
Inactivation of VCP/ter94 Suppresses Retinal Pathology Caused by Misfolded Rhodopsin in
The most common Rhodopsin (Rh) mutation associated with autosomal dominant retinitis pigmentosa (ADRP) in North America is the substitution of proline 23 by histidine (RhP23H). Unlike the wild-type Rh, mutant RhP23H exhibits folding defects and forms intracellular aggregates. The mechanisms responsible for the recognition and clearance of misfolded RhP23H and their relevance to photoreceptor neuron (PN) degeneration are poorly understood. Folding-deficient membrane proteins are subjected to Endoplasmic Reticulum (ER) quality control, and we have recently shown that RhP23H is a substrate of the ER–associated degradation (ERAD) effector VCP/ter94, a chaperone that extracts misfolded proteins from the ER (a process called retrotranslocation) and facilitates their proteasomal degradation. Here, we used Drosophila, in which Rh1P37H (the equivalent of mammalian RhP23H) is expressed in PNs, and found that the endogenous Rh1 is required for Rh1P37H toxicity. Genetic inactivation of VCP increased the levels of misfolded Rh1P37H and further activated the Ire1/Xbp1 ER stress pathway in the Rh1P37H retina. Despite this, Rh1P37H flies with decreased VCP function displayed a potent suppression of retinal degeneration and blindness, indicating that VCP activity promotes neurodegeneration in the Rh1P37H retina. Pharmacological treatment of Rh1P37H flies with the VCP/ERAD inhibitor Eeyarestatin I or with the proteasome inhibitor MG132 also led to a strong suppression of retinal degeneration. Collectively, our findings raise the possibility that excessive retrotranslocation and/or degradation of visual pigment is a primary cause of PN degeneration.
Published in the journal:
. PLoS Genet 6(8): e32767. doi:10.1371/journal.pgen.1001075
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001075
Summary
The most common Rhodopsin (Rh) mutation associated with autosomal dominant retinitis pigmentosa (ADRP) in North America is the substitution of proline 23 by histidine (RhP23H). Unlike the wild-type Rh, mutant RhP23H exhibits folding defects and forms intracellular aggregates. The mechanisms responsible for the recognition and clearance of misfolded RhP23H and their relevance to photoreceptor neuron (PN) degeneration are poorly understood. Folding-deficient membrane proteins are subjected to Endoplasmic Reticulum (ER) quality control, and we have recently shown that RhP23H is a substrate of the ER–associated degradation (ERAD) effector VCP/ter94, a chaperone that extracts misfolded proteins from the ER (a process called retrotranslocation) and facilitates their proteasomal degradation. Here, we used Drosophila, in which Rh1P37H (the equivalent of mammalian RhP23H) is expressed in PNs, and found that the endogenous Rh1 is required for Rh1P37H toxicity. Genetic inactivation of VCP increased the levels of misfolded Rh1P37H and further activated the Ire1/Xbp1 ER stress pathway in the Rh1P37H retina. Despite this, Rh1P37H flies with decreased VCP function displayed a potent suppression of retinal degeneration and blindness, indicating that VCP activity promotes neurodegeneration in the Rh1P37H retina. Pharmacological treatment of Rh1P37H flies with the VCP/ERAD inhibitor Eeyarestatin I or with the proteasome inhibitor MG132 also led to a strong suppression of retinal degeneration. Collectively, our findings raise the possibility that excessive retrotranslocation and/or degradation of visual pigment is a primary cause of PN degeneration.
Introduction
Collapse of protein homeostasis (proteostasis) due to protein misfolding and aggregation is the central pathogenic event in neurodegenerative disease [1]. Retinitis pigmentosa (RP) represents a group of disorders characterized by progressive loss of photoreceptor neurons (PNs) and blindness [2]–[4], and mutations in the visual pigment Rhodopsin (Rh) are the most prevalent genetic defects causing RP [5], [6]. Rh is a G protein-coupled receptor that initiates the phototransduction cascade and more than 120 Rh point mutations have been associated with RP; most of these Rh mutations act dominantly to cause autosomal dominant RP (ADRP), while some mutations cause recessive RP [7]. Based on their biochemical and cellular properties, Rh mutations have been grouped into several classes [7]. Substitution of proline 23 by histidine (RhP23H) is the most common genetic defect associated with ADRP in North America and is arguably the best characterized Rh mutation to date [3], [7], [8]. Unlike wild-type (WT) Rh, which is properly delivered to the plasma membrane, mutant RhP23H fails to fold properly [3], [5], [9], [10], appears to exhibit enhanced retention within the endoplasmic reticulum (ER) [3], [5], [7], [11]–[13] and forms intracellular aggregates [14]–[16].
The presence of misfolded proteins in the ER activates the unfolded protein response (UPR), an adaptive process which helps reduce the load of unfolded proteins; chronic and excessive UPR activation might be pro-apoptotic [17]–[19], although a moderate increase in UPR activation (via the Ire1/Xbp1 pathway) is protective for PNs [20]. In order to reduce protein misfolding-induced stress within the ER, proteins with folding defects are identified and cleared during a process called ER-associated degradation (ERAD), which involves their export from the ER to the cytosol (retrotranslocation) and degradation by the proteasome [21], [22]. The ATP-dependent chaperone Valosin-containing protein VCP/ter94/p97/cdc48 is the driving force for the retrotranslocation of misfolded proteins [23]–[25]. Two ATPase domains (D1 and D2) generate the energy necessary for VCP to promote extraction of misfolded substrates from the ER and delivery to the proteasome [26], [27]. Increased ERAD/VCP activity leads to degradation of the cystic fibrosis-linked mutant ΔF508-CFTR, which retains some functionality despite being misfolded [28]. VCP also promotes degradation of the human V2 vasopressin receptor in X-linked nephrogenic diabetes insipidus [29]. VCP co-localizes with aggregated Ataxin-3 [30], TDP-43 [31] or with ubiquitinated inclusions in Alzheimer's and Parkinson's disease [32] suggesting that modulation of VCP activity might have a broad relevance for protein clearance (including neurodegenerative) disorders.
The mechanisms linking mutant RhP23H to PN degeneration in ADRP are incompletely understood. RhP23H and other misfolded Rh mutants were found to recruit the endogenous WT Rh into aggregates (dominant-negative [DN] effect) leading to decreased levels of mature WT Rh [15], [33]–[36]. The mutant RhP23H might acquire novel properties (gain-of-function [GOF] mechanism) such as: aggregate formation or inclusion formation in the cytosol; generation of ER stress and activation of the UPR pathways; other toxic effects on diverse cellular processes [7], [11], [15], [18], [37]. Therefore, a better understanding of the cellular and molecular mechanisms mediating dominance in RhP23H-linked RP is required for the development of effective therapies [7].
We have recently shown that misfolded RhP23H forms a complex with the ERAD effector VCP in mammalian cell culture systems. The interaction is maximal when both the N-terminal and the D1 ATPase domains of VCP are present; furthermore, VCP uses its D2 ATPase domain to promote RhP23H retrotranslocation and proteasomal delivery [16]. It remained unclear, however, how the activity of VCP impacts on the RhP23H-mediated retinal degeneration and blindness.
To address this question, we have used the previously established Drosophila model of RhP23H-linked RP, in which overexpression of Rh1P37H (the equivalent of mammalian RhP23H) in photoreceptor neurons leads to age - and light-dependent retinal degeneration and blindness [11]. Here, we show that the degeneration of PNs requires the presence of both mutant Rh1P37H and endogenous WT Rh1. Genetic inactivation of the ERAD retrotranslocator VCP increased the levels of misfolded Rh1P37H but surprisingly, conferred protection against Rh1P37H-mediated retinal degeneration. Pharmacological inactivation of VCP/ERAD or proteasome activities also led to a robust suppression of Rh1P37H-mediated neurodegeneration. Our results suggest that excessive retrotranslocation and/or proteasomal degradation of visual pigment is pathogenic for PNs.
Results
Proteostasis Defects and Retinal Degeneration in Rh1P37H-Expressing Flies
To understand how targeting and clearance of misfolded Rh impact on the maintenance of PNs in RP, we used a previously established Drosophila RP model, in which mutant Rh1P37H (the equivalent of mammalian RhP23H) is ectopically expressed in PNs R1-6, under the control of a promoter identical to the endogenous Rh1 promoter [11]. Flies overexpressing mutant Rh1P37H (genotype: Rh1P37H;Rh1+/+), in contrast to flies overexpressing WT Rh1 (genotype: Rh1WT;Rh1+/+) and to control (Rh1-Gal4) flies display a dramatic loss of PNs after 20 and 30 days of light exposure (dle) (Figure 1A–1D). After 30 dle, ommatidia from Rh1P37H;Rh1+/+ retinas displayed 1.7 photoreceptors on average, compared to 5.5 in Rh1WT;Rh1+/+ and 6.5 in Rh1-Gal4 flies (n>7 eyes/genotype and at least 150 ommatidia were analyzed/eye; ** p<0.01 and *** p<0.001 student's t-test). No degeneration was seen in Rh1P37H;Rh1+/+ flies reared at light at postnatal day 1 (P1) or reared in the dark at P30 (Figure S1). Thus, Rh1P37H induces age - and light-dependent retinal degeneration in Drosophila PNs.

In order to characterize the misfolded Rh1-induced proteostasis defects in our transgenic flies, we collected fly heads of defined genotypes and ages and following lysis in detergent-containing RIPA modified buffer (see Materials and Methods), we separated the lysates into detergent-soluble and insoluble fractions. We expected most of the mature (rhabdomeric) Rh1 to be contained within the detergent-soluble fractions, while aggregation-prone Rh1 should be enriched in the detergent-insoluble fraction [14], [15]. To specifically detect the overexpressed protein, we used flies in which ectopically expressed Rh1P37H (or Rh1WT) was hsv-tagged [11], [35]; in the following, all Rh1P37H-hsv;Rh1+/+ and Rh1WT-hsv;Rh1+/+ flies are named Rh1P37H;Rh1+/+ and Rh1WT;Rh1+/+ respectively, unless otherwise stated. When analyzing detergent-soluble fractions, we found that 1-day-old Rh1WT;Rh1+/+ flies displayed 55±7% more Rh1 than Rh1-Gal4 flies, both when reared in the dark or at light (Figure S2A and Figure S3A and S3B) indicating that the Rh1 promoter induces a moderate transgene expression (also seen by [11]). Similar to Rh1WT-expressing flies, 1-day-old Rh1P37H;Rh1+/+ flies displayed 51±8% more Rh1 than Rh1-Gal4 flies when reared in the dark (Figure S3B); interestingly, the same flies reared at light displayed only 9±1% more Rh1 than Rh1-Gal4 flies (Figure S3A). Moreover, light exposure (in contrast to dark rearing) led to loss of the hsv signal in Rh1P37H;Rh1+/+ but not Rh1WT;Rh1+/+ flies (Figure S3C), suggesting that light exposure exacerbates the Rh1P37H maturation defects. We also evaluated Rh1P37H transgene expression on a Rh1 null background. Flies expressing one copy of the Rh1P37H transgene on a complete Rh1 null background (Rh1P37H;Rh1−/−), or two copies of the mutant transgene on a complete Rh1 null background (Rh1P37H/Rh1P37H;Rh1−/−) had significantly higher levels of mature Rh1P37H when reared at light; Rh1 labeling revealed that Rh1P37H/Rh1P37H;Rh1−/− flies (in which all Rh1 is Rh1P37H) exhibited levels of mature Rh1P37H that were comparable to those of the endogenous Rh1 in Rh1P37H;Rh1+/+ flies (Figure S3D), reflecting the increased dosage of the Rh1P37H transgene. This suggests that the mutant Rh1P37H is able to traffic through the secretory pathway and this process is impaired by light exposure.
We then performed a time-course analysis of Rh1 levels in our transgenic and control flies exposed to light and found a dramatic loss of mature Rh1 (endogenous and ectopic) in Rh1P37H;Rh1+/+ but not Rh1WT;Rh1+/+ flies after 20 dle (Figure 1E). We used an hsv-specific antibody to label the ectopic protein and found a complete loss of hsv signal in the Rh1P37H;Rh1+/+ soluble fraction after 10 dle, while no loss of mature Rh1 occurred in Rh1WT;Rh1+/+ flies. As mentioned above, this loss of hsv signal might be due to Rh1P37H maturation defects, and/or to excessive Rh1P37H degradation. We next assessed the situation of Rh1-containing species in the detergent-insoluble fraction. We found a significant increase in Rh1-containing oligomeric species that migrated at approximately 250–300 kDa, in both Rh1WT;Rh1+/+ and Rh1P37H;Rh1+/+ flies after 20 dle (Figure 1F; whole WB scans can be seen in Figure S4). These species represent small Rh1 oligomers (probably containing 6–10 Rh1 molecules) that were extracted using our detergent treatment protocol (and supposedly found as such in vivo), although they might also have assembled ex-vivo [14], [38]. Control (Rh1-Gal4) retinas displayed steady levels of mature Rh1 in the detergent-soluble fraction, while some traces of oligomeric Rh1 were detected in the detergent-insoluble fraction (Figure S2). Since RhP23H-containing oligomeric species were previously found to be ubiquitinated [14], [36], we used an Ubiquitin-specific antibody to label Rh1-containing oligomeric species in Drosophila, and found a robust signal after 20 dle in Rh1P37H;Rh1+/+ flies (Figure 1F). This suggests that Rh1-containing oligomers are ubiquitinated, although it remains possible that other (Rh1-negative) species are also Ubiquitin-positive. Remarkably, hsv labeling was absent in the insoluble fraction from Rh1P37H;Rh1+/+, but not Rh1WT;Rh1+/+ flies, raising the possibility that endogenous Rh1 is the major component of these Rh1 oligomeric species from the Rh1P37H retina (Figure 1F). It remains, however, possible that the lack of hsv signal is due to epitope unavailability (due to oligomerization) or that the hsv tag was cleaved. Accumulation of endogenous Rh1 in the insoluble fraction from the Rh1P37H;Rh1+/+ retina might be a consequence of WT Rh1 recruitment by the mutant Rh1P37H [15], [33]–[36]. To determine whether the loss of mature Rh1 is the cause or the consequence of retinal degeneration in Rh1P37H;Rh1+/+ flies, we compared the time-dependent decrease in mature (rhabdomeric) Rh1 levels, the decrease of the rhabdomeric marker TRP [39] levels and the percentage of surviving PNs in the Rh1P37H;Rh1+/+ retina (Figure 1G; results from 3 independent crosses were averaged). As expected, death of PNs parallels the decrease in TRP levels, consistent with the rhabdomeres being lost during retinal degeneration; moreover, we found that no loss of mature Rh1 precedes the loss of PNs/TRP (Figure 1G). Therefore, the onset of retinal degeneration in Rh1P37H-expressing flies is not caused by loss of mature Rh1; the reverse is true, i.e. loss of mature Rh1 is the result of rhabdomere loss, which takes place during retinal degeneration. We also used INAD as an independent rhabdomeric marker and found a similar profile to TRP (data not shown). Furthermore, given the similar amounts of Rh1 oligomers in Rh1WT;Rh1+/+ and Rh1P37H;Rh1+/+ flies – despite marked differences in retinal integrity – the formation of Rh1 oligomers is probably not a primary cause of retinal degeneration in Rh1P37H;Rh1+/+ flies.
The Endogenous Rh1 Is Required for Rh1P37H Toxicity
The recruitment of endogenous Rh1 into insoluble aggregates prompted us to investigate the relevance of endogenous Rh1 to the retinal pathology initiated by Rh1P37H. To reduce the dosage of the endogenous Rh1 in Rh1P37H;Rh1+/+ flies, we used the Rh1 null allele ninaEI17 (here called Rh1−). We found, remarkably, that PN degeneration was strongly suppressed in Rh1P37H;Rh1+/− flies (Figure 2A–2C; n>7 eyes/genotype), suggesting that the endogenous Rh1 is required for Rh1P37H toxicity. Consistent with the rescue of PN degeneration, the levels of TRP and of total mature Rh1, but not P37H ectopic Rh1 (hsv blot), were partially (51±5% and 59±6,5% respectively) restored (Figure 2D), indicating that rescue of retinal degeneration in Rh1P37H;Rh1+/− flies correlated with decreased degradation of endogenous Rh1. Interestingly, increased dosage of the mutant Rh1P37H transgene relative to endogenous Rh1 led to an increased level of Rh1 oligomers in the detergent-insoluble fraction (that contained a large fraction of endogenous Rh1; Figure 2E, compare Rh1 and hsv blots) in Rh1P37H;Rh1+/− flies. We then wondered whether the mutant Rh1P37H is toxic by itself, i.e. if the presence of Rh1P37H in PNs completely deprived of endogenous Rh1 is sufficient to cause a strong PN degeneration. For this, we generated Rh1P37H;Rh1−/− flies (carrying one copy of the Rh1P37H transgene on a Rh1 null background) and Rh1P37H/Rh1P37H;Rh1−/− flies (carrying two copies of the Rh1P37H transgene on a Rh1 null background). As mentioned above, Rh1P37H maturation was significantly improved in these mutants, (Figure S3D). When analyzing the retinal integrity after 25 dle, we found, remarkably, that PN degeneration was largely suppressed in Rh1P37H;Rh1−/− and Rh1P37H/Rh1P37H;Rh1−/− flies (Figure 2F–2I; n = 7 eyes/genotype). Analysis of Rh1 levels at 25 dle also confirmed the rescue of retinal degeneration (Figure S5). Therefore, the Rh1P37H allele is not toxic by itself, as it is insufficient to cause retinal degeneration when expressed in the absence of the endogenous Rh1. Retinal degeneration in Rh1P37H;Rh1+/+ flies therefore depends on a cellular environment containing both misfolded Rh1P37H and endogenous WT Rh1.

VCP Is Required In Vivo for Clearance of Misfolded Rh1P37H
To investigate the relevance of Rh1P37H retrotranslocation and clearance to retinal degeneration, we focused our attention on the ERAD effector VCP/ter94/p97/cdc48, the driving force for extraction of misfolded proteins from the ER and delivery to the proteasome [23], [24], [27]. To test whether VCP activity is required for clearance of misfolded Rh1, we reduced VCP function using the hypomorphic allele ter9426-8 [40] that we refer to as VCP26-8 (Figure S6A). Using Rh1 - and Ubiquitin-specific antibodies to label oligomeric species, we found that Rh1P37H;VCP26-8/+;Rh1+/+ flies, in contrast to Rh1P37H;Rh1+/+ and control flies, displayed a strong increase in total levels of oligomeric Rh1 at 10 dle (Figure 3A) and similarly at 20 (Figure S6B) and 30 dle (Figure 3B). Flies carrying the VCP26-8 allele in an otherwise WT background (genotype: VCP26-8/+;Rh1+/+) also displayed slightly more oligomeric Rh1 relative to control flies (Figure 3A and 3B) suggesting that VCP is involved in the quality control of endogenous Rh1, which might be a client of ERAD under physiological conditions [41]. When using hsv antibody to label the ectopic Rh1P37H, we found an increased signal in the Rh1P37H;VCP26-8/+;Rh1+/+ retina as compared to the Rh1P37H;Rh1+/+ retina (Figure 3C and 3D and Figure S6C). Thus, partial VCP inactivation prevents the loss of misfolded Rh1P37H. Importantly, this finding also indicates that we are able to detect oligomeric species containing the mutant Rh1P37H. Therefore, the absence of Rh1P37H from the insoluble fractions of Rh1P37H;Rh1+/+ flies (Figure 1F) suggests that the mutant Rh1P37H was degraded, in a process that required VCP activity. Since the increase in Rh1-containing oligomeric species was observed before the onset of retinal degeneration (at 10 dle), we can rule out that this effect was indirectly caused by differences in retinal integrity. Thus, VCP activity is required in vivo for degradation of misfolded Rh1P37H.

To determine the levels of mature (soluble) Rh1 in retinas derived from Rh1P37H;VCP26-8/+;Rh1+/+, Rh1P37H;Rh1+/+ or control flies, we used detergent-soluble fractions and a Rh1-specific antibody to reveal their content of mature Rh1. While the levels of mature total (endogenous and ectopic) Rh1 were similar for all groups after 10 dle (Figure 3E), we found a dramatic loss of mature Rh1 in Rh1P37H;Rh1+/+ flies after 20 dle (Figure S6D) and 30 dle (Figure 3F and 3I; see also Figure 1E). Remarkably, partial VCP inactivation led to an almost complete rescue of mature Rh1 levels (Figure 3F and 3I; results from 3 independent crosses were averaged) and similarly, restored the levels of the rhabdomeric marker TRP (Figure 3F and 3J), suggesting decreased retinal degeneration in the Rh1P37H;VCP26-8/+;Rh1+/+ retina after 30 dle. We observed a similar, although moderate, rescue of the mature ectopic Rh1P37H (hsv-labeled) after 20 dle (Figure S6E) and 30 dle (Figure 3H), but also after 10 dle (Figure 3G) and 1 dle (data not shown). The increased levels of mature Rh1P37H in the intact (1–10 dle) Rh1P37H;VCP26-8/+;Rh1+/+ retina suggests that partial VCP inactivation not only prevents Rh1P37H degradation but also allows some of the mutant Rh1P37H to traffic through the secretory pathway.
Increased Ire1/Xbp1 Pathway Activation in Rh1P37H-Expressing Flies with Decreased VCP Function
Accumulation of misfolded proteins in the ER activates the UPR; the unconventional splicing of xbp1 mRNA is favored during the UPR, generating a transcription factor that activates stress response genes, including the ER stress sensor and chaperone Hsc3/BiP [37], [42], [43]. We tested for the occurrence of xbp1 mRNA unconventional splicing by inducing ubiquitous expression of an UAS-xbp1-EGFP construct, which activates EGFP expression only after unconventional splicing [37]. We stained retinas using a GFP-specific antibody [20], [37] and determined the density of GFP-positive profiles for the different genotypes. We found that Xbp1-EGFP activation is increased in Rh1P37H;Rh1+/+ retinas relative to Rh1WT;Rh1+/+ and control retinas (Figure 4A–4C and 4G). Rh1P37H;Rh1+/+ flies carrying VCP26-8, Rh1− or the stronger VCP loss-of -function (LOF) allele VCPk15502 [40] displayed an increased density of GFP positive profiles in the retina (Figure 4D–4G; n>5 eyes/genotype), consistent with them having increased levels of misfolded oligomeric Rh1 (see Figure 2E and Figure 3A–3D). We then determined the levels of the Hsc3/BiP chaperone in the retina, as an independent read-out for ER stress [18], [20], [37]. Using a Hsc3-specific antibody [37], we found that Rh1P37H;Rh1+/+retinas display a 50% increase in Hsc3 levels relative to Rh1WT;Rh1+/+ retinas, starting from P1 (Figure S6F and S6G). Moreover, as previously seen for Xbp1-EGFP activation, Rh1P37H;Rh1+/+ flies carrying VCP26-8, Rh1− or VCPk15502 alleles displayed increased Hsc3 levels compared to control flies and to Rh1P37H;Rh1+/+ carrying WT alleles of VCP or Rh1 (Figure 4H and 4I and data not shown; results from 3 independent crosses were averaged). Therefore, the increased levels of misfolded Rh1 seen after partial removal of VCP or endogenous Rh1 function were associated with increased activation of the Ire1/Xbp1 UPR pathway.

Suppression of Rh1P37H-Induced Retinal Degeneration by VCP Loss-of-Function Alleles
We next investigated the effect of decreasing VCP function on Rh1P37H-induced PN degeneration. We found that Rh1P37H;Rh1+/+ and Rh1P37H;VCP26-8/+;Rh1+/+ retinas showed a similar degree of PN degeneration at P20 (Figure 5A, 5B, and 5G), while further degeneration (at P30) in the Rh1P37H;Rh1+/+ retina was prevented in the VCP26-8 background (Figure 5D, 5E, and 5G). Remarkably, PN degeneration was strongly suppressed in Rh1P37H;Rh1+/+ flies carrying the stronger VCP LOF allele VCPk15502 (genotype: Rh1P37H;VCPk15502/+;Rh1+/+), both at P20 and P30 (Figure 5A, 5C, 5D, 5F, and 5G). Thus, VCP inactivation exerts a protective role on Rh1P37H-mediated retinal degeneration. We then used a VCP-specific antibody to assess the levels of VCP in Rh1P37H;Rh1+/+ versus Rh1WT;Rh1+/+ flies. We found a small (2-fold) but consistent increase in VCP levels in Rh1P37H;Rh1+/+ flies at P1, which was not maintained at P10 (Figure 5H and 5I; results from 3 independent crosses were averaged). Although an initial increase in VCP levels might induce long-term cellular changes that contribute to PN degeneration, and changes in VCP activation status or localization might be pro-apoptotic, our results suggest that differences in VCP levels play a minor, if any, direct pro-apoptotic role in Rh1P37H-mediated retinal degeneration.
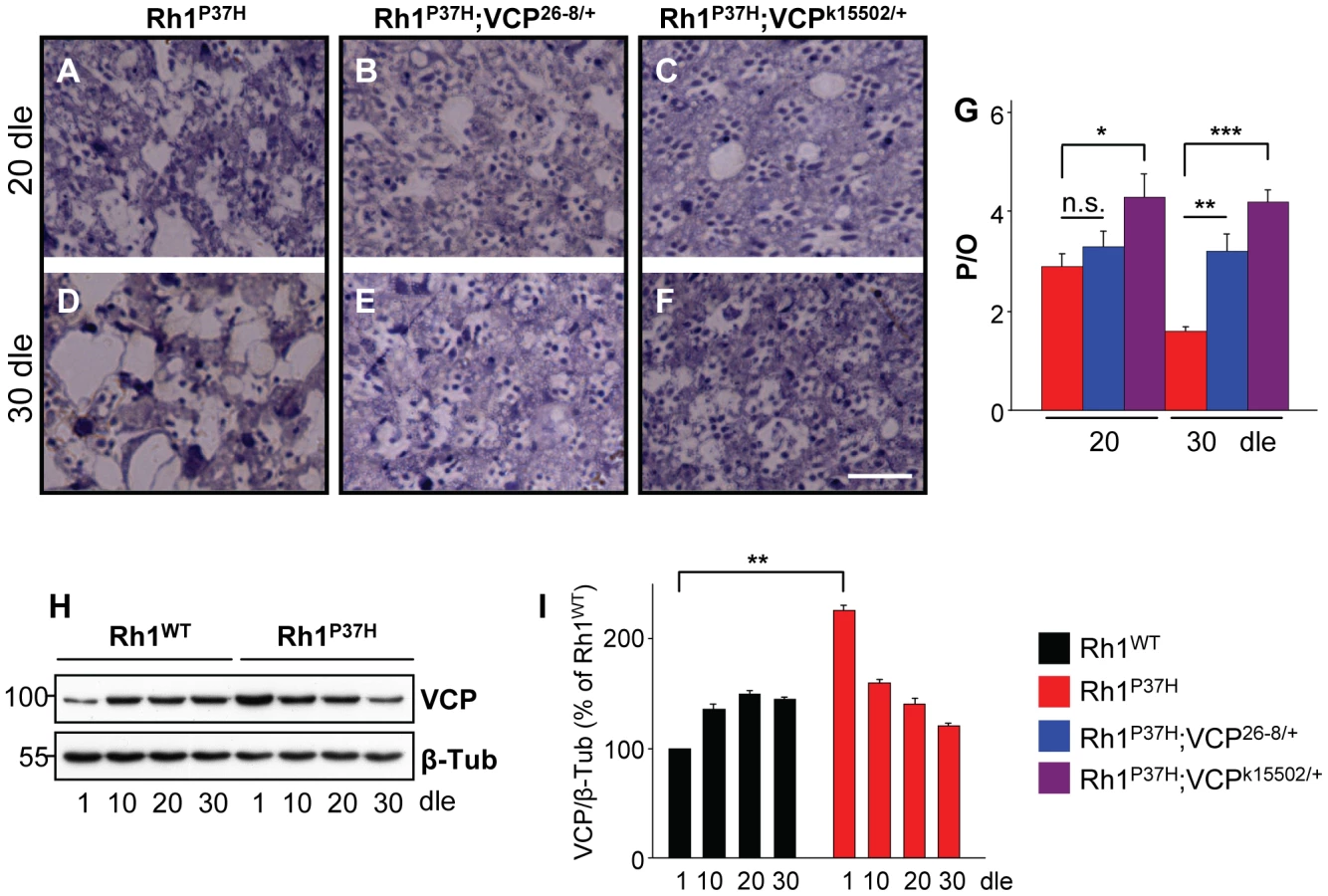
Pharmacological Inhibition of the VCP/ERAD/Proteasome Axis Suppresses Retinal Degeneration in Rh1P37H-Expressing Flies
Our finding that VCP inhibition rescues retinal degeneration in Rh1P37H;Rh1+/+ flies suggests that inhibition of the ERAD activity might exert long-term protective effects in Rh1P37H PNs. The tight coupling between VCP and proteasome activities is essential for substrate delivery and clearance during ERAD [23] and interestingly, proteasome activity is required for proper substrate retrotranslocation [15]. To independently test whether inhibition of the VCP/ERAD/proteasome axis is protective for Rh1P37H-expressing PNs, we used the VCP/ERAD inhibitor Eeyarestatin I (EerI) and the proteasome inhibitor MG132 that we dissolved in fly food. EerI acts on deubiquitinating enzymes that function downstream of VCP during ERAD and therefore inhibits ERAD-associated VCP functions [44], [45]. MG132 is a classical proteasome inhibitor and potently inhibits the proteasome in Drosophila S2 cells [46], [47]. We measured the proteasome activity on head extracts (see Text S1) from control flies to which MG132 was added after lysis and determined that MG132 potently inhibits the activity of the proteasome (Figure S7).
Using two doses of EerI (1 mM and 10 mM), we found that partial VCP/ERAD inhibition potently suppressed retinal degeneration in Rh1P37H;Rh1+/+ flies after 30 dle (Figure 6B–6D and 6G; n>7 flies/genotype). Both doses displayed the same (partial) suppression of PN degeneration, suggesting that a higher dose is required to achieve a more complete rescue or that EerI only inhibits some, but not all, ERAD-related VCP functions. Rh1P37H;Rh1+/+ flies transferred to food containing 5 µM MG132 displayed a partial but robust suppression of PN degeneration after 30 dle (Figure 6B, 6E, and 6G). Remarkably, treatment of Rh1P37H;Rh1+/+ flies with 50 µM MG132 led to a very dramatic rescue of retinal degeneration, the average number of photoreceptors/ommatidium reaching 5.5 (Figure 6B, 6F, and 6G; n>7 flies/genotype and >150 ommatidia scored/animal). Taken together, these results indicate that inhibition of the VCP/ERAD/proteasome axis is protective for Rh1P37H-expressing PNs, further suggesting that excessive retrotranslocation and/or degradation of visual pigment is a critical pro-apoptotic event in the Rh1P37H retina.

Partial VCP Inactivation Restores Visual Processing in Rh1P37H-Expressing Flies
The almost complete restoration of mature Rh1 levels in Rh1P37H;VCP26-8/+;Rh1+/+ flies prompted us to investigate visual processing in these flies. To assess the capacity of different mutants to process visual information, we performed fast phototaxis measurements [11], [48], [49]. This analysis reflects the average visual functioning of a fly population and scores the percentage of flies that successfully respond to five consecutive light stimulations. Control flies are normally attracted by light, and the analysis of light responses in different groups of mutants serves as an indicator of their retinal integrity. After 20 dle, we found that Rh1WT;Rh1+/+, Rh1WT;VCP26-8/+;Rh1+/+ and VCP26-8/+;Rh1+/+ flies displayed a normal and highly reproducible response to light, i.e. most of these flies moved towards the light source (positive phototaxis) in all five consecutive light stimulations (Figure 7A; n = 277–451 flies/genotype). In contrast, Rh1P37H;Rh1+/+ flies were seriously impaired in their light processing capability (Figure 7A) and had a significantly lower phototactic score (PS, defined in Materials and Methods) as compared to Rh1WT;Rh1+/+ and control flies (Figure 7B) [11]. Remarkably, in Rh1P37H;VCP26-8/+;Rh1+/+ flies, positive phototaxis was dramatically improved, consistent with these flies having normal levels of mature Rh1 (Figure 7A and 7B; see Figure S6D). We obtained similar results after 30 dle (Figure 7C and 7D). To rule out that other non-visual related impairments account for the performance of Rh1P37H;Rh1+/+ flies in the phototaxis test, we used geotaxis to measure motor functioning in these and all other fly groups, and found that all had a similar motor ability after 20 and 30 dle (Figure 7E and 7F). We found a similar rescue of visual processing in Rh1P37H-expressing flies when carrying the stronger LOF allele, VCPk15502 (Figure S8H, S8I, S8J); moreover, Rh1P37H-expressing flies carrying one Rh1 null allele (Rh1−) also showed improved phototaxis (Figure S8K, S8L, S8M) although the recovery was partial, reflecting their Rh1 content (Figure 2D). To evaluate whether the protective effect achieved by partial VCP inhibition also extends to other class II Rh1 mutations, we used another class II mutant, ninaED1 (Rh1S137F), which also displays a progressive decline in visual acuity [34]. We found, similarly, that reducing VCP activity partially suppressed the loss of PNs in the ninaED1/+ retina (Figure S8A, S8B, S8C, S8D) and restored the vision deficits displayed by the ninaED1/+ flies (Figure S8E, S8F, S8G). Thus, reducing VCP activity rescues retinal degeneration and blindness induced by a second class II mutant, ninaED1.

Finally, to obtain more direct evidence for a functional rescue of visual processing in Rh1P37H;VCP26-8/+;Rh1+/+ PNs, we performed electroretinogram (ERG) measurements [11], [39], [50]. We determined that, in contrast to Rh1-Gal4, Rh1WT;Rh1+/+, VCP26-8/+;Rh1+/+ or Rh1WT;VCP26-8/+;Rh1+/+ retinas, which displayed a normal photoreceptor depolarization following light stimulation, Rh1P37H-expressing photoreceptors had a decreased amplitude of photoreceptor depolarization (plateau), both after 30 and 45 dle, consistent with their retinal degeneration phenotype (Figure 8; n = 12–20 flies/genotype). In contrast, light processing in Rh1P37H;VCP26-8/+;Rh1+/+ photoreceptors was largely rescued, the ERG amplitude values for these flies reaching control levels, both after 30 dle (Figure 8A and 8B) and after 45 dle (Figure 8C and 8D). Taken together, the deleterious effects of mutant Rh1P37H on light transduction and photoreceptor depolarization are almost completely suppressed by inactivating the function of the ERAD effector VCP, indicating that VCP activity promotes neurodegeneration in the Rh1P37H retina.

Discussion
In our recent study [16] we found that the ERAD effector VCP interacts with misfolded RhP23H in mammalian cells and promotes its ATP-dependent retrotranslocation and proteasomal clearance. Here, we have used Drosophila transgenics in which Rh1P37H (the equivalent of mammalian RhP23H) induces progressive light - and age-dependent neurodegeneration in PNs [11] and manipulated the levels of the endogenous Rh1, as well as the activity of VCP. We found that misfolded Rh1P37H induces deleterious effects in PNs only in the presence of its WT endogenous counterpart. These deleterious effects include an excessive retrotranslocation and/or degradation of Rh1, mediated by VCP. We suggest that the VCP/ERAD/proteasome axis is a component of Rh1P37H-induced toxicity in PNs.
Proteostasis Defects in the Rh1P37H Retina
As a first step toward a detailed characterization of Rh1P37H proteostasis defects and their relevance to PN integrity, we evaluated the detergent solubility of Rh1 species extracted from transgenic Rh1P37H;Rh1+/+ and Rh1WT;Rh1+/+ retinas. In detergent-soluble fractions, we observed an early depletion of the misfolded Rh1P37H (already after 1 dle), which suggests maturation defects. These maturation defects were only seen at light (Figure S3C) and were alleviated after partial VCP inactivation (Figure 3G), suggesting that light-stimulated visual activity might impact on the maturation of Rh1P37H by promoting VCP-mediated ERAD of Rh1P37H.
The detergent-insoluble fractions from transgenic retinas were enriched in Rh1 oligomeric species that migrated at approximately 250–300 kDa - thus presumably containing 6–10 Rh1 molecules. After 20 dle, Rh1P37H and Rh1WT-expressing flies displayed high levels of Rh1 oligomers, and oligomer traces were also detected in control (Rh1-Gal4) flies; this might be potentially due to reduced folding capacity of the ER during aging (that can be further impaired by protein overexpression or misfolding). Since VCP inactivation (which suppressed degeneration) also increased the levels of Rh1 oligomers and both Rh1WT and Rh1P37H-expressing flies (at P20-P30) displayed high levels of oligomers (despite clear differences in retinal integrity), the presence of these Rh1 oligomers is probably not the cause of retinal degeneration in the Rh1P37H;Rh1+/+ retina. We found a complete absence of mutant Rh1P37H from Rh1-containing oligomers in Rh1P37H flies, while VCP inactivation led to accumulation of Rh1P37H-containing oligomers (Figure 3C and 3D and Figure S6C). The fact that we detected Rh1P37H oligomers (hsv positive; Figure 3C and 3D) in Rh1P37H;VCP26-8/+;Rh1+/+ retina suggests that the absence of Rh1P37H from Rh1 oligomeric species in the Rh1P37H;Rh1+/+ retina is due to Rh1P37H degradation, and not to failure to detect the hsv tag. The increased levels of hsv-labeled Rh1P37H oligomers in the insoluble fraction from the Rh1P37H;VCP26-8/+;Rh1+/+ retina and the activation of the Ire1/Xbp1 ER stress pathway under these conditions (suggesting that these species accumulated in the ER; Figure 4) suggest that VCP activity promotes degradation of Rh1P37H, probably by mediating its retrotranslocation from the ER into the cytosol [16], [24], [27]. However, VCP might also promote autophagic degradation of proteins [51]–[53] and it remains to be determined whether Rh1P37H is also degraded by autophagy and whether VCP is involved in this process.
Our experimental protocol for extraction and detection of Rh1-containing insoluble species might not allow us to infer the true nature of Rh1-containing insoluble species in vivo. First, the detergent used during extraction might perturb higher-order aggregates (e.g. protofibrils, fibrils) causing them to disassemble during extraction; in addition, different detergents can lead to the extraction of different Rh oligomeric species [54], [55]. Second, Rh1 is able to form oligomers ex vivo, during sample preparation. Third, insoluble higher-order aggregates might not enter the SDS-PAGE gel, and remain undetected. Another limitation of our biochemical approach is the lack of information concerning the subcellular localization of Rh1-containing oligomers or aggregates. Further studies using different extraction protocols and/or more powerful biophysical approaches (e.g. spectroscopy using fluorescent probes, fluorescence resonance energy transfer, atomic force microscopy [54], [56]) but also the development of antibodies highly specific to various Rh1 epitopes [57] might help to precisely determine the composition and subcellular localization of Rh1-containing species in the diseased retina.
Endogenous Rh1 Is Required for Rh1P37H Toxicity
We found that PN degeneration in the Rh1P37H;Rh1+/+ requires an environment in which both mutant Rh1P37H and endogenous Rh1 are present. Thus, the Rh1P37H-mediated neurodegeneration was strongly suppressed in Rh1 null background, suggesting that the Rh1P37H allele is not toxic by itself nor is it sufficient to cause cell death in the absence of its WT counterpart. Transgenic Rh1WT;Rh1+/+ flies displayed minor degenerative signs in the retina, suggesting that WT Rh1 alone is not sufficient to cause degeneration. Similar conclusions were also reached with another class II Rh1 mutant, ninaED1 [34].
Experiments performed in transgenic mice overexpressing a triple (P23H, V20G, P27L) Rh mutant (RhGHL) indicated that decreased levels of endogenous Rh accelerated the rate of retinal degeneration [58]. These findings are in apparent conflict with our present observations and with those made using ninaED1 flies [34]. Both RhGHL mice and Rh1P37H flies express the mutant Rh at moderate levels (10–25% and 51% of normal levels, respectively), suggesting that toxicity was not due to overexpression. Several differences between the two models might explain these findings. First, RhGHL is a triple Rh mutant and might display different properties when compared to RhP23H; the analysis of RhP23H transgenic mice [59] will address this possibility. Second, the subcellular localization of the mutant Rh was different, RhGHL being properly targeted to the membrane (in the RhGHL;Rh+/+ retina), while Rh1P37H showed severe maturation defects (Rh1P37H;Rh1+/+ retina). Interestingly, removal of endogenous Rh function impacted (in opposite ways) on the maturation of misfolded Rh, and Rh maturation defects caused a more severe pathology in both cases. It would be thus interesting to evaluate the effect of VCP heterozygosity in the RhGHL retina. Third, the most notable difference is that RhGHL-mediated degeneration does not require exposure to light (but can be further accelerated by light [8], [60], [61]), while the Rh1P37H-induced degeneration is strictly light-dependent. Interestingly, we also found that light exposure impairs the maturation of Rh1P37H, suggesting a possible interaction between visual processing and the protein quality control machinery in controlling Rh1 homeostasis. Future studies examining RhP23H RP patients will establish the role played by light in RP.
VCP Activity Promotes Retinal Degeneration and Blindness in the Rh1P37H Retina
Partial VCP inactivation rescued retinal degeneration and blindness in Rh1P37H flies; moreover, the rescuing effect of VCP inactivation also extends to another class II Rh1 mutation, ninaED1 (Figure S8). Two lines of evidence suggest that the effect of VCP on the Rh1P37H pathology involves its activity as ERAD effector. First, the levels of VCP were found to be increased very early (after 1dle) in Rh1P37H-expressing flies, well before the onset of retinal degeneration; this suggests that VCP has no direct cell death-promoting activity. Since UPR activation might induce ERAD genes [19], the increase in VCP levels might have been caused by the early activation of the Ire1/Xbp1 pathway. Second, our pharmacological treatment with the EerI inhibitor potently suppressed retinal degeneration in Rh1P37H flies. EerI was reported to inhibit the activity of deubiquitinating enzymes acting downstream of VCP during ERAD, therefore inhibiting an ERAD-associated VCP activity [44], [45]. Our pharmacological treatment with the proteasome inhibitor MG132 also led to a dramatic suppression of retinal degeneration in the Rh1P37H retina and the proteasome was shown to be tightly coupled to substrate retrotranslocation during ERAD [15], [62], [63]. The very potent rescuing effect on PN degeneration after genetic inactivation of VCP, or after pharmacological treatment with EerI or MG132 suggests that excessive retrotranslocation and/or degradation of visual pigment is pathogenic for Rh1P37H PNs.
Our results suggest that mechanism underlying Rh1P37H dominance in the retina is not a DN effect on the maturation of WT Rh1; the endogenous Rh1 matured normally in Rh1P37H flies (see also [11]) and no decrease in the levels of mature Rh1 preceded PN degeneration in Rh1P37H flies (Figure 1G). Excessive VCP-mediated degradation of visual pigment might be an underlying mechanism of Rh1P37H dominance in PNs. The Rh1P37H mutant is not toxic by itself (Figure 2) but requires the endogenous Rh1, VCP activity and light exposure to cause massive PN degeneration. All components of this quadruple interaction network are necessary for toxicity, as elimination of any of them mitigates neurodegeneration. How do these network components relate to each other? The hypothesis we favor is that light-initiated visual processing affects the maturation and stability of the mutant Rh1P37H, leading to its enhanced VCP-dependent degradation, with deleterious consequences for the cell. During visual processing, the rhabdomeric WT Rh1 undergoes repeated cycles of internalization [64] and new Rh1 synthesis might re-supply the rhabdomeres. The synthesis and trafficking of WT Rh1 through the ER might be enhanced following light exposure, and this might saturate the ER folding machinery. Excess WT Rh1 might fail to fold, and could self-assemble (to form oligomers, as seen in Figure 1F). At the same time, misfolded Rh1P37H would undergo ERAD at a higher rate, which might be pathogenic. The competition between WT Rh1 and Rh1P37H for the ER folding machinery might thus control PN survival. It would be interesting to determine how light exposure and dark rearing impact on the activity of ERAD, in the presence of WT and/or P37H Rh1.
How do enhanced retrotranslocation and/or degradation of visual pigment lead to PN degeneration in the Rh1P37H retina? Excessive retrotranslocation might activate pro-apoptotic pathways, under conditions of chronic ERAD activity. Excessive Rh1P37H retrotranslocation might also increase the amount of cytosolic Rh1P37H, which might aggregate if left undegraded. Cytosolic Rh1P37H-containing aggregates might be more toxic than Rh1P37H mono/oligomers located in the ER and might generate yet unidentified pro-apoptotic signals if the cells cannot adapt to aggregate-induced stress; interestingly, several molecules and mechanisms were suggested to mediate the transition from an adaptive stress response to apoptosis, including the CHOP transcription factor, caspase activation, Ca2+ release and mitochondrial signaling [7].
A recent study [65] investigated another Rh1 allele (ninaEG69D) which, although not found in RP patients, has been classified as class II Rh mutation [33]. Overexpression (Rh1 promoter driven) of ERAD members Hrd1 and EDEM2 partially rescued late-onset PN degeneration and loss of mature Rh1 in ninaEG69D flies. Interestingly, reduced Hrd1 or EDEM2 function (by RNAi) also rescued mature Rh1 levels, although its effect on PN degeneration was not assessed. These observations might hint at different mechanisms of dominance or different levels of UPR activation among the different Rh1 alleles (see [7]). Another possibility is that early clearance of the mutant Rh1 (via enhanced ERAD) might have long-term protective effects [65]. Therefore, the manipulation of ERAD activity for therapeutic purposes should take into account the temporal profile of Rh aggregation and the relative contribution of endogenous Rh to oligomeric species/aggregates associated with each individual Rh dominant mutation.
Recent evidence suggests that VCP is also a mediator of autophagy, the second major route for protein clearance in cells [51]–[53]. Autophagy and the ubiquitin-proteasome system (UPS) appear to be coordinated processes, with the autophagy being upregulated upon proteasome impairment [66]. The enhanced autophagy that follows UPS inactivation requires the cytoplasmic histone deacetylase HDAC6 [67] and interestingly, HDAC6 interacts with VCP to determine the fate of ubiquitinated misfolded proteins [68]. Inhibition of VCP might decrease autophagic degradation of Rh1 and confer neuroprotection in our Rh1P37H flies; however, proteasomal inhibition (that should cause enhanced autophagy) was neuroprotective for the Rh1P37H retina (Figure 6) and we detected no significant impairment of proteasomal function in Rh1P37H flies (A.Gr. and M.U., unpublished observations). These conflicting scenarios and the recent failure of autophagy induction to suppress Rh1RH27-mediated retinal pathology in Drosophila [69] argue for additional studies to address the relevance of autophagy-promoting VCP activity to retinal degeneration caused by misfolded Rh.
Promoting Neuroprotection in the RhP23H Retina
Inhibition of VCP activity or reduced dosage of the endogenous Rh1 conferred neuroprotection for Rh1P37H-expressing PNs, and were associated with increased UPR activation, via the Ire1/Xbp1-mediated Hsc3/BiP production (Figure 2, Figure 4, and Figure 5). Since VCP/ERAD/proteasome inactivation might upregulate other pro-survival pathways, it remains to be determined whether Ire1/Xbp1/Hsc3 activation mediates neuroprotection in the Rh1P37H retina. It is interesting to note that Rh1 accumulation within the ER was recently shown to induce a moderate activation of the Ire1/Xbp1 pathway which induced long-term pro-survival effects, via inhibition of caspase activation and induction of an antioxidant response [20]. Another study found that subretinal delivery of adeno-associated viruses expressing BiP prevented RhP23H-mediated retinal degeneration in rats by suppressing the production of the pro-apoptotic protein CHOP [70]. However, it should be noted that, besides activating Ire1/Xbp1, UPR can also promote apoptosis via two independent pathways [18]. Thus, a better understanding of the differential regulation of UPR pathways during ER stress might provide further clues about the regulation of cellular survival versus apoptosis in PNs expressing misfolded RhP23H.
Is partial VCP inactivation able to protect against retinal degeneration in RhP23H-linked RP? We found that VCP inactivation prevents neurodegeneration in Rh1P37H flies and VCP inhibition is also sufficient to rescue mutant CFTR from degradation and to partially restore CFTR function in a cellular model of cystic fibrosis [28]. However, mutations that impair VCP function are associated with inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia (IBMPFD) [71], [72]. It remains therefore to be determined whether changes in VCP function suppress some pathologies and enhance others. One interesting experiment is the evaluation of the effect of VCP and proteasome inactivation in the RhP23H mouse retina, preferably using local delivery. An improved understanding of the molecular machinery linking Rh quality control and the networks regulating PN maintenance might uncover cellular targets acting specifically in the retina, thus facilitating the therapeutic approaches for RP.
Materials and Methods
Drosophila Stocks
Drosophila lines p(w+ Rh1-Rh1WT), p(w+ Rh1-Rh1P37H), p(w+ Rh1-Rh1P37H-hsv) and p(w+ Rh1-Gal4) lines referred to as Rh1WT;Rh1+/+, Rh1P37H;Rh1+/+, Rh1P37H-hsv;Rh1+/+ and Rh1-Gal4 were previously described [11]. ninaED1 (ninaES137F) and p(ry+ rh1-Rh1WT-hsv) referred to as Rh1WT-hsv;Rh1+/+ were a gift kind from J.E. O'Tousa. ter9426-8 hypomorphic allele, referred to as VCP26-8, was kindly provided by E. Goldstein. UAS-xbp1-EGFP was a generous gift from H.D. Ryoo. Actin-Gal4, ninaE-null I17 (referred to as Rh1−), ter94k15502 (referred to as VCPk15502, strong LOF allele) and w1118 flies were from the Bloomington stock center. Flies were raised on standard cornmeal agar medium, under moderate continuous illumination at 25°C. Moderate illumination was obtained by using photosynthetic fluorescent tubes (in total 170 cd/m2). Fly progeny having same eye pigmentation was used during the study.
Western Blotting
WB was performed as described [15], [73], [74]. Briefly, 30 fly heads were homogenized in 60 µl of RIPA modified buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% SDS and 0.5% sodium deoxycholate) supplemented with protease inhibitors (Roche) and phosphatase inhibitors (Sigma-Aldrich). Lysates were centrifuged at 16.000 g for 15 minutes at 4°C. For detergent-insoluble factions, pellets were solubilised in 50 µl solution containing 10 mM Tris-HCl pH 7.5, 1% SDS and protease inhibitor cocktail for 8 minutes at room temperature. 100 µl of RIPA modified buffer containing protease inhibitors was added, and the pellets were then sonicated six times (10 seconds each) at 4°C. Following sonication, the fractions were incubated 30 minutes at 4°C. Detergent-insoluble fractions were then centrifuged at 100 g for 10 minutes at 4°C and tissue debris were discarded. For Western Blotting, fractions were normalized for total protein using the Dc protein assay (Bio-Rad). An equal volume of 2× Laemmli sample buffer was added to fractions separated by 10% SDS-PAGE for detergent-soluble and by 8% SDS-PAGE for detergent-insoluble fractions and electroblotted onto PVDF membranes (GE Healthcare). Immunodetection was performed according to standard techniques using the following primary antibodies: anti-Rh1 (rabbit polyclonal, 1/5000, gift from D.F. Ready) for detecting Rh1 in detergent-insoluble fractions, 4C5 (mouse monoclonal, 1/5000, DSHB) that detects Rh1 in detergent-soluble fractions, anti-ter94/VCP (rat, 1/5000, gift from D. McKearin), anti-hsv (rabbit polyclonal, 1/8000, Sigma), anti-Hsc3 (guinea pig, 1/2000, gift from H.D. Ryoo), anti-Ubiquitin (mouse monoclonal, 1/300, Invitrogen), anti-TRP (1/10000, gift from A. Huber) and anti-β-Tubulin (mouse monoclonal, 1/4000, Chemicon). Secondary antibodies were horseradish peroxidase-coupled (1/8000, Jackson Immunoresearch). Quantification of band intensity after ECL detection was performed using Image Quant TL software.
Histology and Immunohistochemistry
Heads were dissected and fixed for 24 h in 2.5% glutaraldehyde, phosphate buffered saline (PBS, pH 7.4) and post-fixed in 1% osmium tetroxide in PBS. After a series of ethanol and propylene oxide dehydration, heads were embedded in epoxy resin (Sigma-Aldrich). For toluidine blue staining, semithin (2 µm) sections were stained with 1% toluidine blue [75]. To determine the average number of photoreceptors/ommatidium (P/O), at least l50 ommatidia were scored per animal from at least 4 animals per genotype.
Immunohistochemistry was performed as described [11], [37], heads were dissected and fixed for 15 minutes in 4% formaldehyde, incubated in 10% sucrose during 2 hours and then in 25% sucrose overnight at 4°C, then embedded in cryomedium. 16 µm-thick cryostat sections were fixed for 15 minutes in 4% formaldehyde, permeated with 0.3% Triton-X 100 in PBS (PBST) during 30 minutes and blocked during 1h with 5% normal goat serum in PBST. To detect non-conventional xbp1 mRNA splicing of the xbp1-EGFP construct, we used an anti-GFP antibody (rabbit polyclonal, 1/400, Molecular Probes) diluted in blocking solution. After overnight incubation with primary antibody, an alkaline phosphatase-linked anti-rabbit secondary antibody (1/200, Sigma-Aldrich) was added for 2 hours at room temperature. Sections were washed with alkaline phosphatase buffer (100 mM NaCl, 50 mM MgCl2, 100 mM Tris-HCl pH 9.5, 0.1% Tween 20). Then, sections were incubated with BCIP/NBT substrate (Sigma-Aldrich) during 30 minutes, washed in water, air-dried and mounted in a non-aqueous mounting medium (Clarion, Sigma-Aldrich).
Pharmacological Treatments
Flies were treated with EerI (ChemBridge Corporation, catalog n° 5138427) or MG132 (Sigma-Aldrich) inhibitors dissolved in their food. We used 2 doses of EerI (1 mM and 10 mM) and 2 doses of MG132 (5 µM and 50 µM). These compounds were first dissolved in DMSO and the solution was then added to cooled cornmeal agar fly food. The food was then dispensed into empty vials and allowed to solidify. It was kept at 4°C in the dark for maximum 3 days. The above-mentioned concentrations correspond to the final concentrations in fly food. Flies were transferred in vials containing this modified food right after birth, were reared as described above and transferred to fresh vials every day. The control food contained all the ingredients (including DMSO) except the active compound. After 30 days of light exposure, flies fed on control food (2% DMSO) and on drug food (EerI or MG132 in 2% DMSO) were sacrificed and their retinal integrity was assessed histologically.
Behavioral Assays
Fast phototaxis was performed as previously described [11]. Briefly, 20 flies were placed in tube 1 of a countercurrent apparatus with six tubes and gently tapped to the bottom of the tube. The apparatus was placed horizontally, the light source was switched on and the flies were allowed to walk toward the light during 30 seconds. The flies that moved toward the light were shifted to the second tube and this procedure was repeated five times. At the end of the test, the number of flies was counted in each of the six tubes. Between 277–451 flies were scored per genotype, in 3 independent experiments. The phototactic score (PS) quantifies the visual activity following a weighted equation (ΣiNi)/ΣNi, where N is the number of flies in the ith tube.
Geotaxis was used to assess motor performance in flies chronically exposed to light. About 20 flies/tube were gently tapped to the bottom of the tube and then allowed to climb during 20 seconds. We then scored the number of flies that climbed 4 cm or above. Each tube was tested twice. The results represent the% of flies that reached 4 cm or above after 20 seconds.
Electroretinogram
Electroretinogram analysis was performed as previously described [11]. A tungsten electrode was introduced in the back of the fly head and a glass electrode filled with 3M KCl was introduced through the cornea. Flashes were delivered using a white LED (Nichia, NSPW510BS, emitting from 425 to 750 nm with two peaks at 460 and 560 nm) with a 45° viewing angle, placed at 1.5 cm from the fly eye and controlled via an Analog Out port of a Digidata 1322A (Molecular Devices, USA). After 5 minutes of dark-adaptation, six 1 second light pulses were used to stimulate the eye and their responses averaged. The light intensity reaching the eye (∼300 µW/cm2) was chosen as the minimal intensity that consistently produced maximal ERG plateaus.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. PowersET
MorimotoRI
DillinA
KellyJW
BalchWE
2009 Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem 78 959 991
2. DaigerSP
BowneSJ
SullivanLS
2007 Perspective on genes and mutations causing retinitis pigmentosa. Arch Ophthalmol 125 151 158
3. KennanA
AherneA
HumphriesP
2005 Light in retinitis pigmentosa. Trends Genet 21 103 110
4. FarrarGJ
KennaPF
HumphriesP
2002 On the genetics of retinitis pigmentosa and on mutation-independent approaches to therapeutic intervention. EMBO J 21 857 864
5. ChappleJP
GraysonC
HardcastleAJ
SalibaRS
van der SpuyJ
2001 Unfolding retinal dystrophies: a role for molecular chaperones? Trends Mol Med 7 414 421
6. DaigerSP
2004 Identifying retinal disease genes: how far have we come, how far do we have to go? Novartis Found Symp 255 17 27; discussion 27–36, 177–8
7. MendesHF
van der SpuyJ
ChappleJP
CheethamME
2005 Mechanisms of cell death in rhodopsin retinitis pigmentosa: implications for therapy. Trends Mol Med 11 177 185
8. NaashMI
HollyfieldJG
al-UbaidiMR
BaehrW
1993 Simulation of human autosomal dominant retinitis pigmentosa in transgenic mice expressing a mutated murine opsin gene. Proc Natl Acad Sci USA 90 5499 5503
9. KosmaoglouM
SchwarzN
BettJS
CheethamME
2008 Molecular chaperones and photoreceptor function. Prog Retin Eye Res 27 434 449
10. KaushalS
KhoranaHG
1994 Structure and function in rhodopsin. 7. Point mutations associated with autosomal dominant retinitis pigmentosa. Biochemistry 33 6121 6128
11. GalyA
RouxMJ
SahelJA
LéveillardT
GiangrandeA
2005 Rhodopsin maturation defects induce photoreceptor death by apoptosis: a fly model for RhodopsinPro23His human retinitis pigmentosa. Hum Mol Genet 14 2547 2557
12. MendesHF
CheethamME
2008 Pharmacological manipulation of gain-of-function and dominant-negative mechanisms in rhodopsin retinitis pigmentosa. Hum Mol Genet 17 3043 3054
13. SungCH
SchneiderBG
AgarwalN
PapermasterDS
NathansJ
1991 Functional heterogeneity of mutant rhodopsins responsible for autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci USA 88 8840 8844
14. IllingME
RajanRS
BenceNF
KopitoRR
2002 A rhodopsin mutant linked to autosomal dominant retinitis pigmentosa is prone to aggregate and interacts with the ubiquitin proteasome system. J Biol Chem 277 34150 34160
15. SalibaRS
MunroPM
LuthertPJ
CheethamME
2002 The cellular fate of mutant rhodopsin: quality control, degradation and aggresome formation. J Cell Sci 115 2907 2918
16. GriciucA
AronL
PiccoliG
UeffingM
2010 Clearance of Rhodopsin(P23H) aggregates requires the ERAD effector VCP. Biochim Biophys Acta 1803 424 434
17. BernalesS
PapaFR
WalterP
2006 Intracellular signaling by the unfolded protein response. Annu Rev Cell Dev Biol 22 487 508
18. LinJH
LiH
YasumuraD
CohenHR
ZhangC
2007 IRE1 signaling affects cell fate during the unfolded protein response. Science 318 944 949
19. RonD
WalterP
2007 Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8 519 529
20. MendesCS
LevetC
ChatelainG
DourlenP
FouilletA
2009 ER stress protects from retinal degeneration. EMBO J 28 1296 1307
21. RömischK
2005 Endoplasmic reticulum-associated degradation. Annu Rev Cell Dev Biol 21 435 456
22. VembarSS
BrodskyJL
2008 One step at a time: endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol 9 944 957
23. DaiRM
LiCC
2001 Valosin-containing protein is a multi-ubiquitin chain-targeting factor required in ubiquitin-proteasome degradation. Nat Cell Biol 3 740 744
24. YeY
MeyerHH
RapoportTA
2001 The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 414 652 656
25. DeLaBarreB
ChristiansonJC
KopitoRR
BrungerAT
2006 Central pore residues mediate the p97/VCP activity required for ERAD. Mol Cell 22 451 462
26. SongC
WangQ
LiCC
2003 ATPase activity of p97-valosin-containing protein (VCP). D2 mediates the major enzyme activity, and D1 contributes to the heat-induced activity. J Biol Chem 278 3648 3655
27. WangQ
SongC
LiCC
2004 Molecular perspectives on p97-VCP: progress in understanding its structure and diverse biological functions. J Struct Biol 146 44 57
28. VijN
FangS
ZeitlinPL
2006 Selective inhibition of endoplasmic reticulum-associated degradation rescues DeltaF508-cystic fibrosis transmembrane regulator and suppresses interleukin-8 levels: therapeutic implications. J Biol Chem 281 17369 17378
29. SchwiegerI
LautzK
KrauseE
RosenthalW
WiesnerB
2008 Derlin-1 and p97/valosin-containing protein mediate the endoplasmic reticulum-associated degradation of human V2 vasopressin receptors. Mol Pharmacol 73 697 708
30. BoeddrichA
GaumerS
HaackeA
TzvetkovN
AlbrechtM
2006 An arginine/lysine-rich motif is crucial for VCP/p97-mediated modulation of ataxin-3 fibrillogenesis. EMBO J 25 1547 1558
31. GitchoMA
StriderJ
CarterD
Taylor-ReinwaldL
FormanMS
2009 VCP mutations causing frontotemporal lobar degeneration disrupt localization of TDP-43 and induce cell death. J Biol Chem 284 12384 12398
32. MizunoY
HoriS
KakizukaA
OkamotoK
2003 Vacuole-creating protein in neurodegenerative diseases in humans. Neurosci Lett 343 77 80
33. ColleyNJ
CassillJA
BakerEK
ZukerCS
1995 Defective intracellular transport is the molecular basis of rhodopsin-dependent dominant retinal degeneration. Proc.Natl Acad Sc USA 92 3070 3074
34. KuradaP
O'TousaJE
1995 Retinal degeneration caused by dominant rhodopsin mutations in Drosophila. Neuron 14 571 579
35. KuradaP
ToniniTD
SerikakuMA
PicciniJP
O'TousaJE
1998 Rhodopsin maturation antagonized by dominant rhodopsin mutants. Vis Neurosci 15 693 700
36. RajanRS
KopitoRR
2005 Suppression of wild-type rhodopsin maturation by mutants linked to autosomal dominant retinitis pigmentosa. J Biol Chem 280 1284 1291
37. RyooHD
DomingosPM
KangMJ
StellerH
2007 Unfolded protein response in a Drosophila model for retinal degeneration. EMBO J 26 242 252
38. CrowMK
KarasavvasN
SarrisAH
2001 Protein aggregation mediated by cysteine oxidation during the stacking phase of discontinuous buffer SDS-PAGE. Biotechniques 30 311 316
39. LeeSJ
MontellC
2004 Suppression of constant-light-induced blindness but not retinal degeneration by inhibition of the rhodopsin degradation pathway. Curr Biol 14 2076 2085
40. RudenDM
SollarsV
WangX
MoriD
AltermanM
2000 Membrane fusion proteins are required for oskar mRNA localization in the Drosophila egg chamber. Dev Biol 218 314 325
41. KosmaoglouM
KanugaN
AguilàM
GarrigaP
CheethamME
2009 A dual role for EDEM1 in the processing of rod opsin. J Cell Sci 122 4465 7442
42. YoshidaH
MatsuiT
YamamotoA
OkadaT
MoriK
2001 XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107 881 891
43. SzegezdiE
LogueSE
GormanAM
SamaliA
2006 Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep 7 880 885
44. FiebigerE
HirschC
VyasJM
GordonE
PloeghHL
2004 Dissection of the dislocation pathway for type I membrane proteins with a new small molecule inhibitor, eeyarestatin. Mol Biol Cell 15 1635 1646
45. WangQ
LiL
YeY
2008 Inhibition of p97-dependent protein degradation by Eeyarestatin I. J Biol Chem 283 7445 7454
46. MuroI
HayBA
ClemRJ
2002 The Drosophila DIAP1 protein is required to prevent accumulation of a continuously generated, processed form of the apical caspase DRONC. J Biol Chem 277 49644 49650
47. LundgrenJ
MassonP
MirzaeiZ
YoungP
2005 Identification and characterization of a Drosophila proteasome regulatory network. Mol Cell Biol 25 4662 4675
48. HirschJ
BoudreauJC
1958 Studies in experimental behaviour genetics. I. The heritability of phototaxis in a population of Drosophila melanogaster. J Comp Physiol Psychol 51 647 651
49. BenzerS
1967 Behavioral mutants of Drosophila isolated by countercurrent distribution. Proc Natl Acad Sci USA 58 1112 1119
50. PakWL
1979 Study of photoreceptor function using Drosophila mutants. 67 99 In Neurogenetics: Genetic Approaches to the Nervous System, XO Breakefield, ed. (New York: Elsevier)
51. JuJS
FuentealbaRA
MillerSE
JacksonE
Piwnica-WormsD
2009 Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J Cell Biol 187 875 888
52. JuJS
WeihlCC
2010 Inclusion body myopathy, Paget's disease of the bone and fronto-temporal dementia: a disorder of autophagy. Hum Mol Genet 19 R38 45
53. TresseE
SalomonsFA
VesaJ
BottLC
KimonisV
2010 VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy 6 217 227
54. JastrzebskaB
FotiadisD
JangGF
StenkampRE
EngelA
2006 Functional and structural characterization of rhodopsin oligomers. J Biol Chem 281 11917 11922
55. ShukolyukovSA
2009 Aggregation of frog rhodopsin to oligomers and their dissociation to monomer: application of BN - and SDS-PAGE. Biochemistry (Mosc) 74 599 604
56. LindgrenM
HammarströmP
2010 Amyloid oligomers: spectroscopic characterization of amyloidogenic protein states. FEBS J 277 1380 1388
57. GoldeTE
EckmanCB
YounkinSG
2000 Biochemical detection of Abeta isoforms: implications for pathogenesis, diagnosis, and treatment of Alzheimer's disease. Biochim Biophys Acta 1502 172 187
58. FrederickJM
KrasnoperovaNV
HoffmannK
Church-KopishJ
RütherK
2001 Mutant rhodopsin transgene expression on a null background. Invest Ophthalmol Vis Sci 42 826 833
59. OlssonJE
GordonJW
PawlykBS
RoofD
HayesA
1992 Transgenic mice with a rhodopsin mutation (Pro23His): a mouse model of autosomal dominant retinitis pigmentosa. Neuron 9 815 830
60. NaashML
PeacheyNS
LiZY
GryczanCC
GotoY
1996 Light-induced acceleration of photoreceptor degeneration in transgenic mice expressing mutant rhodopsin. Invest Ophthalmol Vis Sci 37 775 782
61. WangM
LamTT
TsoMO
NaashMI
1997 Expression of a mutant opsin gene increases the susceptibility of the retina to light damage. Vis Neurosci 14 55 62
62. ChillarónJ
HaasIG
2000 Dissociation from BiP and retrotranslocation of unassembled immunoglobulin light chains are tightly coupled to proteasome activity. Mol Biol Cell 11 217 226
63. ManciniR
FagioliC
FraAM
MaggioniC
SitiaR
2000 Degradation of unassembled soluble Ig subunits by cytosolic proteasomes: evidence that retrotranslocation and degradation are coupled events. FASEB J 14 769 778
64. SatohAK
ReadyDF
2005 Arrestin1 mediates light-dependent rhodopsin endocytosis and cell survival. Curr Biol 15 1722 1733
65. KangMJ
RyooHD
2009 Suppression of retinal degeneration in Drosophila by stimulation of ER-associated degradation. Proc Natl Acad Sci USA 106 17043 17048
66. NedelskyNB
ToddPK
TaylorJP
2008 Autophagy and the ubiquitin-proteasome system: collaborators in neuroprotection. Biochim Biophys Acta 1782 691 699
67. PandeyUB
NieZ
BatleviY
McCrayBA
RitsonGP
2007 HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447 859 863
68. BoyaultC
GilquinB
ZhangY
RybinV
GarmanE
2006 HDAC6-p97/VCP controlled polyubiquitin chain turnover. EMBO J 25 3357 3366
69. WangT
LaoU
EdgarBA
2009 TOR-mediated autophagy regulates cell death in Drosophila neurodegenerative disease. J Cell Biol 186 703 711
70. GorbatyukMS
KnoxT
LaVailMM
GorbatyukOS
NoorwezSM
2010 Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc Natl Acad Sci USA 107 5961 5966
71. KimonisVE
WattsGD
2005 Autosomal dominant inclusion body myopathy, Paget disease of bone, and frontotemporal dementia. Alzheimer Dis Assoc Disord Suppl 1 S44 47
72. KimonisVE
FulchieroE
VesaJ
WattsG
2008 VCP disease associated with myopathy, Paget disease of bone and frontotemporal dementia: review of a unique disorder. Biochim Biophys Acta 1782 744 748
73. KyptaRM
SuH
ReichardtLF
1996 Association between a transmembrane protein tyrosine phosphatase and the cadherin-catenin complex. J Cell Biol 134 1519 1529
74. OrmeMH
AlrubaieS
BradleyGL
WalkerCD
LeeversSJ
2006 Input from Ras is required for maximal PI(3)K signalling in Drosophila. Nat Cell Biol 8 1298 1302
75. SatohA
TokunagaF
KawamuraS
OzakiK
1997 In situ inhibition of vesicle transport and protein processing in the dominant negative Rab1 mutant of Drosophila. J Cell Sci 110 2943 2953
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 8
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Identification of the Bovine Arachnomelia Mutation by Massively Parallel Sequencing Implicates Sulfite Oxidase (SUOX) in Bone Development
- Common Inherited Variation in Mitochondrial Genes Is Not Enriched for Associations with Type 2 Diabetes or Related Glycemic Traits
- A Model for Damage Load and Its Implications for the Evolution of Bacterial Aging
- Did Genetic Drift Drive Increases in Genome Complexity?
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy