 2−ΔΔCT. Data are presented as mean values ± S.D. from six technical replicates. ANOVA was used to identify statistically significant differences between genotype groups (P<0.05).
Western blot analyses
Proteins were extracted from E13.5 mouse inner ears or whole heads with NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce Biotechnology) in the presence of Halt Protease Inhibitor Cocktail (Thermo Fisher Scientific Inc.). Proteins were separated by SDS-PAGE in 4–20% NuPage Bis-Tris gels followed by transfer to PVDF membranes (Millipore Corp., Billerica, MA). Proteins were detected with primary antibodies for Zeb1 (ab64098, Abcam, 1∶200) and β-actin (A2228, Sigma-Aldrich, 1∶1000). Secondary antibodies were conjugated with Cy 3 or Cy 5 (GE Healthcare) and detected with a Typhoon Trio+ (GE Healthcare). Band density was measured using ImageQuant TL software. β-actin levels were used for normalization. ANOVA analysis of two to six biological replicates from each genotype group was used to identify statistically significant differences (P<0.05).
Immunohistochemistry
Mouse inner ear sections were harvested, processed and immunostained with anti-Zeb1 or anti-CD326 antibodies as described in Hertzano et al. [13]. CD326 is also known as epithelial cell adhesion/activating molecule (EpCAM) that serves as a specific antigenic marker for epithelial cells [13].
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. LyonMF 1958 Twirler: a mutant affecting the inner ear of the house mouse. J Embryol Exp Morphol 6 105 116
2. LanePWSearleAGBeecheyCVEicherE 1981 Chromosome 18 of the house mouse. J Hered 72 409 412
3. TingCNKohrmanDBurgessDLBoyleAAltschulerRA 1994 Insertional mutation on mouse chromosome 18 with vestibular and craniofacial abnormalities. Genetics 136 247 254
4. LiuHLiuWMaltbyKMLanYJiangR 2006 Identification and developmental expression analysis of a novel homeobox gene closely linked to the mouse Twirler mutation. Gene Expr Patterns 6 632 636
5. SekidoRMuraiKFunahashiJKamachiYFujisawa-SeharaA 1994 The delta-crystallin enhancer-binding protein delta EF1 is a repressor of E2-box-mediated gene activation. Mol Cell Biol 14 5692 5700
6. PostigoAADeanDC 1997 ZEB, a vertebrate homolog of Drosophila Zfh-1, is a negative regulator of muscle differentiation. EMBO J 16 3935 3943
7. ChamberlainEMSandersMM 1999 Identification of the novel player deltaEF1 in estrogen transcriptional cascades. Mol Cell Biol 19 3600 3606
8. PostigoAA 2003 Opposing functions of ZEB proteins in the regulation of the TGFbeta/BMP signaling pathway. EMBO J 22 2443 2452
9. NishimuraGManabeITsushimaKFujiuKOishiY 2006 DeltaEF1 mediates TGF-beta signaling in vascular smooth muscle cell differentiation. Dev Cell 11 93 104
10. TakagiTMoribeHKondohHHigashiY 1998 DeltaEF1, a zinc finger and homeodomain transcription factor, is required for skeleton patterning in multiple lineages. Development 125 21 31
11. SaykallyJNDoganSClearyMPSandersMM 2009 The ZEB1 transcription factor is a novel repressor of adiposity in female mice. PLoS One 4 e8460
12. PeinadoHOlmedaDCanoA 2007 Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer 7 415 428
13. HertzanoRElkonRKurimaKMorrissonAChanS-L 2011 Zeb1 and miR-200b regulate mesenchymal and epithelial cell identities in the developing mouse inner ear. Manuscript in preparation
14. KrafchakCMPawarHMoroiSESugarALichterPR 2005 Mutations in TCF8 cause posterior polymorphous corneal dystrophy and ectopic expression of COL4A3 by corneal endothelial cells. Am J Hum Genet 77 694 708
15. LiuYPengXTanJDarlingDSKaplanHJ 2008 Zeb1 mutant mice as a model of posterior corneal dystrophy. Invest Ophthalmol Vis Sci 49 1843 1849
16. HoweKMWatsonRJ 1991 Nucleotide preferences in sequence-specific recognition of DNA by c-myb protein. Nucleic Acids Res 19 3913 3919
17. KennedyAJEllacottKLKingVLHastyAH 2010 Mouse models of the metabolic syndrome. Dis Model Mech 3 156 166
18. ZhongZWanBQiuYNiJTangW 2007 Identification of a novel human zinc finger gene, ZNF438, with transcription inhibition activity. J Biochem Mol Biol 40 517 524
19. ChaykaOKintscherJBraasDKlempnauerKH 2005 v-Myb mediates cooperation of a cell-specific enhancer with the mim-1 promoter. Mol Cell Biol 25 499 511
20. KalueffAVIshikawaKGriffithAJ 2008 Anxiety and otovestibular disorders: linking behavioral phenotypes in men and mice. Behav Brain Res 186 1 11
21. CalderonADerrAStagnerBBJohnsonKRMartinG 2006 Cochlear developmental defect and background-dependent hearing thresholds in the Jackson circler (jc) mutant mouse. Hear Res 221 44 58
22. Stenzel-PooreMPCameronVAVaughanJSawchenkoPEValeW 1992 Development of Cushing's syndrome in corticotropin-releasing factor transgenic mice. Endocrinology 130 3378 3386
23. ShinaharaMNishiyamaMIwasakiYNakayamaSNoguchiT 2009 Plasma adiponectin levels are increased despite insulin resistance in corticotropin-releasing hormone transgenic mice, an animal model of Cushing syndrome. Endocr J 56 879 886
24. WangJScullyKZhuXCaiLZhangJ 2007 Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature 446 882 887
25. HagerJDinaCFranckeSDuboisSHouariM 1998 A genome-wide scan for human obesity genes reveals a major susceptibility locus on chromosome 10. Nat Genet 20 304 308
26. HinneyAZieglerAOeffnerFWedewardtCVogelM 2000 Independent confirmation of a major locus for obesity on chromosome 10. J Clin Endocrinol Metab 85 2962 2965
27. HsuehWCMitchellBDSchneiderJLSt JeanPLPollinTI 2001 Genome-wide scan of obesity in the Old Order Amish. J Clin Endocrinol Metab 86 1199 1205
28. PriceRALiWDBernsteinACrystalAGoldingEM 2001 A locus affecting obesity in human chromosome region 10p12. Diabetologia 44 363 366
29. CrynsKvan AlphenAMvan SpaendonckMPvan de HeyningPHTimmermansJP 2004 Circling behavior in the Ecl mouse is caused by lateral semicircular canal defects. J Comp Neurol 468 587 595
30. AdamsMEHurdEABeyerLASwiderskiDLRaphaelY 2007 Defects in vestibular sensory epithelia and innervation in mice with loss of Chd7 function: implications for human CHARGE syndrome. J Comp Neurol 504 519 532
31. BajpaiRChenDARada-IglesiasAZhangJXiongY 2010 CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature 463 958 962
32. TellierALCormier-DaireVAbadieVAmielJSigaudyS 1998 CHARGE syndrome: report of 47 cases and review. Am J Med Genet 76 402 409
33. JongmansMCAdmiraalRJvan der DonkKPVissersLEBaasAF 2006 CHARGE syndrome: the phenotypic spectrum of mutations in the CHD7 gene. J Med Genet 43 306 314
34. ChangWLinZKulessaHHebertJHoganBL 2008 Bmp4 is essential for the formation of the vestibular apparatus that detects angular head movements. PLoS Genet 4 e1000050
35. VervoortRCeulemansHVan AerschotLD'HoogeRDavidG 2010 Genetic modification of the inner ear lateral semicircular canal phenotype of the Bmp4 haplo-insufficient mouse. Biochem Biophys Res Commun 394 780 785
36. MiyazonoKMaedaSImamuraT 2005 BMP receptor signaling: transcriptional targets, regulation of signals, and signaling cross-talk. Cytokine Growth Factor Rev 16 251 263
37. BeltranMPuigIPenaCGarciaJMAlvarezAB 2008 A natural antisense transcript regulates Zeb2/Sip1 gene expression during Snail1-induced epithelial-mesenchymal transition. Genes Dev 22 756 769
38. MizuguchiGKanei-IshiiCTakahashiTYasukawaTNagaseT 1995 c-Myb repression of c-erbB-2 transcription by direct binding to the c-erbB-2 promoter. J Biol Chem 270 9384 9389
39. TannoBSestiFCesiVBossiGFerrari-AmorottiG 2010 Expression of Slug is regulated by c-Myb and is required for invasion and bone marrow homing of cancer cells of different origin. J Biol Chem 285 29434 29445
40. DavisNYoffeCRavivSAntesRBergerJ 2009 Pax6 dosage requirements in iris and ciliary body differentiation. Dev Biol 333 132 142
41. FunkeBEpsteinJAKochilasLKLuMMPanditaRK 2001 Mice overexpressing genes from the 22q11 region deleted in velo-cardio-facial syndrome/DiGeorge syndrome have middle and inner ear defects. Hum Mol Genet 10 2549 2556
42. VitelliFViolaAMorishimaMPramparoTBaldiniA 2003 TBX1 is required for inner ear morphogenesis. Hum Mol Genet 12 2041 2048
43. VandewalleCVan RoyFBerxG 2009 The role of the ZEB family of transcription factors in development and disease. Cell Mol Life Sci 66 773 787
44. MountfordPZevnikBDuwelANicholsJLiM 1994 Dicistronic targeting constructs: reporters and modifiers of mammalian gene expression. Proc Natl Acad Sci U S A 91 4303 4307
45. TybulewiczVLCrawfordCEJacksonPKBronsonRTMulliganRC 1991 Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell 65 1153 1163
46. KontgenFSussGStewartCSteinmetzMBluethmannH 1993 Targeted disruption of the MHC class II Aa gene in C57BL/6 mice. Int Immunol 5 957 964
47. HughesEDQuYYGenikSJLyonsRHPachecoCD 2007 Genetic variation in C57BL/6 ES cell lines and genetic instability in the Bruce4 C57BL/6 ES cell line. Mamm Genome 18 549 558
48. Ben-YosefTBelyantsevaIASaundersTLHughesEDKawamotoK 2003 Claudin 14 knockout mice, a model for autosomal recessive deafness DFNB29, are deaf due to cochlear hair cell degeneration. Hum Mol Genet 12 2049 2061
49. MorsliHChooDRyanAJohnsonRWuDK 1998 Development of the mouse inner ear and origin of its sensory organs. J Neurosci 18 3327 3335
50. BokJDolsonDKHillPRutherUEpsteinDJ 2007 Opposing gradients of Gli repressor and activators mediate Shh signaling along the dorsoventral axis of the inner ear. Development 134 1713 1722
51. NoguchiYKurimaKMakishimaTde AngelisMHFuchsH 2006 Multiple quantitative trait loci modify cochlear hair cell degeneration in the Beethoven (Tmc1Bth) mouse model of progressive hearing loss DFNA36. Genetics 173 2111 2119
52. KimHPennisiPAGavrilovaOPackSJouW 2006 Effect of adipocyte beta3-adrenergic receptor activation on the type 2 diabetic MKR mice. Am J Physiol Endocrinol Metab 290 E1227 1236
53. BorkJMPetersLMRiazuddinSBernsteinSLAhmedZM 2001 Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am J Hum Genet 68 26 37
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 9
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Retrotransposon-Induced Heterochromatin Spreading in the Mouse Revealed by Insertional Polymorphisms
- The Evolutionarily Conserved Longevity Determinants HCF-1 and SIR-2.1/SIRT1 Collaborate to Regulate DAF-16/FOXO
- Genome-Wide Analysis of Heteroduplex DNA in Mismatch Repair–Deficient Yeast Cells Reveals Novel Properties of Meiotic Recombination Pathways
- Association of eGFR-Related Loci Identified by GWAS with Incident CKD and ESRD
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy