
Tuberous Sclerosis Complex 1 Regulates dE2F1 Expression during Development and Cooperates with RBF1 to Control Proliferation and Survival
Previous studies in Drosophila melanogaster have demonstrated that many tumor suppressor pathways impinge on Rb/E2F to regulate proliferation and survival. Here, we report that Tuberous Sclerosis Complex 1 (TSC1), a well-established tumor suppressor that regulates cell size, is an important regulator of dE2F1 during development. In eye imaginal discs, the loss of tsc1 cooperates with rbf1 mutations to promote ectopic S-phase and cell death. This cooperative effect between tsc1 and rbf1 mutations can be explained, at least in part, by the observation that TSC1 post-transcriptionally regulates dE2F1 expression. Clonal analysis revealed that the protein level of dE2F1 is increased in tsc1 or tsc2 mutant cells and conversely decreased in rheb or dTor mutant cells. Interestingly, while s6k mutations have no effect on dE2F1 expression in the wild-type background, S6k is absolutely required for the increase of dE2F1 expression in tsc2 mutant cells. The canonical TSC/Rheb/Tor/S6k pathway is also an important determinant of dE2F1-dependent cell death, since rheb or s6k mutations suppress the developmentally regulated cell death observed in rbf1 mutant eye discs. Our results provide evidence to suggest that dE2F1 is an important cell cycle regulator that translates the growth-promoting signal downstream of the TSC/Rheb/Tor/S6k pathway.
Published in the journal:
. PLoS Genet 6(8): e32767. doi:10.1371/journal.pgen.1001071
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001071
Summary
Previous studies in Drosophila melanogaster have demonstrated that many tumor suppressor pathways impinge on Rb/E2F to regulate proliferation and survival. Here, we report that Tuberous Sclerosis Complex 1 (TSC1), a well-established tumor suppressor that regulates cell size, is an important regulator of dE2F1 during development. In eye imaginal discs, the loss of tsc1 cooperates with rbf1 mutations to promote ectopic S-phase and cell death. This cooperative effect between tsc1 and rbf1 mutations can be explained, at least in part, by the observation that TSC1 post-transcriptionally regulates dE2F1 expression. Clonal analysis revealed that the protein level of dE2F1 is increased in tsc1 or tsc2 mutant cells and conversely decreased in rheb or dTor mutant cells. Interestingly, while s6k mutations have no effect on dE2F1 expression in the wild-type background, S6k is absolutely required for the increase of dE2F1 expression in tsc2 mutant cells. The canonical TSC/Rheb/Tor/S6k pathway is also an important determinant of dE2F1-dependent cell death, since rheb or s6k mutations suppress the developmentally regulated cell death observed in rbf1 mutant eye discs. Our results provide evidence to suggest that dE2F1 is an important cell cycle regulator that translates the growth-promoting signal downstream of the TSC/Rheb/Tor/S6k pathway.
Introduction
Retinoblastoma (Rb) family proteins are important regulators of cell cycle progression and survival (reviewed in [1], [2]). Orthologs of Rb exist in all metazoans where their functions are evolutionarily conserved (reviewed in [3]). Their best-known molecular function is to physically interact with E2F family proteins and transcriptionally repress E2F-regulated target genes. Genome-wide expression studies revealed that genes involved in various biological processes, such as cell cycle progression, survival, and development, are regulated by E2F family proteins [4]–[6]. As a consequence, the loss of Rb family genes in mice results in developmental defects that are often associated with uncontrolled S-phase entry and ectopic cell death [7]–[9]. Importantly, reducing E2F activity largely suppresses the Rb mutant phenotypes, indicating that deregulated E2F activity is responsible for the defects [10], [11]. Overall, E2F family proteins are the key molecular targets of Rb family proteins and responsible for the developmental consequence of Rb inactivation.
The long-term consequence of Rb inactivation in mammals is tumorigenesis. In humans, the loss of Rb is believed to be a critical step for retinoblastoma development. Moreover, Rb is believed to be functionally inactivated in most, if not all, cancers (reviewed in [12]). In mice, Rb heterozygosity (Rb+/−) results in the formation of pituitary and thyroid tumors [7], [13]–[16]. The wild type copy of the Rb gene is lost in these tumors, illustrating the importance of Rb as a tumor suppressor gene. Moreover, conditional knockout of Rb and an additional member of the Rb family gene, p107 or p130, in mouse retina is sufficient to promote retinoblastoma development [17]–[20]. Similar to the developmental phenotype, deregulated E2F plays a major role during tumorigenesis in Rb mutant mice. In a pituitary tumor model, the loss of E2f-1 or E2f-3 reduces the frequency of tumor development [21], [22]. More recently, the importance of E2F family proteins in human cancer is further illustrated by the findings that E2F family proteins themselves are often deregulated in many types of cancers (reviewed in [23]). Clearly, E2F family proteins play a critical role during tumorigenesis and also contribute to the developmental defects observed in Rb mutant animals.
Although it is clear that studying the function of E2F is crucial to understand the biology of Rb mutant animals and cancers, it has been difficult to dissect the in vivo roles of E2F family genes in mammals. One of the difficulties is the fact that E2F family proteins can functionally compensate for each other, which is particularly true for the subset of E2F proteins called “activator E2Fs” (reviewed in [24]). This is best demonstrated by a recent study showing that a single “activator E2F”, E2F-3a, is sufficient to support embryonic and post-natal development in mice, and the expression of E2F-3b or E2F-1 under the control of E2F-3a promoter can perform the role of E2F-3a [25]. This study suggests that the unique developmental functions of “activator E2Fs” are largely determined by their expression patterns and not by the differences of their protein sequences. Interestingly, Drosophila melanogaster has only a single “activator E2F”, dE2F1. The function of dE2F1 is evolutionarily conserved and represents the three “activator E2Fs” in mammals. dE2F1 is required for cellular proliferation and controls DNA damage-induced cell death, activities that are shared by the three “activator E2Fs” in mammals (reviewed in [3]). Since dE2F1 is the sole member carrying out the function of three E2Fs in mammals, it is possible that the regulation of dE2F1 expression is more complex and tightly controlled in flies. However, the regulatory mechanism that controls dE2F1 expression in Drosophila is poorly understood.
Like Rb, RBF1 is the major regulator of dE2F1 in flies. Most of the rbf1 mutant phenotypes are believed to be due to deregulated dE2F1 and can be rescued by a hypomorphic mutant allele of de2f1 [26]. Because of its simplicity and conserved developmental function, the Drosophila Rb/E2F is considered as a simplified version of mammalian Rb/E2F. Although rbf1 mutations are not sufficient to promote tumor phenotype in Drosophila, recent genetic studies revealed that RBF1/dE2F1 plays a crucial role when proliferation and/or survival are compromised by various tumor-promoting mutations. For example, dE2F1 is required by hippo mutant cells to overcome the developmentally regulated cell cycle arrest in eye imaginal discs [27]. Moreover, dE2F1-dependent cell death limits the growth promoting effect of the archipelago mutations in the eye, and cooperates with low EGFR activity to promote cell death [28], [29]. Interestingly, although the Drosophila p53 (dp53) does not genetically interact with rbf1 during development, dE2F1 and p53 cooperate to promote DNA damage - induced cell death as they do in mammalian systems [30]. Overall, RBF1/dE2F1 can either promote and/or limit the proliferation of cells that carry tumor-promoting mutations in flies.
Tuberous Sclerosis Complex 1 (TSC1) is a tumor suppressor gene that is mutated in benign tumors (reviewed in [31]). The in vivo function of TSC1 was first identified in Drosophila melanogaster as a regulator of cell size and proliferation (reviewed in [32]). TSC1 is a negative regulator of the Ras Homolog Enriched in Brain (Rheb), which is an activator of Target of Rapamycin (Tor). The canonical TSC/Rheb/Tor pathway has been established as a central network governing cell size and growth regulation. Although initial reports clearly demonstrated that TSC1 inactivation perturbs the cell cycle profile, less is understood about the mechanism by which TSC1 controls the cell cycle as well as cell size. Here, we demonstrate that tsc1 mutations cooperate with rbf1 mutations to promote both unscheduled S-phase entry and cell death during Drosophila eye development. This cooperative effect between tsc1 and rbf1 mutations can be explained, at least in part, by the observation that dE2F1 expression is post-transcriptionally increased in tsc1 mutant cells. A dE2F-reporter construct, PCNA-GFP, is activated in tsc1 mutant cells, and de2f1 mutations completely suppress the ectopic cell death observed in the rbf1 and tsc1 double mutant cells, indicating that dE2F1 is activated by tsc1 mutations and required for cooperative effect between rbf1 and tsc1 mutations. We further demonstrate that Rheb and Tor control dE2F1 expression, and s6k mutations completely abolish the increase of dE2F1 expression observed in tsc2 mutant cells. These results demonstrate that the TSC/Rheb/Tor/S6k pathway is an important regulator of dE2F1 expression during development and cooperates with RBF1 to regulate cell cycle progression and survival.
Results
tsc1 and rbf1 mutations cooperate to promote S-phase entry and cell death
Ectopic S-phase entry and cell death are well-established Rb loss-of-function phenotypes. To address the question whether growth-promoting mutations could alter the Rb mutant phenotypes, we sought to determine the effects of inactivating the Drosophila ortholog of Tuberous Sclerosis Complex 1 (TSC1) in an rbf1 mutant background. To test this, tsc1 mutant clones were generated in wild type or rbf1 mutant eye discs (Figure 1). Since homozygous rbf1 null flies die at the first instar larval stage, we used an rbf1 hypomorphic allele, rbf1120a. Mitotic tsc1 mutant clones were generated by expressing Flippase (FLP) with an eye-specific driver and marked by the absence of GFP. Thus, GFP negative clones in wild type background have only tsc1 mutations while GFP negative clones in the rbf1120a background have both rbf1 and tsc1 mutations. Third instar larval eye discs were dissected and immunostained with anti-BrdU antibodies. During normal eye development in Drosophila, S - phase cells, which can be labeled with BrdU, are found at the anterior portion of the eye imaginal disc where cells are asynchronously dividing, and immediately posterior to the Morphogenetic Furrow (MF) where some cells undergo an extra S-phase called the Second Mitotic Wave (Figure 1A). At the MF, asynchronously dividing precursor cells arrest in G1 and begin differentiation process. Therefore, normally, there is no BrdU incorporating cells at the MF. Surprisingly, in clones that are double mutant for rbf1 and tsc1, ectopic S-phase cells were readily observed at the MF (Figure 1C). Since we can occasionally detect rbf1 mutant cells entering S-phase at the MF, we compared the number of ectopic BrdU positive cells at the MF between rbf1 single and rbf1 tsc1 double mutant clones. We normalized the number of ectopic BrdU positive cells by the clone size, which is measured by the number of the pixels in images taken at the same magnification. Clones that do not contain ectopic BrdU positive cells are excluded from the analysis. We determined that, on average, 3.7±2.2 ectopic S-phase cells/1000 pixels are present in the rbf1 clones while 12.4±5.6 ectopic S-phase cells/1000 pixels cells are present in the rbf1 tsc1 double mutant clones, showing more than 3 fold increase. This result indicates that RBF1 and TSC1 cooperatively regulate G1 to S - phase transition. Next, we stained for dying cells with anti-cleaved Caspase 3 antibodies (C3). rbf1 mutant cells undergo apoptosis at the anterior region of the MF, and this is not observed in the wild type eye disc (Figure 1B). We had previously reported that this developmentally regulated cell death in rbf1 mutant eye discs is dE2F1-dependent [29]. tsc1 mutant cells also undergo apoptosis just anterior to the MF though the level of cell death is much lower than what is observed in rbf1120a eye discs. However, in clones that are double mutant for rbf1 and tsc1, we observed a great increase in C3 staining at the MF and the anterior region of the eye disc (Figure 1B and 1C). Therefore, we concluded that RBF1 and TSC1 synergistically promote survival as well as G1 arrest during Drosophila eye development.

TSC1 regulates dE2F1 protein expression post-transcriptionally
RBF1 is best characterized as a regulator of dE2F1 transcription factors whose activity promotes both S-phase entry and apoptosis. Since we observed that tsc1 mutations are able to enhance both ectopic S-phase entry and cell death phenotypes in rbf1 mutant cells, we sought to determine if dE2F1 itself is deregulated by tsc1 mutations. Eye discs containing tsc1 mutant clones were generated as described previously and immunostained with an anti-dE2F1 antibody. We observed that the intensity of dE2F1 staining is clearly stronger in tsc1 homozygous mutant clones throughout the eye disc, both in dividing and differentiating cells (Figure 2A and Figure S1A). Moreover we detected similar increase in antenna and wing discs, indicating that the effect on dE2F1 protein expression is not tissue-specific (Figure S1B and S1C). Importantly, the intensity of dE2F2 staining, the only other member of the E2F family in Drosophila, is unchanged in tsc1 mutant cells (Figure 2A), indicating that the effect of tsc1 mutations on dE2F1 expression is specific. To confirm the immunostaining result, we performed immunoblot assays using protein extracts from eye imaginal discs comprised mostly of tsc1 mutant cells (see Materials and Methods). Consistent with the immunostaining experiments, dE2F1 protein level is higher in tsc1 mutant eye discs than in control discs while no difference is detected in dE2F2 protein level (Figure 2B). To determine whether TSC1 regulates the level of de2f1 RNA, we performed real-time quantitative PCR (RTq-PCR). RNA was isolated from eye discs of the same genotypes used for immunoblot. We designed de2f1 specific primers that span an intron and amplified portions of two exons (second and third exons or fourth and fifth exons) to distinguish the PCR products from cDNA and genomic DNA. charybdis (chrb), a previously reported TSC1 regulated gene is used as a positive control [33]. Similar to the published result, we observed that the level of chrb RNA is increased by 11-fold in tsc1 mutant eye discs (Figure 2C). However, we could not detect any significant changes in de2f1 RNA level in tsc1 mutant eye discs (Figure 2C). Therefore, we concluded that TSC1 regulates dE2F1 expression post - transcriptionally.

Transcription of dE2F1 target genes is activated in tsc1 mutant cells
Next, we examined whether the transcription of a dE2F1 target gene is activated in tsc1 mutant cells. To address this question, we used a reporter construct, PCNA-GFP, whose GFP expression is under the control of the PCNA promoter, a well-established dE2F1 target gene. As shown in Figure 3, GFP expression is increased in tsc1 mutant cells in the posterior portion of the eye disc, suggesting that, at least in this region, the increase of dE2F1 protein is sufficient to activate the transcription of a target gene. Importantly, the abnormal BrdU positive cells observed in the same region of tsc1 mutant clones are scarcely present (Figure 1A), indicating that the increase in dE2F1-reporter activity is not an indirect consequence of ectopic S-phase cells. We also sought to determine if tsc1 mutations could further activate dE2F1 target gene expression in rbf1 mutant cells. Our attempt to compare dE2F1 target gene expression between rbf1 single and rbf1 tsc1 double mutant eye discs by RTq-PCR did not provide any conclusive results (data not shown). This was somewhat expected since a substantial number of rbf1 tsc1 double mutant cells, presumably cells with hyperactive dE2F1, undergo cell death (Figure 1B and Figure S2A). Therefore, we decided to perform an in situ hybridization experiment, hoping to detect specific changes in a subset of surviving rbf1 tsc1 double mutant cells. Expression patterns of dE2F1 target genes (rnrS, Cyclin E, and PCNA) were determined using antisense RNA probes. In wild type eye discs, the expression pattern of these target genes resembles that of BrdU staining since their transcription is activated during the G1/S phase transition (Figure 3B left panel). In rbf1 mutant eye discs, dE2F1 target genes are strongly expressed at the MF where dE2F1 protein expression is normally high (Figure 3B middle panel). It is probable that, in rbf1 mutant eye discs, dE2F1 target gene expression is mainly controlled by dE2F1 protein level since cell cycle-dependent regulation by RBF1 is absent. Interestingly, in rbf1 tsc1 double mutant eye discs, dE2F1 target genes are strongly expressed both at the MF and in the anterior region of the eye disc (Figure 3B right panel). We reasoned that, since rbf1 mutant cells at the MF already express a high level of dE2F1 protein (previously shown in [29]), there is only a small margin for dE2F1 target gene expression to be further activated by tsc1 mutations. However, in the anterior region of the eye disc where the dE2F1 protein expression is normally kept low [29], tsc1 mutations can have a greater effect on dE2F1 activity and target gene expression. As a consequence, dE2F1 target genes are strongly expressed both at the MF and in the anterior region of rbf1 tsc1 double mutant eye discs, reaching the threshold of expression before undergoing cell death. Supporting this idea, ectopic cell death in rbf1 tsc1 double mutant eye discs is mainly observed at the MF and in the anterior region of the eye disc (Figure S2A). Interestingly, we could not detect much increase in dE2F1 target gene expression in the posterior region of rbf1 tsc1 double mutant eye discs, somewhat contradicting the result obtained by the PCNA-GFP reporter construct (Figure 3A). One explanation is that the in situ hybridization experiment is not as sensitive and quantitative as the PCNA-GFP reporter construct. We also found that the residual RBF1 proteins in the hypomorpic rbf1120a allele are mostly expressed in the posterior region of the MF, explaining why cells in this region do not show much an increase in dE2F1 target gene expression (Figure S2B). Nevertheless, these results indicate that tsc1 mutations can activate dE2F1 target gene expression in the wild type and rbf1 mutant backgrounds.

dE2F1 is required for the ectopic cell death induced by rbf1 and tsc1 mutations
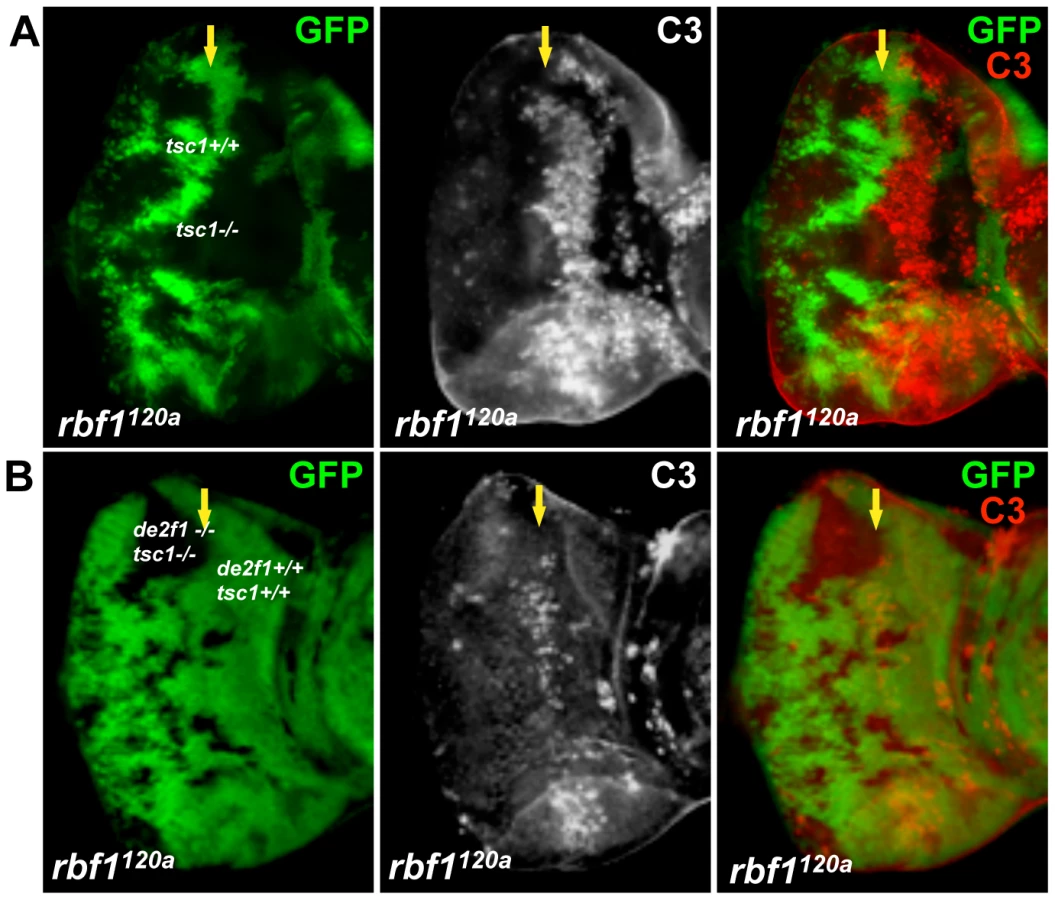
To determine if the cooperative effect on cell death by rbf1 and tsc1 mutations is dE2F1 - dependent, we generated an allele with an FRT chromosome carrying both tsc1 and de2f1 mutations. For this allele, we used the tsc1f01910 allele that contains a piggyBac transposable element inserted in the intron 6 of the tsc1 locus. Generating tsc1f01910 clones in rbf1120a eye discs produces a similar increase in the level of ectopic cell death observed in Figure 1 (Figure 4A). When tsc1f01910 and de2f1729 double mutant clones are generated in rbf1120a eye discs, we noticed that the sizes of tsc1 de2f1 double mutant clones are much smaller than that of tsc1 single mutant clones (compare Figure 4A and 4B). The sizes of tsc1 de2f1 double mutant clones in the wild type background are also small (data not shown), indicating that the loss of de2f1 severely compromises proliferation of tsc1 mutant cells. Occasionally, we were able to obtain rbf1120a mutant eye discs with substantial sizes of the tsc1 de2f1 double mutant clones. We performed C3 staining to measure the level of cell death in rbf1, tsc1, and de2f1 triple mutant cells in these eye discs. Interestingly, the prevailing cell death phenotype observed in rbf1 tsc1 double mutant cells at the MF is no longer present in rbf1 de2f tsc1 triple mutant cells (Figure 4B). This result demonstrates that the increased level of ectopic cell death observed in rbf1 tsc1 double mutant cells is dE2F1-dependent.
Rheb regulates dE2F1 expression and dE2F1-dependent cell death
Next, we asked if the known downstream regulators of TSC1 could regulate dE2F1 expression. We first determined the effect of rheb loss-of-function mutations on dE2F1 expression by generating mitotic mutant clones of rheb in the eye disc. Rheb is a Ras superfamily GTPase whose activity is negatively regulated by TSC1. As shown in Figure 5A, dE2F1 protein level is reduced, though not absent, in rheb mutant cells. This is best observed at the MF where dE2F1 expression is normally high [34]. We then asked if Rheb is required for the increased dE2F1 expression in tsc1 mutant cells. dE2F1 protein level is also reduced in tsc1 rheb double mutant cells (Figure 5A), indicating that Rheb is an important downstream regulator of TSC1 controlling dE2F1 expression. We concluded that, although not essential, Rheb regulates dE2F1 expression during eye development, and is clearly required for dE2F1 upregulation in tsc1 mutant cells. Since Rheb controls dE2F1 expression, we next tested if Rheb is also required for dE2F1-dependent cell death. To test this, we generated rheb mutant clones in the rbf1120a mutant eye disc where deregulated dE2F1 produces a stripe of apoptotic cells at the anterior region of the MF (Figure 1A and [29], [35]). As shown in Figure 5B, this stripe of cell death is interrupted by rheb mutant clones. Moreover, the ectopic cell death observed in rbf1 tsc1 double mutant cells is completely suppressed by rheb mutations. These results indicate that Rheb is an important regulator of dE2F1-dependent cell death as well as dE2F1 expression.

Tor, but neither S6k nor 4E-BP, is required for dE2F1 expression during Drosophila eye development
Rheb activates the Tor serine/threonine kinase, which through phosphorylation, can either inhibit 4EBP or activate S6k. We examined whether these proteins downstream of Rheb also participate in dE2F1 regulation. To address this question, Tor, s6k, and 4ebp mutant clones were generated in the eye disc. Similar to what is observed in rheb mutant clones, dE2F1 expression is reduced, but not absent, in Tor mutant clones, indicating that Tor participates in regulating dE2F1 expression during eye development (Figure 6A). Importantly, dE2F2 expression is unchanged in Tor mutant clones (data not shown). Based on this observation, we had hypothesized that dE2F1 expression levels would decrease in s6k mutant clones and/or increase in 4ebp mutant clones. Surprisingly, dE2F1 expression is unchanged in either 4ebp or s6k mutant clones (Figure 6B). These results suggest that Tor is required for proper dE2F1 expression during eye development while 4EBP and S6k are dispensable.

S6k is required for the effect of TSC inactivation on dE2F1 expression and dE2F1 - dependent cell death
The fact that the loss of neither 4ebp nor s6k has an effect on dE2F1 expression might indicate a functional redundancy between the two genes. Alternatively, an unknown factor downstream of Tor might regulate dE2F1 expression during development. Nevertheless, we assessed whether S6k is required for the increase of dE2F1 expression observed when TSC1 is inactivated. We aimed to generate mitotic clones that are double mutants for tsc1 and s6k. However, because tsc1 and s6k are on the opposite arms of the third chromosome, we used a mutant allele of tsc2 (or gig in Drosophila), which is on the same chromosomal arm as s6k. TSC1 and TSC2 function together as a heterodimer, and mutations of tsc1 or tsc2 yield very similar phenotypes [36]–[38]. As expected, dE2F1 expression is elevated in gig mutant clones (Figure 7A). Furthermore, similar to what was observed in tsc1 mutant clones in the rbf1120a mutant background, the level of ectopic cell death was increased in gig mutant clones generated in rbf1120a mutant eye discs (Figure 7B). Surprisingly, the effects of gig mutations on dE2F1 expression and ectopic cell death are completely suppressed by s6k loss-of-function mutations. We observed that the level of dE2F1 expression in s6k gig double mutant clones is unchanged compared to the control (Figure 7A), and the ectopic cell death observed in rbf1 gig double mutant cells is completely absent in rbf1 gig s6k triple mutant cells (Figure 7B). Moreover, we observed that the basal level of dE2F1-dependent cell death normally present in the rbf1120a mutant eye disc (the stripe of cell death, Figure 1B) is also suppressed (Figure 7B). These results indicate that s6k is required for both the elevation of dE2F1 expression upon TSC inactivation and the increased level of cell death in rbf1 gig double mutant cells. In summary, our genetic studies led us to conclude that TSC1 and TSC2 regulate dE2F1 expression and dE2F1-dependent cell death via the canonical Rheb/Tor/S6k pathway during Drosophila eye development.

Discussion
The loss of Rb leads to hyperactivation of E2F family proteins, which is a crucial event during tumorigenesis. Here, we demonstrate that the Drosophila ortholog of TSC1 tumor suppressor cooperates with RBF1 to regulate dE2F1 activity during development. TSC1 post - transcriptionally regulates dE2F1 expression, and the loss of tsc1 cooperates with rbf1 mutations to promote unscheduled S-phase entry and cell death. This effect of tsc1 mutations on dE2F1 expression requires the components of canonical TSC/Rheb/Tor pathway that are major regulators of cellular growth. Our study provides evidence to suggest that dE2F1 is an important protein that couples growth signals to cell cycle progression.
Recent studies have identified that pro-proliferative and pro-apoptotic activities of dE2F1 are engaged by various Drosophila tumor suppressor genes, such as hippo and archipelago [27], [28]. Our findings add tsc1/2 tumor suppressor genes to this list. Previously, dE2F1 or Cyclin E overexpression is shown to bypass starvation induced G1 arrest at least in endoreduplicating tissues [39]. Moreover, similar to dE2F1, expression of Cyclin E is elevated in tsc1 mutant cells in eye imaginal discs. [36]–[38]. Perhaps, restricting the expression of cell cycle regulators, such as dE2F1 and Cycline E, is a part of the molecular mechanisms by which nutrient deprivation induces G1 arrest. Interestingly, overexpression of dE2F1 or Cycline E does not overcome starvation-induced G1 arrest in larval neuroblasts, indicating that, in mitotic cells, neither dE2F1 nor Cycline E is the limiting factor [39]. Consistent with this observation, we could not observe any appreciable increase in the size of rheb or Tor mutant clones in rbf1 mutant background, suggesting that multiple factors contribute to the proliferative defect observed in rheb or Tor mutant cells in imaginal discs.
Interestingly, despite the elevated level of dE2F1 and Cyclin E, tsc1 mutant clones have relatively normal patterns of BrdU staining at the MF and a limited amount of ectopic cell death. We believe that the activity of dE2F1 in tsc1 mutant cells is normally restricted by the presence of RBF1. The fact that the increase in ectopic S-phase entry and apoptosis by tsc1 mutations can be only observed in the rbf1 mutant background supports this idea. We propose that the TSC/Rheb/Tor pathway during development modulates the amount of dE2F1 needed for cellular division in proportion to the cell size. Supporting this idea, previous studies have demonstrated that tsc1 or tsc2 mutant cells spend less time in G1, a phenotype commonly observed in cells with elevated dE2F1 activity [36]–[38], [40]. It is conceivable that the elevated level of dE2F1 proteins in tsc1 or tsc2 mutant cells allows them to go through G1 to S-phase transition faster where RBF1 is normally inactivated by Cyclin Dependent Kinases (CDKs).
Despite being the only “activator E2F” in Drosophila, it is still unclear how dE2F1 expression is regulated during development. A recent study reported that Cul4(Cdt2) E3 ubiquitin ligase mediates destruction of dE2F1 in S-phase, a mechanism that regulates dE2F1 expression in a cell cycle dependent manner [41]. Our findings here suggest that the expression of dE2F1 is also regulated by a growth-controlling network. However, at this point, we do not know the exact molecular mechanism by which dE2F1 protein level is post - transcriptionally controlled by the TSC/Rheb/Tor pathway. The finding that S6k is involved in this process supports the idea of translational control since S6k directly phosphorylates and regulates proteins involved in translation, such as RpS6, eIF4B, and eEF2K to list a few (reviewed in [42]). However, it is also equally possible that the TSC/Rheb/Tor pathway controls dE2F1 protein stability. In S2 cells, neither tsc1 RNAi nor Rapamycin (Tor inhibitor) treatment in S2 cells had the same effect on dE2F1 expression observed in imaginal discs (Figure S3). It is probable that S2 cells lack factors necessary for dE2F1 regulation that are present in vivo. Nevertheless, it is important to note that this effect on dE2F1 expression is specific since dE2F2 expression is unchanged in tsc1, rheb or Tor mutant cells (Figure 2A and data not shown). Curiously, the requirement of S6k to regulate dE2F1 is limited to the context in which TSC is inactivated. The loss of s6k in the wild type background has no effect on dE2F1 expression while rheb or Tor mutations reduce the level of dE2F1 proteins in the eye disc (Figure 5A and Figure 6). In mammals, it has been demonstrated that the translation of specific mRNA can be mTor-dependent but not S6k - dependent [43]. The molecular mechanism in which S6k promotes dE2F1 expression only when TSC is inactivated is presently unclear and warrants further investigation.
Another interesting finding from our study is that s6k mutations suppress the dE2F1-dependent cell death normally present in rbf1 mutant eye discs (Figure 7). s6k mutations alone did not alter the dE2F1 expression level at least in the wild type background. Although it is not formally tested, this raises a possibility that the TSC/Tor/S6k pathway controls dE2F1-dependent cell death without altering dE2F1 expression. Interestingly, the crosstalk between the InR/Tor and the EGFR signaling pathways during Drosophila eye development has been recently established [44]. InR/Tor signaling regulates the timing of neuronal differentiation in the eye disc by modulating EGFR activity. Since the EGFR pathway is an important determinant of dE2F1-dependent cell death [29], S6k might promote dE2F1-dependent cell death by modulating the EGFR pathway. We speculate that the cooperative effect between tsc1 and rbf1 mutations is the consequence of multiple changes that include the increase in dE2F1 expression.
In cancer cells, it is generally thought that the loss of Rb function is the most common mechanism of deregulating E2F activity. However, in some types of cancers, amplification of E2F genes or overexpression of E2F family proteins have been observed (reviewed in [23]). Moreover, in a subtype of human retinoblastoma where Rb is already deficient, E2f-3 proteins are also overexpressed [45]. These observations suggest that E2F family genes themselves can be directly targeted and deregulated during tumorigenesis. It will be interesting to investigate if TSC1/2 or other tumor suppressors and oncogenes regulate the expression of E2F family proteins to promote tumorigenesis.
Materials and Methods
Fly stocks
All crosses have been performed at 25°C. The rbf1 mutant allele, rbf1120a, and de2f1 allele, de2f1729, are described previously [15], [16]. The tsc1 alleles used in this study are tsc1R453X, a gift from Dr. Hariharan [38], and tsc1f01910 (Exelixis collection, Harvard Medical School). The mutant alleles of the TSC/Rheb/Tor pathway used in this study are as follows: Tor2L19 FRT40A and 4ebpnull are gifts from P. Lasko [46], [47]. s6k l-1 FRT80B is a gift from D.J. Pan [48]. The gig56 FRT80B, FRT82B rheb2D1, and s6kl-1 gig192FRT80B alleles were kindly provided by J.M. Bateman [44]. The 4ebpnull FRT40A, FRT82B de2f1729 tsc1f01910, and FRT82B rheb2D1 tsc1R453X alleles were generated by meiotic recombination. For the double mutant alleles, presence of both mutations is verified by genetic complementation tests using multiple mutant alleles. For example, presence of both s6k and gig mutations in s6k l-1gig192 FRT80B alleles were verified by crossing the alleles to gig52, gig192, s6k l-1 and s6kp{PZ}07084 alleles individually.
Clonal analysis
Flippase (FLP) was expressed from the eyeless promoter to generate mitotic clones in the eye. To examine clones in rbf1 mutant animals, the X chromosome carrying rbf1120a and an ey-FLP transgene was used. Followings are the full genotypes of larvae analysed.
Mutant clones in the wild-type background
y w eyFlp/+ or Y; FRT82B GFPubi/FRT82B tsc1R453X
y w eyFlp/+ or Y; FRT82B GFPubi/FRT82B rheb2D1
y w eyFlp/+ or Y; FRT82B GFPubi/FRT82B rheb2D1 tsc1R453X
y w eyFlp/+ or Y; GFPubi FRT40A/Tor2L19 FRT40A
y w eyFlp/+ or Y; GFPubi FRT80B/s6k l-1 FRT80B
y w eyFlp/+ or Y; GFPubi FRT40A/4ebpnull FRT40A
y w eyFlp/+ or Y; GFPubi FRT80B/gig56 FRT80B
y w eyFlp/+ or Y; GFPubi FRT80B/s6kl-1 gig192 FRT80B
Mutant clones in the rbf1120a background
w rbf1120a eyFlp/Y; FRT82B GFPubi/FRT82B tsc1R453X
w rbf1120a eyFlp/Y; FRT82B GFPubi/FRT82B tsc1f01910
w rbf1120a eyFlp/Y; FRT82B GFPubi/FRT82B de2f1729 tsc1f01910
w rbf1120a eyFlp/Y; FRT82B GFPubi/FRT82B rheb2D1
w rbf1120a eyFlp/Y; FRT82B GFPubi/FRT82B rheb2D1 tsc1R453X
w rbf1120a eyFlp/Y; GFPubi FRT80B/gig56 FRT80B
w rbf1120a eyFlp/Y; GFPubi FRT80B/s6kl-1 gig192 FRT80B
Immunoblot, real-time quantitative PCR, and in situ hybridization
y w eyFlp/Y; FRT82B [W+] l(3)cl-R3/FRT82B (controls)
y w eyFlp/Y; FRT82B [W+] l(3)cl-R3/FRT82B tsc1R453X
w rbf1120a eyFlp/Y; [W+] l(3)cl-R3/FRT82B
w rbf1120a eyFlp/Y; [W+] l(3)cl-R3/FRT82B tsc1R453X
PCNA-GFP in tsc1 mutant clones
y w eyFlp/PCNA-GFP; FRT82B LacZarm/FRT82B tsc1R453X
Immunostaining and microscopy
The antibodies used in this study are: anti-dE2F1 (1/1000) [29], anti-dE2F2 (1/1000) [34], anti-RBF1 (1/100) from Dyson Lab, anti-C3 (1/200, Cell Signaling), anti-GFP-FITC (1/200, abcam), anti-β-galactosidase (Developmental Studies Hybridoma Banks [DSHB]), and anti - ELAV (DSHB). For immunostaining, third-instar eye discs were fixed in 4% formaldehyde for 20 minutes at room temperature (eye discs immunostained for anti-dE2F1 were fixed at 4°C for 30 minutes) and washed twice with 0.3% PBST (0.3% Triton X-100 in PBS) and once with 0.1% PBST (0.1% Triton X-100 in PBS). Fixed eye discs were incubated in primary antibody with 0.1% PBST and 5% normal goat serum (NGS) at room temperature for 3 hours. After four washes with 0.1% PBST, eye discs were incubated in secondary antibody with 0.3% PBST and 5% NGS at room temperature for 2 hours. Immunostained eye discs were then washed five times with 0.1% PBST at room temperature and mounted for confocal microscopy imaging (Zeiss LSM).
In situ hybridization
For in situ hybridization experiments, eye-antennal discs were prepared as described previously [26]. Anti-sense RNA probes were generated using cDNA clones LD41588, LD17578, and LD45889 for rnrS, CycE, and PCNA respectively. After hybridization, Alkaline Phosphatase conjugated anti-DIG antibodies were used to detect DIG labeled anti - sense RNA probes. For each target genes, more than 20 eye antennal discs were analyzed and the representative images were chosen to be presented.
Immunoblotting
40 eye discs of tsc1 mutant and control animals were dissected and used for Western blot as previously described [29].
Real-Time Reverse Transcriptase PCR
The average of three independent experiments of triplicate-PCR reaction is presented. Total RNA was isolated from 40 eye-antenna eye discs with RNeasy Mini kit (QIAGEN) according to manufacturer's protocol, and reverse transcribed using DyNAmo cDNA Synthesis Kit (Finnzymes) according to manufacturer's instructions. Quantitative PCR reactions were performed with DyNAmo Flash SYBR Green qPCR Kit (Finnzymes). Quantification was determined by comparative threshold cycle method (CT) on Bio-Rad CFX Manager software. Both rp49 and β-tubulin were used as normalization controls in a single experiment. All primers were designed with Primer3 (Whitehead Institute fozr Biomedical Research primer3 shareware [http://frodo.wi.mit.edu/primer3/]). Primer pairs used are:
chrb1F (AACTGCAGGCTCAGCTACG)
chrb1R (CGCTCTCGAACTCAATGAAG)
de2f12-3F (CAGCACCACCACCAAAATC)
de2f12-3R (ACTGCTAGCCGTATGCTTCTG)
de2f15-6F (TACAGCCATGACCGCAAC)
de2f15-6R (GTTCAGCGCATACGGATAGTC)
tubulin-F (ACATCCCGCCCCGTGGTC)
tubulin-R (AGAAAGCCTTGCGCCTGAACATAG)
Rp49-F (TACAGGCCCAAGATCGTGAAG)
Rp49-R (GACGCACTCTGTTGTCGATACC)
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. DysonN
1998 The regulation of E2F by pRB-family proteins. Genes Dev 12 2245 2262
2. PolagerS
GinsbergD
2008 E2F - at the crossroads of life and death. Trends Cell Biol 18 528 535
3. van den HeuvelS
DysonNJ
2008 Conserved functions of the pRB and E2F families. Nat Rev Mol Cell Biol 9 713 724
4. MullerH
BrackenAP
VernellR
MoroniMC
ChristiansF
2001 E2Fs regulate the expression of genes involved in differentiation, development, proliferation, and apoptosis. Genes Dev 15 267 285
5. DimovaDK
StevauxO
FrolovMV
DysonNJ
2003 Cell cycle-dependent and cell cycle - independent control of transcription by the Drosophila E2F/RB pathway. Genes Dev 17 2308 2320
6. IshidaS
HuangE
ZuzanH
SpangR
LeoneG
2001 Role for E2F in control of both DNA replication and mitotic functions as revealed from DNA microarray analysis. Mol Cell Biol 21 4684 4699
7. JacksT
FazeliA
SchmittEM
BronsonRT
GoodellMA
1992 Effects of an Rb mutation in the mouse. Nature 359 295 300
8. ClarkeAR
MaandagER
van RoonM
van der LugtNM
van der ValkM
1992 Requirement for a functional Rb-1 gene in murine development. Nature 359 328 330
9. LeeEY
ChangCY
HuN
WangYC
LaiCC
1992 Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature 359 288 294
10. TsaiKY
HuY
MacleodKF
CrowleyD
YamasakiL
1998 Mutation of E2f-1 suppresses apoptosis and inappropriate S phase entry and extends survival of Rb - deficient mouse embryos. Mol Cell 2 293 304
11. ZieboldU
RezaT
CaronA
LeesJA
2001 E2F3 contributes both to the inappropriate proliferation and to the apoptosis arising in Rb mutant embryos. Genes Dev 15 386 391
12. SherrCJ
1996 Cancer cell cycles. Science 274 1672 1677
13. HarrisonDJ
HooperML
ArmstrongJF
ClarkeAR
1995 Effects of heterozygosity for the Rb-1t19neo allele in the mouse. Oncogene 10 1615 1620
14. HuN
GutsmannA
HerbertDC
BradleyA
LeeWH
1994 Heterozygous Rb-1 delta 20/+mice are predisposed to tumors of the pituitary gland with a nearly complete penetrance. Oncogene 9 1021 1027
15. DuW
DysonN
1999 The role of RBF in the introduction of G1 regulation during Drosophila embryogenesis. EMBO J 18 916 925
16. DuronioRJ
O'FarrellPH
XieJE
BrookA
DysonN
1995 The transcription factor E2F is required for S phase during Drosophila embryogenesis. Genes Dev 9 1445 1455
17. MacPhersonD
SageJ
KimT
HoD
McLaughlinME
2004 Cell type-specific effects of Rb deletion in the murine retina. Genes Dev 18 1681 1694
18. Robanus-MaandagE
DekkerM
van der ValkM
CarrozzaML
JeannyJC
1998 p107 is a suppressor of retinoblastoma development in pRb-deficient mice. Genes Dev 12 1599 1609
19. ChenD
Livne-barI
VanderluitJL
SlackRS
AgochiyaM
2004 Cell-specific effects of RB or RB/p107 loss on retinal development implicate an intrinsically death - resistant cell-of-origin in retinoblastoma. Cancer Cell 5 539 551
20. ZhangJ
SchweersB
DyerMA
2004 The first knockout mouse model of retinoblastoma. Cell Cycle 3 952 959
21. YamasakiL
BronsonR
WilliamsBO
DysonNJ
HarlowE
1998 Loss of E2F-1 reduces tumorigenesis and extends the lifespan of Rb1(+/−)mice. Nat Genet 18 360 364
22. ZieboldU
LeeEY
BronsonRT
LeesJA
2003 E2F3 loss has opposing effects on different pRB-deficient tumors, resulting in suppression of pituitary tumors but metastasis of medullary thyroid carcinomas. Mol Cell Biol 23 6542 6552
23. ChenHZ
TsaiSY
LeoneG
2009 Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer 9 785 797
24. StevauxO
DysonNJ
2002 A revised picture of the E2F transcriptional network and RB function. Curr Opin Cell Biol 14 684 691
25. TsaiSY
OpavskyR
SharmaN
WuL
NaiduS
2008 Mouse development with a single E2F activator. Nature 454 1137 1141
26. DuW
2000 Suppression of the rbf null mutants by a de2f1 allele that lacks transactivation domain. Development 127 367 379
27. NicolayBN
FrolovMV
2008 Context-dependent requirement for dE2F during oncogenic proliferation. PLoS Genet 4 e1000205 doi:10.1371/journal.pgen.1000205
28. NicholsonSC
GilbertMM
NicolayBN
FrolovMV
MobergKH
2009 The archipelago tumor suppressor gene limits rb/e2f-regulated apoptosis in developing Drosophila tissues. Curr Biol 19 1503 1510
29. MoonNS
Di StefanoL
DysonN
2006 A gradient of epidermal growth factor receptor signaling determines the sensitivity of rbf1 mutant cells to E2F-dependent apoptosis. Mol Cell Biol 26 7601 7615
30. MoonNS
Di StefanoL
MorrisEJ
PatelR
WhiteK
2008 E2F and p53 induce apoptosis independently during Drosophila development but intersect in the context of DNA damage. PLoS Genet 4 e1000153 doi:10.1371/journal.pgen.1000153
31. InokiK
CorradettiMN
GuanKL
2005 Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet 37 19 24
32. PanD
DongJ
ZhangY
GaoX
2004 Tuberous sclerosis complex: from Drosophila to human disease. Trends Cell Biol 14 78 85
33. HarveyKF
MattilaJ
SoferA
BennettFC
RamseyMR
2008 FOXO-regulated transcription restricts overgrowth of Tsc mutant organs. J Cell Biol 180 691 696
34. FrolovMV
MoonNS
DysonNJ
2005 dDP is needed for normal cell proliferation. Mol Cell Biol 25 3027 3039
35. Tanaka-MatakatsuM
XuJ
ChengL
DuW
2009 Regulation of apoptosis of rbf mutant cells during Drosophila development. Dev Biol 326 347 356
36. GaoX
PanD
2001 TSC1 and TSC2 tumor suppressors antagonize insulin signaling in cell growth. Genes Dev 15 1383 1392
37. PotterCJ
HuangH
XuT
2001 Drosophila Tsc1 functions with Tsc2 to antagonize insulin signaling in regulating cell growth, cell proliferation, and organ size. Cell 105 357 368
38. TaponN
ItoN
DicksonBJ
TreismanJE
HariharanIK
2001 The Drosophila tuberous sclerosis complex gene homologs restrict cell growth and cell proliferation. Cell 105 345 355
39. BrittonJS
EdgarBA
1998 Environmental control of the cell cycle in Drosophila: nutrition activates mitotic and endoreplicative cells by distinct mechanisms. Development 125 2149 2158
40. NeufeldTP
de la CruzAF
JohnstonLA
EdgarBA
1998 Coordination of growth and cell division in the Drosophila wing. Cell 93 1183 1193
41. ShibutaniST
de la CruzAF
TranV
TurbyfillWJ3rd
ReisT
2008 Intrinsic negative cell cycle regulation provided by PIP box - and Cul4Cdt2-mediated destruction of E2f1 during S phase. Dev Cell 15 890 900
42. RuvinskyI
MeyuhasO
2006 Ribosomal protein S6 phosphorylation: from protein synthesis to cell size. Trends Biochem Sci 31 342 348
43. PendeM
UmSH
MieuletV
StickerM
GossVL
2004 S6K1(−/−)/S6K2(−/−) mice exhibit perinatal lethality and rapamycin-sensitive 5'-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol Cell Biol 24 3112 3124
44. McNeillH
CraigGM
BatemanJM
2008 Regulation of neurogenesis and epidermal growth factor receptor signaling by the insulin receptor/target of rapamycin pathway in Drosophila. Genetics 179 843 853
45. OrlicM
SpencerCE
WangL
GallieBL
2006 Expression analysis of 6p22 genomic gain in retinoblastoma. Genes Chromosomes Cancer 45 72 82
46. OldhamS
MontagneJ
RadimerskiT
ThomasG
HafenE
2000 Genetic and biochemical characterization of dTOR, the Drosophila homolog of the target of rapamycin. Genes Dev 14 2689 2694
47. TettweilerG
MironM
JenkinsM
SonenbergN
LaskoPF
2005 Starvation and oxidative stress resistance in Drosophila are mediated through the eIF4E-binding protein, d4E-BP. Genes Dev 19 1840 1843
48. GaoX
ZhangY
ArrazolaP
HinoO
KobayashiT
2002 Tsc tumour suppressor proteins antagonize amino-acid-TOR signalling. Nat Cell Biol 4 699 704
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 8
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Identification of the Bovine Arachnomelia Mutation by Massively Parallel Sequencing Implicates Sulfite Oxidase (SUOX) in Bone Development
- Common Inherited Variation in Mitochondrial Genes Is Not Enriched for Associations with Type 2 Diabetes or Related Glycemic Traits
- A Model for Damage Load and Its Implications for the Evolution of Bacterial Aging
- Did Genetic Drift Drive Increases in Genome Complexity?
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy