
Extracellular Dopamine Potentiates Mn-Induced Oxidative Stress, Lifespan Reduction, and Dopaminergic Neurodegeneration in a BLI-3–Dependent Manner in
Parkinson's disease (PD)-mimicking drugs and pesticides, and more recently PD-associated gene mutations, have been studied in cell cultures and mammalian models to decipher the molecular basis of PD. Thus far, a dozen of genes have been identified that are responsible for inherited PD. However they only account for about 8% of PD cases, most of the cases likely involving environmental contributions. Environmental manganese (Mn) exposure represents an established risk factor for PD occurrence, and both PD and Mn-intoxicated patients display a characteristic extrapyramidal syndrome primarily involving dopaminergic (DAergic) neurodegeneration with shared common molecular mechanisms. To better understand the specificity of DAergic neurodegeneration, we studied Mn toxicity in vivo in Caenorhabditis elegans. Combining genetics and biochemical assays, we established that extracellular, and not intracellular, dopamine (DA) is responsible for Mn-induced DAergic neurodegeneration and that this process (1) requires functional DA-reuptake transporter (DAT-1) and (2) is associated with oxidative stress and lifespan reduction. Overexpression of the anti-oxidant transcription factor, SKN-1, affords protection against Mn toxicity, while the DA-dependency of Mn toxicity requires the NADPH dual-oxidase BLI-3. These results suggest that in vivo BLI-3 activity promotes the conversion of extracellular DA into toxic reactive species, which, in turn, can be taken up by DAT-1 in DAergic neurons, thus leading to oxidative stress and cell degeneration.
Published in the journal:
. PLoS Genet 6(8): e32767. doi:10.1371/journal.pgen.1001084
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001084
Summary
Parkinson's disease (PD)-mimicking drugs and pesticides, and more recently PD-associated gene mutations, have been studied in cell cultures and mammalian models to decipher the molecular basis of PD. Thus far, a dozen of genes have been identified that are responsible for inherited PD. However they only account for about 8% of PD cases, most of the cases likely involving environmental contributions. Environmental manganese (Mn) exposure represents an established risk factor for PD occurrence, and both PD and Mn-intoxicated patients display a characteristic extrapyramidal syndrome primarily involving dopaminergic (DAergic) neurodegeneration with shared common molecular mechanisms. To better understand the specificity of DAergic neurodegeneration, we studied Mn toxicity in vivo in Caenorhabditis elegans. Combining genetics and biochemical assays, we established that extracellular, and not intracellular, dopamine (DA) is responsible for Mn-induced DAergic neurodegeneration and that this process (1) requires functional DA-reuptake transporter (DAT-1) and (2) is associated with oxidative stress and lifespan reduction. Overexpression of the anti-oxidant transcription factor, SKN-1, affords protection against Mn toxicity, while the DA-dependency of Mn toxicity requires the NADPH dual-oxidase BLI-3. These results suggest that in vivo BLI-3 activity promotes the conversion of extracellular DA into toxic reactive species, which, in turn, can be taken up by DAT-1 in DAergic neurons, thus leading to oxidative stress and cell degeneration.
Introduction
Mn is the twelfth most prevalent natural element in the Earth's crust [1] and is an essential transition metal required for normal growth, development and cellular homeostasis [2], [3]. It acts as a cofactor for multiple enzymes (Mn superoxide dismutase, pyruvate carboxylase, arginase, and glutamine synthase) [4], [5], [6], [7], [8] and can substitute for magnesium (Mg) in many enzymatic reactions catalyzed by kinases. Although dietary Mn intake by the gastrointestinal tract (GIT) and excretion via the bile are tightly regulated [9], inhalation of toxic concentrations of Mn can lead to nasal and pulmonary inflammation, renal dysfunction and neurodegeneration [7], [10], [11], [12]. A recent study also suggests that high levels of Mn in drinking water (>300 µg/liter) are associated with reduced intellectual function in children [13]. Mn mining, steel manufacturing and welding represent occupational exposures linked to increased risk for parkinsonian syndrome [14], . In addition, Mn is used in other industrial and agricultural applications. Fungicides, such as Maneb or Mancozeb, increase the risk of environmental Mn exposure in agricultural workers [16]. An organic Mn compound, methylcyclopentadienyl Mn tricarbonyl (MMT), used as an octane booster or anti-knock agent in gasoline, has also been shown to cause adverse health effects [17], [18], [19], [20].
Exposure to high levels of Mn in occupational or environmental settings or disease conditions (hepatic encephalopathy) [21] is accompanied by Mn accumulation in specific brain regions that are highly sensitive to oxidative injury, namely the substantia nigra (SN), globus pallidus (GP) and striatum [22]. Excessive Mn deposition in these regions leads to dopaminergic (DAergic) neuronal loss accompanied by an extrapyramidal syndrome referred to as manganism. Manganism patients exhibit rigidity, tremor, dystonic movements and bradykinesia, all of which are also characteristic features of Parkinson's disease (PD) [23], [24], [25]. Exposure to Mn also represents a risk factor for PD [26], [27], [28]. Indeed, the strongest correlation between any type of environmental exposure and PD is noted in Mn-exposed human cohorts [29], and occupational exposure to Mn for >20 years or combined long-term exposures to Mn and Al (>30 years) are associated with an increased occurrence of PD [30]. Parkinsonism in welders (vs. non-welders) is clinically distinguishable only by age of onset (46 vs. 63 years, respectively) [28], and the prevalence of PD is higher among welders as compared with age-standardized individuals in the general population [31].
Appraisal of the literature strongly suggests that in addition to targeting similar brain areas and causing similar clinical syndromes, DAergic neurodegeneration associated with PD or PD-mimicking drugs (6-hydroxydopamine/6-OHDA, 1-methyl-4-phenylpyridium/MPP+, rotenone, paraquat) and Mn neurotoxicity share multiple common effector mechanisms, namely mitochondrial dysfunction, ATP depletion, aberrant signal transduction, oxidative stress, protein aggregation and the activation of cell death pathways [32]. Damage to DAergic nigral neurons that is induced by MPP+ and rotenone involves oxidative stress [33]. Oxidative damage also plays a significant role in 6-OHDA-induced DAergic neuronal cell death. In cell cultures, hydrogen peroxide (H2O2), superoxide ions and hydroxyl radicals [34] generated by the non-enzymatic breakdown of 6-OHDA and the direct inhibition of complex-I activity, lead to lipid peroxidation, protein denaturation and a decrease in glutathione (GSH), all hallmark features of post-mortem PD [35], [36], [37].
Intrastriatal Mn injections result in the loss of DAergic neurons, a process in which oxidative stress plays a significant role [38], [39], [40], resembling toxicity caused by the mitochondrial poisons, aminooxyacetic acid and MPP+ [41]. Similarly to mitochondrial inhibitors such as MPP+ [42], Mn increases in vivo synaptic glutamate concentrations, which leads to excitotoxic and oxidative injury [43] and interfers with ATP synthesis [44]. Analogous to MPP+ and 6-OHDA, Mn elevates intracellular H2O2 and related peroxides [45] and reduces tyrosine hydroxylase (TH) activity and intracellular antioxidant levels (GSH, thiols, catalase) in DAergic neurons [43], [46], [47], [48]. Intracellular Mn2+ inhibits the mitochondrial complex-I, a feature inherent to PD and its experimental models (MPP+, 6-OHDA, rotenone, paraquat) [43]. A link between mitochondrial impairment, oxidative stress and increased α-synuclein aggregation is well documented for Mn and in various models of PD [37], [43], [49], [50], [51]. Studies have also confirmed that treatment with Mn in a pre-parkinsonian state (6-OHDA) significantly exacerbates neurobehavioral impairment in the rat, not only suggesting that Mn exposure may increase the risk of injury in subpopulations that are in a pre-parkinsonism state, but also pointing to the convergence of signaling pathways that lead to such injury [52].
MPP+ and 6-OHDA exposures as well as wild-type or mutant α-synuclein overexpression cause specific DAergic neurodegeneration in the worm [53], , a process which involves ATP depletion and oxidative stress [36], [57], [58], analogous to vertebrate models of PD. It was further confirmed that C. elegans orthologues of PD-associated genes play a role in α-synuclein toxicity and DAergic neurodegeneration. Additionally, conserved genetic networks were identified in C. elegans that potentiate or protect against α-synuclein toxicity, such as the torsin pathway [36], [59], [60], [61], [62], [63], [64]. A link between α-synuclein and Mn toxicity was also demonstrated in the worm [59], [65]. Furthermore, we previously established that Mn uptake and toxicity pathways in C. elegans relate to those described in vertebrates, which involves the NRAMP/DMT family of metal transporters and leads to defects in the developmental and excretory systems [66]. Here we show that at a sub-lethal range of concentrations, acute Mn exposure leads to a specific and dose-dependent neurodegeneration of all C. elegans DAergic neurons, while sparing other neurotransmitter systems, findings that corroborate the specificity of DAergic sensitivity shared with vertebrate models. We investigated the causes of this DAergic specificity, and we demonstrated that endogenous extracellular, but not intracellular DA potentiates Mn toxicity and that Mn-induced neurodegeneration requires the DAergic neuron-specific dopamine re-uptake transporter, DAT-1. We also found that Mn toxicity in the worm is associated with increased reactive-oxygen species (ROS), lipid peroxidation and lifespan reduction, all of which were dependent on extracellular DA concentrations. Additionally, we observed a relocation of the oxidant-responsive transcription factor, SKN-1, in ASI nuclei upon Mn exposure, whereas SKN-1 overexpression afforded protection against Mn-induced toxicity. Finally, we identified the NADPH dual-oxidase, BLI-3, as a key mediator of the DA-dependency of Mn toxicity, suggesting that BLI-3 potentiates the formation of ROS from DA-derived species obtained through the reaction of divalent Mn and extracellular DA.
Results
C. elegans DAergic neurons degenerate in a specific and concentration-dependent manner upon exposure to Mn
We first ascertained the suitability of Mn-exposed C. elegans as an in vivo model for manganism and PD and examined whether features inherent to mammalian DAergic neurodegeneration can be convincingly recapitulated in this model. We took advantage of the BY200 strain, which expresses the green fluorescent protein (GFP) under the control of the DAergic-specific dopamine re-uptake transporter 1 promoter, dat-1::GFP(vtIs1) (Figure 1A). After acute exposure to Mn, a dose-dependent neurodegeneration was observed in all DAergic neurons, namely the 4 CEP, 2 ADE, 2 PDE and the male specific R5A, R7A and R9A pairs of neurons (Figure 1A and 1B). Typically, the primary defects were observed in neuron extensions, such as CEP mechanosensory processes, resulting in discontinued and punctuated GFP labeling (Figure 1A, 1B, 1D, arrowheads). With an increased dose (Figure 1D, upper to lower panels) or longer exposure to Mn (data not shown), these defects were exacerbated, leading to shortening or disappearance of the neuronal extensions (Figure 1D, lower panels) and eventually neuronal death as revealed by the shrinkage of the cell body, and ultimately, complete loss of GFP (data not shown, Figure 2A left panel). This effect was specific to DAergic neurons, since it was not observed in GABAergic, cholinergic, glutamatergic (Figure 1C) or in other biogenic-amine systems (data not shown). Despite the direct exposure of chemosensory neurons to the Mn-containing solution, DiI staining in these neurons failed to reveal any degeneration (Figure 1C), confirming the specificity of the Mn-induced DAergic neurodegeneration.


DAergic neurodegeneration requires the DA re-uptake transporter DAT-1
The selectivity of Mn-induced neurodegeneration to DAergic neurons suggests that some factor(s) specific to these neurons sensitize(s) them to Mn-induced toxicity. To determine which factor(s) may account for this effect, a candidate gene approach was employed, starting with a gene known to be specific to the DAergic neurons, namely, dat-1. DAT-1 is the C. elegans orthologue of the vertebrate DAT, which is a highly conserved member of a family of transporters involved in neurotransmitter clearance, including the GABA re-uptake transporters, the GATs, the serotonin re-uptake transporter, SERT, and the excitatory amino-acid transporters, the EAATs. DAT is specifically responsible for DA clearance at the synapse, removing excessive extracellular DA into presynaptic DAergic termini [67], [68], [69]. Accordingly, chemical inhibition of DAT or DAT loss-of-function leads to high extracellular DA levels [70], [71]. In C. elegans, the dat-1(ok157) loss-of-function mutant displays a swimming-induced paralysis (SWIP) phenotype, likely due to the hyper-activation of DA-responsive motorneurons exposed to excessive synaptic DA concentrations [71]. C. elegans dat-1 is also required for 6-OHDA DAergic neuron toxicity as noted in dat-1::GFP transgenic worms [56]. Given the specific neurodegeneration of dat-1::GFP-expressing neurons upon 6-OHDA or Mn exposure, we hypothesized that dat-1 is required for Mn-induced DAergic degeneration. Accordingly, dat-1(ok157) worms expressing dat-1::GFP were exposed to graded Mn doses in parallel with wild-type worms expressing the same dat-1::GFP array, and both strains were scored for DAergic neuron defects. Mn treatment was associated with marked DAergic neurodegeneration in wild-type worms, while dat-1 mutant DAergic neurons were not significantly affected even at the highest doses of Mn (Figure 2A) exposure for which fewer than 5% of the worms survived (Figure 2B). Accordingly, dat-1 mutant worms exposed to Mn most likely died from osmoregulation defects before displaying any neurodegeneration in DAergic neurons. These results established that DAergic neurodegeneration in Mn-exposed worms requires a functional DAT-1 transporter.
dat-1(ok157) mutant worms are hypersensitive to Mn-induced toxicity
Additional studies showed that the protective effect of dat-1 loss-of-function on DAergic neurodegeneration was not due to an indirect effect, such as an adaptive mechanism resulting in reduced Mn-uptake. First, the lethal dose 50 at 24 h (LD50, dose of Mn exposure at which 50% of the animals die from the treatment) for both transgenic strains, dat-1(ok157); dat-1::GFP and dat-1(+); dat-1::GFP, was determined. Notably, the dat-1(ok157); dat-1::GFP strain showed significant hypersensitivity (p<0.001) to Mn-induced toxicity with a LD50 = 20 mM, whereas the dat-1(+); dat-1::GFP strain exhibited a LD50 = 74 mM (Figure 2B). Next, the non-transgenic dat-1(ok157) and the N2 wild-type strains were treated with the same range of Mn doses (0.001 mM to 1 M). Both non-transgenic strains were slightly more sensitive than the corresponding dat-1::GFP-expressing strains (Figure 2C). This “protective effect” of the transgene expression was not specific to the dat-1::GFP construct and was systematically observed with any type of transcriptional GFP-expressing construct (data not shown). Transgenic worms may experience higher basal levels of stress associated with increased stress-response protein levels, enabling them to better cope with environmental stress. Nevertheless, dat-1(ok157) mutants exhibited hypersensitivity to Mn-induced lethality (p<0.001), which was characterized by a LD50 = 9 mM as compared to a LD50 = 47 mM for wild-type worms (Figure 2C). This observation rules out the possibility that dat-1(ok157) takes up less Mn. Moreover, it shows that upon Mn exposure, DAT-1 loss-of-function is detrimental to worm survival, though protective to DAergic neurons.
Sub-lethal exogenous DA pre-treatment potentiates Mn toxicity
The importance of a functional DAT-1 in conferring selective neurodegeneration in C. elegans DAergic neurons upon acute Mn exposure reflected upon earlier work with the PD-mimicking drug and DA analogue, 6-OHDA. Indeed, 6-OHDA induced a dose-dependent DAergic neurodegeneration in C. elegans, which was prevented by DAT-1 mutations [56], [72]. The fact that both Mn and 6-OHDA induce neurotoxicity through DAT-1 raises a question regarding the relationship between Mn and DA analogues upstream of DAT-1 in the DAergic neurodegeneration pathway. It has been hypothesized that Mn, in its divalent or trivalent cationic form, like iron, reacts with biogenic amines, such as DA, to generate ROS [73], [74], [75]. It is also known that DA and its derivatives, such as L-DOPA and 6-OHDA, can be toxic to the mammalian DAergic system and can lead to intracellular oxidative stress alone or in combination with metals and ensuing DAergic neurodegeneration [46], [76], [77]. Accordingly, we hypothesized that DA and Mn have a synergistic toxic effect in the worm. To test this hypothesis, worms were acutely treated with 10 mM DA prior to Mn exposure. DA pre-treatment led to a significant leftward shift in the Mn dose-response survival curve (p<0.001) with a LD50 = 25 mM (Figure 3A), while 10 mM acute DA treatment alone did not affect the worms' survival (Figure 3B). These results establish that exogenously applied DA and Mn can act synergistically in vivo to promote increased toxicity in the worm.

Endogenous DA potentiates Mn toxicity
In certain mutant backgrounds, such as vertebrate DAT mutants, DA levels were reported to be abnormally elevated due to a lack of DA clearance at the DAergic synapses [71]. Therefore, the effect of DAT-1 loss-of-function on Mn sensitivity may reflect increased endogenous levels of DA (Figure 4A). To test this hypothesis, we measured DA levels in the C. elegans dat-1(ok157) mutants. We found that dat-1(ok157) endogenous DA levels were significantly higher when compared with wild-type worms (Figure 4B). To confirm the role of endogenous DA in Mn toxicity, we tested the effect of endogenous DA depletion on Mn sensitivity using the cat-2(e1112) loss-of-function mutant, in which the tyrosine hydroxylase (TH) activity is abolished, resulting in the absence of DA synthesis (Figure 4A and 4B). cat-2(e1112) mutants exposed to Mn revealed significant hyper-resistance (p<0.001), with a LD50 = 95 mM (Figure 4C). Moreover, the e1112 deletion rescued the dat-1(ok157) hypersensitivity (LD50 = 9 mM, Figure 3A) and led to hyper-resistance to Mn (p<0.001) with a LD50 = 83 mM for cat-2(e1112);dat-1(ok157) worms (Figure 4C). The LD50 difference between cat-2(e1112) and cat-2(e1112);dat-1(ok157) worm strains was not statistically significant (p>0.05), suggesting that the cat-2(e1112) effect takes place upstream of dat-1(ok157) in the same pathway (Figure 4A). Finally, we supplemented cat-2(e1112) mutants with 10 mM DA exposure prior Mn treatment, which rendered those mutants hypersensitive (p<0.001) to Mn with a LD50 = 34 mM (Figure 4C). Pre-exposure to 10 mM DA of cat-2(e1112);dat-1(ok157) worms led to a comparable LD50 = 40 mM, not significantly different from cat-2(e1112) mutants pre-exposed to 10 mM DA, but significantly different from cat-2(e1112);dat-1(ok157) without DA pre-exposure (Figure 4C). These data further confirm that endogenous DA level regulation plays a key role in Mn toxicity and that DA itself is required for the full extent of Mn toxicity.

Extracellular DA, and not intracellular DA, is involved in Mn-induced lethality
Next, we determined which step along the DAergic metabolic pathway contributes to the synergistic toxic effect of Mn. To address this issue, endogenous DA levels at the L1 stage were measured in a combination of deletion mutants (Figure 4A). First, we sought to determine if DA release was necessary for the DA-dependent Mn-induced lethality. Accordingly, cat-1(e1111) mutants in which the vesicular monoamine transporter 2 (VMAT2) is defective, were exposed to Mn (Figure 4B). This mutant is unable to pack DA in secretory vesicles, thus abolishing DA synaptic release, while still producing normal levels of DA, at least at the L1 stage (Figure 4A). Upon exposure to Mn, cat-1(e1111) mutants were more resistant to Mn-induced lethality than wild-type worms (p<0.001), with a LD50 = 108 mM (Figure 4D), which was statistically insignificant when compared to the cat-2(e1112) mutant LD50 (p>0.05). These data establish that synaptic DA release is necessary for DA-dependent Mn-induced lethality and that extracellular DA, and not presynaptic DA, mediates this effect.
Furthermore, the data imply that blockage of extracellular DA receptor activity (and the ensuing increase in extracellular DA levels) should exacerbate Mn sensitivity. C. elegans expresses three DA receptors, namely DOP-1, DOP-2 and DOP-3 (Figure 4A). A triple knock-out was generated [78], and as expected, the dop-2(vs105); dop-1(vs100) dop-3(vs106) mutant exhibited higher levels of DA (Figure 4B) and was hypersensitive (p<0.001 vs. wild-type) to Mn-induced lethality with a LD50 = 27 mM (Figure 4D). However, this mutant was not as sensitive to Mn as the dat-1(ok157) mutant, probably because the DA clearance activity of the wild-type DAT-1 limited the extent of the extracellular DA accumulation in the dop-2(vs105); dop-1(vs100) dop-3(vs106) triple mutant. These results confirm that extracellular, and not intracellular DA is responsible for the DA-dependent Mn-induced lethality.
DA-dependent Mn toxicity is associated with decreased longevity
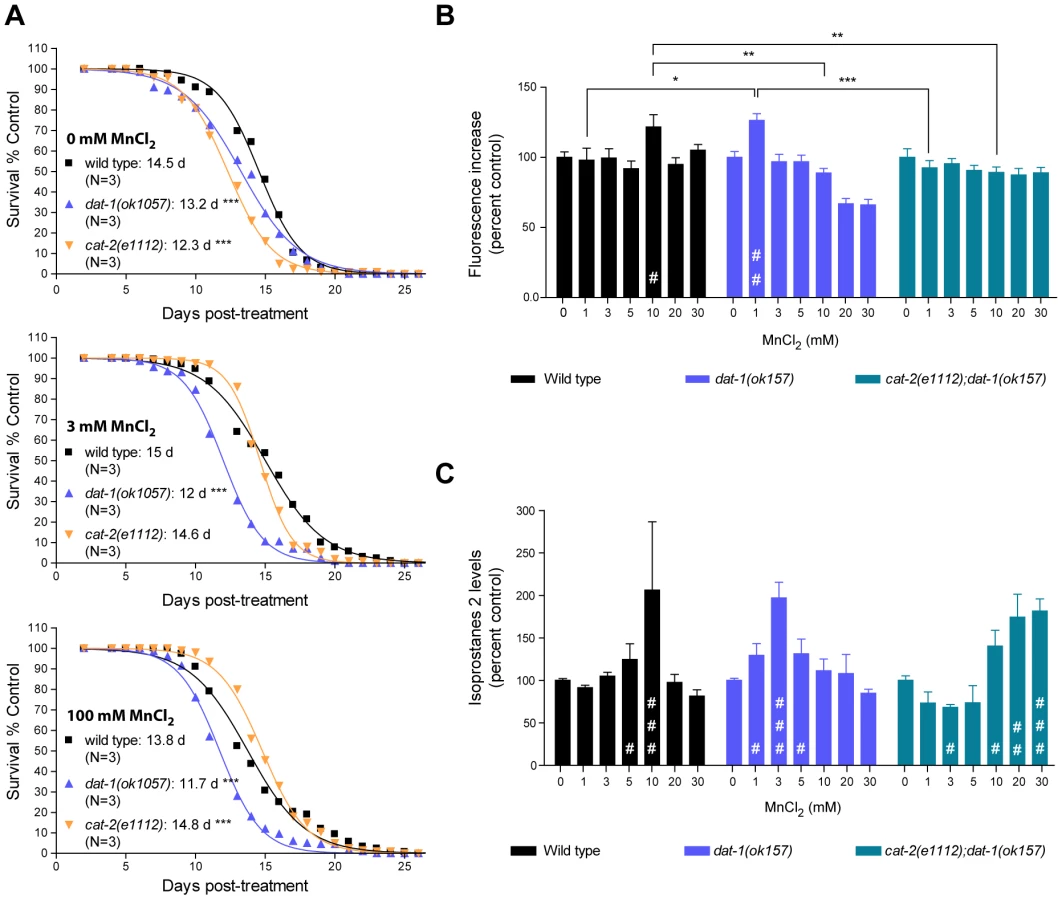
To further investigate the pathways involved in the DA-dependent Mn toxicity, and especially its long-term effects, we scored the survival rate of worms which survived acute Mn-exposure. For each strain tested, we picked healthy-looking young adults homogeneous in stage at 48 h post-exposure (for all strains tested these animals were representative of more than 90% of the surviving population), and disregarded sick-looking or developmentally delayed worms. Accordingly, all animals selected survived at least 4 days in adulthood, regardless of their genetic background. This bias was deemed necessary to ensure that any significant differences potentially observed between strains could not be attributed to early toxicity events. In accordance with previous reports [79], low doses of Mn (3 mM for 30 min) led to a minor (albeit insignificant) increase in the surviving wild-type lifespan from 14.5 days post-treatment (dpt) to 15 dpt (Figure 5A, upper and middle graphs). Higher doses of Mn (100 mM for 30 min) led to a decrease in lifespan from 14.5 dpt to 13.8 dpt (p<0.001, Figure 5A, bottom graph). dat-1(ok1057) mutants exhibited a significantly shorter lifespan compared with wild-type worms in the absence of Mn treatment (13.2 dpt, p<0.001), which was further reduced (p<0.001) by both low - and high-dose acute Mn treatments (12 dpt and 11.7 dpt, respectively). Conversely, the lifespan of short-lived cat-2(e1112) mutants (12.3 dpt in absence of Mn, p<0.001) was significantly increased with increased Mn dosing (p<0.001, 14.6 dpt upon 3 mM and 14.8 dpt upon 100 mM acute Mn exposure). Moreover, the lifespan of the cat-2(e1112) mutants was significantly extended as compared to wild-type worms upon 100 mM Mn exposure (14.8 dpt vs 13.8 dpt, p<0.001), reaching untreated wild-type worm average lifespan (14.8 dpt vs 14.5 dpt). These observations made on worms, all of which survived for at least 5 days post-treatment, mirror the results obtained for lethality rates at 24 h post-treatment, indicating that the DA-dependent Mn toxicity observed upon acute Mn treatment has short - and long-term components, affecting both developmental (see also [66]) and aging processes.
DA-dependent Mn-induced toxicity is associated with increased oxidative stress
Toxicity mechanisms implicated in neurodegenerative diseases, such as PD and Alzheimer's disease, involve oxidative stress [32], [35], [37], [80], [81], [82], [83]. In particular, DAergic cell loss in PD patients and experimental PD models invokes excessive ROS production [77], [84], [85], [86], [87], [88]. Moreover, oxidizing metals such as copper, Mn and iron in their 2+ or 3+ states are known to be sources of ROS via the Fenton reaction [89]. In C. elegans, pre-treatment with anti-oxidants such as ebselen, affords protection against acutely-induced Mn toxicity (Avila and Aschner, unpublished data). To determine if oxidative stress plays a role in DA-dependent Mn toxicity in C. elegans, a double-pronged approach was undertaken. First, the presence of ROS in response to acute Mn treatment was determined with the fluorescent dye, 2′7′ dichlorodihydrofluorescein diacetate (H2DCF-DA). As shown in Figure 5B, Mn-treated wild-type worms showed a significant (p<0.05) increase in fluorescence at sub-lethal Mn doses (10 mM). Interestingly, dat-1(ok157) mutants showed a significant increase in fluorescence (p<0.01) after 1 mM exposure, whereas cat-2(e1112); dat-1(ok157) double mutants did not show any significant increase in fluorescence (p>0.05) upon exposure up to 30 mM Mn (Figure 5B). To confirm these results, we also used a more quantitative method involving the measurement of lipid peroxidation. Isoprostanes F2 (F2IP) and F3 (F3IP) are oxidation products of arachidonic acid (AA), which is released from membranes upon oxidative injury [90], [91], [92]. A new protocol was developed to extract and measure F2IP and F3IP from C. elegans. Corroborating the H2DCF-DA results (Figure 5B), dat-1(ok157) mutants exhibited significantly higher levels of F2IP (Figure 5C) and F3IP (data not shown) upon 1, 3 and 5 mM Mn exposure (p<0.05, p<0.001, p<0.05, respectively), as compared to wild-type worms, which showed a significant increase in F2IP content upon 5 and 10 mM Mn exposure (p<0.05 and p<0.001, respectively) (Figure 5C). The double mutant cat-2(e1112);dat-1(ok157) consistently displayed decreased F2IP levels between 1 and 5 mM Mn, with significantly lower levels at 3 mM (p<0.05), and exhibited significantly higher F2IP levels upon 10, 20 and 30 mM acute Mn exposure (p<0.05, p<0.01, p<0.001, respectively) (Figure 5C). Thus, to attain maximal levels of F2IP (corresponding to a 2-fold increase), higher doses of Mn were required in cat-2(e1112);dat-1(ok157) mutants compared to wild-type and in wild-type compared to dat-1(ok157). Taken together, the H2DCF-DA and isoprostane measurements show that acute Mn treatments induce oxidative stress in C. elegans. In addition, the degree of oxidative stress depends on the extracellular DA content. These experiments strongly suggest that the DA-dependent Mn-induced toxicity involves oxidative stress.
The anti-oxidant transcription factor, SKN-1, protects against Mn toxicity and relocalizes in amphide sensilla ASI neuron nuclei upon Mn exposure
An alternative functional way to establish oxidative stress, especially in its earliest stages, is to demonstrate a physiological response to it. Sodium arsenite exposure in C. elegans has previously been shown to induce a strong intestinal expression of the anti-oxidant response gene, skn-1, the orthologue of the vertebrate gene, Nrf2 [93]. SKN-1 initiates the development of the digestive system and feeding during the earliest embryonic stages, and post-embryonically, is required for a normal lifespan and adequate resistance to stress [94], [95], [96]. Consistent with these observations, skn-1 deletions or loss-of-function mutations suppress oxidative stress resistance [97]. Aging in C. elegans is delayed when SKN-1 is transgenically expressed, and a mutant skn-1 form that constitutively localizes to nuclei increases the worm's lifespan [94], [97], [98], [99]. To determine if SKN-1 can afford protection against Mn toxicity and to confirm that oxidative stress plays an important role in Mn-induced toxicity in C. elegans, SKN-1::GFP over-expressing worms were exposed to Mn. These worms exhibited a strong hyper-resistant phenotype to Mn exposure with a LD50 = 114 mM (Figure 6A). This effect was significantly greater in comparison to wild-type (p<0.001) or GFP expression alone (p<0.01) under various promoters (cf. dat-1::GFP, Figure 2; data not shown) despite the aforementioned protective effect of GFP expression, suggesting that SKN-1 directly protects against Mn toxicity. Moreover, a mixed population of two-third heterozygous and one-third homozygous deletion-mutant skn-1(ok2315) expressing a truncated SKN-1 protein, exhibited increased sensitivity to Mn exposure with a LD50 = 34 mM (Figure 6A), confirming that SKN-1 is required for mediating optimal resistance to Mn exposure. Unexpectedly, no obvious increase in SKN-1::GFP intestinal expression was noted upon Mn exposure, possibly because SKN-1 was already over-expressed or because it is not activated by Mn. However, a notable change in the SKN-1::GFP localization pattern in the ASI neuron was associated with Mn exposure, where SKN-1::GFP relocalized in discrete nuclear puncta, a result distinctly different from the diffuse pattern exhibited in non-Mn exposed worms (Figure 6B). This change was associated with a significantly increased average nuclear density of SKN-1::GFP (p<0.05, Figure 6C) but a non-significant increase in ASI expression, as revealed by the integral GFP intensity measurements (Figure 6D). These results support the notion that Mn exposure triggers an increase in ROS levels, thus activating the antioxidant response pathway, at least in ASI neurons. The data also indicate that the nuclear relocation of SKN-1, rather than its increased expression, is responsible for this protective effect against Mn toxicity.

The dual-oxidase mutant bli-3(e767) is resistant to DA-dependent Mn toxicity
Given that Mn induces DA-dependent oxidative stress, we next sought to identify genetic factors involved in this process. In addition to the mitochondrial electron-transport chain (ETC), plasma membrane NADPH oxidases are major contributors to ROS production in rotenone, paraquat and MPP+-induced toxicity [86], [100], [101], [102], [103]. The bli-3 gene encodes a C. elegans dual-oxidase orthologue to vertebrates DUOX1 and DUOX2, which is involved in the di-tyrosine bond formation in the worm cuticle [104] and pathogen-induced ROS production [105], [106]. Di-tyrosine bonds maintain cuticle integrity, and bli-3(e767) mutants display a blistered cuticle and a mild dumpy phenotype. This phenotype renders worms more sensitive to toxicants due to increased absorbance of the toxicants through the damaged cuticle [104]. Interestingly, bli-3(e767) worms displayed hyper-resistance to acute Mn treatments when compared to wild-type worms (p<0.001), with a LD50 = 83 mM (Figure 7A), suggesting that BLI-3 is involved in mediating Mn-toxicity, most likely by potentiating ROS production and oxidative stress. Notably, sub-lethal (Figure 7B) DA pre-treatment did not affect bli-3(e767) worm sensitivity to Mn: LD50 = 84mM (Figure 7A). Moreover, similar to cat-2(e1112) mutants (Figure 5B), bli-3(e767) mutants did not show any dose-dependent increase in ROS production from 1 mM to 30 mM Mn exposures (Figure 7C). ANOVA (comparing data from Figure 5B and Figure 7C) reveals that bli-3(e767) mutants show a significant difference in ROS production compared to dat-1(ok157) worms at 3 mM (p<0.001) and compared to wild-type worms at 10 mM (p<0.01), but no difference compared to cat-2(e1112) worms at any Mn concentration tested.

These observations strongly support the notion that BLI-3 is required for the ROS-associated potentiating effect of DA in Mn toxicity. Given the structural analogy between DA and tyrosine, DA or DA-derived species formed upon reaction with Mn may serve as substrates for BLI-3, facilitating their conversion to highly oxidized species, and in turn, potentiating oxidative stress.
Discussion
This study establishes that Mn induces a specific and dose-dependent neurodegeneration of DAergic neurons in C. elegans, corroborating findings in rodents and non-human primates. This neurodegeneration was proven to require the DA re-uptake transporter, DAT-1, which is also the case in 6-OHDA-induced neurotoxicity. Combined, these observations suggest that upon Mn exposure, DAT-1 facilitates the intracellular transport of toxic species into DAergic neurons. Mn toxicity was associated with increased ROS generation and elevated F2IP levels as well as reduced longevity, all of which were aggravated by high endogenous DA levels and were attenuated by DA depletion. Consistent with a Mn-induced increase in oxidative stress, nuclear relocation of the anti-oxidant transcription factor, SKN-1, was noted in ASI neurons, and SKN-1 overexpression afforded protection against Mn-induced toxicity, while skn-1 deletion rendered worms more vulnerable to Mn toxicity. Increased extracellular, but not intracellular DA levels resulting from direct DA exposure or genetic manipulation potentiated Mn-induced lethality, oxidative-stress and reduction in lifespan. Finally, our studies establish that the dual-oxidase, BLI-3, is involved in the DA-dependent Mn-induced toxicity and that bli-3 loss-of-function suppresses the DA-dependency of Mn toxicity and associated oxidative stress. Combined, these data suggest that oxidation of extracellular DA promoted by the BLI-3 dual-oxidase activity triggers DA-derived ROS generation, which, in turn, are taken up into the DAergic terminals by DAT-1 and lead to specific DAergic neurodegeneration (Figure 8).

Relevance of our findings to the study of manganism and PD in mammalian models
Mn-treated C. elegans recapitulate essential physiological aspects of parkinsonism, namely: the importance of NRAMP/DMT orthologues in the toxicity process [107], [108], [109]; the specificity and dose-dependency of the DAergic neurodegeneration [110], [111]; the involvement of DAT [112], [113], [114]; the synergy between DA and Mn [115]; and the associated oxidative stress [116], [117], [118].
As many divalent metallic cations are able to oxidize biogenic amines (amongst which DA and serotonin) in vitro via the Fenton's reaction, we carried out two control experiments to ascertain the specificity of the relationship between DA and Mn in vivo. Unlike dat-1 mutation, deletion of the serotonin transporter (mod-5 in C. elegans) did not affect Mn-induced lethality (LD50 = 45 mM, Figure S1), and DA-depleted cat-2(e1112);dat-1(ok157) mutants revealed greater susceptibility to iron-induced ROS production (p<0.05) than wild-type worms (Figure S2), supporting the fact that in vivo, metal-induced DA-dependent toxicity is specific to Mn. An additional novelty resides in the finding of the early and central role played by endogenous extracellular DA and the NADPH dual-oxidase in Mn-induced toxicity (Figure 4, Figure 5, and Figure 7). The centrality of extracellular DA in Mn-induced toxicity was further exemplified by the level of resistance to Mn conferred by mutations in cat-1 (LD50 = 108 mM) and cat-2 (LD50 = 95 mM), which approach those conferred by deletion of the Mn transporter, smf-3 (LD50 = 126 mM) [66]. Moreover, Mn in its normal toxic range had a strong beneficial effect on the lifespan of DA-depleted cat-2(e1112) mutants (Figure 5A), further confirming that the DA-dependent component of Mn-induced toxicity accounts for a significant share of the overall toxicity. Importantly, the hypersensitivity to Mn of the DA receptor triple-knockout (Figure 4D), shows that Mn-induced neurodegeneration does not require DA receptors. Combined with the absence of post-synaptic neurodegeneration in dop-1::GFP and dop-3::RFP-expressing worms upon Mn exposure (data not shown) and despite the excitotoxic potential of other DA-related compounds, such as L-DOPA and 6-OHDA [119], the results establish that DA-dependent Mn toxicity does not involve excitotoxicity, in contrary to the glutamate-induced neurodegeneration involved in amyotrophic lateral sclerosis (ALS) [120]. Together, these results also provide a novel explanation for the requirement of DAT in the DAergic neurodegeneration in vivo. Rather than being responsible for cytosolic DA accumulation followed by downstream ROS production, which was considered as the reason for the sensitivity of DAergic neurons to PD-mimicking drugs (MPP+, 6-OHDA) [56], [114], [121], [122], DAT may facilitate the transport of extracellulary oxidized DA-derived species into DAergic neurons. In man and rat, subchronic Mn exposure has been shown to reduce DAT expression levels [123], [124]. Although it is unclear if it reflected a decline of DAergic neuron activity or a specific down-regulation of DAT, Mn interference with DAT activity should be further investigated. The importance of the NADPH dual-oxidase, BLI-3, in the DA-dependent Mn toxicity in C. elegans corroborates the up-regulation of its mammalian orthologue in PD patients and MPP+-exposed mice, as well as the protective effect conferred by its loss-of-function against oxidative stress and DAergic neurodegeneration in MPP+-exposed mutant mice [86]. Finally, DA-dependent Mn-induced toxicity has both short and long-term components as revealed by survival rate and oxidative stress measurements at 24 h (Figure 4, Figure 5B and 5C) and by lifespan data (Figure 5A). The protective effect afforded by SKN-1 upregulation upon Mn exposure and its nuclear relocation in ASI neurons (Figure 6) emphasizes the influence of acute Mn treatments on aging. ASI neurons which strongly express SKN-1, were shown to play a key role in lifespan modulation through the well-studied diet-restriction pathway [125]. Hence, further examination of Mn-induced toxicity and lifespan reduction in ASI neuron-ablated worms, skn-1(RNAi) worms and mutants of the diet-restriction pathway may provide essential insights in the relationship between aging and PD, as well as on the role of specific brain area (corresponding to the C. elegans ASI neurons) on aging and PD.
Despite the absence of an obvious PD-like behavior in the worm, DAergic neurodegeneration in C. elegans induced either by PD-drug treatment (6-OHDA, DA, MPP+) or by Mn involves the same key genes and molecular pathways as in vertebrates [32], [36], [54], [59], [60], [64], [65], [66], [126], [127], [128], has both short-term and long-term components and therefore represents a powerful model to investigate genetic and environmental causes of PD and manganism. To further demonstrate the relevance of our findings to vertebrate physiology, it would be interesting to test (1) whether co-treatments of DA and Mn lead to the same synergy in DAergic neurodegeneration in rodents; (2) whether such synergy is dependent on DAT; and (3) whether tyrosine hydroxylase (TH/CAT-2) and VMAT inhibition affords neuroprotection. Establishing the contribution of vertebrate NADPH dual-oxidases to the oxidation state of DA in the absence or presence of Mn would also be important. The present study also revealed DA-dependent mechanisms of Mn-induced toxicity by focusing on a whole-organism approach, while acknowledging that further investigation should also focus specifically on DAergic neuron physiology. For instance, determining if upon Mn exposure, cat-2, cat-1 and cat-2;dat-1 mutants display less or no neurodegeneration and if DA supplementation can reverse this effect, would allow clearer understanding of the role of DAT, and intracellular and extracellular DA in the demise of the DAergic system. It would also be important to develop full-length GFP-tagged protein markers of the DAergic neurons as the dat-1::GFP probe can only reveal relatively advanced stages of neuronal decay, and not more subtle functional changes. This point is exemplified by the observation of SKN-1::GFP relocation in ASI neurons (Figure 6), which would have been missed if only a transcriptional GFP reporter approach had been used. Finally, exploring the effect of aging and diet-restriction modulated by the ASI neurons and the Nrf2-like transcription factor, SKN-1, would enhance our understanding of the pathophysiology and the progressive nature of PD.
Further developments and implications for the treatment of DAergic neurodegeneration
Given the similarities between Mn toxicity in C. elegans and in vertebrates [66] as well as the great conservation of most PD-related genes involved in DAergic neurodegeneration in rodents and humans [32], [36], [59], [60], [64], [129], it is likely that the DA-dependent toxicity revealed herein plays an important role in diseases associated with DAergic neuron loss in mammals. The importance of extracellular DA levels in the toxicity mechanisms described here, if corroborated in vertebrates, could bear important implications for the treatment of DAergic neurodegeneration, as modulating extracellular vs. intracellular DA levels requires different strategies. For instance, L-DOPA is prescribed in PD and manganism patients to treat the tremor and bradykinesia arising from the loss of DAergic activity. If L-DOPA, like DA, led to the excessive generation of oxidized reactive species in vivo, L-DOPA treatment, although compensating for the DA loss, could accelerate or exacerbate the DAergic neurodegeneration over a longer term. If the involvement of dual-oxidases in DA oxidation was confirmed in vertebrates and if L-DOPA was shown to be easily oxidized by dual-oxidases, it would be important to design alternative DA analogues, which maintain high affinity for DA receptors and DAT but are poor substrates for the dual-oxidases. Another strategy to limit the extent of the DAergic neurodegeneration could be the direct inhibition of the dual-oxidases. Finally, the protective effect of SKN-1 overexpression on Mn-induced toxicity (Figure 6A), which mirrors the neuroprotection afforded by astrocytic overexpression of Nrf2 in mice [130], suggests that promoting Nrf2 activity may be beneficial in limiting Mn-induced toxicity [131], [132]. Such an approach would be particularly relevant to welding and smelting activities in which workers exposed to metal fumes have an increased risk of developing parkinsonian syndromes [28], [133], [134], [135], [136].
Nature of the DA-derived oxidized species and oxidative-stress pathways involved in PD
The current literature provides a conceptual framework for addressing the synergistic mechanism of Mn and DA in DAergic neurodegeneration [76], [137], [138], [139], [140], [141]. Accordingly, Mn may enter DAergic neurons via DAT as a complex with DA-derivatives, such as dopaminochrome [138], [141], [142], [143], [144], [145], [146], [147]. This hypothesis provides explanations both for the specificity of Mn toxicity towards DAergic neurons as well as for the synergistic toxicity of extracellular DA and Mn, while remaining consistent with studies that report a significant Mn uptake by the DAergic neurons [148], [149], [150], [151]. Purification and identification of the DA-derived reactive species (using HPLC-ED [152]) are essential for proving these suggestions. Further quantification of those DA-derived species in Mn-, MPP+-, 6-OHDA - and DA-exposed worms or in the rodent brain and cerebrospinal fluid should help identify the key toxic species in DAergic neurodegeneration as well as point out candidate enzymes possibly involved in PD. However, in biochemical approaches, the timing of extraction is critical to robustly detecting and measuring those species. This concern is exemplified by our oxidative stress marker measurements in wild-type and dat-1(ok157) mutant worms (Figure 5B and 5C). Hence, above the Mn dose reaching the maximum increase in ROS or F2IP levels, higher Mn doses led to lower oxidative stress marker levels suggesting a decrease in oxidative stress, while the lethality rate at 24 h and lifespan results suggest otherwise (Figure 4, Figure 5A). It is unlikely that oxidative stress decreased past the dose showing the maximum increase in oxidative stress markers. Downstream conversion or degradation of ROS or F2IP associated with a worsening of the condition of the worm may be responsible for this effect. Using genetic backgrounds that stop or delay the oxidative cascade at different steps of the pathway would allow the accumulation of specific ROS and increase their detection. Hence, Mn-resistant C. elegans mutants exposed to medium - to high-dose treatments (in the 20 to 100 mM range) could be very helpful in identifying the early steps of the oxidative cascade(s) leading to DAergic neurodegeneration and could yield new molecular targets for PD treatment. This study identifies a few of the potential candidates (bli-3, SKN-1 overexpressing and maybe cat-1 mutant worms, Figure 4 and Figure 6), but genetic screens to isolate new Mn-resistant mutants would provide powerful means for further investigation.
Role of Mn and DA in C. elegans longevity
In the course of this study, we came across several indications of the influence of the DA-dependent and Mn-induced toxicity on C. elegans longevity, which is unlikely to be a direct consequence of DAergic neurodegeneration, but rather a concomitant effect (dat-1(ok157) mutants did not show DAergic neurodegeneration, whereas their lifespan decreased upon Mn exposure).
First, the fact that SKN-1 protects against Mn-induced toxicity and modifies its nuclear localization pattern upon Mn exposure in ASI neurons (Figure 6), provides a genetic link with the Insulin/IGF-1 and caloric restriction pathway known to be involved in the modulation of lifespan in C. elegans [97]. The nuclear relocation of SKN-1 in ASI neurons in a punctuate pattern is suggestive of binding of this transcription factor to specific chromosomal regions, likely corresponding to the loci of its downstream targets. Chromatine immunoprecipitation experiments could be used to identify those loci, while generation of transgenic worms expressing SKN-1::GFP and tagged sequences corresponding to candidate gene (for instance the superoxide dismutase genes) regulatory sequences would allow immuno-colocalization of SKN-1 and its targets. Nrf2/SKN-1 is known to be a key regulator of antioxidant response from man to worm [94], [99], and its impact on lifespan could as well be due to its ability to reduce oxidative stress naturally occurring with age, without involving caloric restriction. Given the impact of Mn exposure and DA metabolism on oxidative stress (Figure 5B and 5C), SKN-1 overexpression may improve Mn-exposed worm survival merely by compensating for the excessive ROS produced when extracellular DA levels are high and/or when Mn exposure reaches toxicity levels. Interestingly, the survival dose-response curve obtained with a non-null mutation (ok2315) in a mixed population of heterozygous and homozygous animals (Figure 6A) showed that even for very low doses of Mn (0.001 and 0.01 mM), a noticeable fraction of the worms (about 15–20%) died at 24 h post treatment compared to untreated animals, making it difficult to fit the experimental data with a simple sigmoid dose-response curve. As it is a mixed population, it is likely that mostly homozygous mutants died at those lowest doses, suggesting that their LD50 could be in the submillimolar range, making skn-1(ok2315) worms the most Mn-sensitive mutants tested so far. This requires direct confirmation, but it further supports an essential role for SKN-1 in regulating even slight changes in oxidative stress levels.
Second, our lifespan experiments showed that both dat-1 and cat-2 mutants are short-lived in control conditions (no published literature could be identified reporting on the lifespan of these mutants) compared to wild-type worms (Figure 5A). According to our ROS and isoprostane measurements (Figure 5B and 5C), dat-1 mutants show increased oxidative stress, which is known to worsen with age; therefore explaining their shorter lifespan. Conversely, cat-2 mutants seemed much less affected by Mn-induced oxidative stress, as Mn treatment did not lead to detectable increase in ROS and only led to a significant increase in isoprostane levels above 10 mM of Mn exposure. Moreover, high Mn exposure was able to rescue the shorter lifespan of cat-2 mutants (Figure 5A). Mn is naturally required for the antioxidant activities of several enzymes, such as catalase and superoxide dismutase, which activities are critical in the aging brain [153], [154], [155]. Mn also has bactericidal and fungicidal [156] properties that are exploited in pesticides, such as Maneb. Several hypothesis can be formulated: (1) cat-2 mutants may take up less Mn than wild-type worms, possibly below optimal levels, (2) DA depletion in cat-2 mutants could make them feed improperly and somehow normally toxic Mn doses would restore the metabolic balance in those worms, (3) cat-2 worms may be prone to infections and high Mn exposure would help the worms cope with a weak immunity. The first point could involve DA as a regulator of feeding behavior. This aspect can be tested by measuring Mn levels, the basal slowing response [157], sharp angle turns [158] and the pharyngeal pumping rate of dat-1, cat-2, cat-2;dat-1 and wild-type animals upon Mn exposure. Those tests would also allow functional characterization of the DAergic circuit of Mn-exposed worms. The second hypothesis involves energy depletion as a cause of shortened lifespan, whereas the stress of Mn exposure would restrict energy expenditure by inhibiting the Insulin/IGF-1 pathway, possibly involving SKN-1 activation. In this case, Mn intake may not be different from other worms. Mn level measurements, Nile red and oil red O [159] staining to monitor fat stores, as well as RT-PCR and western-blots to measure daf-2, akt-1/-2, sgk-1, daf-16 and skn-1 expression levels would allow to test this idea. The third hypothesis implies that DA-derived ROS naturally play a role in the worm's immunity and that a high Mn dose compensates for their absence in cat-2 mutants. The reasoning behind this idea is detailed in the next paragraph. This could be easily tested by comparing the resistance to infections of wild-type, dat-1, cat-2, cat-2;dat-1 and bli-3 mutants in presence or absence of Mn.
Mn, DA, BLI-3: a link between immunity and neurodegeneration?
Our data show that DA and BLI-3-dependent ROS production is triggered or amplified by Mn, which was shown to be taken up by the NRAMP/DMT orthologues, smf-1, -2 and -3 [66]. Recently, bli-3 and BLI-3-generated ROS have been implicated in C. elegans defense against bacteria and fungi [105], [106]. The NRAMP/DMT family of metal transporters has a well-established role in innate immunity in vertebrates [160], [161], [162], [163]. In C. elegans, NRAMP/DMT orthologue deletion mutants were found to be hypersensitive to Staphyloccocus aureus infection, which was rescued by Mn feeding [164]. Involvement of BLI-3-generated ROS, Mn and NRAMP/DMT in the defense against pathogens in C. elegans, as well as in DA-dependent neurodegeneration, raises the question of a link between immunity and DAergic neurodegeneration. The finding that DA and Mn act synergistically in a BLI-3-dependent ROS production pathway suggests that DA may play a direct role in the worm's immunity. If this mechanism is conserved in vertebrates, perhaps as a relic of a primitive immune system, it could bear interesting implications for brain physiology. For instance, DA-derived ROS could help fight infections when the blood-brain barrier (BBB) is compromised. On the other hand, they could also injure the brain and more specifically the DAT-expressing DAergic neurons. Infections and inflammation have long been suspected to play a role in the etiology of PD, as various infections have been associated with cases of PD [165], [166], [167], [168], [169], [170], [171], [172]. The identification of a DA-derived ROS production mechanism implicating dual oxidases may hold some clues for the understanding of those associations.
Summary
This study confirms the conservation across the animal kingdom of molecular pathways involved in manganism and PD, provides the grounds for further biochemical and genetic investigations using the C. elegans model to tackle the complex issue of environment-gene interactions in age-related DAergic neurodegenerative disorders, and unravels the essential role of extracellular DA oxidation and the NADPH dual-oxidase, BLI-3, upstream of DAT-1 requirement in the neurodegeneration pathway. The data also point to a genetic link with distinct and more general aging processes, such as the diet-restriction pathway through the involvement of SKN-1, and with an innate immunity genetic network involving metal-content regulation via the SMF transporters and oxidative defence mechanisms via BLI-3 in C. elegans.
Materials and Methods
C. elegans strains and handling of the worms
C. elegans strains were handled and maintained at 20°C as previously described [173]. The following strains were used: N2 (+); BY200, dat-1::GFP(vtIs1) V; BZ555, dat-1::GFP(egIs1); VH15, glr-1::GFP(rhIS4) III; LX929, unc-17::GFP(vsIs48); EG1285, lin-15B(n765) unc-47::GFP(oxIs12) X; DA1240, adIs1240[lin-15((+) eat-4::GFP) lin-15B(n765) X; LX734, dop-2(vs105) V; dop-1(vs100) dop-3(vs106) X; LX831, lin-15B(n765) X; DOP-1::GFP(vsIs28) dop-3::RFP (vsIs33); RM2702, dat-1(ok157) III; MT9772, mod-5(n3314) I; CB1111, cat-1(e1111) X; CB1112, cat-2(e1112) II; CB767, bli-3(e767) I; BY602, cat-2(e1112) II; dat-1(ok157) III; BY645, dat-1(ok157) III; dat-1::GFP(vtIs1) V; VC1772, skn-1(ok2315) IV;nT1[qIs51](IV;V). All strains were provided by the Caenorhabditis Genetic Center (CGC, Minnesota), except for the BY602 and BY645 strains, which were generously provided by Randy Blakely (Vanderbilt University Medical Center, TN, USA).
Acute manganese chloride treatments
Acute (30 min) manganese chloride (MnCl2) treatments were performed on 5,000 synchronized L1 per sample, and live worms were scored 24 h later, as previously described [66]. Scores were normalized to percent control (0 mM MnCl2 exposure).
Acute dopamine treatments
Dopamine (Sigma Chemical Co., St Louis, MO) solutions were prepared in M9 buffer. Acute (30 min) treatments on young L1 worms were first tested from 1 mM to 50 mM DA, guiding us in choosing 10 mM as the working sub-lethal dose. Synchronized L1 were acutely pre-treated with 10 mM DA for 10 min, washed 5 times in 85 mM NaCl solution and subjected to MnCl2 acute treatments. Control worms were pre-treated with M9.
Dopamine content measurement
Synchronized L1 worms were collected, washed three times in 85 mM NaCl and distributed in tetraplicates of 200,000 L1. Worms were pelleted and the supernatant was removed. The tubes were then immediately frozen in liquid nitrogen and stored at −80°C. For each tube, the worm pellet was re-suspended in lysis buffer containing EDTA to scavenge free metal ions and was sonicated to disrupt cell membranes. Fifty µL of the lysate was used to perform a BCA assay to measure protein levels. Isoproterenol was added as an internal standard to the remaining 250 µL of the lysate, which was applied to the aluminum membrane to bind DA. Collected DA samples were then processed for High Performance Liquid Chromatography (HPLC). To correct for inter-sample variations in the extraction efficiency, the ratio of DA to the internal standard, isoproternol, was estimated, and the total DA content was calculated relative either to protein levels or to the number of worms.
Isoprostane content measurement
Worms were grown at high densities on 8P-plates. 20,000 synchronized L1 per sample were washed off the plates in 85 mM NaCl, collected in 10 mL 85 mM NaCl and acutely treated (30 min under gentle agitation) with MnCl2. Worms were pelleted and washed three times in 85 mM NaCl and then re-suspended in 85 mM NaCl, 0.5% Triton X-100, 5 mM Tris Buffer pH 6.8, 0.5× protease inhibitor cocktail (Sigma P8340) with zirconia beads, up to 1 mL. Samples were then processed with a Mini beadbeater-16 (Biospec Products, OK, USA) for 7 cycles of 20 s and kept 1 min in ice-cold water after each cycle. 20 µL of supernatant per sample were kept for measurement of protein levels by the Bradford method. 850 µL were added to 10 mL Folch solution and gently shaken every 5 min for 30 min. 2 mL of 0.9% NaCl per tube were added. Tubes were centrifuged at 3,000 rpm for 10 min at 4°C, the aqueous layer was discarded, and the organic phase was dried under nitrogen flow at 37°C. The detailed procedures for the purification and derivatization steps were previously described [174].
ROS measurement
Synchronized L1 were acutely treated with MnCl2 as described earlier and washed 4 additional times in M9 buffer. 2′7′ dichlorodihydrofluorescein diacetate (H2DCF-DA) was added at 1 mM for one hour in the dark. Worms were then washed 4 times in M9 buffer. Worms were frozen and thawed twice and homogenized by sonication and then centrifuged. The supernatants were transferred to a 96-well plate and their fluorescence levels (excitation: 485 nm; emission: 535 nm) were detected using a FLEXstation III (Molecular Devices, Sunnyvale, California) pre-heated at 37°C. The fluorescence from each well was measured every 20 min for up to 2 h. Here, we report values obtained at 1 h. Fluorescence measurements were normalized to time zero values, and rates of increase in fluorescence (reflecting ROS levels) were expressed as percent control. Measurements were repeated 3 times, each condition was performed in triplicate, and the experiment was repeated in three independent worm preparations for each tested strain.
Lifespan experiments
Synchronized L1 worms were acutely exposed to MnCl2 concentrations as described earlier. Live and healthy-looking worms (60–66 per condition) were collected on the same day at the late L4 stage and transferred every five days to new OP50-seeded NGM plates. Survival was assessed every two to three days until all worms had died. All tested C. elegans strains were assessed in parallel, and each experiment was performed three times, yielding qualitatively identical results. Plotted curves represent averages of those triplicate independent experiments.
Epifluorescence, DIC, and confocal microscopy
For each slide, at least 30 worms were mounted on 4% agarose pads in M9 and anaesthetized with 0.2% tricaine/0.02% tetramisole in M9. Fluorescence observations and scoring of neuronal defects were performed with an epifluorescence microscope (Nikon Eclipse 80i, Nikon Corporation, Tokyo, Japan) equipped with a Lambda LS Xenon lamp (Sutter Instrument Company) and Nikon Plan Fluor 20× dry and Nikon Plan Apo 60× 1.3 oil objectives. Confocal images acquired for illustration or GFP intensity measurement purposes were captured through Plan-Neofluar 40×, Plan-Apochromat 63×, or Plan-Neofluar 100× oil objectives with a 1.3, 1.4 and 1.3 apertures, respectively, on a LSM510 confocal microscope (Carl Zeiss MicroImaging, Inc.) scanning every 200 nm for XZ sections. Images were processed with the Zeiss LSM Image Browser 4.0.0.157 software and edited using Photoshop 7.0 (Adobe). Microscopes were housed in air-conditioned rooms (20–22°C). Amphid and phasmid neuron staining was performed following MnCl2 acute treatment by soaking the worms for 2 h in a 10 µg/mL DiI solution prepared with M9, washing off the dye for 1 h in M9 and then recovering the worms on OP50 coated NGM plates.
SKN-1::GFP fluorescence measurements
SKN-1::GFP transgenic worms were acutely treated as previously described, transferred to OP50-1 seeded NGM plates and imaged 1 h post-treatment. Fluorescence measurements of the SKN-1::GFP signal were performed on complete confocal Z-stack maximal projections of L1 C. elegans ASI neurons. Treated and untreated animals were mounted on the same slide and imaged with the same magnification, gain, offset, pinhole and laser power settings. GFP integral intensity and signal density of the maximal projection of the ASI nuclei were measured with the freeware ImageJ (developed by Wayne Rasband, NIMH, Maryland, USA).
Statistics
Dose-response lethality curves, longevity curves and histograms for DA, isoprostane or ROS content measurements were generated with GraphPad Prism (GraphPad Software Inc.). We used a sigmoidal dose-response model with a top constraint at 100% to draw the curves and determine the LD50 or the average lifespan values reported in the graphs. Statistical analysis of significance was carried out by one-way ANOVA for the dose-response curves, longevity curves and dopamine measurements; two-way ANOVA was used to measure isoprostane and ROS content, followed by post-hoc Bonferroni test when the overall p value was less than 0.05. For SKN-1::GFP fluorescence analysis, unpaired two-tailed T-test was used to assess statistical differences in mean values. In all figures, error bars represent SEM; * refers to differences between genotypes; # refers to differences between doses; */# p<0.05; **/## p<0.01; and ***/### p<0.001.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. PostJE
1999 Manganese oxide minerals: crystal structures and economic and environmental significance. Proc Natl Acad Sci U S A 96 3447 3454
2. ProhaskaJR
1987 Functions of trace elements in brain metabolism. Physiol Rev 67 858 901
3. TakedaA
SotogakuN
OkuN
2003 Influence of manganese on the release of neurotransmitters in rat striatum. Brain Res 965 279 282
4. BalyDL
KeenCL
HurleyLS
1985 Pyruvate carboxylase and phosphoenolpyruvate carboxykinase activity in developing rats: effect of manganese deficiency. J Nutr 115 872 879
5. BrockCJ
WalkerJE
1980 Superoxide dismutase from Bacillus stearothermophilus. Complete amino acid sequence of a manganese enzyme. Biochemistry 19 2873 2882
6. TakedaY
AvilaH
1986 Structure and gene expression of the E. coli Mn-superoxide dismutase gene. Nucleic Acids Res 14 4577 4589
7. SaricM
1986 Manganese.
Lars FribergGFN
VelmirBVouk
Handbook on The Toxicology of Metals Amsterdam Elsevier Science Publishers B.V 354 386
8. WedlerFC
DenmanRB
1984 Glutamine synthetase: the major Mn(II) enzyme in mammalian brain. Curr Top Cell Regul 24 153 169
9. AuC
BenedettoA
AschnerM
2008 Manganese transport in eukaryotes: the role of DMT1. Neurotoxicology 29 569 576
10. AschnerM
AschnerJL
1991 Manganese neurotoxicity: cellular effects and blood-brain barrier transport. Neurosci Biobehav Rev 15 333 340
11. ThompsonK
MolinaRM
DonagheyT
SchwobJE
BrainJD
2007 Olfactory uptake of manganese requires DMT1 and is enhanced by anemia. FASEB J 21 223 230
12. DormanDC
McManusBE
ParkinsonCU
ManuelCA
McElveenAM
2004 Nasal toxicity of manganese sulfate and manganese phosphate in young male rats following subchronic (13-week) inhalation exposure. Inhal Toxicol 16 481 488
13. WassermanGA
LiuX
ParvezF
AhsanH
LevyD
2006 Water manganese exposure and children's intellectual function in Araihazar, Bangladesh. Environ Health Perspect 114 124 129
14. MyersJE
teWaterNaudeJ
FourieM
ZogoeHB
NaikI
2003 Nervous system effects of occupational manganese exposure on South African manganese mineworkers. Neurotoxicology 24 649 656
15. PalPK
SamiiA
CalneDB
1999 Manganese neurotoxicity: a review of clinical features, imaging and pathology. Neurotoxicology 20 227 238
16. FerrazHB
BertolucciPH
PereiraJS
LimaJG
AndradeLA
1988 Chronic exposure to the fungicide maneb may produce symptoms and signs of CNS manganese intoxication. Neurology 38 550 553
17. AbbottPJ
1987 Methylcyclopentadienyl manganese tricarbonyl (MMT) in petrol: the toxicological issues. Sci Total Environ 67 247 255
18. CooperWC
1984 The health implications of increased manganese in the environment resulting from the combustion of fuel additives: a review of the literature. J Toxicol Environ Health 14 23 46
19. FrumkinH
SolomonG
1997 Manganese in the U.S. gasoline supply. Am J Ind Med 31 107 115
20. SierraP
LorangerS
KennedyG
ZayedJ
1995 Occupational and environmental exposure of automobile mechanics and nonautomotive workers to airborne manganese arising from the combustion of methylcyclopentadienyl manganese tricarbonyl (MMT). Am Ind Hyg Assoc J 56 713 716
21. McKinneyAM
FiliceRW
TeksamM
CaseyS
TruwitC
2004 Diffusion abnormalities of the globi pallidi in manganese neurotoxicity. Neuroradiology 46 291 295
22. NewlandMC
CecklerTL
KordowerJH
WeissB
1989 Visualizing manganese in the primate basal ganglia with magnetic resonance imaging. Exp Neurol 106 251 258
23. CalneDB
ChuNS
HuangCC
LuCS
OlanowW
1994 Manganism and idiopathic parkinsonism: similarities and differences. Neurology 44 1583 1586
24. CersosimoMG
KollerWC
2006 The diagnosis of manganese-induced parkinsonism. Neurotoxicology 27 340 346
25. OlanowCW
2004 Manganese-induced parkinsonism and Parkinson's disease. Ann N Y Acad Sci 1012 209 223
26. GorellJM
JohnsonCC
RybickiBA
PetersonEL
KortshaGX
1999 Occupational exposure to manganese, copper, lead, iron, mercury and zinc and the risk of Parkinson's disease. Neurotoxicology 20 239 247
27. KimY
KimJW
ItoK
LimHS
CheongHK
1999 Idiopathic parkinsonism with superimposed manganese exposure: utility of positron emission tomography. Neurotoxicology 20 249 252
28. RacetteBA
McGee-MinnichL
MoerleinSM
MinkJW
VideenTO
2001 Welding-related parkinsonism: clinical features, treatment, and pathophysiology. Neurology 56 8 13
29. HudnellHK
1999 Effects from environmental Mn exposures: a review of the evidence from non-occupational exposure studies. Neurotoxicology 20 379 397
30. NormandinL
PanissetM
ZayedJ
2002 Manganese neurotoxicity: behavioral, pathological, and biochemical effects following various routes of exposure. Rev Environ Health 17 189 217
31. RacetteBA
TabbalSD
JenningsD
GoodL
PerlmutterJS
2005 Prevalence of parkinsonism and relationship to exposure in a large sample of Alabama welders. Neurology 64 230 235
32. BenedettoA
AuC
AschnerM
2009 Manganese-induced dopaminergic neurodegeneration: insights into mechanisms and genetics shared with Parkinson's disease. Chem Rev 109 4862 4884
33. ChiuehCC
WuRM
MohanakumarKP
SternbergerLM
KrishnaG
1994 In vivo generation of hydroxyl radicals and MPTP-induced dopaminergic toxicity in the basal ganglia. Ann N Y Acad Sci 738 25 36
34. De IuliisA
GrigolettoJ
RecchiaA
GiustiP
ArslanP
2005 A proteomic approach in the study of an animal model of Parkinson's disease. Clin Chim Acta 357 202 209
35. JennerP
1998 Oxidative mechanisms in nigral cell death in Parkinson's disease. Mov Disord 13 Suppl 1 24 34
36. VedR
SahaS
WestlundB
PerierC
BurnamL
2005 Similar patterns of mitochondrial vulnerability and rescue induced by genetic modification of alpha-synuclein, parkin, and DJ-1 in Caenorhabditis elegans. J Biol Chem 280 42655 42668
37. ShererTB
BetarbetR
TestaCM
SeoBB
RichardsonJR
2003 Mechanism of toxicity in rotenone models of Parkinson's disease. J Neurosci 23 10756 10764
38. KienzlE
PuchingerL
JellingerK
LinertW
StachelbergerH
1995 The role of transition metals in the pathogenesis of Parkinson's disease. J Neurol Sci 134 Suppl 69 78
39. MontgomeryEBJr
1995 Heavy metals and the etiology of Parkinson's disease and other movement disorders. Toxicology 97 3 9
40. OestreicherE
SengstockGJ
RiedererP
OlanowCW
DunnAJ
1994 Degeneration of nigrostriatal dopaminergic neurons increases iron within the substantia nigra: a histochemical and neurochemical study. Brain Res 660 8 18
41. BrouilletEP
ShinobuL
McGarveyU
HochbergF
BealMF
1993 Manganese injection into the rat striatum produces excitotoxic lesions by impairing energy metabolism. Exp Neurol 120 89 94
42. LoschmannPA
LangeKW
WachtelH
TurskiL
1994 MPTP-induced degeneration: interference with glutamatergic toxicity. J Neural Transm Suppl 43 133 143
43. ChunHS
GibsonGE
DeGiorgioLA
ZhangH
KiddVJ
2001 Dopaminergic cell death induced by MPP(+), oxidant and specific neurotoxicants shares the common molecular mechanism. J Neurochem 76 1010 1021
44. SuzukiY
MouriT
SuzukiY
NishiyamaK
FujiiN
1975 Study of subacute toxicity of manganese dioxide in monkeys. Tokushima J Exp Med 22 5 10
45. HaMaiD
BondySC
2004 Oxidative basis of manganese neurotoxicity. Ann N Y Acad Sci 1012 129 141
46. MigheliR
GodaniC
SciolaL
DeloguMR
SerraPA
1999 Enhancing effect of manganese on L-DOPA-induced apoptosis in PC12 cells: role of oxidative stress. J Neurochem 73 1155 1163
47. ParentiM
RusconiL
CappabiancaV
ParatiEA
GroppettiA
1988 Role of dopamine in manganese neurotoxicity. Brain Res 473 236 240
48. Tomas-CamardielM
HerreraAJ
VeneroJL
Cruz Sanchez-HidalgoM
CanoJ
2002 Differential regulation of glutamic acid decarboxylase mRNA and tyrosine hydroxylase mRNA expression in the aged manganese-treated rats. Brain Res Mol Brain Res 103 116 129
49. HsuLJ
SagaraY
ArroyoA
RockensteinE
SiskA
2000 alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am J Pathol 157 401 410
50. ShererTB
BetarbetR
StoutAK
LundS
BaptistaM
2002 An in vitro model of Parkinson's disease: linking mitochondrial impairment to altered alpha-synuclein metabolism and oxidative damage. J Neurosci 22 7006 7015
51. TanakaY
EngelenderS
IgarashiS
RaoRK
WannerT
2001 Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum Mol Genet 10 919 926
52. WitholtR
GwiazdaRH
SmithDR
2000 The neurobehavioral effects of subchronic manganese exposure in the presence and absence of pre-parkinsonism. Neurotoxicol Teratol 22 851 861
53. KuwaharaT
KoyamaA
Gengyo-AndoK
MasudaM
KowaH
2006 Familial Parkinson mutant alpha-synuclein causes dopamine neuron dysfunction in transgenic Caenorhabditis elegans. J Biol Chem 281 334 340
54. BraungartE
GerlachM
RiedererP
BaumeisterR
HoenerMC
2004 Caenorhabditis elegans MPP+ model of Parkinson's disease for high-throughput drug screenings. Neurodegener Dis 1 175 183
55. LaksoM
VartiainenS
MoilanenAM
SirvioJ
ThomasJH
2003 Dopaminergic neuronal loss and motor deficits in Caenorhabditis elegans overexpressing human alpha-synuclein. J Neurochem 86 165 172
56. NassR
HallDH
MillerDM3rd
BlakelyRD
2002 Neurotoxin-induced degeneration of dopamine neurons in Caenorhabditis elegans. Proc Natl Acad Sci U S A 99 3264 3269
57. LockeCJ
FoxSA
CaldwellGA
CaldwellKA
2008 Acetaminophen attenuates dopamine neuron degeneration in animal models of Parkinson's disease. Neurosci Lett 439 129 133
58. WangYM
PuP
LeWD
2007 ATP depletion is the major cause of MPP+ induced dopamine neuronal death and worm lethality in alpha-synuclein transgenic C. elegans. Neurosci Bull 23 329 335
59. GitlerAD
ChesiA
GeddieML
StrathearnKE
HamamichiS
2009 Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat Genet 41 308 315
60. van HamTJ
ThijssenKL
BreitlingR
HofstraRM
PlasterkRH
2008 C. elegans model identifies genetic modifiers of alpha-synuclein inclusion formation during aging. PLoS Genet 4 e1000027 doi:10.1371/journal.pgen.1000027
61. KuwaharaT
KoyamaA
KoyamaS
YoshinaS
RenCH
2008 A systematic RNAi screen reveals involvement of endocytic pathway in neuronal dysfunction in alpha-synuclein transgenic C. elegans. Hum Mol Genet 17 2997 3009
62. IchibangaseT
SaimaruH
TakamuraN
KuwaharaT
KoyamaA
2008 Proteomics of Caenorhabditis elegans over-expressing human alpha-synuclein analyzed by fluorogenic derivatization-liquid chromatography/tandem mass spectrometry: identification of actin and several ribosomal proteins as negative markers at early Parkinson's disease stages. Biomed Chromatogr 22 232 234
63. CaoS
GelwixCC
CaldwellKA
CaldwellGA
2005 Torsin-mediated protection from cellular stress in the dopaminergic neurons of Caenorhabditis elegans. J Neurosci 25 3801 3812
64. HamamichiS
RivasRN
KnightAL
CaoS
CaldwellKA
2008 Hypothesis-based RNAi screening identifies neuroprotective genes in a Parkinson's disease model. Proc Natl Acad Sci U S A 105 728 733
65. SettivariR
LevoraJ
NassR
2009 The divalent metal transporter homologues SMF-1/2 mediates dopamine neuron sensitivity in caenorhabditis elegans models of manganism and Parkinson's disease. J Biol Chem
66. AuC
BenedettoA
AndersonJ
LabrousseA
EriksonK
2009 SMF-1, SMF-2 and SMF-3 DMT1 orthologues regulate and are regulated differentially by manganese levels in C. elegans. PLoS ONE 4 e7792 doi:10.1371/journal.pone.0007792
67. CassWA
ZahniserNR
FlachKA
GerhardtGA
1993 Clearance of exogenous dopamine in rat dorsal striatum and nucleus accumbens: role of metabolism and effects of locally applied uptake inhibitors. J Neurochem 61 2269 2278
68. KiltyJE
LorangD
AmaraSG
1991 Cloning and expression of a cocaine-sensitive rat dopamine transporter. Science 254 578 579
69. ShimadaS
KitayamaS
LinCL
PatelA
NanthakumarE
1991 Cloning and expression of a cocaine-sensitive dopamine transporter complementary DNA. Science 254 576 578
70. HuotariM
SanthaM
LucasLR
KarayiorgouM
GogosJA
2002 Effect of dopamine uptake inhibition on brain catecholamine levels and locomotion in catechol-O-methyltransferase-disrupted mice. J Pharmacol Exp Ther 303 1309 1316
71. McDonaldPW
HardieSL
JessenTN
CarvelliL
MatthiesDS
2007 Vigorous motor activity in Caenorhabditis elegans requires efficient clearance of dopamine mediated by synaptic localization of the dopamine transporter DAT-1. J Neurosci 27 14216 14227
72. NassR
HahnMK
JessenT
McDonaldPW
CarvelliL
2005 A genetic screen in Caenorhabditis elegans for dopamine neuron insensitivity to 6-hydroxydopamine identifies dopamine transporter mutants impacting transporter biosynthesis and trafficking. J Neurochem 94 774 785
73. ArchibaldFS
TyreeC
1987 Manganese poisoning and the attack of trivalent manganese upon catecholamines. Arch Biochem Biophys 256 638 650
74. GrahamDG
1984 Catecholamine toxicity: a proposal for the molecular pathogenesis of manganese neurotoxicity and Parkinson's disease. Neurotoxicology 5 83 95
75. SistrunkSC
RossMK
FilipovNM
2007 Direct effects of manganese compounds on dopamine and its metabolite Dopac: an in vitro study. Environ Toxicol Pharmacol 23 286 296
76. ParisI
Dagnino-SubiabreA
MarcelainK
BennettLB
CaviedesP
2001 Copper neurotoxicity is dependent on dopamine-mediated copper uptake and one-electron reduction of aminochrome in a rat substantia nigra neuronal cell line. J Neurochem 77 519 529
77. PongK
DoctrowSR
BaudryM
2000 Prevention of 1-methyl-4-phenylpyridinium - and 6-hydroxydopamine-induced nitration of tyrosine hydroxylase and neurotoxicity by EUK-134, a superoxide dismutase and catalase mimetic, in cultured dopaminergic neurons. Brain Res 881 182 189
78. ChaseDL
PepperJS
KoelleMR
2004 Mechanism of extrasynaptic dopamine signaling in Caenorhabditis elegans. Nat Neurosci 7 1096 1103
79. LinYT
HoangH
HsiehSI
RangelN
FosterAL
2006 Manganous ion supplementation accelerates wild type development, enhances stress resistance, and rescues the life span of a short-lived Caenorhabditis elegans mutant. Free Radic Biol Med 40 1185 1193
80. SingerTP
RamsayRR
AckrellBA
1995 Deficiencies of NADH and succinate dehydrogenases in degenerative diseases and myopathies. Biochim Biophys Acta 1271 211 219
81. BockelmannR
WolfG
RansmayrG
RiedererP
1994 NADPH-diaphorase/nitric oxide synthase containing neurons in normal and Parkinson's disease putamen. J Neural Transm Park Dis Dement Sect 7 115 121
82. JennerP
1991 Oxidative stress as a cause of Parkinson's disease. Acta Neurol Scand Suppl 136 6 15
83. JennerP
2003 Oxidative stress in Parkinson's disease. Ann Neurol 53 Suppl 3 S26 36; discussion S36–28
84. CassarinoDS
FallCP
SwerdlowRH
SmithTS
HalvorsenEM
1997 Elevated reactive oxygen species and antioxidant enzyme activities in animal and cellular models of Parkinson's disease. Biochim Biophys Acta 1362 77 86
85. YooMS
ChunHS
SonJJ
DeGiorgioLA
KimDJ
2003 Oxidative stress regulated genes in nigral dopaminergic neuronal cells: correlation with the known pathology in Parkinson's disease. Brain Res Mol Brain Res 110 76 84
86. WuDC
TeismannP
TieuK
VilaM
Jackson-LewisV
2003 NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. Proc Natl Acad Sci U S A 100 6145 6150
87. LothariusJ
DuganLL
O'MalleyKL
1999 Distinct mechanisms underlie neurotoxin-mediated cell death in cultured dopaminergic neurons. J Neurosci 19 1284 1293
88. SchapiraAH
1995 Oxidative stress in Parkinson's disease. Neuropathol Appl Neurobiol 21 3 9
89. LinertW
HerlingerE
JamesonRF
KienzlE
JellingerK
1996 Dopamine, 6-hydroxydopamine, iron, and dioxygen–their mutual interactions and possible implication in the development of Parkinson's disease. Biochim Biophys Acta 1316 160 168
90. MilneGL
YinH
BrooksJD
SanchezS
Jackson RobertsL2nd
2007 Quantification of F2-isoprostanes in biological fluids and tissues as a measure of oxidant stress. Methods Enzymol 433 113 126
91. FamSS
MorrowJD
2003 The isoprostanes: unique products of arachidonic acid oxidation-a review. Curr Med Chem 10 1723 1740
92. TaberDF
MorrowJD
RobertsLJ2nd
1997 A nomenclature system for the isoprostanes. Prostaglandins 53 63 67
93. OliveiraRP
Porter AbateJ
DilksK
LandisJ
AshrafJ
2009 Condition-adapted stress and longevity gene regulation by Caenorhabditis elegans SKN-1/Nrf. Aging Cell 8 524 541
94. AnJH
BlackwellTK
2003 SKN-1 links C. elegans mesendodermal specification to a conserved oxidative stress response. Genes Dev 17 1882 1893
95. BowermanB
EatonBA
PriessJR
1992 skn-1, a maternally expressed gene required to specify the fate of ventral blastomeres in the early C. elegans embryo. Cell 68 1061 1075
96. KellA
VenturaN
KahnN
JohnsonTE
2007 Activation of SKN-1 by novel kinases in Caenorhabditis elegans. Free Radic Biol Med 43 1560 1566
97. TulletJM
HertweckM
AnJH
BakerJ
HwangJY
2008 Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell 132 1025 1038
98. AnJH
VranasK
LuckeM
InoueH
HisamotoN
2005 Regulation of the Caenorhabditis elegans oxidative stress defense protein SKN-1 by glycogen synthase kinase-3. Proc Natl Acad Sci U S A 102 16275 16280
99. ParkSK
TedescoPM
JohnsonTE
2009 Oxidative stress and longevity in Caenorhabditis elegans as mediated by SKN-1. Aging Cell 8 258 269
100. DennisKE
AschnerJL
MilatovicD
SchmidtJW
AschnerM
2009 NADPH oxidases and reactive oxygen species at different stages of chronic hypoxia-induced pulmonary hypertension in newborn piglets. Am J Physiol Lung Cell Mol Physiol 297 L596 607
101. Bonneh-BarkayD
ReaneySH
LangstonWJ
Di MonteDA
2005 Redox cycling of the herbicide paraquat in microglial cultures. Brain Res Mol Brain Res 134 52 56
102. GaoHM
LiuB
HongJS
2003 Critical role for microglial NADPH oxidase in rotenone-induced degeneration of dopaminergic neurons. J Neurosci 23 6181 6187
103. AnantharamV
KaulS
SongC
KanthasamyA
KanthasamyAG
2007 Pharmacological inhibition of neuronal NADPH oxidase protects against 1-methyl-4-phenylpyridinium (MPP+)-induced oxidative stress and apoptosis in mesencephalic dopaminergic neuronal cells. Neurotoxicology 28 988 997
104. TheinMC
WinterAD
StepekG
McCormackG
StapletonG
2009 Combined extracellular matrix cross-linking activity of the peroxidase MLT-7 and the dual oxidase BLI-3 is critical for post-embryonic viability in Caenorhabditis elegans. J Biol Chem 284 17549 17563
105. JainC
YunM
PolitzSM
RaoRP
2009 A pathogenesis assay using Saccharomyces cerevisiae and Caenorhabditis elegans reveals novel roles for yeast AP-1, Yap1, and host dual oxidase BLI-3 in fungal pathogenesis. Eukaryot Cell 8 1218 1227
106. ChavezV
Mohri-ShiomiA
GarsinDA
2009 Ce-Duox1/BLI-3 generates reactive oxygen species as a protective innate immune mechanism in Caenorhabditis elegans. Infect Immun 77 4983 4989
107. SalazarJ
MenaN
HunotS
PrigentA
Alvarez-FischerD
2008 Divalent metal transporter 1 (DMT1) contributes to neurodegeneration in animal models of Parkinson's disease. Proc Natl Acad Sci U S A 105 18578 18583
108. SongN
JiangH
WangJ
XieJX
2007 Divalent metal transporter 1 up-regulation is involved in the 6-hydroxydopamine-induced ferrous iron influx. J Neurosci Res 85 3118 3126
109. ZhangS
WangJ
SongN
XieJ
JiangH
2008 Up-regulation of divalent metal transporter 1 is involved in 1-methyl-4-phenylpyridinium (MPP(+))-induced apoptosis in MES23.5 cells. Neurobiol Aging
110. BarzilaiA
MelamedE
2003 Molecular mechanisms of selective dopaminergic neuronal death in Parkinson's disease. Trends Mol Med 9 126 132
111. BetarbetR
ShererTB
GreenamyreJT
2002 Animal models of Parkinson's disease. Bioessays 24 308 318
112. Afonso-OramasD
Cruz-MurosI
de la RosaDA
AbreuP
GiraldezT
2009 Dopamine transporter glycosylation correlates with the vulnerability of midbrain dopaminergic cells in Parkinson's disease. Neurobiol Dis 36 494 508
113. McKinleyET
BaranowskiTC
BlavoDO
CatoC
DoanTN
2005 Neuroprotection of MPTP-induced toxicity in zebrafish dopaminergic neurons. Brain Res Mol Brain Res 141 128 137
114. StorchA
LudolphAC
SchwarzJ
2004 Dopamine transporter: involvement in selective dopaminergic neurotoxicity and degeneration. J Neural Transm 111 1267 1286
115. PrabhakaranK
GhoshD
ChapmanGD
GunasekarPG
2008 Molecular mechanism of manganese exposure-induced dopaminergic toxicity. Brain Res Bull 76 361 367
116. ReaneySH
SmithDR
2005 Manganese oxidation state mediates toxicity in PC12 cells. Toxicol Appl Pharmacol 205 271 281
117. MilatovicD
Zaja-MilatovicS
GuptaRC
YuY
AschnerM
2009 Oxidative damage and neurodegeneration in manganese-induced neurotoxicity. Toxicol Appl Pharmacol 240 219 225
118. MilatovicD
YinZ
GuptaRC
SidorykM
AlbrechtJ
2007 Manganese induces oxidative impairment in cultured rat astrocytes. Toxicol Sci 98 198 205
119. OlneyJW
ZorumskiCF
StewartGR
PriceMT
WangGJ
1990 Excitotoxicity of L-dopa and 6-OH-dopa: implications for Parkinson's and Huntington's diseases. Exp Neurol 108 269 272
120. Van Den BoschL
Van DammeP
BogaertE
RobberechtW
2006 The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim Biophys Acta 1762 1068 1082
121. BerrettaN
FreestonePS
GuatteoE
de CastroD
GeracitanoR
2005 Acute effects of 6-hydroxydopamine on dopaminergic neurons of the rat substantia nigra pars compacta in vitro. Neurotoxicology 26 869 881
122. WersingerC
ProuD
VernierP
SidhuA
2003 Modulation of dopamine transporter function by alpha-synuclein is altered by impairment of cell adhesion and by induction of oxidative stress. FASEB J 17 2151 2153
123. KernCH
StanwoodGD
SmithDR
Preweaning manganese exposure causes hyperactivity, disinhibition, and spatial learning and memory deficits associated with altered dopamine receptor and transporter levels. Synapse 64 363 378
124. McDougallSA
ReichelCM
FarleyCM
FlesherMM
Der-GhazarianT
2008 Postnatal manganese exposure alters dopamine transporter function in adult rats: Potential impact on nonassociative and associative processes. Neuroscience 154 848 860
125. BishopNA
GuarenteL
2007 Two neurons mediate diet-restriction-induced longevity in C. elegans. Nature 447 545 549
126. BerkowitzLA
HamamichiS
KnightAL
HarringtonAJ
CaldwellGA
2008 Application of a C. elegans dopamine neuron degeneration assay for the validation of potential Parkinson's disease genes. J Vis Exp
127. MarvanovaM
NicholsCD
2007 Identification of neuroprotective compounds of caenorhabditis elegans dopaminergic neurons against 6-OHDA. J Mol Neurosci 31 127 137
128. SpringerW
HoppeT
SchmidtE
BaumeisterR
2005 A Caenorhabditis elegans Parkin mutant with altered solubility couples alpha-synuclein aggregation to proteotoxic stress. Hum Mol Genet 14 3407 3423
129. SamannJ
HegermannJ
von GromoffE
EimerS
BaumeisterR
2009 Caenorhabditits elegans LRK-1 and PINK-1 act antagonistically in stress response and neurite outgrowth. J Biol Chem 284 16482 16491
130. ChenPC
VargasMR
PaniAK
SmeyneRJ
JohnsonDA
2009 Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson's disease: Critical role for the astrocyte. Proc Natl Acad Sci U S A 106 2933 2938
131. CuadradoA
Moreno-MurcianoP
Pedraza-ChaverriJ
2009 The transcription factor Nrf2 as a new therapeutic target in Parkinson's disease. Expert Opin Ther Targets 13 319 329
132. van MuiswinkelFL
KuiperijHB
2005 The Nrf2-ARE Signalling pathway: promising drug target to combat oxidative stress in neurodegenerative disorders. Curr Drug Targets CNS Neurol Disord 4 267 281
133. ParkRM
BowlerRM
EggerthDE
DiamondE
SpencerKJ
2006 Issues in neurological risk assessment for occupational exposures: the Bay Bridge welders. Neurotoxicology 27 373 384
134. McMillanG
2005 Is electric arc welding linked to manganism or Parkinson's disease? Toxicol Rev 24 237 257
135. SadekAH
RauchR
SchulzPE
2003 Parkinsonism due to manganism in a welder. Int J Toxicol 22 393 401
136. WennbergA
IregrenA
StruweG
CizinskyG
HagmanM
1991 Manganese exposure in steel smelters a health hazard to the nervous system. Scand J Work Environ Health 17 255 262
137. AndersonJG
CooneyPT
EriksonKM
2007 Inhibition of DAT function attenuates manganese accumulation in the globus pallidus. Environ Toxicol Pharmacol 23 179 184
138. GraumannR
ParisI
Martinez-AlvaradoP
RumanqueP
Perez-PasteneC
2002 Oxidation of dopamine to aminochrome as a mechanism for neurodegeneration of dopaminergic systems in Parkinson's disease. Possible neuroprotective role of DT-diaphorase. Pol J Pharmacol 54 573 579
139. ShenXM
DryhurstG
1998 Iron - and manganese-catalyzed autoxidation of dopamine in the presence of L-cysteine: possible insights into iron - and manganese-mediated dopaminergic neurotoxicity. Chem Res Toxicol 11 824 837
140. ParisI
Martinez-AlvaradoP
CardenasS
Perez-PasteneC
GraumannR
2005 Dopamine-dependent iron toxicity in cells derived from rat hypothalamus. Chem Res Toxicol 18 415 419
141. ZoccaratoF
ToscanoP
AlexandreA
2005 Dopamine-derived dopaminochrome promotes H(2)O(2) release at mitochondrial complex I: stimulation by rotenone, control by Ca(2+), and relevance to Parkinson disease. J Biol Chem 280 15587 15594
142. BaezS
LindersonY
Segura-AguilarJ
1995 Superoxide dismutase and catalase enhance autoxidation during one-electron reduction of aminochrome by NADPH-cytochrome P-450 reductase. Biochem Mol Med 54 12 18
143. ParisI
CardenasS
LozanoJ
Perez-PasteneC
GraumannR
2007 Aminochrome as a preclinical experimental model to study degeneration of dopaminergic neurons in Parkinson's disease. Neurotox Res 12 125 134
144. ZafarKS
SiegelD
RossD
2006 A potential role for cyclized quinones derived from dopamine, DOPA, and 3,4-dihydroxyphenylacetic acid in proteasomal inhibition. Mol Pharmacol 70 1079 1086
145. Diaz-VelizG
MoraS
DossiMT
GomezP
ArriagadaC
2002 Behavioral effects of aminochrome and dopachrome injected in the rat substantia nigra. Pharmacol Biochem Behav 73 843 850
146. De IuliisA
BurlinaAP
BoschettoR
ZambenedettiP
ArslanP
2002 Increased dopamine peroxidation in postmortem Parkinsonian brain. Biochim Biophys Acta 1573 63 67
147. GalzignaL
SchiappelliMP
RigoA
ScarpaM
1999 A rat brain fraction and different purified peroxidases catalyzing the formation of dopaminochrome from dopamine. Biochim Biophys Acta 1427 329 336
148. RothJA
HorbinskiC
HigginsD
LeinP
GarrickMD
2002 Mechanisms of manganese-induced rat pheochromocytoma (PC12) cell death and cell differentiation. Neurotoxicology 23 147 157
149. TjalveH
MejareC
Borg-NeczakK
1995 Uptake and transport of manganese in primary and secondary olfactory neurones in pike. Pharmacol Toxicol 77 23 31
150. SlootWN
GramsbergenJB
1994 Axonal transport of manganese and its relevance to selective neurotoxicity in the rat basal ganglia. Brain Res 657 124 132
151. TholeyG
Megias-MegiasL
WedlerFC
LedigM
1990 Modulation of Mn2+ accumulation in cultured rat neuronal and astroglial cells. Neurochem Res 15 751 754
152. OchsSD
WestfallTC
MacarthurH
2005 The separation and quantification of aminochromes using high-pressure liquid chromatography with electrochemical detection. J Neurosci Methods 142 201 208
153. Del MaestroR
McDonaldW
1987 Distribution of superoxide dismutase, glutathione peroxidase and catalase in developing rat brain. Mech Ageing Dev 41 29 38
154. DumontM
WilleE
StackC
CalingasanNY
BealMF
2009 Reduction of oxidative stress, amyloid deposition, and memory deficit by manganese superoxide dismutase overexpression in a transgenic mouse model of Alzheimer's disease. FASEB J 23 2459 2466
155. PengJ
StevensonFF
DoctrowSR
AndersenJK
2005 Superoxide dismutase/catalase mimetics are neuroprotective against selective paraquat-mediated dopaminergic neuron death in the substantial nigra: implications for Parkinson disease. J Biol Chem 280 29194 29198
156. JainM
NehraS
TrivediPC
SinghRV
2002 Nematicidal, Fungicidal and Bactericidal Activities of Manganese (II) Complexes with Heterocyclic Sulphonamide Imines. Met Based Drugs 9 53 60
157. SawinER
RanganathanR
HorvitzHR
2000 C. elegans locomotory rate is modulated by the environment through a dopaminergic pathway and by experience through a serotonergic pathway. Neuron 26 619 631
158. HillsT
BrockiePJ
MaricqAV
2004 Dopamine and glutamate control area-restricted search behavior in Caenorhabditis elegans. J Neurosci 24 1217 1225
159. O'RourkeEJ
SoukasAA
CarrCE
RuvkunG
2009 C. elegans major fats are stored in vesicles distinct from lysosome-related organelles. Cell Metab 10 430 435
160. Canonne-HergauxF
GruenheidS
GovoniG
GrosP
1999 The Nramp1 protein and its role in resistance to infection and macrophage function. Proc Assoc Am Physicians 111 283 289
161. SearleS
BrightNA
RoachTI
AtkinsonPG
BartonCH
1998 Localisation of Nramp1 in macrophages: modulation with activation and infection. J Cell Sci 111 ( Pt 19) 2855 2866
162. MackenzieB
HedigerMA
2004 SLC11 family of H+-coupled metal-ion transporters NRAMP1 and DMT1. Pflugers Arch 447 571 579
163. ForbesJR
GrosP
2001 Divalent-metal transport by NRAMP proteins at the interface of host-pathogen interactions. Trends Microbiol 9 397 403
164. BandyopadhyayJ
SongHO
ParkBJ
SingaraveluG
SunJL
2009 Functional assessment of Nramp-like metal transporters and manganese in Caenorhabditis elegans. Biochem Biophys Res Commun 390 136 141
165. JangH
BoltzDA
WebsterRG
SmeyneRJ
2009 Viral parkinsonism. Biochim Biophys Acta 1792 714 721
166. Diaz-CorralesFJ
ColasanteC
ContrerasQ
PuigM
SerranoJA
2004 Nocardia otitidiscaviarum (GAM-5) induces parkinsonian-like alterations in mouse. Braz J Med Biol Res 37 539 548
167. CassarinoDS
QuezadoMM
GhatakNR
DurayPH
2003 Lyme-associated parkinsonism: a neuropathologic case study and review of the literature. Arch Pathol Lab Med 127 1204 1206
168. KoutsilieriE
SopperS
SchellerC
ter MeulenV
RiedererP
2002 Parkinsonism in HIV dementia. J Neural Transm 109 767 775
169. WellerC
OxladeN
DobbsSM
DobbsRJ
CharlettA
2005 Role of inflammation in gastrointestinal tract in aetiology and pathogenesis of idiopathic parkinsonism. FEMS Immunol Med Microbiol 44 129 135
170. DobbsRJ
DobbsSM
WellerC
CharlettA
BjarnasonIT
2008 Helicobacter hypothesis for idiopathic parkinsonism: before and beyond. Helicobacter 13 309 322
171. TakahashiM
YamadaT
1999 Viral etiology for Parkinson's disease–a possible role of influenza A virus infection. Jpn J Infect Dis 52 89 98
172. StoesslAJ
1999 Etiology of Parkinson's disease. Can J Neurol Sci 26 Suppl 2 S5 12
173. BrennerS
1974 The genetics of Caenorhabditis elegans. Genetics 77 71 94
174. MilatovicD
AschnerM
2009 Measurement of Isoprostanes as Markers of Oxidative Stress in Neuronal Tissue. Current Protocols in Toxicology unit 12.14 1 12
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 8
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Identification of the Bovine Arachnomelia Mutation by Massively Parallel Sequencing Implicates Sulfite Oxidase (SUOX) in Bone Development
- Common Inherited Variation in Mitochondrial Genes Is Not Enriched for Associations with Type 2 Diabetes or Related Glycemic Traits
- A Model for Damage Load and Its Implications for the Evolution of Bacterial Aging
- Did Genetic Drift Drive Increases in Genome Complexity?
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy