
Assembly of a 40 Mb Eukaryotic Genome from Short Sequence Reads: , a Model Organism for Fungal Morphogenesis
Filamentous fungi are of great importance in ecology, agriculture, medicine, and biotechnology. Thus, it is not surprising that genomes for more than 100 filamentous fungi have been sequenced, most of them by Sanger sequencing. While next-generation sequencing techniques have revolutionized genome resequencing, e.g. for strain comparisons, genetic mapping, or transcriptome and ChIP analyses, de novo assembly of eukaryotic genomes still presents significant hurdles, because of their large size and stretches of repetitive sequences. Filamentous fungi contain few repetitive regions in their 30–90 Mb genomes and thus are suitable candidates to test de novo genome assembly from short sequence reads. Here, we present a high-quality draft sequence of the Sordaria macrospora genome that was obtained by a combination of Illumina/Solexa and Roche/454 sequencing. Paired-end Solexa sequencing of genomic DNA to 85-fold coverage and an additional 10-fold coverage by single-end 454 sequencing resulted in ∼4 Gb of DNA sequence. Reads were assembled to a 40 Mb draft version (N50 of 117 kb) with the Velvet assembler. Comparative analysis with Neurospora genomes increased the N50 to 498 kb. The S. macrospora genome contains even fewer repeat regions than its closest sequenced relative, Neurospora crassa. Comparison with genomes of other fungi showed that S. macrospora, a model organism for morphogenesis and meiosis, harbors duplications of several genes involved in self/nonself-recognition. Furthermore, S. macrospora contains more polyketide biosynthesis genes than N. crassa. Phylogenetic analyses suggest that some of these genes may have been acquired by horizontal gene transfer from a distantly related ascomycete group. Our study shows that, for typical filamentous fungi, de novo assembly of genomes from short sequence reads alone is feasible, that a mixture of Solexa and 454 sequencing substantially improves the assembly, and that the resulting data can be used for comparative studies to address basic questions of fungal biology.
Published in the journal:
. PLoS Genet 6(4): e32767. doi:10.1371/journal.pgen.1000891
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1000891
Summary
Filamentous fungi are of great importance in ecology, agriculture, medicine, and biotechnology. Thus, it is not surprising that genomes for more than 100 filamentous fungi have been sequenced, most of them by Sanger sequencing. While next-generation sequencing techniques have revolutionized genome resequencing, e.g. for strain comparisons, genetic mapping, or transcriptome and ChIP analyses, de novo assembly of eukaryotic genomes still presents significant hurdles, because of their large size and stretches of repetitive sequences. Filamentous fungi contain few repetitive regions in their 30–90 Mb genomes and thus are suitable candidates to test de novo genome assembly from short sequence reads. Here, we present a high-quality draft sequence of the Sordaria macrospora genome that was obtained by a combination of Illumina/Solexa and Roche/454 sequencing. Paired-end Solexa sequencing of genomic DNA to 85-fold coverage and an additional 10-fold coverage by single-end 454 sequencing resulted in ∼4 Gb of DNA sequence. Reads were assembled to a 40 Mb draft version (N50 of 117 kb) with the Velvet assembler. Comparative analysis with Neurospora genomes increased the N50 to 498 kb. The S. macrospora genome contains even fewer repeat regions than its closest sequenced relative, Neurospora crassa. Comparison with genomes of other fungi showed that S. macrospora, a model organism for morphogenesis and meiosis, harbors duplications of several genes involved in self/nonself-recognition. Furthermore, S. macrospora contains more polyketide biosynthesis genes than N. crassa. Phylogenetic analyses suggest that some of these genes may have been acquired by horizontal gene transfer from a distantly related ascomycete group. Our study shows that, for typical filamentous fungi, de novo assembly of genomes from short sequence reads alone is feasible, that a mixture of Solexa and 454 sequencing substantially improves the assembly, and that the resulting data can be used for comparative studies to address basic questions of fungal biology.
Introduction
Fungi are heterotrophic eukaryotes found in nearly all ecosystems. About 100,000 fungi have been described to date, but conservative estimates predict at least 1.5 million different species [1],[2]. Fungi exhibit a wide range of different lifestyles, particularly as saprobes, pathogens or symbionts. As saprobes, fungi acquire nutrients from dead organic matter and are among the main recyclers on the planet. They play important roles in the degradation of cellulose and lignin, contributing greatly to the global carbon cycle. However, their saprotrophic activities also cause severe problems with the degradation of man-made products and in causing food spoilage. Mortality from human fungal pathogens has increased in recent years, especially in immunocompromised patients. In plants, ∼90% of diseases are caused by fungi, and these result in massive losses in crop yield worldwide, with often profound socio-economic effects, sometimes resulting in severe famines [3]. Nevertheless, fungi also have beneficial effects in symbioses, such as mycorrhiza (fungus/plant root) and lichen (fungus/algae) associations. Greater than 80% of terrestrial plants have mycorrhizal relationships with fungi that allow the plants to access key nutrients such as nitrogen and phosphorus from the soil [4]. Fungi are also of great importance in biotechnology, e.g. in the production of drugs and enzymes [5],[6]. In addition, many fungi can be easily cultured and are amenable to microbiological, genetic, and molecular techniques. Therefore, fungi were some of the earliest model organisms for the study of genetics, biochemistry, cell and developmental biology. It is thus not surprising that the first eukaryotic organism for which a complete genome sequence was obtained is a fungus, the budding yeast Saccharomyces cerevisiae [7]. Today, fungi are the eukaryotic group with the greatest number of completely, or nearly completely, sequenced genomes (http://www.ncbi.nlm.nih.gov/genomes/leuks.cgi, [2]). This is not only owing to their ecological, medical, agricultural, biotechnological and economic significance, but also due to the fact that with a size of 10–90 Mb and 4,700–17,000 predicted genes, fungal genomes are among the smallest and most compact eukaryotic genomes known.
The sequences for almost all sequenced eukaryotic genomes have been obtained by conventional Sanger sequencing technology. Over the past five years “next-generation sequencing” techniques have revolutionized large-scale sequencing projects because of massively increased throughput, resulting in much reduced costs per base [8]. One major disadvantage of the current techniques is that none of them delivers read lengths that approach conventional Sanger technology: whereas Sanger sequencing routinely yields 900 nt, the longest next-generation reads obtained are in the range of ∼450 nt for Roche/454 pyrosequencing (from now on abbreviated as 454 sequencing), and the techniques with the highest throughput are with 36–80 nt still well below this. Short reads, e.g. as obtained by Illumina/Solexa sequencing (from now on abbreviated as Solexa sequencing) cause severe difficulties for the assembly of genome sequences that contain repetitive sequences, as is the case for many higher eukaryotes. Thus, next-generation sequencing techniques have so far mostly been used for the de novo sequencing of prokaryotic genomes or the re-sequencing of eukaryotic species with reference genomes, where the next-generation reads can be mapped on an existing genome sequence [8]–[11]. Recent improvements, e.g. paired-end sequencing (reads from matched ends of longer DNA fragments) and a steady increase in read length should make the de novo assembly of high-quality eukaryotic genomes possible. For example, the genome of the filamentous fungus Grosmannia clavigera was assembled from a combination of Sanger, 454, and Solexa sequence data [12] and a first draft of the 2.4 Gb Giant Panda genome has been assembled from Solexa sequence reads alone [13]. Because of their small size, fungal genomes are perfectly suited for the task of optimizing de novo assembly approaches to generate high-quality or even finished larger eukaryotic genomes.
Here, we present the de novo assembly and annotation of the genome sequence of the filamentous fungus Sordaria macrospora. The genome was sequenced solely by next-generation techniques (Solexa sequencing by synthesis and 454 pyrosequencing). S. macrospora is an ascomycete with a long-standing history as a model organism for fungal sexual development and meiosis [14]–[18] (Figure 1). Development of a large set of genetic tools for this fungus [19]–[24] resulted in the discovery of novel proteins involved in central events of meiosis and organogenesis [25]–[32]. Similar to its close relative Neurospora crassa, S. macrospora is haploid with a nuclear genome of seven chromosomes and an estimated 39.5 Mb of DNA sequence [24], [33]–[35]. Previous studies found ∼90% nucleic acid identity within coding regions of orthologous genes from S. macrospora and N. crassa as well as a high degree of synteny over large genomic regions [36],[37]. Despite their close phylogenetic relationship, S. macrospora is homothallic (self-fertile) in contrast to the heterothallic (self-sterile) N. crassa. The natural habitat of S. macrospora is herbivore dung in temperate climates, whereas N. crassa is usually found on burned vegetation and the soil throughout the world [14], [38]–[41]. Thus, these two closely related fungi have evolved different life styles and inhabit different ecological niches. These differences may be at least partially reflected in their genomes.

The S. macrospora genome sequencing project had two aims: (1) to assemble a first, high-quality draft of the genome sequence after next-generation sequencing to show that this approach is feasible for filamentous fungi in general, and (2) to annotate the genome sequence by a community effort, with the goal of a better understanding of S. macrospora biology and the idea of improving its value as a model organism for fungal development.
Results/Discussion
Sequencing and assembly of the S. macrospora genome
The genome of the S. macrospora strain k-hell was sequenced by a combination of Solexa and 454 sequencing. First, a total of 3.4 Gb of DNA sequence in 95,153,034 Solexa 36-nt reads were obtained from one single-read lane (9,688,226 reads), four lanes of paired-end reads (55,337,284 reads) from a 300-bp insert library, and three lanes (30,172,524 reads) of paired-end reads from a 500-bp insert library (Table 1, Figure S1). This represents 85-fold coverage of the S. macrospora genome. Assembly of the Illumina/Solexa data with the Velvet assembler [42] resulted in 38.7 Mb of sequence data in 3,344 contigs with an N50 size of 51 kb (Table 2). As expected, these contigs contained a substantial number of internal gaps (17,956 gaps, Table 2), because paired-end data allows contigs to be scaffolded by inferred physical linkage of the matched pairs in the absence of contiguous coverage of intervening segments. Despite the internal gaps in some of the contigs, we decided not to call them scaffolds to differentiate between the Velvet output (referred to as contigs even when containing gaps) and a subsequent scaffolding step (see below). When compared with the N. crassa genome, we were able to map 8,350 of ∼10,000 predicted proteins to the 10,066 predicted N. crassa genes (e-value ≤10−20) which is only slightly lower than the number obtained with the final high-quality draft (8,519 proteins, see below). Thus, even this preliminary assembly covered most of the protein-coding genome.

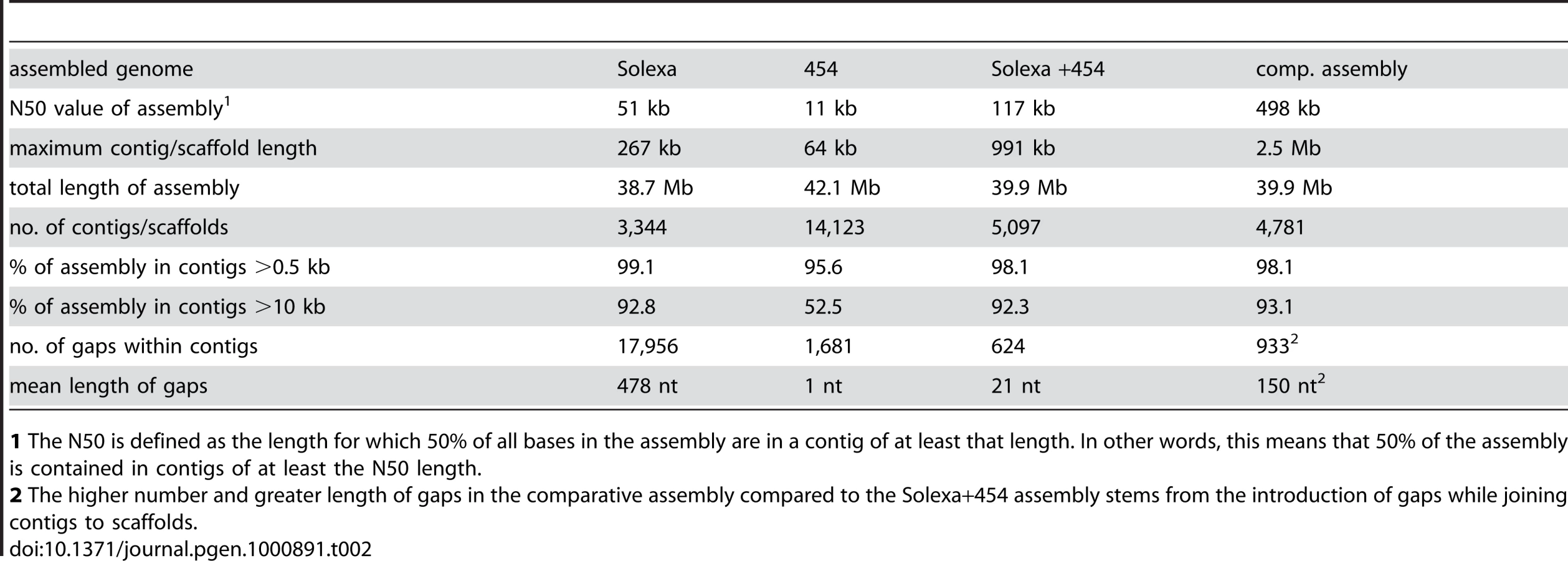
To close most gaps, we obtained additional sequence data by 454 sequencing. Because of longer reads, a relatively low coverage with 454 reads in combination with the previously obtained Solexa reads was expected to allow assembly with a higher N50 value and close internal gaps in the contigs. We obtained 415 Mb (∼10-fold coverage) of single-end 454 reads with an average read length of 367 bp (Table 1, Figure S1). Assembly of 454 reads only (with the Celera Assembler 5.3; Eurofins MWG GmbH, Ebersberg, Germany) yielded 14,123 contigs (N50 size 11 kb; 1,681 internal gaps; Table 2). Gaps in this assembly were primarily caused by sequencing ambiguities.
The combined raw data (Solexa and 454 reads) and the pre-assembled 454 data were used for constructing an assembly with the Velvet assembler version 0.7.31 [42] (Figure S1). This resulted in an assembly of 39.9 Mb of sequence data (5,097 contigs with an N50 size of 117 kb) and only 624 internal gaps within the contigs (Table 2). Thus, the combination of Solexa paired-end reads with 454 reads resulted in an increase of the N50 value and a drastic reduction in the number of gaps compared to assemblies where each data set was used alone. With a size of 39.9 Mb, this combined assembly corresponds well to previous analyses of the S. macrospora genome by pulsed-field gel electrophoresis that estimated the genome size at 39.5 Mb [24].
To determine whether similar results might be obtained with fewer sequence reads, thereby further decreasing sequencing costs, we generated test assemblies with different combinations of coverage levels (Figure S2, Table S1). The addition of 454 reads had the most drastic effect on the number and length of gaps whereas addition of paired-end reads improved mostly the N50 value. The inclusion of fewer sequence reads resulted in suboptimal assemblies; however, at the number of reads used for our assembly, bench mark values were no longer changing dramatically, suggesting that a plateau had been reached where addition of this type of sequence reads did not significantly improve assemblies. Further improvement might be achieved by sequencing paired-end libraries with longer inserts. The genome sequence of the filamentous ascomycete Grosmannia clavigera was assembled from a combination of Sanger paired-end reads (0.3-fold coverage), 454 single reads (7.7-fold coverage), and Solexa paired-end reads (100-fold coverage) [12], resulting in a high-quality draft genome sequence of 32.5 Mb with an N50 size of 164 kb. Our data show that similar values can be obtained even without including Sanger sequencing data thereby drastically decreasing sequencing costs.
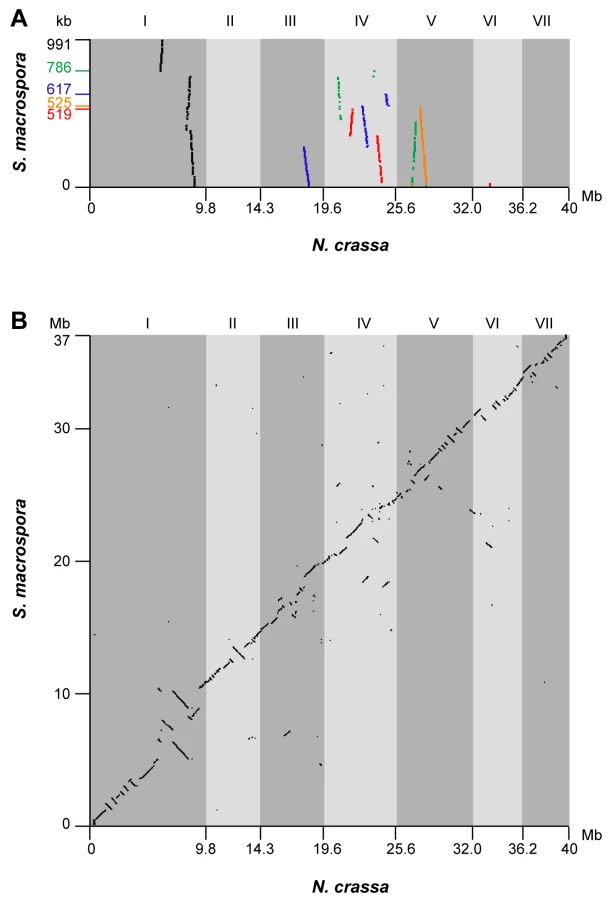
It has been previously demonstrated that several regions of up to 50 kb of the S. macrospora genome are syntenic to N. crassa [36],[37]. To extend this analysis to the newly assembled S. macrospora contigs, the five largest contigs from the Velvet assembly (519–991 kb) were compared to contigs of the N. crassa finished genome that have been assigned to specific linkage groups by mapped genetic markers (Assembly 9; http://www.broadinstitute.org/annotation/genome/neurospora/Regions.html). The results were visualized as dot plot (Figure 2A), and show that each contig maps to one or two linkage groups with only one to three breaks of synteny. Thus, the high degree of synteny between S. macrospora and N. crassa that was expected from previous studies was reflected in the Velvet assembly. To make use of this high degree of synteny and further improve the S. macrospora assembly, we generated a comparative assembly with Mercator by using the scaffolded chromosomes of the draft N. crassa genome (assembly 7, [43]) and the draft-sequences of the Neurospora discreta (http://genome.jgi-psf.org/Neudi1/Neudi1.home.html) and Neurospora tetrasperma (http://genome.jgi-psf.org/Neute1/Neute1.home.html) genomes to order and scaffold the S. macrospora contigs [44]. This resulted in a total of 152 scaffolds and 4,629 contigs with an N50 size of 498 kb (Table 2). Syntenic regions between the S. macrospora and N. crassa genomes were analyzed by dot plot analysis (Figure 2B). To verify that the scaffolded contigs represent the correct order within the S. macrospora genome, three regions spanning gaps between contigs on scaffolds 17, 58, and 98, respectively, were amplified by PCR and sequenced. In all cases, sequences between 0.8 and 1.2 kb were retrieved that close the gap between adjacent contigs thereby validating the scaffolding results (data not shown). This assembly represents the first high-quality draft version of the S. macrospora genome (“S. macrospora assembly 1”, acc. no. CABT01000001-CABT01004783, http://gb2.fungalgenomes.org/gb2/gbrowse/sordaria_macrospora).
Neither the rDNA repeat units nor the mitochondrial genome was represented in the Velvet assembly. We therefore searched the raw data as well as preassembled 454 and Solexa contigs for sequences with significant identity to rDNA or mitochondrial DNA from other fungi (Text S1). These reads were used to assemble both one rDNA unit as well as the mitochondrial DNA using CodonCode Aligner version 3.0.3 (http://www.codoncode.com/aligner/). The rDNA unit shows ∼98% DNA sequence identity to that of N. crassa. Unlike in N. crassa, no additional smaller rDNA regions with point mutations were found by this method. Four shorter contigs had SNPs in various locations when compared to the full-length rDNA region. These SNPs all occurred as part of a homonucleotide run (4–6 nt), suggesting either sequencing errors or true polymorphisms in the rDNA repeats, which are considered to be rare in filamentous fungi but do exist in N. crassa because of the occurrence of RIP (see below; K.M. Smith and M. Freitag, unpublished data).
The mitochondrial genome encompasses 88.4 kb, and thus is larger than the 64.8 kb mitochondrial genome of N. crassa and smaller than the 94.2 kb mitochondrial genome of Podospora anserina. With 33.6%, the GC content of the mitochondrial genome is in the same range as that of N. crassa (36.1%) and P. anserina (29.9%) (Text S1, Figure S3). Our data show that not only the single copy regions of the nuclear genome can be assembled from the next-generation sequencing data, but also multi-copy regions like the rDNA unit and the mitochondrial genome, even if they are not initially recovered in typical Velvet runs.
Comparisons between closely related species reduce the number of orphan genes
Gene models for the first draft of the S. macrospora genome were predicted with four independent ab initio gene prediction programs trained on N. crassa and evidence-based predictions with N. crassa proteins (see Materials and Methods). The results were integrated with Evigan [45] to yield ∼12,000 gene models. Additionally, 455 tRNA genes were predicted, similar to the 424 tRNA genes predicted for N. crassa [43]. The initially predicted ∼12,000 protein coding genes were screened for ORFs with internal stops, lack of initiation or termination codons, unusually long introns and insufficient support by sequence similarity. Such ORFs were corrected or removed resulting in a refined gene set of 10,789 genes with an average length of 1,432 bp for all predicted coding sequences (CDS, Table 3, Table S2). The overall GC content of the genome is 52.4%. This is changed to 56.5% in coding regions, which represent 38.4% of the genome, and 49.8% in non-coding regions, which make up 61.6% of the genome.

To address the question of sequencing errors, we PCR-amplified and resequenced coding regions for six predicted genes (SMAC_01188, SMAC_01198, SMAC_6009, SMAC_07685, SMAC_07776, SMAC_09680) with frameshifts or internal stops. These were confirmed by resequencing in four cases, whereas in two cases, insertions or deletions of 1 nt were found in the assembled sequence which when corrected led to the prediction of functional open reading frames. In total, we tested 21 kb of coding sequence by resequencing and found four insertion/deletion errors (0.02%). Although it is difficult to compare errors and error rates, this rate is similar to the 0.1–0.001% error rates achieved in microbial draft genomes sequenced by Sanger technology [46],[47].
With 10,789 predicted and partially curated genes, the gene count in S. macrospora is similar to that of N. crassa (10,066 community-annotated and centrally curated genes). To determine how many predicted proteins in these two closely related species are orthologs, reciprocal BLASTP analysis was performed: At an e-value of ≤10−20, 8,519 S. macrospora proteins have at least one homolog among the N. crassa proteins; vice versa, 8,179 N. crassa proteins have at least one homolog among the S. macrospora proteins. In total, 7,855 proteins (72.7% of all S. macrospora proteins) have reciprocal best hits in both searches identifying them as likely orthologs (Table S3).
Sequencing of the first few eukaryotic genomes revealed relatively high frequencies of “orphan genes” (i.e. genes without apparent homologs in any of the already known sequence databases and proteomes). As more genomes become available, this number has been rapidly decreasing, e.g. for N. crassa from ∼41% [43] to currently 22% (2,219/10,066 [48]). Because S. macrospora is more closely related to N. crassa than any other previously sequenced filamentous fungus, we compared the N. crassa orphan genes with the S. macrospora genome using TBLASTN and BLASTP to assess how many proteins are lineage-specific (Table S4). Of 2,112 N. crassa orphan genes that were retrieved from the current N. crassa MIPS protein list (http://mips.helmholtz-muenchen.de/genre/proj/ncrassa/), 870 do not have significant hits in the S. macrospora genome at an e-value of ≤10−20. Orphan genes might comprise more quickly evolving genes [48], and we therefore repeated our analysis at an e-value ≤10−5. This analysis still left 471 (4.7%) genes without significant hits, suggesting that these genes may constitute the remaining true orphan genes that separate the genus Sordaria from Neurospora (Table S4). The recent sequencing of additional Neurospora species is expected to further reduce the number of genus-specific genes.
In addition to assessing the conservation of protein-coding gene regions, we sought to investigate the conservation of non-coding regions between S. macrospora and its closest relatives. Therefore, we performed comparisons of 5′ upstream regions in 1 kb blocks from 1 kb to 4 kb as well as comparisons of introns and coding regions for S. macrospora, N. crassa, N. discreta and N. tetrasperma (Figure 3, Figure S4 and Table S5). We observed that introns are more conserved than upstream regions. Among the upstream regions, pairwise identity is slightly but significantly higher in the 1 kb upstream regions than in any of the other tested upstream regions (Table S5). This suggests that most regulatory (and therefore putatively conserved) elements in 5′ UTRs and promoters reside within the 1 kb upstream regions.

We also compared the predicted S. macrospora proteins to the non-redundant GenBank and Swissprot databases (Table S2). Approximately 6% (631/10,789) of all predicted proteins did not have a significant hit against the non-redundant database at an e-value ≤10−5. This number is only slightly higher than that for N. crassa (4.7%, 471 genes, see above). Taking into account that no other Sordaria species have been sequenced yet, we suggest that the number of true orphan genes in ascomycetes might be less than 5% or 500 genes per genome.
A search for conserved protein domains in the predicted S. macrospora proteins was performed with the HMMER program hmmpfam [49],[50] and with the InterProScan function from Blast2GO [51],[52]. With HMMER, one or more conserved domains were found in 5,471 predicted proteins (50.7%, Tables S2 and S6), the InterProScan found domains in 7,099 predicted proteins (65.7%, Table S2). These values might seem rather low when compared to the more than 10,000 proteins that have a hit in the non-redundant database, but it reflects the fact that many (predicted, hypothetical or conserved hypothetical) proteins have not yet been functionally characterized; therefore many domains remain to be identified.
In addition to a comparison to N. crassa, an analysis of the predicted proteins from S. macrospora, N. crassa, N. discreta, P. anserina, and Chaetomium globosum was performed with OrthoMCL, a software that clusters orthologs and “recent” paralogs [53]. We identified 9,971 orthogroups, and among these 5,428 (54.4%) comprise single genes from each of the five species, i.e. single-copy genes that are conserved among all species investigated (Tables S7 and S8). 31 orthogroups contain genes with three or more paralogs in S. macrospora, but fewer or no paralogs in other fungi, and these were investigated further. Some of these orthogroups contained proteins suggestive of transposon activity (see below), whereas others have no homology to transposons or pseudogenes. Phylogenetic analysis of two orthogroups (99 and 79) indicates evolutionary histories of ancient gene family expansion and subsequent differential gene loss (Figure 4). Orthogoup 99 comprises three genes from S. macrospora and two genes from P. anserina, whereas in the Neurospora species and C. globosum, only one gene is present. The genes from this orthogroup encode putative P450 oxygenases, and one might speculate that these proteins are beneficial for a coprophilic lifestyle, because only the coprophilic fungi S. macrospora and P. anserina have retained more than one copy. A similar case of duplication and subsequent loss can be postulated for orthogroup 79, which contains genes encoding chitin binding and glycosyl hydrolase domains.

In contrast, orthogroups 49 and 180 contain one or no gene for the Neurospora species, P. anserina, and C. globosum, but six and four members, respectively, in S. macrospora; and the S. macrospora genes cluster together in a phylogenetic tree (Figure 4). Thus, these genes seem to represent recent duplication events in S. macrospora. Both orthogroups are part of larger gene families, and to verify that placement of these subfamilies in different orthogroups was correct, an independent phylogenetic analysis was performed (Figure S5). This analysis supports the grouping by OrthoMCL. To test whether these genes are expressed genes and not simply annotation errors, quantitative real time PCR experiments were performed for ten S. macrospora genes from orthogroups 49 and 180 (Figure 4). For eight of the ten genes, transcripts were found under conditions of sexual development and/or vegetative growth, and all eight genes are upregulated during sexual development. In contrast, the homologous N. crassa genes are downregulated or not differentially regulated (Figure 4). Thus, the S. macrospora genes that are expressed under the conditions investigated might have gained developmental regulation after the split of the Neurospora and Sordaria lineages, probably as a result of gene family diversification after gene duplications. Whether these genes have a function during sexual morphogenesis in S. macrospora remains to be determined.
Repeated sequences, transposons, and genome integrity
Transposons and repeat elements have been identified in all eukaryotic groups investigated so far, and they can comprise large portions of a genome, e.g. 85% of the recently published maize genome [54],[55]. In fungi they usually make up only a comparatively small part of the genome (usually ≤10%), because effective defense mechanisms against repeated sequences are in place and because smaller genomes are more streamlined [56]. Eukaryotic transposons can be divided into two classes, class I elements that transpose via an RNA intermediate, and class II elements that transpose at the DNA level by excision and reintegration [57]. To analyze the transposon content of the S. macrospora genome, several approaches were used. First, amino acid sequences of known transposon open reading frames were used for comparison with the predicted S. macrospora peptides as described previously [58]. Second, DNA sequences of randomly selected scaffolds were compared to Repbase data [59]. These two approaches will identify only those repeated sequences or transposons that are similar to previously described elements. Third, DNA sequences of randomly selected scaffolds were compared to the complete genome sequence in order to identify new repeated sequences without similar entities in the databases.
Most interesting is the presence of five ORFs with amino acid sequence similarity to the N. crassa Tad LINE-like transposon [60] (Table 4). In addition, there are ∼20 ORFs with sequence similarity to gypsy-type retrotransposons. However, these ORFs exhibit rather diverse sequences and do not form element families. In contrast to these class I elements, there are only three ORFs with similarities to class II eukaryotic transposons; two of these represent a hAT-like element [61] that we called “Scarce”, and one ORF with amino acid similarity to the Fot1 transposon from Fusarium oxysporum [62]. As the only full-length Scarce ORF SMAC_09680 contains a nonsense codon, it is likely that the element is no longer active, thus explaining the low copy number. Overall, the transposon load of S. macrospora is very low, much more resembling that of another homothallic fungus Gibberella zeae (anamorph Fusarium graminearum) [63] than that of N. crassa. This is also reflected in a search for regions of high similarity within the S. macrospora genome by performing a BLASTN analysis of the genome sequence versus itself (Figure S6). In this analysis, the prevalence of regions with high intragenomic similarity in S. macrospora is between those of N. crassa [43] and F. graminearum [63], all of which have significantly fewer intragenomic regions of high similarity than the repeat-rich genome of Magnaporthe grisea [64]. This finding correlates well with the low transposon count of the S. macrospora genome. Taken together with the fact that we were able to assemble long contigs and that the assembly size correlates well with the genome size determined by pulsed-field gel electrophoresis, this suggests that the low repeat content is not an assembly artefact.

In addition to class I and II transposable elements, five different non-coding short repeat sequences (Smini1 to Smini5, 150–670 bp, Table 4) were detected. Two of these have partial sequence identity (Smini3 and Smini4) because of overlapping sequences. To verify that these repeats are real and not due to assembly problems, at least two copies for each repeat were PCR amplified and sequenced. For all tested repeats, their presence within the predicted genomic context was confirmed. Ten copies of repeat Smini5 are within ORFs, two are outside of ORFs, and another two overlap with ORFs. At least six of these ORFs show similarities to retrotransposon sequences. Some Smini1 and Smini3 repeats possess 5 bp target site duplications suggesting that they may be transposons or integrated elements caused by transposition. In some cases, point mutations may have modified target site duplications, or recombination may have occurred as has been shown for Aspergillus niger [65]. As Smini1 and Smini3 are both uniform in size (with the exception of some truncated elements), they may be solo-LTRs rather than mini-transposons such as the guest element of N. crassa [66]. Altogether, these five short repeat types cover only 56.8 kb of the genome (0.14%).
In N. crassa and a few other ascomycetes (e.g. P. anserina [67],[68], M. grisea [69], F. graminearum [63], and Leptosphaeria maculans [70]), the RIP machinery detects pairs of repeated segments during premeiosis, introduces C∶G to T∶A mutations and can trigger DNA methylation of the mutated repeats in the vegetative cells resulting from ascospores, presumably by virtue of the increased AT content [71]. We analyzed the entire S. macrospora genome sequence for the presence of RIP footprints by calculating RIP indices [72] on the concatenated contigs and scaffolds (Figure S7). In contrast to the situation in N. crassa, where large regions mutated by RIP make up the centromeric DNA (K.M. Smith, L.R. Connolly and M. Freitag, unpublished data), we found no large blocks of AT-rich regions with the typical RIP bias (e.g., TpA/ApT >1.0). The only large region with atypical dinucleotide distribution was scaffold 0, which contains the mtDNA. Here, both TpA/ApT and (CpA+TpG)/(ApC+GpT) were close to 1, suggesting DNA composition more reminiscent of bacteria or budding yeast. Our results suggest the absence of large regions mutated by RIP in the S. macrospora genome. Previous analyses have shown that there is no active RIP in S. macrospora (Kück et al., unpublished data). However, an ortholog of the N. crassa rid gene, the only gene known to be important for RIP [73], is present in the S. macrospora genome (Table S9), indicating that S. macrospora might have been able to undergo RIP during some time of its evolution; alternatively, RIP may occur at such low levels that it is difficult to detect in typical transformation and selfing experiments. RID homologs are involved in sexual development in two other fungi, Ascobolus immersus [74] and Aspergillus nidulans [75], suggesting that the S. macrospora protein may carry out a function independent of RIP.
In N. crassa, two other genome defense mechanisms in addition to RIP have been identified, namely meiotic silencing by unpaired DNA (MSUD or “meiotic silencing”) and a form of RNAi (“quelling”) [76],[77]. All N. crassa genes identified in these processes have orthologs in S. macrospora suggesting that S. macrospora might be able to perform different varieties of genome defense (Table S9). The fact that endogenous genes can be silenced via introduction of transgenic constructs that result in double-stranded RNA molecules indicates an active RNAi-like mechanism [78]. Nevertheless, transformants with ectopically integrated copies for genes involved in meiosis (which might be subject of MSUD) or other processes (which might be subject to RNAi) have been successfully generated in different laboratories working with S. macrospora for years. Silencing of the resident and/or ectopically located gene functions has never been observed or described (e.g. [21],[25],[30],[79],[80]). This suggests that S. macrospora might possess gene silencing mechanisms but that they are perhaps less active, at least with respect to transgenes, than in N. crassa.
Apart from genome defense mechanisms, there are a number of conserved processes in eukaryotes that are involved in maintaining genome integrity and regulating genome activity at the chromatin level [81]. We annotated chromatin-associated proteins, histone modification proteins, genes involved in the structural maintenance of chromosomes as well as centromere and kinetochore proteins and found that S. macrospora contains essentially the same set of genes as N. crassa (Table S9). Like its close relative, S. macrospora has single genes for the histone H3 K9 methyltransferase (DIM5), the heterochromatin protein 1 (HP1) and the DNA methyltransferase DIM-2, suggesting that heterochromatin formation and DNA methylation in S. macrospora are similar to what has been observed in N. crassa [43]. Taken together, these data indicate that S. macrospora contains the typical, conserved eukaryotic machinery for genome maintenance. Despite the absence of active RIP, this fungus appears to prevent the spreading of transposons and other repeated sequences as indicated by the low content of these elements within the genome.
Genes for regulatory networks, signaling, meiosis, and development
Since the 1950s, S. macrospora has been used as a model system for the analysis of fungal sexual development and meiosis, and a number of developmental genes have been characterized at the molecular level [14],[82]. We searched for genes known to be involved in development or in signaling cascades in S. macrospora and other fungi and found that S. macrospora contains homologs to all conserved genes as expected, further confirming the quality of the genome sequence.
Specifically, we looked for orthologs to known genes for fungal sexual development, meiosis, GTP-, phospholipid - and calcium-signaling, motor proteins, senescence, photoreceptors and light signaling (Tables S10, S11, S12, S13, S14). In the case of photoreceptor-coding genes, it was found that S. macrospora contains homologs to known or putative fungal photoreceptors (Table S10). S. macrospora is able to undergo sexual development both in the dark as well as under white light [83]; however, in the light perithecial necks of Sordaria and Neurospora species exhibit positive phototropism in order to aim the active discharge of ascospores away from the growth substrate [84],[85]. In N. crassa, this photoresponse is mediated by the blue light photoreceptor WC-1 [84],[86]–[88]. Photoresponses often involve multiple photoreceptors, e.g. photoreceptors for red and blue light are present in one protein complex in A. nidulans [89],[90]. To test whether wavelengths other than blue light also play a role in regulating neck phototropism, we tested the photoresponse of S. macrospora to green and red light. Under red light, perithecial necks were oriented in random directions similar to that of perithecia grown in complete darkness, but perithecial necks showed a strong positive phototropism in response to green light (Figure S8). Our results suggest that perithecial neck phototropism in S. macrospora is regulated by blue light, similar to photoresponses in N. crassa [91], and additionally by green light, a response not yet observed in other fungi. The photoreceptors responsible for this phenotype remain to be uncovered; possible candidates are two putative rhodopsin-like green light photoreceptors (SMAC_02424 and SMAC_06025) that are orthologs of ORP-1 and NOP-1 in N. crassa, respectively [81],[92].
Senescence in fungi has been observed in the model organism P. anserina, in strains of N. crassa and N. intermedia [93],[94], but not in S. macrospora. A search for homologs to genes that are known to be involved in the aging process in P. anserina revealed that for the majority of the genes clear homologs are present in S. macrospora (Table S11). This includes genes that are required for mitochondrial protein quality control, programmed cell death, DNA repair, ROS scavenging, mitochondrial dynamics, and respiration, among other processes. Two genes not identified in S. macrospora are the apoptosis-related genes PaAif1 and PaAmid2. PaAIF1 (apoptosis-inducing factor) and PaAMID2 (AIF-like mitochondrion-associated inducer of death) are putative NADH oxidoreductases. In mammals, AMID is present in mitochondria, and its overexpression induces cell death [95]. The third protein that is missing in S. macrospora is the SAM-dependent O-methyltransferase PaMth1. An accumulation of this protein was detected in the mitochondria and in total protein extracts of senescent P. anserina wild type strains [96],[97]. Investigation of substrate-specificity of the protein hints to a protecting role of this methyltransferase against the generation of reactive oxygen species [98],[99]. While PaMth1 overexpressing strains show a significantly elongated life span, PaMth1 deletion strains are short-lived. However, S. macrospora does not show a restricted lifespan despite the lack of a PaMth1 homolog, indicating that the aging process in P. anserina is not conserved in other members of the Sordariales, and that the P. anserina aging genes that are present in S. macrospora may function in other cellular pathways.
Fungi have long been used as model systems to study the molecular mechanisms of meiosis, and S. macrospora has played a prominent role in these investigations due to its simple sexual life cycle, large meiotic products (ascospores) and the production of an ordered tetrad of ascospores that allows the differentiation between pre - and postreduction segregation of alleles [14],[82]. Comparison of the predicted S. macrospora genes with the S. cerevisiae and Schizosaccharomyces pombe genomes [100],[101] allowed the identification of 92 “meiotic” genes. Reciprocal best hit BLASTP similarity searches against the predicted ORFs of S. macrospora, N. crassa and P. anserina showed that the 92 genes display orthologs in all three species (Table S15) [81],[102]. Nine of the genes were already characterized in S. macrospora (Table S15). The most conserved proteins include enzymes that are implicated in the recombination process and the proteins involved in sister-chromatid cohesion. In contrast, structural proteins like the components of the synaptonemal complex (SC) are poorly conserved despite the fact that the SC is as conserved during evolution as meiosis itself. This is similar to findings in other groups of organisms, e.g. mammals and plants [103]. Remarkably, S. macrospora, N. crassa, and P. anserina, like other filamentous fungi [104] possess only the RecA ortholog RAD51 and lack a recognizable DMC1, the meiosis-specific homolog of RAD51, thought to play an essential role in strand invasion [105]. The meiotic regulators are also poorly conserved (Table S15): among the three meiotic-specific transcription factors in yeast (Abf1p, Ume6p and Ndt80p) only an Ndt80p homolog is identifiable. Thus, S. macrospora has a conserved set of meiotic core genes whereas the regulators are more diverged, probably indicating life style-specific adaptations.
We also searched for genes that may be involved in GTP-dependent and/or phospholipid or calcium signaling as well as known fungal developmental genes and genes encoding motor proteins, and found for all groups that the gene content of the S. macrospora genome is similar to that of N. crassa, and thus in most cases larger than that of S. cerevisiae (Tables S12, S13, S14). This shows that S. macrospora is a useful model organism for studying developmental processes because it contains the full repertoire of higher eukaryotic genes involved in signaling and regulatory networks. Nevertheless, there are several groups of genes where S. macrospora differs from other fungi and that warrant a closer look because they allow insights into fungal evolution and biology. These are described below.
Genes for conidiation and nonself recognition: a case of “cryptic” incompatibility?
Two features in which S. macrospora differs from its close relative N. crassa are the lack of both asexual spores (“mitospores” or conidia) and heterokaryon incompatibility reactions. Searches in the S. macrospora genome for conserved genes that are involved in these processes revealed that homologs for conidiation genes are present (Table S16). These homologs seem to encode functional proteins, as they are not enriched in missense or nonsense mutations. Furthermore, quantitative real time PCR analysis for orthologs of six genes involved in conidiation in N. crassa revealed that these genes are expressed both during vegetative growth and sexual development in S. macrospora (Figure S9). Of course, additional unknown genes that are essential for conidiation may be missing or mutated in S. macrospora. Another possibility is that S. macrospora is able to conidiate, but does not do so under laboratory conditions. This would be analogous to the situation of Aspergillus fumigatus, which was recently shown to undergo sexual development when grown under suitable conditions [106],[107]. A third possibility, discussed below, might be that S. macrospora no longer produces conidia due to an unfavorable combination of heterokaryon incompatibility genes.
Filamentous fungi can undergo hyphal fusion (anastomosis, [108]) between individuals of different genotypes leading to the formation of a mycelium containing genetically different nuclei (heterokaryon). In many ascomycetes such as N. crassa, P. anserina, and A. nidulans, the viability of these heterokaryons is genetically controlled by a set of heterokaryon incompatibility (het) loci. A het locus can be defined as a locus at which heteroallelism cannot be tolerated in a heterokaryon [109], thus a fusion between two individuals that differ genetically at a het locus results in a nonself recognition reaction which leads to phenotypes ranging from inhibited, abnormal growth to cell death [110]. Heterokaryon incompatibility (HI) has been shown to prevent the spread of viruses and the exploitation of aggressive phenotypes and is believed to reduce the risk of resource plundering between individuals [111]–[114]. However, heterokaryon formation can also have benefits for the individuals involved, e.g. the formation of functional diploids and mitotic genetic exchange in the parasexual cycle [115].
Several het loci have been characterized at the molecular level, and a conserved region of about 150 residues has been identified within various HI proteins. This domain is termed the HET domain [116]. The parts of het genes not encoding the HET domain are highly polymorphic; they ensure nonself recognition and are evolving very rapidly whereas the HET domain triggers cell death [117]. In addition to het domain genes, several other genes function as het loci, among them the mating-type genes in N. crassa, which act as het genes during vegetative cell fusion but are required to be different during sexual cell fusion [118]–[120].
Vegetative incompatibility has not been observed in S. macrospora [35]. Nevertheless, S. macrospora harbors genes for homologs to known het genes in other fungi (Table S17). A rather surprising finding was that in the case of het-c, pin-c, and a tol-related HET domain gene, not one, but two closely linked copies for each of these genes are present in the S. macrospora genome (Figure 5, Table S17). This is in contrast to all other filamentous ascomycetes which encode only one homolog of the het-c gene [121]. In addition to het-c, a second, closely linked HET domain-encoding gene named pin-c is essential for the HI reaction in N. crassa. It was shown that nonallelic genetic interactions between het-c and pin-c mediate nonself recognition while the severity of the HI depends on allelic interactions at the het-c locus [122]. In S. macrospora, the genomic region that is orthologous to the het-c/pin-c locus in N. crassa contains two copies of pin-c (SMAC_07217 and SMAC_07219) and one full-length (SMAC_07220) and one partial (SMAC_07218) copy of het-c (Figure 5). BLASTP comparison shows that the two PIN-C proteins from S. macrospora differ from each other to about the same degree as the N. crassa PIN-C allelic variants differ from each other (data not shown). The het-c/pin-c region is inverted in S. macrospora, and the genes at the ends of the inverted region, het-c and pin-c, are duplicated. To exclude the possibility that this is an assembly error, we amplified by PCR and end-sequenced DNA fragments spanning the regions between SMAC_07217 and SMAC_07218, between SMAC_07218 and SMAC_07219, between SMAC_07219 and SMAC_07220, and between SMAC_07228 and SMAC_07229. In all cases, we obtained PCR fragments of the expected size and sequence thereby validating that this gene order is not an assembly error but represents the wild type situation. Interestingly, the intergenic region between SMAC_07217 and SMAC_07218 contains two copies of the Smini1 repeat; thus, the duplication in this region may have originated from a transposition event. Phylogenetic analysis of the duplicated PIN-C homologs and the duplicated TOL-related proteins indicates that for pin-c, the duplication arose after the divergence of Sordaria from Neurospora, because the two pin-c copies are more similar to each other than to either of the three known pin-c alleles from N. crassa (Figure 5).

In N. crassa, two copies of het-c are only present in one cytoplasm after heterokaryon formation, and it has been shown that HET-C proteins encoded by different het-c alleles form a heterodimer complex at the plasma membrane during the HI reaction [123]. Thus, with respect to het-c and pin-c, the genomic situation in S. macrospora resembles that of a heterokaryon in N. crassa (Figure 6), but no obvious signs of HI, e.g. compartmentalization and cell death, are evident in S. macrospora. However, mild HI reactions in N. crassa can lead to less severe phenotypes, e.g. aconidial strains [124]–[127]. In S. macrospora, the second het-c copy is incomplete and the ortholog of het-6, another gene involved in HI in N. crassa, contains internal stop codons so that a full HI reaction might be prevented by only partially functional het genes. Thus, we hypothesize that the lack of conidiation in S. macrospora may be due to “cryptic” or “mild” HI caused by the presence of more than one copy of putative HI genes in the genome (Figure 6). However, as indicated above, this is just one of several hypotheses to explain the fact that S. macrospora is aconidiate despite possessing orthologs to all known conidiation genes.

Another point worth considering is that S. macrospora is homothallic and encodes mating type genes in one locus that are present in separate mating-type idiomorphs in N. crassa [128]. This situation would result in severe HI in vegetative cells of N. crassa mediated by the TOL protein. Only in tol mutants both mating type idiomorphs are tolerated in one vegetative cytoplasm [120]. Introgression of the N. crassa tol into N. tetrasperma caused HI and disrupted the pseudohomothallic nature of this fungus indicating that the native N. tetrasperma tol does not mediate HI [129]. Interestingly, the S. macrospora TOL, SMAC_08253, has only 40% amino acid identity to its N. crassa ortholog, an extremely low value compared to the average 89% identity in coding regions at the DNA level [37]. Probably this very divergent TOL does not mediate HI and allows co-existence of all mating type genes within vegetative cells. Thus, HI in S. macrospora might be attenuated (“cryptic” HI) or abolished by mutations in critical HI-mediating genes to cope with or allow the presence of otherwise incompatible genes within one genome.
A second genomic locus that is important for HI in N. crassa and N. tetrasperma contains the het-6 and un-24 (rnr-1) genes. In this case, the two known alleles, Oak Ridge (OR) and Panama (PA), of both genes in both species differ not only in the sequences of the alleles, but also in the gene order within the het-6/un-24 locus, which was caused by an inversion of a block of five genes including un-24 [116],[130],[131]. An analysis of the orthologous region in S. macrospora revealed the same gene order as in the OR allele (Figure S10A). Phylogenetic analysis of both genes showed that the different allelic versions of N. crassa and N. tetrasperma cluster together as has been shown previously [131], while the S. macrospora genes occupy a basal position relative to the two Neurospora species (Figure S10B). This suggests that the OR allele represents the ancient gene order, and that the PA allele arose from an inversion after separation of Sordaria and Neurospora, but before speciation of N. crassa and N. tetrasperma; otherwise one would have to postulate two independent inversion events of the same genomic region leading to the OR gene order which is rather unlikely.
Some genes for secondary metabolism may have been acquired by horizontal gene transfer
Polyketides and non-ribosomal peptides are the most prominent classes of fungal secondary metabolites [6]. They comprise a wide variety of chemical structures, and a number of them have pharmaceutical applications, but their biological functions remain largely unknown [132],[133]. Most filamentous fungi harbor several genes encoding polyketide synthases (PKS) as well as non-ribosomal peptide synthases (NRPS) in their genomes. Apart from the pks and nrps genes, the biosynthesis of a polyketide or non-ribosomal peptide usually requires additional genes that encode, for example, enzymes that modify the products of the PKSs and NRPSs. These genes are often clustered together with the corresponding pks or nrps gene within the genome [134]. In order to determine the potential of S. macrospora for the biosynthesis of secondary metabolites, we searched the predicted proteins for the occurrence of typical domains associated with PKS or NRPS proteins, and additionally also for fatty acid synthase (FAS) proteins as these have structural similarity to PKS proteins (Table S18). S. macrospora contains three putative nrps genes, three genes that fall into the fas class, and eleven putative pks genes. The numbers of nrps and fas genes are the same as in N. crassa, and the corresponding genes in the two fungi are orthologs. However, of the predicted eleven pks genes, only seven have an ortholog in N. crassa, whereas four PKS proteins have a higher sequence identity to other, more distantly related fungi. The N. crassa genome contains only eight putative pks genes one of which has no ortholog in S. macrospora [36],[135]. Thus, with respect to pks genes and putative polyketides, S. macrospora appears to possess a greater potential for the production of secondary metabolites than its close relative N. crassa (Table S18, Figure S11).
Most of the S. macrospora polyketide biosynthesis genes that have been studied previously have been found to be upregulated during sexual development, and polyketides may play a role in fruiting body formation in S. macrospora [36],[78]. Therefore, we determined the expression of the remaining five pks as well as the three nrps genes during sexual development (Figure 7). The nrps genes as well as eight of the eleven pks genes are transcriptionally upregulated during sexual development. The three pks genes that are not upregulated comprise the single type III pks gene as well as two pks genes without orthologs in N. crassa. These two pks genes, SMAC_01188 and SMAC_01198, are organized in a cluster of putative polyketide biosynthesis genes (Figure 8). Despite the fact that polyketide biosynthesis genes are often clustered in filamentous fungi [134], in S. macrospora only one such cluster has been found [36], and the genome sequence shows that most pks genes of S. macrospora do not occur clustered with other polyketide biosynthesis genes.

Apart from them being clustered, the two pks genes SMAC_01188 and SMAC_01198 are interesting because they do not have orthologs in N. crassa or any of the other sequenced Sordariomycete genomes (P. anserina, C. globosum, F. graminearum, M. grisea). This is true for most of the genes from the cluster spanning the region from SMAC_01188 to SMAC_01201 (Table S19). With the exception of SMAC_01192 and SMAC_01197, the clustered genes do not have identifiable homologs within the Sordariomycetes, rather their most similar homologs are found within the Eurotiomycetes (Aspergillus, Neosartorya, Penicillium) or Dothideomycetes (Phaeosphaeria). In the center of the cluster, six genes are orthologs to genes from a putative polyketide biosynthesis cluster of Phaeosphaeria nodorum (Figure 8, syn. Stagonospora nodorum, http://www.broadinstitute.org/annotation/genome/stagonospora_nodorum/Home.html [136]). There are two likely explanations for these findings: (1) the cluster originated through gene duplication in a common ancestor of the Sordariomycetes and Dothideomycetes, and later on, massive gene loss occurred in the Sordariomycetes with the exception of S. macrospora; (2) S. macrospora acquired the cluster through horizontal gene transfer (HGT). To examine these two possibilities, we determined the sequence identity between the S. macrospora cluster proteins and their orthologs in the P. nodorum cluster as well as the sequence identity between all homologous S. macrospora and P. nodorum proteins, and found that the sequence identity between the proteins from the cluster is significantly higher (Figure 8B). This is also the case when looking at the sequence identity of proteins with the same domains as the orthologs in the cluster.

A phylogenetic analysis was performed with the cluster protein SMAC_01196 that encodes a putative phenylalanine ammonia lyase (PAL), a second PAL protein SMAC_05651 present in S. macrospora, and the homologs from seven other fungi (Figure 9). As expected, SMAC_05651 groups with the corresponding proteins from the Sordariales N. crassa, C. globosum, and P. anserina, each of which encodes only one PAL protein in their genomes. However, the “additional” PAL protein SMAC_01196 from the cluster groups among the Leotiomycetes/Dothideomycetes proteins and is closest to the P. nodorum cluster protein SNOG09914. Phylogenetic analysis of the cluster protein SMAC_01190 that encodes a putative member of the UbiA prenyltransferase family, its two other S. macrospora paralogs, SMAC_02313 and SMAC_06375, and the homologs from eleven other fungi gives a similar picture: SMAC_02313 and SMAC_06375 group within the Sordariales, whereas SMAC_01190 groups with the P. nodorum protein SNOG_09915 within a section of the tree that contains proteins from the Dothideomycetes, Eurotiomycetes, and Leotiomycetes, but not Sordariomycetes (Figure S12).

The findings of (1) a conserved cluster of genes with closest homologs from the Dothideomycete P. nodorum instead of members of the Sordariomycetes, (2) the significantly higher sequence similarity between S. macrospora and P. nodorum proteins from the cluster compared to the overall sequence similarity between other proteins from these species, and (3) the phylogenetic positioning of two of the clustered proteins within the Dothideomycetes rather than the Sordariomycetes are more consistent with HGT than with the hypothesis of gene duplication and subsequent gene loss even though the latter cannot be excluded [137]. Recent studies, made possible by the increasing number of fungal genome sequences, have indicated that HGT may be more common in fungi than previously thought, and that genes for secondary metabolism are especially prone to HGT [138]–[141]. Even though in many cases “non-canonic” phylogenetic tree topologies can be explained by a combination of duplication, diversification, and differential gene loss [138],[142], that still leaves a number of cases where a HGT model best fits the observed data [140],[141],[143],[144]. HGT may be one way for fungi to increase their biochemical repertoire, thereby increasing their ability to adapt to new ecological niches [137].
In the case of the S. macrospora cluster presented here, it is interesting to note that it contains two putative pks genes (SMAC_01188 and SMAC_01198), one of which (SMAC_01198) has acquired 16 frame shifts/stop codons that interrupt the open reading frame whereas the other pks gene SMAC_01188 as well as the additional ten genes that comprise the putative polyketide biosynthesis cluster represent functional genes. For seven of the twelve genes from the cluster (SMAC_01188 to SMAC_01991, SMAC_01194, SMAC_01196 and SMAC_01198), transcriptional expression was verified by cDNA sequencing, and spliced cDNAs were obtained for all of the genes including SMAC_01198 which is unlikely to yield a functional protein due to the frameshifts (data not shown). Thus, this cluster might represent a case of an evolutionary recent acquisition that was introduced into the S. macrospora genome since its divergence from the last common ancestor with N. crassa. While part of the cluster appears to be retained and under purifying selection in S. macrospora, the gene SMAC_01198 has drifted and accumulated frameshift/nonsense mutations, even though it is still transcribed. Further analyses are necessary to determine the function of this putative polyketide biosynthesis cluster in S. macrospora.
Conclusions
Due to their high throughput and low costs, next-generation sequencing techniques have greatly changed the way large-scale sequencing projects are done. This includes e.g. re-sequencing of existing genomes for the discovery of variations, “RNA-sequencing” for transcriptome analysis, or “ChIP-Seq” for the genome-wide analysis of DNA-protein interactions [8]. Until recently, de novo genome assembly from next-generation sequences has been restricted to prokaryotic genomes [10],[11]. This is due to the fact that eukaryotic genomes are larger and often contain high amounts of repetitive sequences that cannot be assembled from read lengths that are smaller than the length of the repeats. With the recent release of the Giant Panda genome [13] it has become obvious that even more complex eukaryotic genomes can be sequenced and assembled from short reads. Here, we present a high-quality draft of the S. macrospora genome, assembled solely from next-generation sequences, showing that de novo assembly from Solexa paired-end reads in combination with 454 sequence reads is feasible, cost-effective and fast, at least for compact eukaryotic genomes with few repetitive sequences.
Additionally, the S. macrospora genome revealed several features that are of interest with respect to fungal evolution, namely its complement of het genes as well as polyketide biosynthesis genes. In the case of the closely linked het-c and pin-c genes, it was found that S. macrospora contains additional copies that might have arisen from inversion/duplication events. In other fungi, the presence of non-identical het alleles within one cytoplasm leads to HI, which in its extreme results in cell death [109],[115]. In contrast, S. macrospora is able to cope with this situation as no obvious HI phenotypes are observed in this fungus. However, we suggest that the aconidial phenotype of S. macrospora may be the result of “cryptic HI” caused by the presence of incompatible het genes within a single genome. Furthermore, analysis of a second het gene locus shows how the analysis of closely related genome sequences can help to pinpoint evolutionary events, in this case the occurrence of an inversion after separation of Sordaria and Neurospora but before speciation of N. crassa and N. tetrasperma. The analysis of predicted polyketide biosynthesis genes showed that S. macrospora contains more pks genes than its close relative N. crassa, and therefore probably has a wider biochemical repertoire available. One putative polyketide biosynthesis cluster might have been acquired through HGT, and this fits with previous results that show that HGT is probably rather widespread in fungi both for the transfer of single genes, clustered genes like polyketide biosynthesis genes, or even larger stretches of DNA up to whole chromosomes as was found in the phytopathogenic fungus Nectria haematococca [139]–[141],[143],[144]. These findings support the theory that HGT plays a role in fungal evolution and might be a source of genetic variation that allows fungi to adapt to different ecological niches [137].
Materials and Methods
Strains and culture conditions
The sequenced reference strain is Sordaria macrospora k-hell from the strain collection of the Department of General and Molecular Botany at the Ruhr-Universität Bochum. The strain was grown on cornmeal medium as previously described [38].
DNA preparation for sequencing
Genomic DNA from S. macrospora was prepared by following a modified previously published method [145]. Mycelium was frozen in liquid nitrogen, pulverized, and incubated in equal volumes of lysis buffer (0.2 M sodium borate, 30 mM EDTA, 1% SDS, pH 9.0) and phenol at 60°C for 5 min. After centrifugation, the supernatant was treated with RNase, and afterwards with an equal volume phenol/chloroform (1∶1). After centrifugation, genomic DNA was purified from the supernatant by cesium chloride density gradient centrifugation.
Illumina/Solexa sequencing by synthesis
To construct libraries of two different insert sizes, 5 µg DNA each were sonicated with a Branson sonicator. Sonicated DNA was separated through 2% NuSieve agarose gels and fragments of ∼300 and ∼500 bp were purified. After generation of blunt-end fragments, A-overhangs were added, adaptors ligated, and the fragments were PCR amplified [146]. The resulting libraries were sequenced on an Illumina Genome Analyzer with a paired-end module generating reads of 36 bases. Four lanes from the 300 bp library and three lanes from the 500 bp library resulted in 3.4 Gb of sequence data (Table 1, Figure S1).
Roche/454 pyrosequencing
Roche/454 sequencing was performed with 50 µg genomic DNA at Eurofins MWG GmbH (Ebersberg, Germany). This resulted in 415 Mb of sequence data with an average read length of 367 bp (Table 1). The 454 raw data were extracted from the sff file and converted to a fasta file using sff_extract.py (written by Jose Blanca and Bastien Chevreux, http://bioinf.comav.upv.es/sff_extract/index.html).
Assembly
Assembly of the Solexa reads only as well as the combined Solexa and 454 reads was carried out with the Velvet assembler [42]. A description of the parameters used with Velvet can be found in Text S1 and Figure S1. An assembly of only the 454 data with the Celera Assembler 5.3 was performed by Eurofins MWG GmbH (Ebersberg, Germany). Comparison of the S. macrospora genome with the N. crassa genome [43] was done with BLAST [147] and visualized with Combo [148]. Comparative assembly of the S. macrospora genome along the genome sequences of N. crassa, N. discreta (http://genome.jgi-psf.org/Neudi1/Neudi1.home.html) and N. tetrasperma (http://genome.jgi-psf.org/Neute1/Neute1.home.html) genomes was done with Mercator [44]. Assembly of the mitochondrial genome and the rDNA unit was done with CodonCode Aligner version 3.0.3 (http://www.codoncode.com/aligner/), details can be found in Text S1 and Figure S3.
Annotation
Gene models were predicted independently with the ab initio predictors AUGUSTUS, GeneMark+ES, SNAP, and the evidence-based predictor Genewise [149]–[153]. The ab initio SNAP and AUGUSTUS parameters were trained on all N. crassa gene models while GeneMark performs an iterative self-training procedure. The Genewise predictions were generated from N. crassa proteins aligned to the genome by first aligning the proteins with TBLASTN, choosing the S. macrospora locus with only the best alignment for each protein and then refining the alignment and splice-sites with Genewise. The processing of outputs from these tools was completed with custom scripts utilizing tools from the BioPerl toolkit [154]. The resulting GFF annotation from each of the prediction programs was used as input to Evigan, a program that integrates the four sources of gene evidence [45].
For each of the predicted proteins, the protein with the highest sequence identity in GenBank was determined using BLASTP [147] (Table S2). Additionally, putative domains were predicted with the HMMER (version 2.3.2) program hmmpfam using the hidden Markov models from the pfam database [49],[50] and with the InterProScan function from Blast2GO [51],[52]. The resulting data can be found in Table S8. Putative localization of the predicted proteins was determined with WoLF PSORT [155], putative signal peptides and signal anchors were predicted with SignalP 3.0 [156], and transmembrane domains with HMMTOP [157] and TMHMM [158] (Table S2, Text S2). tRNAs were predicted using a combination of Infernal 1.0, tRNAscan-SE, and TFAM 1.0 [159]–[161]. Orthologous groups of genes among the five fungal species S. macrospora, N. crassa [43], N. discreta (http://genome.jgi-psf.org/Neudi1/Neudi1.home.html), P. anserina [102], and C. globosum (http://www.broadinstitute.org/annotation/genome/chaetomium_globosum, Chaetomium globosum Sequencing Project, Broad Institute of Harvard and MIT http://www.broad.mit.edu) were identified with OrthoMCL [53]. Searches for transposons and repeat elements were done with BLAST [147] and by searches in Repbase (http://www.girinst.org/) [59].
For comparison of different genomic regions (CDSs, introns and upstream regions) from S. macrospora, N. crassa, N. discreta and N. tetrasperma, a Mercator alignment [44] of the genome sequences was performed and the parts of the alignment corresponding to the genomic regions were used to compute pairwise identities and evolutionary distances. Only those upstream regions were used that do not overlap with a protein coding region, and each region was used only once even if it is upstream of two divergently transcribed genes to avoid double-counting.
Accession numbers
The sequence and annotation data are available under the accession numbers CABT01000001-CABT01004783. The sequence reads that were used for the assembly of the S. macrospora genome were submitted to the NCBI sequence read archive (accession number SRA010462).
RNA preparation and expression analysis
For comparison of vegetative growth versus sexual development, growth and harvesting of S. macrospora and N. crassa, RNA preparation, reverse transcription and quantitative real time PCR were as described previously [19],[162].
Phylogenetic analysis
Multiple alignments were created in CLUSTALX [163] and trimmed with Jalview [164], and the same alignment was used for analysis by distance-matrix (DM), maximum parsimony (MP) or Bayesian methods. Phylogenetic analyses were made with PAUP version 4.0b10 for Windows (D.L. Swofford, distributed by Sinauer Associates, copyright 2001 Smithsonian Institution) for DM and MP analyses, and with MrBayes [165],[166]. DM and MP analyses were performed as described using 10,000 bootstrap replicates, Bayesian analysis was performed with at least 250,000 generations [167]. Consensus trees were graphically displayed with TREEVIEW or Dendroscope [168],[169].
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. HawksworthDL
2001 The magnitude of fungal diversity: the 1.5 million species estimate revisited. Mycol Res 105 1422 1432
2. StajichJE
BerbeeML
BlackwellM
HibbettDS
JamesTY
2009 The fungi. Curr Biol 19 R840 R845
3. BuckleyM
2008 The fungal kingdom - diverse and essential roles in earth's ecosystem Washington DC American Academy of Microbiology
4. SmithSE
ReadDJ
2000 Mycorrhizal symbiosis London, San Diego Academic Press
5. ArcherDB
ConnertonIF
MacKenzieDA
2008 Filamentous fungi for production of food additives and processing aids. Adv Biochem Eng Biotechnol 111 99 147
6. HoffmeisterD
KellerNP
2007 Natural products of filamentous fungi: enzymes, genes, and their regulation. Nat Prod Rep 24 393 416
7. GoffeauA
BarrellBG
BusseyH
DavisRW
DujonB
1996 Life with 6000 genes. Science 274 546 567
8. ShendureJ
LiH
2008 Next-generation DNA sequencing. Nature Biotechnol 26 1135 1145
9. WhitefordN
HaslamN
WeberG
Prügel-BennettA
EssexJW
2005 An analysis of the feasibility of short read sequencing. Nucl Acids Res 33 e171
10. MarguliesM
EgholmM
AltmanWE
AttiyaS
BaderJS
2005 Genome sequencing in microfabricated high-density picolitre reactors. Nature 437 376 380
11. ReinhardtJA
BaltrusDA
NishimuraMT
JeckWR
JonesCD
2009 De novo assembly using low-coverage short read sequence data from the rice pathogen Pseudomonas syringae pv. oryzae. Genome Res 19 294 305
12. DiGuistiniS
LiaoN
PlattD
RobertsonG
SeidelM
2009 De novo genome sequence assembly of a filamentous fungus using Sanger, 454 and Illumina sequence data. Genome Biol 10 R94
13. LiR
FanW
TianG
ZhuH
HeL
2009 The sequence and de novo assembly of the giant panda genome. Nature advance online publication doi:10.1038/nature08696
14. KückU
PöggelerS
NowrousianM
NoltingN
EnghI
2009 Sordaria macrospora, a model system for fungal development.
AnkeT
WeberD
The Mycota XV, Physiology and Genetics. 1st ed Berlin, Heidelberg Springer 17 39
15. ZicklerD
2005 Meiosis in mycelial fungi.
KüesU
FischerR
The Mycota I Growth, differentiation and sexuality Berlin, Heidelberg Springer 415 438
16. ZicklerD
2006 From early homologue recognition to synaptonemal complex formation. Chromosoma 115 158 174
17. PöggelerS
NowrousianM
KückU
2006 Fruiting-body development in ascomycetes.
KüesU
FischerR
The Mycota I Berlin, Heidelberg Springer 325 355
18. EsserK
StraubJ
1958 Genetische Untersuchungen an Sordaria macrospora Auersw., Kompensation und Induktion bei genbedingten Entwicklungsdefekten. Z Vererbungsl 98 729 746
19. NowrousianM
RingelbergC
DunlapJC
LorosJJ
KückU
2005 Cross-species microarray hybridization to identify developmentally regulated genes in the filamentous fungus Sordaria macrospora. Mol Genet Genomics 273 137 149
20. PöggelerS
KückU
2006 Highly efficient generation of signal transduction knockout mutants using a fungal strain deficient in the mammalian ku70 ortholog. Gene 378 1 10
21. PöggelerS
MasloffS
HoffB
MayrhoferS
KückU
2003 Versatile EGFP reporter plasmids for cellular localization of recombinant gene products in filamentous fungi. Curr Genet 43 54 61
22. PöggelerS
NowrousianM
JacobsenS
KückU
1997 An efficient procedure to isolate fungal genes from an indexed cosmid library. J Microbiol Meth 29 49 61
23. RechC
EnghI
KückU
2007 Detection of hyphal fusion in filamentous fungi using differently fluorescene-labeled histones. Curr Genet 52 259 266
24. WalzM
KückU
1995 Transformation of Sordaria macrospora to hygromycin B resistance: characterization of transformants by electrophoretic karyotyping and tetrad analysis. Curr Genet 29 88 95
25. EnghI
WürtzC
Witzel-SchlömpK
ZhangHY
HoffB
2007 The WW domain protein PRO40 is required for fungal fertility and associates with Woronin bodies
26. MasloffS
PöggelerS
KückU
1999 The pro1+ gene from Sordaria macrospora encodes a C6 zinc finger transcription factor required for fruiting body development. Genetics 152 191 199
27. NowrousianM
FrankS
KoersS
StrauchP
WeitnerT
2007 The novel ER membrane protein PRO41 is essential for sexual development in the filamentous fungus Sordaria macrospora. Mol Microbiol 64 923 937
28. PöggelerS
KückU
2004 A WD40 repeat protein regulates fungal cell differentiation and can be replaced functionally by the mammalian homologue striatin. Eukaryot Cell 3 232 240
29. StorlazziA
TesséS
GarganoS
JamesF
KlecknerN
2003 Meiotic double-strand breaks at the interface of chromosome movement, chromosome remodeling, and reductional division. Genes Dev 17 2675 2687
30. StorlazziA
TesseS
Ruprich-RobertG
GarganoS
PöggelerS
2008 Coupling meiotic chromosome axis integrity to recombination. Genes Dev 22 796 809
31. van HeemstD
JamesF
PöggelerS
Berteaux-LecellierV
ZicklerD
1999 Spo76p is a conserved chromosome morphogenesis protein that links the mitotic and meiotic programs. Cell 98 261 271
32. NowrousianM
MasloffS
PöggelerS
KückU
1999 Cell differentiation during sexual development of the fungus Sordaria macrospora requires ATP citrate lyase activity. Mol Cell Biol 19 450 460
33. McClintockB
1945 Neurospora. I. Preliminary observations of the chromosomes of Neurospora crassa. Am J Bot 32 671 678
34. OrbachM
VollrathD
DavisRW
YanofskyC
1988 An electrophoretic karyotype of Neurospora crassa. Mol Cell Biol 8 1469 1473
35. PöggelerS
MasloffS
JacobsenS
KückU
2000 Karyotype polymorphism correlates with intraspecific infertility in the homothallic ascomycete Sordaria macrospora. J Evol Biol 13 281 289
36. NowrousianM
2009 A novel polyketide biosynthesis gene cluster is involved in fruiting body morphogenesis in the filamentous fungi Sordaria macrospora and Neurospora crassa. Curr Genet 55 185 198
37. NowrousianM
WürtzC
PöggelerS
KückU
2004 Comparative sequence analysis of Sordaria macrospora and Neurospora crassa as a means to improve genome annotation. Fungal Genet Biol 41 285 292
38. EsserK
1982 Cryptogams - Cyanobacteria, Algae, Fungi, Lichens London Cambridge University Press
39. PerkinsDD
1992 Neurospora: the organism behind the molecular revolution. Genetics 130 687 701
40. JacobsonDJ
DettmanJR
AdamsRI
BoeslC
SultanaS
2006 New findings of Neurospora in Europe and comparisons of diversity in temperate climates on continental scales. Mycologia 98 550 559
41. JacobsonDJ
PowellAJ
DettmanJR
SaenzGS
BartonMM
2004 Neurospora in temperate forests of western North America. Mycologia 96 66 74
42. ZerbinoDR
BirneyE
2008 Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18 821 829
43. GalaganJE
CalvoSE
BorkovichKA
SelkerEU
ReadND
2003 The genome sequence of the filamentous fungus Neurospora crassa. Nature 422 859 868
44. DeweyCN
2007 Aligning multiple whole genomes with Mercator and MAVID.
BergmanNH
Comparative genomics Totowa Humana Press
45. LiuQ
MackeyAJ
RoosDS
PereiraFCN
2008 Evigan: a hidden variable model for integrating gene evidence for eukaryotic gene predictions. Bioinformatics 24 597 605
46. BranscombE
PredkiP
2002 On the high value of low standards. J Bacteriol 194 6406 6409
47. FraserCM
EisenJA
NelsonKE
PaulsenIT
SalzbergSL
2002 The value of complete microbial genome sequencing (you get what you pay for). J Bacteriol 184 6403 6405
48. KasugaT
MannhauptG
GlassNL
2009 Relationship between phylogenetic distribution and genomic features in Neurospora crassa. PLOS ONE 4 e5286 doi:10.1371/journal.pone.0005286
49. EddySR
1998 Profile Hidden Markov Models. Bioinformatics 14 755 763
50. FinnRD
TateJ
MistryJ
CoggillPC
SammutSJ
2008 The Pfam protein families database. Nucl Acids Res 36 D281 288
51. ConesaA
GotzS
Garcia-GomezJM
TerolJ
TalonM
2005 Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21 3674 3676
52. ZdobnovEM
ApweilerR
2001 InterProScan - an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17 847 848
53. LiL
StoeckertCJJ
RoosDS
2003 OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res 13 2178 2189
54. JurkaJ
1998 Repeats in genomic DNA: mining and meaning. Curr Opin Struct Biol 8 333 337
55. SchnablePS
WareD
FultonRS
SteinJC
WeiF
2009 The B73 maize genome: complexity, diversity, and dynamics. Science 326 1112 1115
56. LynchM
2007 The origins of genome architecture Sunderland, MA Sinauer Assocs. Inc
57. KempkenF
KückU
1998 Transposons in filamentous fungi - facts and perspectives. Bioessays 20 652 659
58. BraumannI
van den BergM
KempkenF
2007 Transposons in biotechnologically relevant strains of Aspergillus niger and Penicillium chrysogenum. Fungal Genet Biol 44 1399 1414
59. KapitonovVV
JurkaJ
2008 A universal classification of eukaryotic transposable elements in Repbase. Nat Rev Genet 9 411 412
60. KinseyJ
HelberJ
1989 Isolation of a transposable element from Neurospora crassa. Proc Nat Acad Sci USA 86 1929 1933
61. KempkenF
WindhoferF
2001 The hAT family: a versatile transposon group common to plants, fungi, animals, and man. Chromosoma 110 1 9
62. DaboussiMJ
Langint
BrygooY
1992 Fot1, a new family of fungal transposable elements. Mol Gen Genet 232 12 16
63. CuomoCA
GüldenerU
XuJR
TrailF
TurgeonBG
2007 The Fusarium graminearum genome reveals a link between localized polymorphism and pathogen specialization. Science 317 1400 1402
64. DeanRA
TalbotNJ
EbboleDJ
FarmanML
MitchellTK
2005 The genome sequence of the rice blast fungus Magnaporthe grisea. Nature 434 980 986
65. BraumannI
van den BergM
KempkenF
2008 Strain-specific retrotransposon-mediated recombination in a commercially used Aspergillus niger strain. Mol Genet Genomics 280 319 325
66. RasmussenJP
TaylorAH
MaLJ
PurcellS
KempkenF
2004 Guest, a transposable element belonging to the Tc1/mariner superfamily is an ancient invader of Neurospora genomes. Fungal Genet Biol 41 52 61
67. GraiaF
LespinetO
RimbaultB
Dequard-ChablatM
CoppinE
2001 Genome quality control: RIP (repeat-induced point mutation) comes to Podospora. Mol Microbiol 40 586 595
68. HamannA
FellerF
OsiewaczHD
2000 The degenerate DNA transposon Pat and repeat-induced point mutation (RIP) in Podospora anserina. Mol Gen Genet 263 1061 1069
69. IkedaK
NakayashikiH
KataokaT
TambaH
HashimotoY
2002 Repeat-induced point mutation (RIP) in Magnaporthe grisea: implications for its sexual cycle in the natural field context. Mol Microbiol 45 1355 1364
70. IdnurmA
HowlettBJ
2003 Analysis of loss of pathogenicity mutants reveals that repeat-induced point mutations can occur in the Dothideomycete Leptosphaeria maculans. Fungal Genet Biol 39 31 37
71. GalaganJE
SelkerEU
2004 RIP: the evolutionary cost of genome defense. Trends Genet 20 417 423
72. MargolinBS
Garrett-EngelePW
StevensJN
FritzDY
Garrett-EngeleC
1998 A methylated Neurospora 5S rRNA pseudogene contains a transposable element inactivated by repeat-induced point mutation. Genetics 149 1787 1797
73. FreitagM
WilliamsRL
KotheGO
SelkerEU
2002 A cytosine methyltransferase homologue is essential for repeat-induced point mutation in Neurospora crassa. Proc Nat Acad Sci USA 99 8802 8807
74. MalagnacF
WendelB
GoyonC
FaugeronG
ZicklerD
1997 A gene essential for de novo methylation and development in Ascobolus reveals a novel type of eukaryotic DNA methyltransferase structure. Cell 91 281 290
75. LeeDW
FreitagM
SelkerEU
AramayoR
2008 A cytosine methyltransferase homologue is essential for sexual development in Aspergillus nidulans. PLoS ONE 3 e2531 doi:10.1371/journal.pone.0002531
76. FulciV
MacinoG
2007 Quelling: post-transcriptional gene silencing guided by small RNAs in Neurospora crassa. Curr Opin Microbiol 10 199 203
77. ShiuPKT
RajuNB
ZicklerD
MetzenbergRL
2001 Meiotic silencing by unpaired DNA. Cell 107 905 916
78. EnghI
NowrousianM
KückU
2007 Regulation of melain biosynthesis via the dihydroxynaphtalene pathway is dependent on sexual development in the ascomycete Sordaria macrospora. FEMS Microbiol Lett 275 62 70
79. NowrousianM
PiotrowskiM
KückU
2007 Multiple layers of temporal and spatial control regulate accumulation of the fruiting body-specific protein APP in Sordaria macrospora and Neurospora crassa. Fungal Genet Biol 44 602 614
80. KückU
2005 A Sordaria macrospora mutant lacking the leu1 gene shows a developmental arrest during fruiting body formation. Mol Genet Genomics 274 307 315
81. BorkovichKA
AlexLA
YardenO
FreitagM
TurnerGE
2004 Lessons from the genome sequence of Neurospora crassa: tracing the path from genomic blueprint to multicellular organism. Microbiol Mol Biol Rev 68 1 108
82. ZicklerD
2009 Observing meiosis in filamentous fungi: Sordaria and Neurospora. Methods Mol Biol 558 91 114
83. NowrousianM
KückU
2006 Comparative gene expression analysis of fruiting body development in two filamentous fungi. FEMS Microbiol Lett 257 328 335
84. HardingRW
MellesS
1983 Genetic analysis of phototropism of Neurospora crassa perithecial beaks using white collar and albino mutants. Plant Physiol 72 996 1000
85. ReadND
1983 A scanning electron microscopic study of the external features of perithecium development in Sordaria humana. Can J Bot 61 3217 3229
86. FroehlichAC
LiuY
LorosJJ
DunlapJC
2002 White Collar-1, a circadian blue light photoreceptor, binding to the frequency promoter. Science 297 815 819
87. HeQ
ChengP
YangY
WangL
GardnerK
2002 White collar-1, a DNA binding transcription factor and a light sensor. Science 297 840 843
88. BallarioP
VittoriosoP
MagrelliA
TaloraC
CabibboA
1996 White collar-1, a central regulator of blue-light responses in Neurospora, is a zinc finger protein. EMBO J 15 1650 1657
89. PurschwitzJ
MüllerS
KastnerC
SchöserM
HaasH
2008 Functional and physical interaction of blue - and red-light sensors in Aspergillus nidulans. Curr Biol 18 255 259
90. BlumensteinA
VienkenK
TaslerR
PurschwitzJ
VeithD
2005 The Aspergillus nidulans phytochrome FphA represses sexual development in red light. Current Biology 15 1833 1838
91. LindenH
BallarioP
ArpaiaG
MacinoG
1999 Seeing the light: News in Neurospora blue light signal transduction. Adv Genet 41 35 54
92. BieszkeJA
SpudichEN
ScottKL
BorkovichKA
SpudichJL
1999 A eukaryotic protein, NOP-1, binds retinal to form an archaeal rhodopsin-like photochemically reactive pigment. Biochemistry 38 14138 14145
93. GriffithsAJ
1992 Fungal senescence. Annu Rev Genet 26 351 372
94. OsiewaczHD
1990 Molecular analysis of aging processes in fungi. Mut Res 237 1 8
95. WuM
XuL-G
LiX
ZhaiZ
ShuH-B
2002 AMID, an apoptosis-inducing factor-homologous mitochondrion-associated protein, induces caspase-independent apoptosis. J Biol Chem 277 25617 25623
96. AverbeckNB
JensenON
MannM
SchäggerH
OsiewaczHD
2000 Identification and characterization of PaMTH1, a putative O-methyltransferase accumulating during senescence of Podospora anserina cultures. Curr Genet 37 200 208
97. GroebeK
KrauseF
KunstmannB
UnterluggauerH
ReifschneiderNH
2007 Differential proteomic profiling of mitochondria from Podospora anserina, rat and human reveals distinct patterns of age-related oxidative changes. Exp Gerontol 42 887 898
98. KunstmannB
OsiewaczHD
2008 Over-expression of an S-adenosylmethionine-dependent methyltransferase leads to an extended lifespan of Podospora anserina without impairments in vital functions. Aging Cell 7 651 662
99. KunstmannB
OsiewaczHD
2009 The S-adenosylmethionine dependent O-methyltransferase PaMTH1: a longevity assurance factor protecting Podospora anserina against oxidative stress. Aging 1 328 334
100. MataJ
LyneR
BurnsG
BählerJ
2002 The transcriptional program of meiosis and sporulation in fission yeast. Nat Genet 32 143 147
101. SchlechtU
PrimigM
2003 Mining meiosis and gametogenesis with DNA microarrays. Reproduction 225 447 456
102. EspagneE
LespinetO
MalagnacF
Da SilvaC
JaillonO
2008 The genome sequence of the model ascomycete fungus Podospora anserina. Genome Biol 9 R77
103. PageSL
HawleyRS
2004 The genetics and molecular biology of the synaptonemal complex. Annu Rev Cell Dev Biol 20 525 558
104. MalikSB
PightlingAW
StefaniakLM
SchurkoAM
LogsdonJMJ
2008 An expanded inventory of conserved meiotic genes provides evidence for sex in Trichomonas vaginalis. PLOS ONE 3 e2879 doi:10.1371/journal.pone.0002879
105. HunterN
2006 Meiotic recombination.
AguileraA
RothsteinR
Molecular genetics of recombination Berlin, Heidelberg Springer 381 443
106. O'GormanCM
FullerHT
DyerPS
2009 Discovery of a sexual cycle in the opportunistic fungal pathogen Aspergillus fumigatus. Nature 457 471 475
107. PöggelerS
2002 Genomic evidence for mating abilities in the asexual pathogen Aspergillus fumigatus. Curr Genet 42 153 160
108. ReadND
FleissnerA
RocaMG
GlassNL
2010 Hyphal fusion.
BorkovichKA
EbboleD
Molecular biology of filamentous fungi Am. Soc. Microbiol 260 273
109. SaupeS
2000 Molecular genetics of heterokaryon incompatibility in filamentous ascomycetes. Microbiol Mol Biol Rev 64 489 502
110. MicaliOC
SmithML
2006 A nonself recognition gene complex in Neurospora crassa. Genetics 173 1991 2004
111. CortesiP
McCullochCE
SongH
LinH
MilgroomMG
2001 Genetic control of horizontal virus transmission in the chestnut blight fungus, Cryphonectria parasitica. Genetics 159 107 118
112. DebetsA
GriffithsAJ
1998 Poymorphism of het genes prevents resource plundering in Neurospora crassa. Mycol Res 102 1343 1349
113. DebetsF
YangX
GriffithsAJ
1994 Vegetative incompatibility in Neurospora: its effect on horizontal transfer of mitochondrial plasmids and senescence in natural populations. Curr Genet 26 113 119
114. van DiepeningenAD
DebetsAJ
HoekstraRF
1997 Heterokaryon incompatibility blocks virus transfer among natural isolates of black Aspergilli. Curr Genet 32 209 217
115. GlassNL
KanekoI
2003 Fatal attraction: nonself recognition and heterokaryon incompatibility in filamentous fungi. Eukaryot Cell 2 1 8
116. SmithML
MicaliOC
HubbardSP
Mir-RashedN
JacobsonDJ
2000 Vegetative incompatibility in the het-6 region of Neurospora crassa is mediated by two linked genes. Genetics 155 1095 1104
117. PaolettiM
SaupeSJ
ClavéC
2007 Genesis of a fungal non-self recognition repertoire. PLoS ONE 2 e283 doi:10.1371/journal.pone.0000283
118. BeadleGW
CoonradtVL
1944 Heterocaryosis in Neurospora crassa. Genetics 29 291 308
119. EspagneE
BalhadèreP
PeninML
BarreauC
TurcqB
2002 HET-E and HET-D belong to a new subfamily of WD40 proteins involved in vegetative incompatibility specificity in the fungus Podospora anserina. Genetics 161 71 81
120. ShiuPK
GlassNL
1999 Molecular characterization of tol, a mediator of mating-type-associated vegetative incompatibility in Neurospora crassa. Genetics 151 545 555
121. FedorovaN
BadgerJ
RobsonG
WortmanJ
NiermanW
2005 Comparative analysis of programmed cell death pathways in filamentous fungi. BMC Genomics 6 177
122. KanekoI
DementhonK
XiangQ
GlassNL
2006 Nonallelic interactions between het-c and a polymorphic locus, pin-c, are essential for nonself recognition and programmed cell death in Neurospora crassa. Genetics 172 1545 1555
123. SarkarS
IyerG
WuJ
GlassNL
2002 Nonself recognition is mediated by HET-C heterocomplex formation during vegetative incompatibility. EMBO J 21 4841 4850
124. JacobsonDJ
BeurkensK
KlomarensKL
1998 Microscopic and ultrastructural examination of vegetative incompatibility in partial diploids heterozygous at het loci in Neurospora crassa. Fungal Genet Biol 23 45 56
125. MylekOM
1975 Heterokaryon incompatibility genes in Neurospora crassa detected using duplication-producing chromosome rearrangements. Genetics 80 107 124
126. PerkinsDD
1975 The use of duplication-generating rearrangements for studying heterokaryon incompatibility genes in Neurospora. Genetics 80 87 105
127. WuJ
GlassNL
2001 Identification of specificity determinants and generation of alleles with novel specificity at the het-c heterokaryon incompatibility locus of Neurospora crassa. Mol Cell Biol 21 1045 1057
128. PöggelerS
RischS
KückU
OsiewaczHD
1997 Mating-type genes from the homothallic fungus Sordaria macrospora are functionally expressed in a heterothallic ascomycete. Genetics 147 567 580
129. JacobsonDJ
1992 Control of mating type heterokaryon incompatibility by the tol gene in Neurospora crassa and N. tetrasperma. Genome 35 347 353
130. Mir-RashedN
JacobsonDJ
DehghanyMR
MicaliOC
SmithML
2000 Molecular and functional analyses of incompatibility genes at het-6 in a population of Neurospora crassa. Fungal Genet Biol 30 197 205
131. PowellAJ
JacobsonDJ
NatvigDO
2007 Ancestral polymorphism and linkage disequilibrium at the het-6 region in pseudohomothallic Neurospora tetrasperma. Fungal Genet Biol 44 896 904
132. CoxRJ
2007 Polyketides, proteins and genes in fungi: programmed nano-machines begin to reveal their secrets. Org Biomol Chem 5 2010 2026
133. WalshCT
2004 Polyketide and nonribosomal peptide antibiotics: modularity and versatility. Science 303 1805 1810
134. KellerNP
HohnTM
1997 Metabolic pathway gene clusters in filamentous fungi. Fungal Genet Biol 21 17 29
135. KrokenS
GlassNL
TaylorJW
YoderOC
TurgeonBG
2003 Phylogenomic analyis of type I polyketide synthase genes in pathogenic and saprobic ascomycetes. Proc Nat Acad Sci USA 100 15670 15675
136. HaneJK
LoweRGT
SolomonPS
TanK-C
SchochCL
2007 Dothideomycete plant interactions illuminated by genome sequencing and EST analysis of the wheat pathogen Stagonospora nodorum. Plant Cell 19 3347 3368
137. RosewichUL
KistlerHC
2000 Role of horizontal gene transfer in the evolution of fungi. Annu Rev Phytopathol 38 326 363
138. KhaldiN
WolfeKH
2008 Elusive origins of the extra genes in Aspergillus oryzae. PLOS ONE 3 e3036 doi:10.1371/journal.pone.0003036
139. PatronNJ
WallerRF
CozijnsenAJ
StraneyDC
GardinerDM
2007 Origin and distribution of epipolythiodioxopiperazine (ETP) gene clusters in filamentous ascomycetes. BMC Evol Biol 7 174
140. KhaldiN
CollemareJ
LebrunM-H
WolfeKH
2008 Evidence for horizontal transfer of a secondary metabolite gene cluster between fungi. Genome Biol 9 R18
141. FriesenTL
StukenbrockEH
LiuZ
MeinhardtS
LingH
2006 Emergence of a new disease as a result of interspecific virulence gene transfer. Nat Genet 38 953 956
142. FedorovaND
KhaldiN
JoardarVS
MaitiR
AmedeoP
2008 Genomic islands in the pathogenic filamentous fungus Aspergillus fumigatus. PLOS Genet 4 e1000046 doi:10.1371/journal.pgen.1000046
143. Garcia-VallvéS
RomeuA
PalauJ
2000 Horizontal gene transfer of glycosyl hydrolases of the rumen fungi. Mol Biol Evol 17 352 361
144. ColemanJJ
RounsleySD
Rodriguez-CarresM
KuoA
WasmannCC
2009 The genome of Nectria haematococca: Contribution of supernumerary chromosomes to gene expansion. PLoS Genet 5 e1000618 doi:10.1371/journal.pgen.1000618
145. HogeJHC
SpringerJ
ZantingeB
WesselsJGH
1982 Absence of differences in polysomal RNAs from vegetative monokaryotic and dikaryotic cells of the fungus Schizophyllum commune. Exp Mycol 6 225 232
146. PomraningKR
SmithKM
FreitagM
2009 Genome-wide high throughput analysis of DNA methylation in eukaryotes. Methods 47 142 150
147. AltschulSF
MaddenTL
SchafferAA
ZhangJ
ZhangZ
1997 Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25 3389 3402
148. EngelsR
YuT
BurgeC
MesirovJP
DeCaprioD
2006 Combo: a whole genome comparative browser. Bioinformatics 22 1782 1783
149. KorfI
2004 Gene finding in novel genomes. BMC Bioinformatics 5 59
150. StankeM
SchöffmannO
MorgensternB
WaackS
2006 Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics 7 62
151. StankeM
WaackS
2003 Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19 suppl. 2 ii215 225
152. BesemerJ
BorodovskyM
2005 GeneMark: web software for gene finding in prokaryotes, eukaryotes and viruses. Nucl Acids Res 33 W451 454
153. BirneyE
ClampM
DurbinR
2004 GeneWise and Genomewise. Genome Res 14 988 955
154. StajichJE
BlockD
BoulezK
BrennerSE
ChervitzSA
2002 The Bioperl Toolkit: Perl modules for the life sciences. Genome Res 12 1611 1618
155. HortonP
ParkKJ
ObayashiT
NakaiK
2006 Protein subcellular localization prediction with WoLF PSORT. 39 48 Proceedings of the 4th Annual Asia Pacific Bioinformatics Conference APBC06, Taipei, Taiwan
156. BendtsenJD
NielsenH
von HeijneG
BrunakS
2004 Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 340 783 795
157. TusnadyGE
SimonI
1998 Principles governing amino acid composition of integral membrane proteins: Applications to topology prediction. J Mol Biol 283 489 506
158. KroghA
LarssonB
von HeijneG
SonnhammerELL
2001 Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J Mol Biol 305 567 580
159. LoweTM
EddySR
1997 tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucl Acids Res 25 955 964
160. NawrockiEP
KolbeDL
EddySR
2009 Infernal 1.0: inference of RNA alignments. Bioinformatics 25 1335 1337
161. TaquistH
CuiY
ArdellDH
2007 TFAM 1.0: an online tRNA function classifier. Nucl Acids Res 35 W350 353
162. NowrousianM
CebulaP
2005 The gene for a lectin-like protein is transcriptionally activated during sexual development, but is not essential for fruiting body formation in the filamentous fungus Sordaria macrospora. BMC Microbiol 5 64
163. ThompsonJD
GibsonTJ
PlewniakF
JeanmouginF
HigginsDG
1997 The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 24 4876 4882
164. WaterhouseAM
ProcterJB
MartinDMA
ClampM
BartonGJ
2009 Jalview Version 2 - a multiple sequence alignment editor and analysis workbench. Bioinformatics 25 1189 1191
165. HuelsenbeckJP
RonquistF
2001 Bayesian inference of phylogeny. Bioinformatics 17 754 755
166. RonquistF
HuelsenbeckJP
2003 Bayesian phylogenetic inference under mixed models. Bioinformatics 19 1572 1574
167. HallBG
2004 Phylogenetic trees made easy Sunderland Sinauer Associates
168. HusonDH
RichterDC
RauschC
DezulianT
FranzM
2007 Dendroscope - An interactive viewer for large phylogenetic trees. BMC Bioinformatics 8 460
169. PageR
1996 TREEVIEW: an application to display phylogenetic trees on personal computers. Appl Biosci 12 357 358
170. DementhonK
IyerG
GlassNL
2006 VIB-1 Is required for expression of genes necessary for programmed cell death in Neurospora crassa. Eukaryot Cell 5 2161 2173
171. LiuYJ
HallBD
2004 Body plan evolution of ascomycetes, as inferred from an RNA polymerase II phylogeny. Proc Nat Acad Sci USA 101 4507 4512
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 4
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Whole-Genome SNP Association in the Horse: Identification of a Deletion in Myosin Va Responsible for Lavender Foal Syndrome
- Admixture Mapping Scans Identify a Locus Affecting Retinal Vascular Caliber in Hypertensive African Americans: the Atherosclerosis Risk in Communities (ARIC) Study
- Genetic Tests for Ecological and Allopatric Speciation in Anoles on an Island Archipelago
- Human Telomeres Are Hypersensitive to UV-Induced DNA Damage and Refractory to Repair
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy