
Chromosome Axis Defects Induce a Checkpoint-Mediated Delay and Interchromosomal Effect on Crossing Over during Drosophila Meiosis
Crossovers mediate the accurate segregation of homologous chromosomes during meiosis. The widely conserved pch2 gene of Drosophila melanogaster is required for a pachytene checkpoint that delays prophase progression when genes necessary for DSB repair and crossover formation are defective. However, the underlying process that the pachytene checkpoint is monitoring remains unclear. Here we have investigated the relationship between chromosome structure and the pachytene checkpoint and show that disruptions in chromosome axis formation, caused by mutations in axis components or chromosome rearrangements, trigger a pch2-dependent delay. Accordingly, the global increase in crossovers caused by chromosome rearrangements, known as the “interchromosomal effect of crossing over,” is also dependent on pch2. Checkpoint-mediated effects require the histone deacetylase Sir2, revealing a conserved functional connection between PCH2 and Sir2 in monitoring meiotic events from Saccharomyces cerevisiae to a metazoan. These findings suggest a model in which the pachytene checkpoint monitors the structure of chromosome axes and may function to promote an optimal number of crossovers.
Published in the journal:
. PLoS Genet 6(8): e32767. doi:10.1371/journal.pgen.1001059
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001059
Summary
Crossovers mediate the accurate segregation of homologous chromosomes during meiosis. The widely conserved pch2 gene of Drosophila melanogaster is required for a pachytene checkpoint that delays prophase progression when genes necessary for DSB repair and crossover formation are defective. However, the underlying process that the pachytene checkpoint is monitoring remains unclear. Here we have investigated the relationship between chromosome structure and the pachytene checkpoint and show that disruptions in chromosome axis formation, caused by mutations in axis components or chromosome rearrangements, trigger a pch2-dependent delay. Accordingly, the global increase in crossovers caused by chromosome rearrangements, known as the “interchromosomal effect of crossing over,” is also dependent on pch2. Checkpoint-mediated effects require the histone deacetylase Sir2, revealing a conserved functional connection between PCH2 and Sir2 in monitoring meiotic events from Saccharomyces cerevisiae to a metazoan. These findings suggest a model in which the pachytene checkpoint monitors the structure of chromosome axes and may function to promote an optimal number of crossovers.
Introduction
Meiotic recombination occurs during prophase I when homologous chromosomes are synapsed along their entire length. Synapsis is defined as the close and stable association of homologous chromosomes through a proteinaceous structure called the synaptonemal complex (SC). In most organisms, this complex is composed of two main parts: lateral elements that attach along the axis of each homologous chromosome and transverse elements that span the central region of the SC and function to tether the homologs [1], [2]. At the leptotene/zygotene stages of meiotic prophase, these structural proteins begin to load onto the chromosome axes, and are completely assembled at pachytene, when homologous chromosomes are synapsed along their entire length.
Recombination between the homologous chromosomes initiates with DNA double-strand breaks (DSBs) that are repaired as either crossovers or noncrossovers [3]–[5]. Crossovers establish chromatin linkages called chiasmata, which, along with sister chromatid cohesion, hold homologs together after recombination has been completed and chromosomes have dissociated their SC proteins. Chiasmata help orient the homologous chromosomes on the metaphase I spindle and ensure their proper segregation at anaphase I. The failure to establish a crossover/chiasma can result in the nondisjunction of homologs and lead to aneuploid gametes.
Crossover formation is a tightly regulated process. Mutational analysis has revealed evidence for several mechanisms that control the frequency and position of crossovers along the chromosome arms [6]–[9]. For example, in Drosophila melanogaster, the precondition class of mutants exhibit reduced crossover levels with an altered distribution pattern, suggesting these genes have a role in establishing the number and distribution of crossover sites [10]. Changes in chromosome structure can also affect crossover distribution. Heterozygous inversions suppress crossing over near the breakpoints, yet enhance the frequency of exchange on the remaining chromosome pairs, a phenomenon referred to as the “interchromosomal effect” [11].
Crossing over may also be regulated by surveillance mechanisms that coordinate the sequence of critical events throughout prophase. In Drosophila, the process of repairing meiotic DSBs is monitored by at least two checkpoints: the canonical DSB repair checkpoint that responds to DNA damage [12], [13] and another that monitors DSB-independent events leading to crossover formation, hereafter referred to as the “pachytene checkpoint” [14]. The pachytene checkpoint induces a delay in response to defects in DSB repair genes required to repair all meiotic DSBs and genes encoding an endonuclease complex required for crossover formation (exchange class). Pachytene checkpoint activity requires a group of MCM-related genes that promote crossover formation (precondition class) and the Drosophila homolog of the widely conserved AAA+ ATPase PCH2.
In Saccharomyces cerevisiae and Caenorhabditis elegans, pachytene checkpoint activity has been detected in mutants with disrupted SC formation [15], [16]; however, it remains unclear what the underlying process is that the pachytene checkpoint is monitoring. For instance, yeast carrying a non-null zip1 allele appear to form SC normally, yet still exhibit a Pch2-dependent delay [17]. Mutations that impair SC initiation in C. elegans triggers a Pch2-dependent response [16], although it is unclear whether the defect being monitored is synapsis per se, a prerequisite to synapsis such as homolog pairing and/or DSB repair. Some mutations that exhibit pch2-dependent delays in Drosophila have no obvious defects in SC formation and abolishing synapsis does not elicit any delay phenotypes [14]. Therefore, at least in Drosophila and possibly in these other organisms, it may not be the SC that is being monitored by the pachytene checkpoint. Instead, the pachytene checkpoint may be important to monitor synapsis-independent changes in chromosome structure required for crossover formation [14].
We have investigated the relationship between chromosome structure and the pachytene checkpoint and show that disruptions in chromosome axis components cause pch2-dependent delays. Unlike the delays observed in DSB repair mutants, these delays occur independently of MCM-related genes. Heterozygous chromosome aberrations also result in a MCM-independent pachytene delay and interchromosomal increase in crossovers that require pch2. These findings suggest a model in which the pachytene checkpoint monitors two genetically distinct events: an early function of DSB repair proteins and the structure of chromosome axes. A checkpoint response to both events requires the histone deacetlyase Sir2, showing that a functional connection between PCH2 and Sir2 in monitoring meiotic events is conserved in Saccharomyces cerevisiae and Drosophila. Checkpoint activity is also associated with prolonged PCH2 expression. We propose the pachytene checkpoint may function to promote an optimal number of crossovers by regulating the timing of a crossover determination phase defined by PCH2 expression.
Results
Defects in chromosome axis components cause a pch2-dependent pachytene delay
In the Drosophila germarium, oocytes are born within cysts composed of 16 cells that are connected by ring canals. Two out of the sixteen cells, each with four ring canals, initially contain equivalent levels of SC proteins and are termed the pro-oocytes (Figure 1A). As the developing cysts travel from the anterior (region 2) toward the posterior (region 3) of the germarium, the pro-oocytes proceed through the pachytene stage of meiosis where synapsis is completed and DSB formation and recombination occurs. By region 3 of the wild-type germarium, DSB repair is completed and one of the two pro-oocytes will exit meiosis, lose its SC and become a nurse cell while the other will continue through development and form the oocyte (Figure 1A) [18].

In DSB repair and exchange-defective mutants, the transition through pachytene is delayed by pachytene checkpoint activity [14]. This results in both pro-oocytes persisting into region 3 cysts, referred to as the “two-oocyte phenotype.” Delays can be identified either by the persistence of the SC transverse filament C(3)G in both pro-oocytes [14] or by the concentration of ORB protein in the cytoplasm of two cells rather than one in region 3 cysts (Figure 1B) [19]. ORB staining, however, is less sensitive than C(3)G at detecting pachytene delays, resulting in a different frequency of the two-oocyte phenotype between the two assays [14].
Abolishing synapsis by mutation of c(3)G does not elicit the two-oocyte phenotype (Figure 1D), suggesting the pachytene checkpoint is not monitoring SC formation [14]. We further investigated the relationship between chromosome structure components and the pachytene checkpoint by determining the effects of mutations in two other genes, ord and c(2)M, which encode structural proteins.
ORD localizes to chromosome axes in oocytes independent of synapsis (i.e. in c(3)G mutants) and has roles in meiotic recombination and sister chromatid cohesion [20], [21]. Although ord mutants initially display normal C(3)G and C(2)M localization, only rare structures resembling SC were observed by electron microscopy (EM), suggesting that the ultrastructure of chromosome axes was disorganized [21]. Consistent with this interpretation, C(3)G and C(2)M staining precociously deteriorates in ord mutants as the oocytes progress through pachytene [21]. We found that ord mutants displayed a high frequency of the two-oocyte phenotype (Figure 1D), indicative of a delay in meiotic progression. The two-oocyte phenotype was suppressed in ord; pch2 double mutants, indicating the delay was dependent on the pachytene checkpoint (Figure 1D) and supporting the hypothesis that the pachytene checkpoint is sensitive to defects in axis components.
C(2)M is a component of the SC lateral element and localizes adjacent to the chromosome axes even in the absence of synapsis (in c(3)G mutants), suggesting it may interact with axis components [22], [23]. In c(2)M mutant oocytes, C(3)G protein fails to develop into complete strands along the lengths of each chromosome, but instead appears as small patches (Figure 1C). The most likely explanation is that SC initiates in c(2)M mutants but polymerization is defective. Similar to ord mutants, c(2)M mutants exhibited a high frequency of the two-oocyte phenotype, which was suppressed in c(2)M; pch2 double mutants (Figure 1D). The high frequency of the two-oocyte phenotype observed in c(2)M mutants was not suppressed by mutation of c(3)G (Figure 1D), demonstrating the pachytene checkpoint can signal independently of SC initiation. Together, these results suggest the pachytene checkpoint may monitor a synapsis-independent function of ORD and C(2)M, such as the formation of chromosome axes.
Chromosomal rearrangements disrupt axis integrity and cause a pch2-dependent pachytene delay
If the pachytene checkpoint monitors the integrity of chromosome axes we reasoned that structural rearrangements would also exhibit pachytene delays. Balancers are multiply-inverted chromosomes that fail to cross over with a normal homolog, presumably due to a disruption in the continuity of pairing and/or synapsis [24]–[26]. We characterized the integrity of SC-associated proteins in balancer heterozygotes with antibodies recognizing the SC components C(3)G and C(2)M. Single balancer heterozygotes (TM3/+) had thread-like C(3)G and C(2)M staining that was indistinguishable from wild-type (Figure 2A) [26]. Double balancer heterozygotes (CyO/+; TM3/+)also initially displayed normal C(3)G and C(2)M localization, but the staining became fragmented and sometimes undetectable in region 3 oocytes (Figure 2A). This precocious deterioration of SC proteins during pachytene is similar to what is observed in ord mutant oocytes [21], suggesting that rearrangement breakpoints might disrupt axis stability.

Using C(3)G staining to detect oocytes, we found that FM7, Bwinscy, TM2 and TM3 balancer heterozygotes each exhibited a significantly higher frequency of the two-oocyte phenotype compared to wild-type (Figure 2B), suggestive of a pachytene delay. The high frequency of the two-oocyte phenotype was suppressed to wild-type levels in FM7/+; pch2 and TM3/+; pch2 females, confirming the delays were dependent on the pachytene checkpoint (Figure 2B; P<0.05 compared to either balancer heterozygote alone). pch2 had no effect on the SC morphology in single balancer heterozygotes (data not shown).
Each balancer chromosome contains several inversions. For example, the TM3 chromosome includes 10 breakpoints [27]. To investigate the effects of a more subtle disruption in chromosome organization on the pachytene checkpoint, we tested whether a single aberration, or two breakpoints, would be enough to induce pachytene delays. Remarkably, heterozygotes of single translocations between the 2nd and 3rd chromosomes (T(2;3)DP77/+, T(2;3)dpD/+, and T(2;3)ltX16/+) and a single inversion on the X chromosome (In(1)dl49/+) each exhibited a high frequency of the two-oocyte phenotype, suggesting the threshold to trigger the pachytene checkpoint is low and requires as few as two breaks in axis continuity (Figure 2B). Importantly, the delays were not dependent on C(3)G and were not significantly increased in In(1)dl49 homozygotes compared to wild-type (Figure 2B and 2C), indicating the pachytene checkpoint responds to a break in alignment between homologs in a way that is independent of SC initiation.
Chromosome axis defects cause pachytene checkpoint delays independent of the MCM–related precondition genes
In addition to the delay in oocyte selection, DSB repair and exchange-defective mutants also exhibit a pch2-dependent delay in the response to DSBs [14]. To monitor DSB formation and repair in balancer heterozygotes, we stained ovaries with an antibody to γ-H2AV [28], [29]. In wild-type oocytes, γ-H2AV foci are most abundant in region 2a nuclei (cyst 3 in Figure 2D) and absent by region 3 (cyst 8 in Figure 2D), indicating DSBs have been repaired. Likewise, both FM7/+ and CyO/+ heterozygotes exhibited maximum γ-H2AV foci in region 2a oocytes at a similar cyst age to wild-type (Figure 2D). The same result was also observed in the double heterozygote FM7/+; CyO/+. Therefore, balancer heterozygotes do not cause a delay in the γ-H2AV response to DSBs, revealing a distinction between the effect of DSB repair mutants and chromosomal rearrangements on the pachytene checkpoint. While mutations in DSB repair genes induce two pch2-dependent delays in pachytene, delayed response to DSBs and delayed oocyte selection, chromosomal rearrangements only delay the latter.
If the pachytene checkpoint can cause delays through two distinct pathways, it should be possible to define them genetically. This was tested with mutations in the MCM-related precondition genes mei-218 and rec, which are required for 90% of all crossovers and the pachytene delays caused by mutations in DSB repair and exchange genes [14]. Unexpectedly, the high frequency of the two-oocyte phenotype was still observed in mei-218; TM3/+ and FM7/+; rec (Figure 2B, P<0.05 compared to mei-218 and rec single mutants). Consistent with this finding, the pachytene delay in c(2)M mutants was not suppressed in mei-218; c(2)M double mutants (Figure 1D). These results show that, unlike the DSB repair and exchange-defective mutants, defective and/or misaligned chromosome axes interact with the pachytene checkpoint independent of precondition genes and possibly at a later step (i.e. after the DSB response).
PCH2 can induce interchromosomal effects on crossing over
PCH2 is required for some of the crossovers that occur in the exchange-defective mutant, hdm [14]. Consequently, hdm; pch2 double mutants exhibit an elevated frequency of nondisjunction compared to hdm single mutants. These results suggest a functional link between the pachytene checkpoint and the production of crossovers. To determine if this is a general property of mutants that exhibit pachytene delays, we tested additional double mutants with pch2. Exchange class genes Ercc1 and mei-9 encode components of an endonuclease complex of proteins that includes HDM and is required for normal levels of meiotic crossing over [30], [31]. Loss of ERCC1 function results in 14% X-chromosome nondisjunction, which is elevated to 30% in a pch2 mutant background, suggesting crossovers are further reduced (Table S1). In addition, the low level of crossovers that are generated along the 2nd chromosome in mei-9 mutants are mostly suppressed in mei-9; pch2 double mutants (Figure 3A). These results suggest the residual crossovers in recombination-defective mutants depend on a mechanism facilitated by pch2.
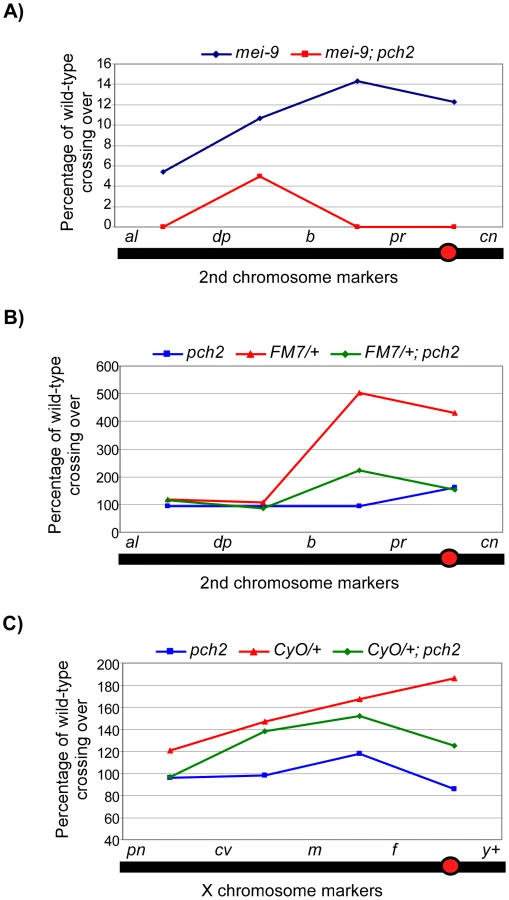

When crossing over is suppressed along a normal chromosome heterozygous to a balancer, there is an interchromosomal effect that increases crossing over on the remaining chromosome pairs [11]. Since PCH2 is responsible for the residual level of crossovers in recombination-defective mutants, we asked if it was also responsible for the increase in crossovers observed in balancer heterozygotes. Consistent with previous work on interchromosomal effects [32], [33], we found that FM7/+ heterozygotes exhibit 151% of wild-type crossing over along the 2nd chromosome with an altered distribution (Table 1). Although there was little deviation from wild-type controls in the distal regions of the chromosome (al-b), the genetic map distance was increased ∼4–5 times that observed in wild-type centromere-proximal intervals (Table 1; Figure 3B). Remarkably, 2nd chromosome crossing over in FM7/+ heterozygotes was reduced to 106% of wild-type in a pch2 mutant background (p<0.00005; Table 1; Figure 3B). Similarly, introduction of the CyO chromosome increased crossing over along the X chromosome to 149% of wild-type, which was reduced to 128% in pch2 mutants (p<0.05; Table 2; Figure 3C). Interestingly, the closer the interval being tested was to the centromere, the greater the interchromosomal effect and pch2 dependence (Table 2; Figure 3C). Since pch2 single mutants exhibited normal levels of crossing over on the X and 2nd chromosome (Table 1; Table 2; Figure 3), these data show that pch2 is required for most of the interchromosomal increase in crossover levels in balancer heterozygotes.
Pachytene checkpoint activity does not lead to an increase in DSB levels
The increased crossing over observed in balancer heterozygotes could be explained by pachytene checkpoint activity increasing DSB levels. However, we failed to observe any significant change in the number of γ-H2AV foci in oocytes single or doubly heterozygous for FM7 and CyO compared to wild-type (Figure 2D). Since asynchrony of DSB formation can complicate measuring the total number of γ-H2AV foci, we repeated the above experiment in a spn-A (Drosophila Rad51) mutant background, in which repair of DSBs is blocked [12]. The number of γ-H2AV foci in region 3 oocytes of these mutants is expected to be close to the total number of DSBs that occurred [28], [34]. spn-A mutant region 3 oocytes displayed an average of 21.0 (+/−1.5) γ-H2AV foci. Similarly, FM7/+; CyO/+; spn-A region 3 oocytes had an average of 24.0 (+/−1.4) γ-H2AV foci.
These results indicate that the ability of the pachytene checkpoint to increase crossing over in the genome is not mediated by a substantial increase in the total number of DSBs. Instead, pachytene checkpoint activity most likely increases the chance of DSBs becoming crossovers, particularly those that occur near centromeres.
PCH2 localizes to the nuclear periphery and persists when pachytene is delayed
To investigate how PCH2 affects crossover frequency, we monitored the protein localization pattern during meiosis. A transgene was constructed containing a hemagluttin (HA) epitope at the N-terminus of the pch2 transcript under the control of an inducible UASP promoter. We expressed PCH2 using the germline specific driver P(Gal4-nos.NGT)40 [35], abbreviated as NGT, known to express in pachytene at moderate levels [36]. The NGT-driven P(HA-pch2)71 transgenic line restored the two-oocyte phenotype in FM7/+; pch2 females to similar levels found in FM7/+ heterozygotes (Figure S1), demonstrating that the NGT-driven pch2 transgene was functional.
PCH2 staining formed foci that localized around the nucleus in zygotene and early pachytene (region 2) cells (Figure 4A). No PCH2 foci were detected in region 3 cells, suggesting the protein is rapidly degraded or no longer produced after early pachytene. Surprisingly, PCH2 foci did not overlap with the DNA stain. To determine if PCH2 foci localized within the nucleus, we counterstained with the nuclear envelope component, Lamin. We found that 73% of PCH2 foci showed a close association (i.e. touching) with the cytoplasmic side of the Lamin staining (n = 368; Figure 4B), indicating they localized adjacent to the nuclear envelope and outside the nucleus. The remaining 27% of PCH2 foci not closely associated with Lamin were found dispersed within the cytoplasm.

To determine if PCH2 localization pattern changes in mutant backgrounds that exhibit pachytene delays, we examined PCH2 expression in mutants that cause checkpoint delays: hdm, mei-9 and in TM3/+ heterozygotes. In hdm and mei-9 mutants, the number of PCH2 foci per oocyte was increased ∼2-fold compared to controls (Figure 4C). In addition, the foci persisted into region 3 oocytes, which was never observed in control germaria (Figure 4A and 4C). However, PCH2 localization was not detected past stage 2 of oogenesis (data not shown), indicating the loss of PCH2 is only delayed in exchange-defective mutants. In TM3/+ heterozygotes, the levels of PCH2 foci in region 2 cells was unchanged compared to controls, but were present in region 3 (Figure 4C), revealing a correlation between the prolonged expression of PCH2 and a delay in pachytene.
The intracellular localization pattern of PCH2 did not change when pachytene was delayed since the foci remained juxtaposed to the nuclear envelope in hdm and mei-9 mutants and in TM3/+ heterozygotes at all stages (Figure 4A and data not shown). Furthermore, mutation of mei-W68, which eliminates DSB formation, showed a normal staining pattern of PCH2, and hdm; mei-W68 double mutants showed the same PCH2 staining pattern as hdm single mutants (Figure 4A and data not shown), consistent with our previous conclusion that the pachytene checkpoint functions independently of DSB formation [14], [16]. These observations provide a connection between the nuclear envelope and pachytene checkpoint activity and suggest that PCH2′s role in nuclear events like crossing over is indirect and at a distance from the chromosomes.
Prolonged PCH2 activity leads to a pachytene delay and altered crossover distribution
To test the significance of the correlation between pachytene delays and prolonged PCH2 expression, we manipulated the timing and expression levels of PCH2 in the germline. PCH2 levels were increased by driving the UASP:pch2 transgene with P(Gal4::VP16-nos.UTR)MVD1 [37], abbreviated as MVD1, known to drive high levels of expression in the germarium. MVD1-driven pch2 caused the protein to persist into region 3 oocytes, which was never observed with the NGT driver in a wild-type background (Figure 4A and 4C). In addition to distinct foci, PCH2 was also distributed more evenly throughout the cytoplasm (Figure 4A). Thus, MVD1-driven pch2 resulted in overproduction and prolonged expression of the protein throughout pachytene.
Pachytene delays were not observed when the pch2 transgenes were expressed using the NGT driver (Figure 5A). In contrast, MVD1-driven pch2 induced a pachytene delay that resulted in a high frequency of the two-oocyte phenotype (Figure 5A). In fact, 100% (n = 10) of the germaria with PCH2 expression in region 3 cysts also contained two oocytes, as viewed by C(3)G staining, suggesting prolonged PCH2 expression is sufficient to induce a delay in pachytene progression.

Since overproducing PCH2 caused a pachytene delay, we determined if crossover frequency or distribution was also affected. We found that PCH2 expression driven by MVD1 altered the distribution of exchange in all 3 transgenic lines tested (Table 1; Figure 5B). The most dramatic increase in crossover frequencies was observed in the centromere proximal interval of chromosome 2, b-pr. Although all the transgenic lines that were tested showed the same change in crossover distribution, the magnitude was different, which probably reflects different transgenic protein levels. In support of this conclusion, the presence of two transgenic copies of P(HA-pch2)71 driven by MVD1 exacerbated the effect on both crossover levels and distribution (Table 1; Figure 5B). These data show that the frequency and distribution of crossing over is sensitive to the timing and level of PCH2 expression during pachytene.
sir2 is required for prolonged PCH2 expression and the pachytene checkpoint
We sought to identify factors that facilitate prolonged PCH2 expression and cause pachytene delays. The first candidate we tested was mei-218 since it is required for the pch2-dependent pachytene delays observed in DSB repair and exchange-defective mutants. mei-218 mutants, however, did not show an effect on the levels or distribution of MVD1-driven PCH2 (Figure S2). Also, the two-oocyte phenotype caused by PCH2 overexpression was not suppressed in mei-218 mutants (Figure 5A), suggesting MEI-218 is not required for PCH2 localization.
The second candidate tested was Sir2, which encodes a histone deacetylase that is required for the nucleolus localization of Pch2 and the pachytene checkpoint during S. cerevisiae meiosis [15]. Five Drosophila genes belong to the Sir2 family. Of these, Sir2 is the closest homolog of the S. cerevisiae Sir2 [38]. Drosophila sir2 null alleles have no obvious effects on viability, but affect position effect variegation, heterochromatic silencing and fly life span [38]–[40]. sir2 mutants were fully fertile with wild-type levels of X-chromosome nondisjunction (Table S1), indicating Sir2 is dispensable for meiotic recombination.
We investigated whether Sir2 is involved in the pachytene checkpoint and found that mutation of sir2 suppressed the high frequency of the two-oocyte phenotype observed when PCH2 is overexpressed with the MVD1 driver (Figure 5A). The high frequency of the two-oocyte phenotypes observed in the exchange-defective mutant hdm and DSB repair mutant spn-B were also suppressed by sir2 (Figure 6A). Likewise, Sir2 was required for the pachytene delay observed in TM3/+ heterozygotes (Figure 6A) and the delayed onset of γ-H2AV staining in spn-B mutants (cyst 2–5 in Figure 6B). Thus, like pch2, sir2 is required for the pachytene checkpoint.

Strikingly, the region 3 localization of MVD1-driven PCH2 was eliminated in a sir2 mutant (Figure 4A and 4C). In contrast, loss of sir2 only slightly reduced the level of PCH2 in region 2 cells and had no effect on the peri-nuclear localization of PCH2 driven by NGT (Figure 4C and data not shown). In addition, expression of a c(2)M transgene driven by MVD1 was not affected, indicating the effect of sir2 on PCH2 was not due to a reduction in the transcription of UAS-driven genes (Figure S3). These results support the hypothesis that high levels of PCH2 are dependent on Sir2 and essential for the pachytene delays.
Discussion
The pachytene checkpoint is sensitive to defects in chromosome axes
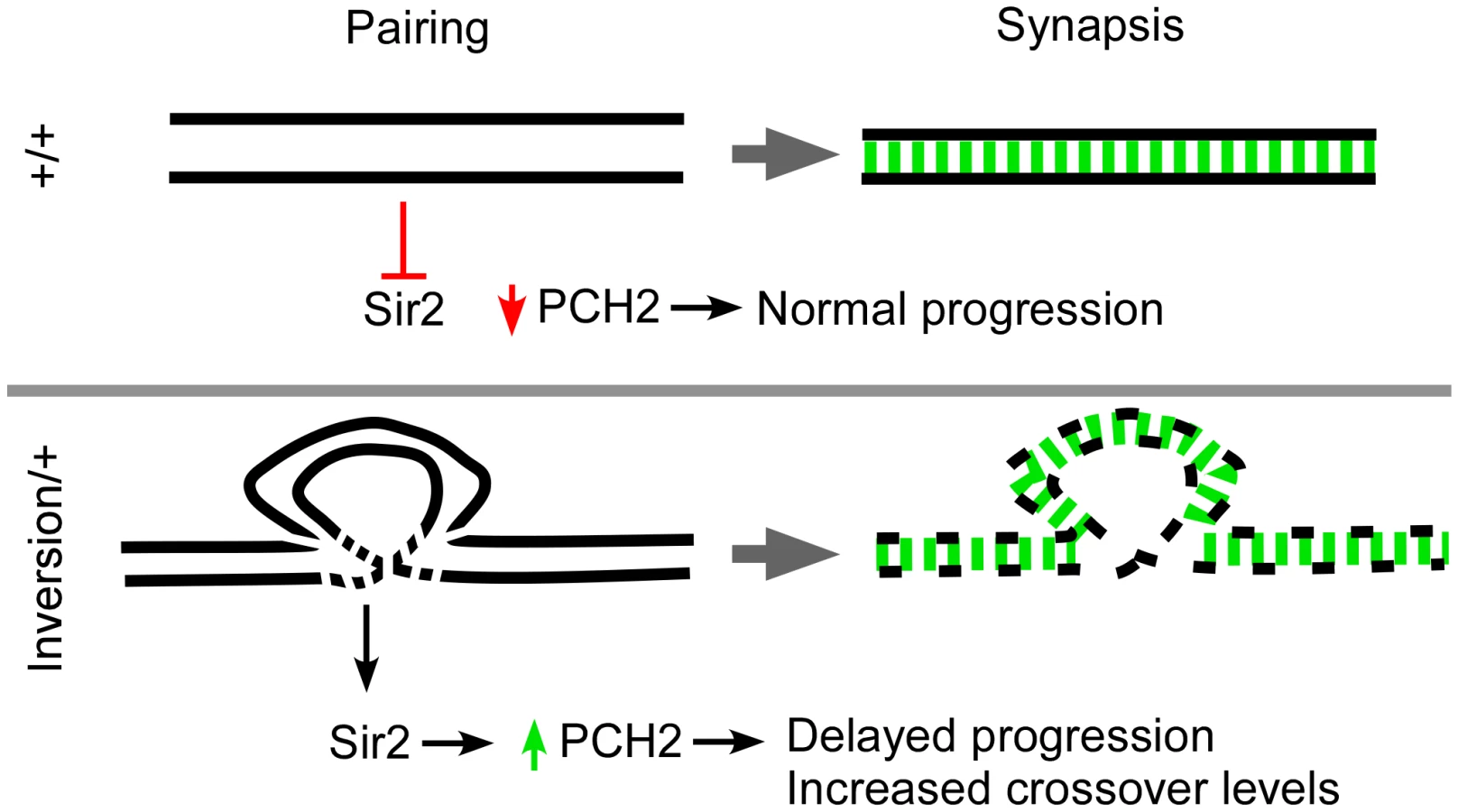
We have previously shown that removing the SC central element component C(3)G does not cause pch2-dependent delays in Drosophila meiotic prophase [14]. Therefore, the pachytene checkpoint is not monitoring the process of synapsis per se. Instead, two lines of evidence suggest the pachytene checkpoint is sensitive to defects in chromosome axes. First, mutations in genes that encode structural axis components, C(2)M and ORD, cause pch2-dependent pachytene delays. Second, heterozygous chromosomal rearrangements also cause a pch2-dependent delay. Homozygous rearrangements do not cause delays; therefore, the pachytene checkpoint is sensitive to any discontinuity in the alignment between homologous chromosomes. Since the delays do not depend on C(3)G, the defect must be prior to or independent of synapsis initiation. The misalignment of homologous sequences could destabilize the integrity of chromosome axes, such as the assembly of ORD or C(2)M, and expose substrates that trigger the pachytene checkpoint. Indeed, females doubly heterozygous for balancer chromosomes show deterioration of C(2)M staining in pachytene oocytes similar to ord mutants [21], suggesting the axial elements are compromised by the heterozygous inversion breakpoints.
Two genetically distinct pathways can trigger the pachytene checkpoint
The delays observed in c(2)M mutants and balancer heterozygotes do not depend on the MCM-related precondition genes such as mei-218, which are required for the pachytene delays in DSB repair and exchange-defective mutants [14]. Balancer heterozygotes also do not cause a delayed response to DSBs or increase in the number of PCH2 foci as observed in DSB repair and exchange-defective mutants. Therefore, two pathways probably lead into a pch2-dependent checkpoint: a mei-218-dependent pathway involving the early function of DSB repair proteins and a mei-218-independent pathway involving the structure of chromosome axes.
Of the two pathways in Drosophila, the pachytene checkpoint in other organisms has similarities to the mei-218-independent pathway involving chromosome structure. Heterozygous inversions and translocations induce a pachytene delay, suggesting a model in which the pachytene checkpoint can respond to breaks in axis continuity between paired homologs. The C. elegans pachytene checkpoint also monitors meiotic chromosome structure since a defect in a SC-nucleating “pairing center” triggers a Pch2-dependent response [16]. Similarly, the budding yeast pachytene checkpoint has been proposed to monitor SC-dependent events that may involve the relationship between recombination complexes and chromosome axes [41]–[43]. Therefore, a common feature of the pachytene checkpoints in these organisms is their sensitivity to the axis continuity between paired homologs with the main difference being SC-dependent defects (yeast and nematodes) versus SC-independent axis defects (Drosophila). Interestingly, both yeast Pch2 and mouse Trip13/Pch2 have been proposed to have a checkpoint-independent role in the organization of chromosome axis proteins [43], [44]. We do not know as of yet, however, if this is related to the sensitivity of paired axes at the Drosophila pachytene checkpoint, although it is tempting to suggest such a model.
Pachytene checkpoint activity in budding yeast is associated with prolonged Pch2 expression that requires Sir2 [15]. As in budding yeast, Drosophila sir2 mutants are defective in the pachytene checkpoint and our overexpression studies suggest Sir2 is also required for the prolonged expression of PCH2. These results provide evidence for an evolutionarily conserved role of Pch2 and Sir2 in monitoring changes in chromosome structure during meiotic prophase from yeast to a metazoan (Figure 7). Drosophila may have evolved an additional mei-218-dependent pachytene checkpoint, not shared with yeast and nematodes, which is sensitive to DSB repair complexes.
Chromosomal rearrangements induce pch2-dependent interchromosomal effects on crossing over
The effect of inversion heterozygosity on the frequency of crossing over has been known since the work of Sturtevant in 1919. Most often these intrachromosomal rearrangements cause an interchromosomal increase in recombination levels. Exhaustive work has been carried out investigating the interchromosomal effect and several models have been proposed in order to account for the increase in crossing over [11]. The most recent and generally accepted model was last described by Lucchesi and Suzuki in 1968 who proposed a timing model where pairing and crossover formation are coupled during the pachytene stage of prophase [11]. They suggested that when the pairing process between one set of homologs is perturbed or delayed by chromosome rearrangements, pachytene was lengthened and the opportunity to make crossovers was prolonged. We propose a modified version of the timing model where breaks in homology cause disruptions in the axis structure, resulting in a checkpoint-mediated delay (Figure 7).
The timing model proposed by Lucchesi and Suzuki predicts that a factor exists which controls the timing of meiotic prophase and can affect the level of exchange [11]. The pachytene checkpoint may regulate this timing mechanism. Although pch2 is dispensable for crossing over in a wild-type background, it is required for most of the residual crossovers that occur in recombination-defective mutants. pch2 is also required for most of the interchromosomal effect and pachytene delays observed in inversion heterozygotes. To our knowledge, pch2 is the first example of a gene in Drosophila required for the interchromosomal effect that is not required for crossing over in general. Prolonged PCH2 expression may facilitate the formation of more crossovers by simply delaying a pachytene transition and extending the crossover determination phase, thereby allowing more crossover sites to be selected. An alternative explanation is that pch2, while not required for crossover formation in wild-type, is required for a crossover mechanism active only in axis-defective situations. Since the interchromosomal effect is not mediated by an increase in DSBs, PCH2 most likely increases the chance of DSBs becoming crossovers at the expense of noncrossovers.
PCH2 function, localization, and mechanism of checkpoint activity
Drosophila PCH2 localizes to peri-nuclear foci in zygotene and early pachytene cells and is rapidly degraded or no longer made at mid-pachytene. In mutants in which pachytene delays are observed, PCH2 expression persists longer than in wild-type. The observation that overexpressing PCH2 induces effects on both timing and crossover levels indicates prolonged PCH2 expression is necessary and sufficient for the pachytene checkpoint's downstream effects. Since the duration of early pachytene correlates with the domain of PCH2 expression, we suggest that degradation of PCH2 turns off checkpoint activity and allows progression through pachytene, which ends the crossover determination phase (Figure 7).
We observed PCH2 localization to the outside of the nuclear envelope. These results were surprising considering the effect a pch2 mutation has on nuclear events like crossing over. While we cannot rule out the possibility that a small undetectable fraction of PCH2 protein enters the nucleus and interacts with the chromosomes, PCH2 may indirectly affect nuclear events by facilitating interactions between the chromosomes and the nuclear envelope. In budding yeast, the pachytene checkpoint requires the localization of Pch2 to the nucleolus [15]. Therefore, like budding yeast, PCH2 in Drosophila may mediate the pachytene checkpoint at a distance from the recombination sites. Intriguingly, the nuclear envelope has been linked to several cellular processes relevant to meiotic recombination, including homolog pairing and DSB repair [45]–[48]. In C. elegans, the pairing of homologous chromosomes first requires the relocation of chromosomal regions known as pairing centers to the nuclear envelope [45]. Chromosome deficiencies that remove the pairing center impair relocation, homolog pairing and synapsis as well as trigger a pch2-dependent response [16]. Therefore, it is possible that in other organisms, the nuclear envelope has a conserved role in transducing pachytene checkpoint effects.
Materials and Methods
Drosophila strains
Drosophila stocks and crosses were maintained on a standard medium at 25°C. The following mutant alleles were used unless otherwise noted - ord10 [20], c(2)MEP, pch2EY01788a (pch2EY), c(3)G68 [18], hdmg7, mei-2181, rec1and rec2 [49], Ercc1X [30], mei-9a, spn-A1, spn-BBu, sir217 [38], and mei-W684572. The deficiency Df(2L)BSC245 deletes cytological bands 33F3-34A9, which includes the sir2 locus. T(2;3)DP77 and T(2;3)dpD translocations were obtained from the Bloomington Stock Center. T(2;3)DP77 breakpoints are at 26E-27A on the 2nd and 85C on the 3rd. T(2;3)dpD breakpoints are at 25A on the 2nd and 95B–D on the 3rd. The T(2;3)ltX16 translocation has breakpoints at 40 (heterochromatin) on the 2nd and 95A3 on the 3rd and was obtained from B. Wakimoto [50].
Genetic techniques
X-chromosome nondisjunction was assayed by crossing females to y w/YBS males. The frequency of X-chromosome nondisjunction is calculated as 2(Bar females + Bar+ males)/[2(Bar females + Bar+ males) + Bar+ females + Bar males]. To estimate wild-type X chromosome crossing over frequency, y/y pn cv m f • y+ female flies were crossed to C(1:Y)1, v f B: [+]; C(4)RM, ci ey males, and X chromosome recombinants were scored in males. Second chromosome crossing over was assayed by crossing al dp b pr cn/+ females to al dp b pr cn/CyO males and scoring for recombinants among the Cy+ progeny. P-values were calculated using the Fisher's exact test.
Cytology and immunofluorescence
For immunolocalization experiments, females were aged at room temperature for about 16 hours and ovaries were dissected and fixed using the “Buffer A” protocol [51]. The antibody to γ-H2AV was described by Mehrotra et al. [28] and used at a 1∶500 dilution. Additional primary antibodies included mouse anti-C(3)G antibody used at 1∶500 [18], rabbit anti-C(2)M antibody used at 1∶400 [52], a combination of two mouse anti-Orb antibodies (4H8 and 6H4) used at 1∶100 [53], mouse anti-Lamin antibody developed by Fisher, P.A. used at 1∶800, and a rat anti-HA antibody (Roche) used at 1∶15.
The secondary antibodies were Cy3 labeled goat anti-rabbit (Jackson Labs) used at 1∶250, Cy3 labeled goat anti-rat (Jackson Labs) used at 1∶100 and Alexa fluor 488 goat anti-mouse (Invitrogen) used at 1∶100. Chromosomes were stained with Hoechst 33342 at 1∶50,000 (10 mg/ml solution) for seven minutes at room temperature. Images were collected using a Leica TCS SP2 confocal microscope with a 63X, N.A. 1.3 lens. In most cases, whole germaria were imaged by collecting optical sections through the entire tissue. These data sets are shown as maximum projections. The analysis of the images, however, was performed by examining one section at a time.
Counting the frequency of the two-oocyte phenotype and calculating P-values
The oocytes were observed using an anti-C(3)G antibody. In some cases, such as when C(3)G staining was not visible, anti-ORB staining was used to identify the oocyte(s). A cell was scored as an oocyte if complete SC filaments were clear and distinct or by a concentration of ORB staining in the cytoplasm of a cell [54]. P-values were calculated using the Fisher's exact test. The P-value from the test compares the ratio of one-oocyte to two-oocyte cysts that were observed in two genotypes.
Counting γ-H2AV foci in repair-proficient and repair-defective backgrounds
The γ-H2AV foci were counted from germaria where the foci were clear and distinct. Foci numbers in wild-type were at a maximum in region 2a (early pachytene) and few foci were visible by region 2b (mid pachytene). Therefore, to compare foci numbers in different genotypes, we used a method that calculates all cysts with γ-H2AV foci, averaging the amount in each pair of pro-oocytes. We compared the average foci in all the pro-oocytes or oocytes of each germarium, starting with the youngest cysts at the anterior end, by examining a full series of optical sections.
For counting γ-H2AV foci in repair-defective backgrounds, ORB staining was used to identify oocytes in region 3. The foci were counted from germaria where the foci were clear and distinct. The foci were counted manually by examining each section in a full series of optical sections containing complete pro-oocyte nucleus.
Plotting γ-H2AV foci as a function of relative cyst age
Since the position of a cyst in the germarium is only a rough estimate of its meiotic stage, the foci were first counted in all the pro-oocytes/oocytes (identified by C(3)G staining) in the germarium. The meiotic stage of each pro-oocyte was then normalized according to the relative position of the entire cyst within the germarium since the relative position is more important than absolute position. The pro-oocytes from 13 wild-type germaria, 4 FM7/+, 4 CyO/+, 5 FM7/+; CyO/+, 5 spn-BBu, and 4 sir217/Df; spn-BBu were arranged according to their relative age. The average number of γ-H2AV foci per pro-oocyte at each stage was then calculated and plotted as a function of relative cyst age.
Construction of PCH2 transgenes
The annotated coding region of pch2 was obtained from Flybase and amplified off the pch2 cDNA clone LD24646 [55] by PCR. The coding region of pch2 was then cloned into the Gateway® pENTRTM4 vector (Invitrogen). An LR ‘clonase’ reaction was then performed to recombine pch2 into the ppHW destination vector (Invitrogen) that contains 3 copies of an N-terminus HA-tag under the control of an inducible UASP promoter. The construct was injected into fly embryos by Model System Genomics at Duke University. To express the transgenic lines, they were crossed to flies expressing Gal4 using either the NGT (P[Gal4-nos.NGT]40) [35] or MVD1 (P[Gal4::VP16-nos.UTR]MVD1) drivers [37].
Counting PCH2 foci
The HA-PCH2 foci were counted from germaria where the foci were clear and distinct. We counted the average foci surrounding nuclei in all the cysts at region 2 and region 3 of each germarium by examining a full series of optical sections.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. PageSL
HawleyRS
2004 The genetics and molecular biology of the synaptonemal complex. Annu Rev Cell Dev Biol 20 525 558
2. ZicklerD
KlecknerN
1999 Meiotic chromosomes: integrating structure and function. Annu Rev Genet 33 603 754
3. McKimKS
Hayashi-HagiharaA
1998 mei-W68 in Drosophila melanogaster encodes a Spo11 homolog: evidence that the mechanism for initiating meiotic recombination is conserved. Genes & Dev 12 2932 2942
4. DernburgAF
McDonaldK
MoulderG
BarsteadR
DresserM
1998 Meiotic recombination in C. elegans initiates by a conserved mechanism and is dispensable for homologous chromosome synapsis. Cell 94 387 398
5. KeeneyS
2001 Mechanism and control of meiotic recombination initiation. Curr Top Dev Biol 52 1 53
6. MullerHJ
1916 The mechanism of crossing-over. American Naturalist 50 193 434
7. SturtevantAH
1915 The behavior of the chromosomes as studied through linkage. Z Induktive Abstammungs Vererbungslehre 13 234 287
8. HillersKJ
2004 Crossover interference. Curr Biol 14 R1036 1037
9. KlecknerN
ZicklerD
JonesGH
DekkerJ
PadmoreR
2004 A mechanical basis for chromosome function. Proc Natl Acad Sci U S A 101 12592 12597
10. CarpenterATC
SandlerL
1974 On recombination-defective meiotic mutants in Drosophila melanogaster. Genetics 76 453 475
11. LucchesiJC
SuzukiDT
1968 The interchromosomal control of recombination. Ann Rev Genet 2 53 86
12. JangJK
SherizenDE
BhagatR
ManheimEA
McKimKS
2003 Relationship of DNA double-strand breaks to synapsis in Drosophila. J Cell Sci 116 3069 3077
13. GhabrialA
SchupbachT
1999 Activation of a meiotic checkpoint regulates translation of Gurken during Drosophila oogenesis. Nat Cell Biol 1 354 357
14. JoyceEF
McKimKS
2009 Drosophila PCH2 Is Required for a Pachytene Checkpoint That Monitors Double-Strand-Break-Independent Events Leading to Meiotic Crossover Formation. Genetics 181 39 51
15. San-SegundoPA
RoederGS
1999 Pch2 links chromatin silencing to meiotic checkpoint control. Cell 97 313 324
16. BhallaN
DernburgAF
2005 A conserved checkpoint monitors meiotic chromosome synapsis in Caenorhabditis elegans. Science 310 1683 1686
17. MitraN
RoederGS
2007 A novel nonnull ZIP1 allele triggers meiotic arrest with synapsed chromosomes in Saccharomyces cerevisiae. Genetics 176 773 787
18. PageSL
HawleyRS
2001 c(3)G encodes a Drosophila synaptonemal complex protein. Genes & Dev 15 3130 3143
19. McCaffreyR
St JohnstonD
Gonzalez-ReyesA
2006 Drosophila mus301/spindle-C Encodes a Helicase With an Essential Role in Double-Strand DNA Break Repair and Meiotic Progression. Genetics 174 1273 1285
20. BickelSE
WymanDW
Orr-WeaverTL
1997 Mutational analysis of the Drosophila sister-chromatid cohesion protein ORD and its role in the maintenance of centromeric cohesion. Genetics 146 1319 1331
21. WebberHA
HowardL
BickelSE
2004 The cohesion protein ORD is required for homologue bias during meiotic recombination. J Cell Biol 164 819 829
22. AndersonLK
RoyerSM
PageSL
McKimKS
LaiA
2005 Juxtaposition of C(2)M and the transverse filament protein C(3)G within the central region of Drosophila synaptonemal complex. Proc Natl Acad Sci U S A 102 4482 4487
23. KhetaniRS
BickelSE
2007 Regulation of meiotic cohesion and chromosome core morphogenesis during pachytene in Drosophila oocytes. J Cell Sci 120 3123 3137
24. NovitskiE
BraverG
1954 An analysis of crossing over within a heterozygous inversion in Drosophila melanogaster. Genetics 39 197 209
25. SherizenD
JangJK
BhagatR
KatoN
McKimKS
2005 Meiotic recombination in Drosophila females depends on chromosome continuity between genetically defined boundaries. Genetics 169 767 781
26. GongWJ
McKimKS
HawleyRS
2005 All Paired Up with No Place to Go: Pairing, Synapsis, and DSB Formation in a Balancer Heterozygote. PLoS Genet 1 e67 doi:10.1371/journal.pgen.0010067
27. LindsleyDL
ZimmGG
1992 The Genome of Drosophila melanogaster. San Diego, CA Academic Press, Inc
28. MehrotraS
McKimKS
2006 Temporal analysis of meiotic DNA double-strand break formation and repair in Drosophila females. PLoS Genet 2 e200 doi:10.1371/journal.pgen.0020200
29. MadiganJP
ChotkowskiHL
GlaserRL
2002 DNA double-strand break-induced phosphorylation of Drosophila histone variant H2Av helps prevent radiation-induced apoptosis. Nucleic Acids Res 30 3698 3705
30. RadfordSJ
GoleyE
BaxterK
McMahanS
SekelskyJ
2005 Drosophila ERCC1 is required for a subset of MEI-9-dependent meiotic crossovers. Genetics 170 1737 1745
31. JoyceEF
TannetiSN
McKimKS
2009 Drosophila Hold'em Is Required for a Subset of Meiotic Crossovers and Interacts With the DNA Repair Endonuclease Complex Subunits MEI-9 and ERCC1. Genetics 181 335 340
32. SzauterP
1984 An analysis of regional constraints on exchange in Drosophila melanogaster using recombination-defective meiotic mutants. Genetics 106 45 71
33. CarpenterATC
1988 Thoughts on Recombination nodules, meiotic recombination, and chiasmata.
KucherlapatiR
SmithG
Genetic recombination Washington, D.C. American Society of Microbiology 529 548
34. McKimKS
JangJK
ManheimEA
2002 Meiotic recombination and chromosome segregation in Drosophila females. Annu Rev Genet 36 205 232
35. TraceyWDJr
NingX
KlinglerM
KramerSG
GergenJP
2000 Quantitative analysis of gene function in the Drosophila embryo. Genetics 154 273 284
36. RorthP
1998 Gal4 in the Drosophila female germline. Mech Dev 78 113 118
37. Van DorenM
WilliamsonAL
LehmannR
1998 Regulation of zygotic gene expression in Drosophila primordial germ cells. Curr Biol 8 243 246
38. AstromSU
ClineTW
RineJ
2003 The Drosophila melanogaster sir2+ gene is nonessential and has only minor effects on position-effect variegation. Genetics 163 931 937
39. RosenbergMI
ParkhurstSM
2002 Drosophila Sir2 is required for heterochromatic silencing and by euchromatic Hairy/E(Spl) bHLH repressors in segmentation and sex determination. Cell 109 447 458
40. NewmanBL
LundbladJR
ChenY
SmolikSM
2002 A Drosophila homologue of Sir2 modifies position-effect variegation but does not affect life span. Genetics 162 1675 1685
41. BornerGV
KlecknerN
HunterN
2004 Crossover/noncrossover differentiation, synaptonemal complex formation, and regulatory surveillance at the leptotene/zygotene transition of meiosis. Cell 117 29 45
42. BornerGV
BarotA
KlecknerN
2008 Yeast Pch2 promotes domainal axis organization, timely recombination progression, and arrest of defective recombinosomes during meiosis. Proc Natl Acad Sci U S A 105 3327 3332
43. JoshiN
BarotA
JamisonC
BornerGV
2009 Pch2 links chromosome axis remodeling at future crossover sites and crossover distribution during yeast meiosis. PLoS Genet 5 e1000557 doi:10.1371/journal.pgen.1000557
44. WojtaszL
DanielK
RoigI
Bolcun-FilasE
XuH
2009 Mouse HORMAD1 and HORMAD2, two conserved meiotic chromosomal proteins, are depleted from synapsed chromosome axes with the help of TRIP13 AAA-ATPase. PLoS Genet 5 e1000702 doi:10.1371/journal.pgen.1000702
45. PhillipsCM
DernburgAF
2006 A Family of Zinc-Finger Proteins Is Required for Chromosome-Specific Pairing and Synapsis during Meiosis in C. elegans. Dev Cell 11 817 829
46. PenknerA
TangL
NovatchkovaM
LadurnerM
FridkinA
2007 The nuclear envelope protein Matefin/SUN-1 is required for homologous pairing in C. elegans meiosis. Dev Cell 12 873 885
47. DingX
XuR
YuJ
XuT
ZhuangY
2007 SUN1 is required for telomere attachment to nuclear envelope and gametogenesis in mice. Dev Cell 12 863 872
48. NagaiS
DubranaK
Tsai-PflugfelderM
DavidsonMB
RobertsTM
2008 Functional targeting of DNA damage to a nuclear pore-associated SUMO-dependent ubiquitin ligase. Science 322 597 602
49. BlantonHL
RadfordSJ
McMahanS
KearneyHM
IbrahimJG
2005 REC, Drosophila MCM8, Drives Formation of Meiotic Crossovers. PLoS Genet 1 e40 doi:10.1371/journal.pgen.0010040
50. WakimotoBT
HearnMG
1990 The effects of chromosome rearrangements on the expression of heterochromatic genes in chromosome 2L of Drosophila melanogaster. Genetics 125 141 154
51. McKimK
JoyceEF
JangJK
2008 Cytological analysis of meiosis in fixed Drosophila ovaries Totowa, NJ Humana Press
52. ManheimEA
McKimKS
2003 The Synaptonemal Complex Component C(2)M Regulates Meiotic Crossing over in Drosophila. Curr Biol 13 276 285
53. LantzV
ChangJS
HorabinJI
BoppD
SchedlP
1994 The Drosophila ORB RNA-binding protein is required for the formation of the egg chamber and establishment of polarity. Genes & Dev 8 598 613
54. Gonzalez-ReyesA
ElliotH
Johnston DSt
1997 Oocyte determination and the origin of polarity in Drosophila: the role of the spindle genes. Development 124 4927 4937
55. StapletonM
LiaoG
BroksteinP
HongL
CarninciP
2002 The Drosophila gene collection: identification of putative full-length cDNAs for 70% of D. melanogaster genes. Genome Res 12 1294 1300
56. KingRC
1970 Ovarian development in Drosophila melanogaster. New York Academic Press, Inc
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 8
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Identification of the Bovine Arachnomelia Mutation by Massively Parallel Sequencing Implicates Sulfite Oxidase (SUOX) in Bone Development
- Common Inherited Variation in Mitochondrial Genes Is Not Enriched for Associations with Type 2 Diabetes or Related Glycemic Traits
- A Model for Damage Load and Its Implications for the Evolution of Bacterial Aging
- Did Genetic Drift Drive Increases in Genome Complexity?
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy