
HOX geny a vývoj končetin v klinické medicíně i v experimentu
HOX genes and the limb development in the clinical praxis and in the experiment
In human, congenital malformations of the limbs are ranked among the most prevalent of all congenital birth defects. Substantial portion of these defects has genetic origin. Increasing knowledge about the particular mutations responsible for limb malformations in human results in the increasing availability of DNA diagnostic procedures for confirmation of clinical diagnosis and family counselling. Increasing understanding of the underlying developmental processes revealed by studying limb defects both in human and animal models may offer better therapeutic options in the future.
This review concentrates on the role of Hox genes in limb development. Man, as well as other mammals, has 39 HOX genes, divided into 4 complexes (clusters). HOX genes play a major role in body plan layout and development of many organ systems. Experimental data show that during the limb development, HOX genes influence patterning along the proximodistal and anteroposterior (thumb-little finger) axis of the limb bud. In human, limb malformation was described in patients with mutations in HOXA11, HOXA13, HOXD10, and HOXD13 genes. Most frequent among these malformations are hand-foot-genital syndrome caused by HOXA13 gene mutation, and synpolydactyly caused by HOXD13 mutation. Severity of the phenotype manifestation of these diseases is variable, and depends on the particular mutation type, where point mutations, polyalanine expansions and deletions can take part.
Key words:
HOX, congenital birth defect, limb development, hand-foot-genital syndrome, synpolydactyly.
Autoři:
Pavel Šnajdr 1; Miloš Grim 1; František Liška 2
Působiště autorů:
Univerzita Karlova v Praze, 1. lékařská fakulta, Anatomický ústav
1; Univerzita Karlova v Praze, 1. lékařská fakulta a VFN, Ústav biologie a lékařské genetiky
2
Vyšlo v časopise:
Čas. Lék. čes. 2010; 149: 4-9
Kategorie:
Přehledový článek
Souhrn
Vrozené vady končetin patří mezi nejčastější vrozené vady u člověka. Významná část těchto vad má genetický původ. Rostoucí znalost konkrétních mutací podmiňujících malformace končetin u člověka umožňuje větší dostupnost diagnostického vyšetření DNA pro potvrzení klinické diagnózy a pro genetické poradenství. Přibývající znalosti vývojových procesů získané studiem vad končetin u člověka i na experimentálních modelech mohou v budoucnosti nabídnout i lepší terapeutické možnosti.
Tento článek se soustřeďuje na roli Hox genů ve vývoji končetiny. Člověk má stejně jako ostatní savci 39 HOX genů, rozdělených do čtyř komplexů. Tyto geny hrají zásadní roli při organizaci tělního plánu a vývoji mnoha orgánových systémů. Z experimentálně získaných údajů víme, že při vývoji končetiny ovlivňují její proximodistální a palcomalíkovou osu. U člověka byly v souvislosti s postižením končetin popsány mutace v genech HOXA11, HOXA13, HOXD10 a HOXD13. Nejčastější z těchto onemocnění jsou syndrom hand-foot-genital podmíněný mutací genu HOXA13 a synpolydaktylie podmíněná mutací genu HOXD13. Závažnost fenotypových projevů těchto onemocnění je variabilní v závislosti na konkrétním typu mutace daného genu, kdy se mohou uplatnit bodové mutace, polyalaninové expanze či delece.
Klíčová slova:
HOX, vrozená vada, vývoj končetiny, hand-foot-genital syndrom, synpolydaktylie.
ÚVOD
Vrozené vady končetin jsou vedle vad srdce a močového systému jedněmi z nejčastějších vrozených vad. Podle údajů Národního registru vrozených vad České republiky – ÚZIS ČR (1) bylo v naší republice v roce 2008 nově zaznamenáno 154 případů polydaktylie, 142 případů syndaktylie a 41 případů redukčních defektů končetin. Vrozené vady končetiny se objevují samostatně i jako součásti syndromů a tvoří zhruba 10 % ze všech vrozených vad (2).
Příčiny vzniku vrozených vad se tradičně dělí na genetické a epigenetické (zevní). Předpokládá se, že genetické faktory se uplatňují při vzniku asi 20 % vrozených vad, izolované zevní faktory asi v 10 %, u zbývajících 70 % vad není jednoznačná příčina známa a jedná se patrně o kombinaci genetických a epigenetických faktorů.
Embryonální vývoj končetin a jeho regulace
Končetina všech čtyřnožců včetně člověka se skládá ze třech základních částí: z proximálního stylopodia, středního zeugopodia a distálního autopodia.
Vývoj končetiny lze zhruba rozdělit do třech fází: inicializace končetinového pupenu, fáze časného vývoje pupenu a pozdní fáze morfogeneze končetiny. Po fázi inicializace, kdy je indukován jeho vznik, roste končetinový pupen pod vlivem signalizačních center a vytvářejí se tři osy budoucí končetiny: proximodistální, palcomalíková (ve srovnávací embryologické literatuře bývá označováno jako anteroposteriorní) a dorzoventrální (obr. 1). V pozdní fázi morfogeneze se vytvářejí kosti, svaly se šlachami, cévy, nervy a další struktury končetiny.

Základ končetiny, končetinový pupen, se u člověka začíná vyvíjet koncem 4. týdne po oplození. Epitel na povrchu pupenu vytváří na jeho konci tzv. apikální ektodermový hřeben produkující růstové faktory z rodiny FGF. Ty udržují subektodermovou zónu pupenu v proliferační aktivitě, která je klíčová pro proximodistální růst končetiny. Anteroposteriorní (palcomalíkové) uspořádání základu končetiny je řízeno buňkami při bázi zadního okraje končetinového pupenu, z tzv. zóny polarizující aktivity produkující faktor SHH. Pro dorzoventrální osu končetiny je klíčová rozdílná exprese genů WNT7a a LMX1 v dorzální části, respektive ENGRAILED-1 ve ventrální části končetinového pupenu.
Tvarové změny vyvíjející se končetiny jsou podmíněny lokálně rozdílnou proliferační aktivitou a odumíráním buněk. Zvláště při vývoji autopodia při tvorbě prstů se vznikem meziprstních štěrbin se uplatňuje apoptóza, která proběhne v nedostatečném rozsahu u syndaktylie. Rozsah apotózy je změněn také při polydaktylii či oligodaktylii (3). Koncem 8. týdne od oplození jsou všechny části končetiny včetně plně oddělených prstů již vytvořeny.
Vývoj horní a dolní končetiny je podobný (4), dolní končetina je ve vývoji o 1–2 dny zpožděna. Geny zodpovědné za identitu hornía dolní končetiny jsou TBX5 instruující vývoj horní a PITX1 a TBX4 vývoj dolní končetiny.
Z mezenchymu základu končetiny se diferencují buňky všech typů pojiva. Další buněčné populace do končetin migrují z axiálních struktur embrya. Základ končetiny je tak osidlován myogenními buňkami ze somitů, jejichž migrace je řízena interakcí signálů PAX-3, HGH/SF, c-MET. Například mutace genu PAX-3 u myši způsobuje specificky redukci svalů končetin, zatímco svaly hlavy a trupu se vyvíjejí normálně (5). Krevní cévy končetiny vznikají převážně prorůstáním intersomitických cév, v menší míře z angioblastů, které se rekrutují a diferencují in situ z materiálu končetiny (6). Do končetiny vrůstají dále z míchy axony motoneuronů a migrují do ní buňky neurální lišty, ze kterých vznikají Schwannovy buňky, melanocyty a Merkelovy buňky (7).
HOX geny
Hox geny patří v posledních dvou desetiletích doslova k celebritám vývojové biologie. Vzhledem k jejich klíčové roli pro evoluci tělního plánu živočichů je jejich role zkoumána na mnoha klasických (Drosophila melanogaster, myš, potkan), i méně klasických (žahavci, ostnokožci, pláštěnci) experimentálních modelech. S dnes již rutinní dostupností diagnostických metod analýzy těchto genů u člověka přibývá v posledních letech také množství údajů o onemocněních, která u člověka způsobují.
Hox geny obsahují vysoce konzervovaný, mezidruhově se jen minimálně lišící, 180 bp dlouhý úsek zvaný homeobox, kódující tzv. homeodoménu o délce 60 aminokyselin. Homeodoména je zodpovědná za rozpoznávání a interakci s regulačními sekvencemi DNA. Hox proteiny tak fungují jako transkripční faktory, které regulují funkci mnoha „downstream“ genů.
Přítomnost sekvence homologické sekvenci homeoboxu definuje nadrodinu homeotických genů, Hox geny jsou tedy jen jednou konkrétní skupinou z této nadrodiny.
U obratlovců jsou Hox geny exprimovány již časně v průběhu gastrulace a definují identitu segmentů podél kraniokaudální osy těla. V pozdějších etapách vývoje řídí tyto geny kromě růstu končetin také například vývoj urogenitálního systému, trávicí trubice, plic a jsou důležité pro krvetvorbu. Změněná funkce Hox genů pak podmiňuje vrozené vady těchto systémů. U člověka je exprese HOX genů změněna také u mnoha krevních malignit i solidních nádorů (8), jakou roli však hrají v jejich etiopatogenezi, není vždy zcela jasné.
Na počátku poznávání funkce Hox genů stála analýza dvou mutací (9) u octomilky (Drosophila melanogaster), které způsobují homeotickou přeměnu jednoho segmentu těla v jiný, podobající se segmentu nacházejícímu se normálně v jiné úrovni těla (homoios = podobný) (obr. 2).

Popis těchto i dalších homeotických mutací vedl k úvaze, že geny za ně zodpovědné musí nějakým způsobem určovat identitu jednotlivých tělních segmentů, jejich poziční informaci.
U Drosophily je celkem osm Hox genů, organizovaných ve dvou od sebe oddělených komplexech (úsecích, klastrech). Uspořádání a pozice genů v komplexu má však jednu zvláštnost, typickou pro Hox geny obecně. Pozice genu v komplexu (a tím pádem na chromozomu) není náhodná, reflektuje funkční uspořádání. Geny lokalizované blíže 3’konci jsou exprimovány v částech embrya blíže hlavy a v časnější fázi vývoje, geny lokalizované blíže 5’konci jsou exprimovány v kaudálnějších částech a v pozdější fázi vývoje. To se nazývá prostorová a časová kolinearita. Tyto principy kolinearity jsou zakonzervovány v Hox komplexech drtivé většiny živočichů.
Jak HOX geny definují poziční informaci?
Každý segment těla živočicha nemůže mít svůj vlastní „specifický“ Hox gen, neboť těch je obvykle mnohem méně než segmentů, které je třeba definovat (např. osm Hox genů u Drosophily vs. jejích 17 tělních segmentů). Svou roli hraje obvykle kombinace více Hox genů pravděpodobně spolu s dalšími geny z jiných rodin. Obvykle je (skoro) každý HOX gen exprimován ve více segmentech, v každém segmentu je pak zároveň exprimováno více Hox genů. Každý segment je tak definován pro něj unikátním kombinačním kódem a zároveň i „dávkou“ exprese Hox genů. Podle pravidla kolinearity jsou 5’geny exprimovány v kaudálnějších částech, 3’geny v kraniálnějších částech těla. Jedno z možných (a relativně častých) uspořádání exprese Hox genů je schematicky zobrazeno na obrázku 3.
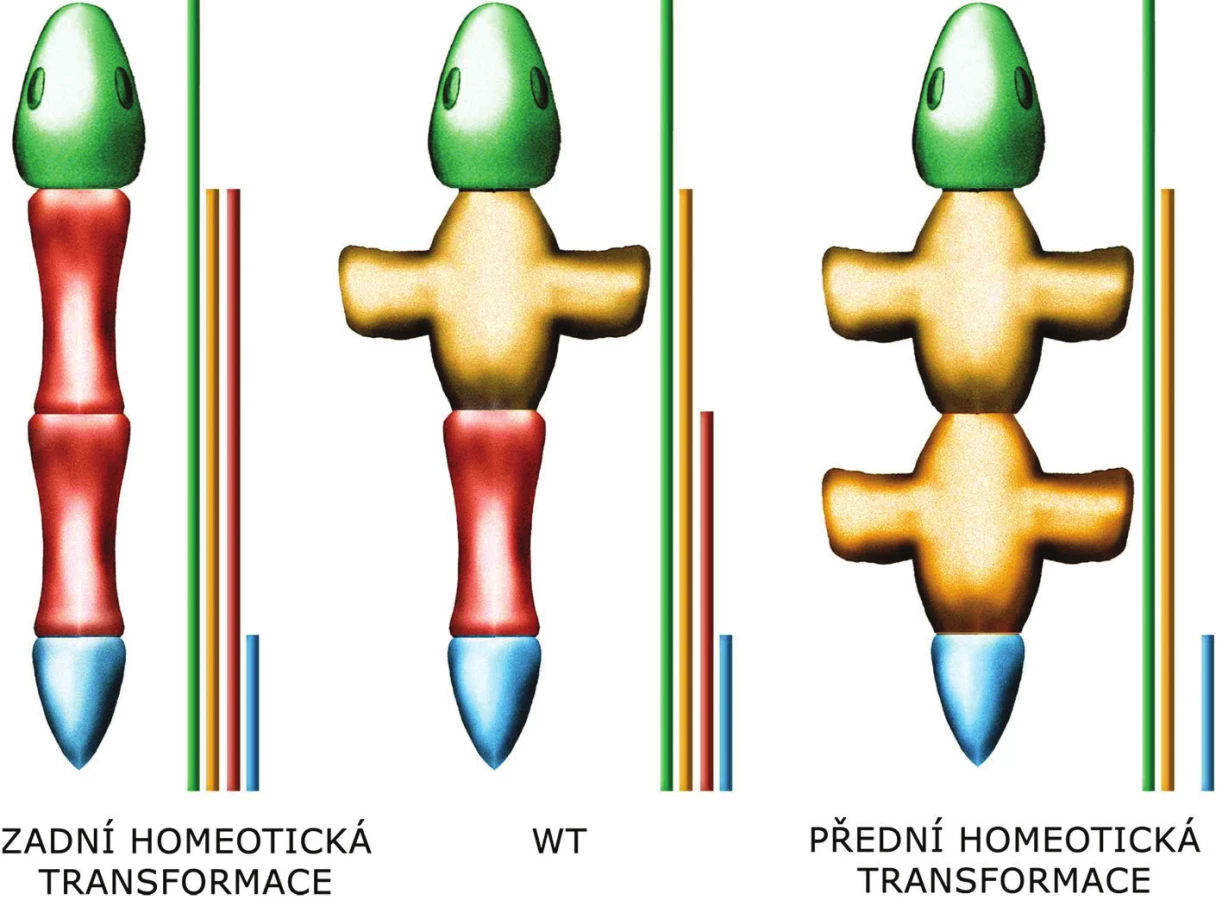
Obvykle platí, že funkce „předních“ 3’HOX genů je inhibována „zadními“ 5’geny. Tato inhibice se odehrává na úrovni proteinů, 5’HOX proteiny inhibují funkci 3’HOX proteinů. Mutace HOX genu tak většinou ovlivní jen ten nejkraniálnější segment, kam exprese příslušného genu (a tedy překlad/produkce příslušného proteinu) dosahuje, přestože gen je exprimován (protein je překládán) i v dalších, kaudálnějších segmentech. Tomuto efektu se říká zadní prevalence.
Pokud se změněný segment těla podobá segmentu před ním, jedná se o tzv. přední homeotickou transformaci, pokud se podobá segmentu za ním, hovoříme o zadní homeotické transformaci. Z principu zadní prevalence vyplývá, že pokud chybí jeden z Hox genů, změněný segment těla se podobá segmentu před ním, dojde tedy k přední homeotické transformaci. Naopak exprese ve zvětšeném rozsahu vede k zadní homeotické transformaci.
Jak bylo již řečeno, typickým znakem Hox genů je prostorová a časová kolinearita jejich exprese. Ta odpovídá pozicím jednotlivých genů v komplexu na chromozomu. Toto uspořádání umožňuje „úspornou“ regulaci, kdy jeden globální regulátor je schopen regulovat více genů komplexu najednou (10–12).
Exprese Hox genů je regulována různými strategiemi, jako je remodelace chromatinu, aktivita cis regulačních sekvencí DNA v blízkosti, i ve velké vzdálenosti od Hox komplexu. Významnou roli v posttranskripční regulaci Hox genů hrají také miRNA (13). Funkce Hox proteinů je pak zásadně ovlivněna interakcí s jinými proteiny. Specifické funkce Hox proteinů jsou tak definovány přítomností mnoha funkčních domén v rámci proteinu, které jim umožňují heterodimerizovat s různými kofaktory.
HOX geny u obratlovců včetně člověka
Všichni savci mají čtyři komplexy HOX genů na čtyřech různých chromozomech, dohromady 39 HOX genů uspořádaných do 13 paralogních skupin (obr. 4). Žádný komplex však neobsahuje všech 13 možných genů a jen tři paralogní skupiny obsahují všechny čtyři geny. U člověka jsou HOX komplexy lokalizovány na chromozomech 7p15, 17q 21, 12q13 a 2q31 (14).

Díky experimentálním delecím genů u myší víme, že funkce paralogních genů je (alespoň zčásti) redundantní, kompenzující vyřazení jednotlivých genů skupiny. Vyřazení více paralogních genů má pak kumulativně závažnější fenotypový projev. Efekt genů se tak sčítá a záleží na celkovém množství (dávce) exprimované RNA a následně překládaného proteinu. Například mutace vždy jednoho z paralogních genů ze skupiny Hox4 způsobí transformace pouze jednoho z krčních obratlů, vyřazení tří genů ze 4 (Hoxa4, Hoxb4, Hoxd4) způsobí transformaci čtyř krčních obratlů v rozsahu C2–C5.
Žádné experimentální vyřazení Hox genu či jejich kombinací však nevede u axiálního skeletu myší ke změně v počtu obratlů (16). Zdá se tedy, že Hox geny neregulují počet vytvořených obratlů, ale pouze jejich morfologii. Hoxgeny tedy v případě axiálního skeletu zprostředkují poziční informaci, neovlivní však vznik nebo zánik obratle.
HOX geny a vývoj končetin v experimentu
Experimenty na myších modelech ukazují, že pro vývoj končetin jsou klíčové geny Hoxa9-13 a Hoxd9-13 (17). Hoxa9-13 definují spíše proximodistální, Hoxd9-13 pak anteroposteriorní osu končetiny. Jejich exprese probíhá ve dvou vlnách a každá z nich je řízena transkripčně nezávisle. První vlna exprese určuje vývoj končetiny zhruba do úrovně karpu/tarsu (tedy vývoj stylopodia a zeugopodia). Druhá vlna exprese je nutná pro vývoj nejdistálnější části končetiny, autopodia (ruky/nohy) (obr. 5). Geny jsou exprimovány v sekvenci odpovídající místní i časové kolinearitě typické pro Hox rodinu. Geny skupiny 9 jsou exprimovány homogenně již v časném končetinovém pupenu, postupně s růstem končetinového pupenu nastupuje exprese skupin 10,11, exprese skupiny 12 a 13 je pak omezena na distální část vyvíjející se končetiny. Na rozdíl od situace na trupu nezpůsobuje vyřazení Hox genu na končetině klasickou homeotickou transformaci, ale redukci toho segmentu končetiny, ve kterém je za vývoje gen exprimován. Delece genů blíže 3’konci chromozomu (Hox 9,10) způsobuje postižení časněji se vyvíjející proximální etáže končetiny (stylopodia), zatímco delece 5’genů (Hox 12,13) ovlivňuje později se vyvíjející distální etáž (autopodium). Delece se projevuje oboustranně (na pravé i levé končetině) shodným fenotypem, někdy pouze na jednom z párů končetin (horním či dolním), někdy na obou párech končetin.
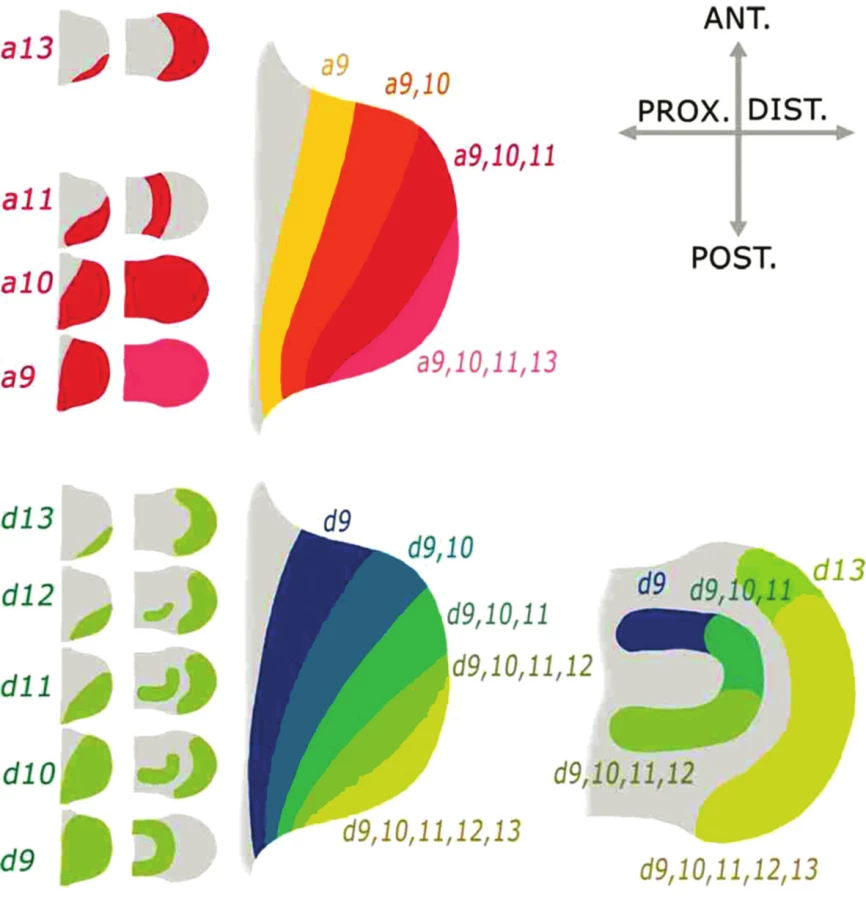
Jedním z proteinů, které regulují expresi Hoxd genů v končetině je protein Plzf (promyelocytic leukemia zinc finger). Funguje jako transkripční represor, kdy brání expresi Hoxd11–13 genů v přední, palcové části autopodia. Vyřazení jeho funkce u myši (18) či spontánní mutace jeho regulační sekvence u Lx potkana (19, 20) vede k posunu exprese Hoxd genů do přední části autopodia (obr. 6). To vede k vytvoření nadpočetného prstu na palcové straně, k preaxiální polydaktylii. Plzf protein brání expresi přímou interakcí se sekvencemi Hox komplexu.

HOX geny a vývoj končetin u člověka
Není překvapením, že i některé z vrozených vad končetin u člověka jsou způsobeny mutacemi HOX genů. Zatím byly u člověka v souvislosti s postižením končetin popsány mutace v genech HOXA11, HOXA13, HOXD10 a HOXD13.
U dvou rodin byla popsána delece jednoho nukleotidu ve druhém exonu genu HOXA11 vedoucí k posunu čtecího rámce a deleci proteinu, projevující se jako proximální radioulnární synostóza (OMIM #605432). Hlavice radia je srostlá s ulnou, zatímco distální radioulnární skloubení je normálně vytvořeno (21). Mutace HOXA13 byla identifikována jako příčina syndromu hand-foot-genital (22) (OMIM #140000). Jedná se o autozomálně dominantní onemocnění s postižením autopodií s typickou hypoplazií palců a malformacemi močopohlavního traktu (u žen uterus bicornis či bipartitus, malpozice ostium urethrae, u mužů hypospadie). Odlišná závažnost fenotypového projevu v různých rodinách závisí na konkrétní mutaci HOXA13, jsou již popsány různé bodové mutace či expanze některého ze tří polyalaninového úseků na N-konci HOXA13 proteinu (23). Bodová mutace HOXA13 způsobuje také vzácný Guttmacherův syndrom (OMIM #176305), který kromě hypoplazie palců a postižení genitálu shodného jako u hand-food-genital syndromu vykazuje pro něj unikátní postaxiální polydaktylii a zkrácení druhého prstu s pouze jedním článkem (24).
Bodová mutace genu HOXD10 (25) vedoucí k záměně methioninu za lyzin byla prokázána u jedné z forem dominantně dědičné deformity nohy vedoucí ke změně anatomické pozice talu. To vede k vykloubení talonavikulárního kloubu a následně ke vzniku pes planus (vrozená vertikalita talu, OMIM #192950).
Mutace HOXD13 způsobuje synpolydaktylii (SPD) (OMIM #186000). Synpolydaktylie je dominantně (s neúplnou penetrancí) dědičná malformace s kombinovanými příznaky syndaktylie a polydaktylie. Typicky se objevuje srůst 3. a 4. prstu na ruce a/nebo 4. a 5. prstu na noze, nadbytečný prst je většinou vytvořen právě v místě srůstu prstů. U normálních zdravých jedinců obsahuje první exon genu HOXD13 nukleotidovou sekvenci kódující polyalaninový úsek o délce 15 aminokyselin, u postižených rodin dochází k expanzi tohoto úseku o dalších 7–15 aminokyselin. Je zajímavé, že čím delší expanze, tím těžší fenotypový projev nemoci; délka expanze je však v rodinách zakonzervovaná a z generace na generaci se nemění (26).
U syndaktylie, stejně jako u hand-foot -genital syndromu, se jako jeden z typů mutace uplatňuje expanze polyalaninového úseku. Pokud expanze překročí určitý práh, změní se zásadně prostorové uspořádání (terciární struktura) proteinu, která vede ke ztrátě funkce proteinu nebo naopak k jeho abnormální aktivitě. Obdobné polyalaninové expanze způsobující u člověka vrozené syndromy byly popsány ještě u minimálně sedmi dalších genů, které ale nepatří do HOX rodiny (27). Obecně patří tyto expanze do širší rodiny onemocnění způsobených expanzí tandemových repetic, které ovlivňují buď strukturu proteinu – jako např. polyglutaminové expanze u Huntingtonovy nemoci, nebo regulaci exprese proteinu – jako např. expanze v promotorové oblasti u Friedreichovy ataxie.
U HOXA13 dochází expanzí ke ztrátě aktivity proteinu (28) a projevy hand-foot-genital syndromu se, alespoň na končetinách podobají fenotypu po vyřazení genu Hoxa13 u myši. V případě HOXD13 je polyalaninovou expanzí zesílen dominantně negativní efekt proteinu blokující funkci proteinů HOXD11 a 12. Projevy synpolydaktylie u člověka se tak spíše než myšímu fenotypu po vyřazení funkce Hoxd13 podobají myšímu fenotypu kombinovaného vyřazení Hoxd11-13 (29). Synpolydaktylii podobné postižení má i delece genů HOXD9-13 popsaná u otce a dcery (30).
Vedle polyalaninové expanze byl popsána řada dalších mutací genu HOXD13, např. bodové mutace v oblasti homeodomény, delece v kódujícím úseku genu i přilehlých nekódujících, avšak pro regulaci exprese důležitých úseků DNA. Ty způsobují malformace autopodia poněkud odlišné než u synpolydaktylie, s fenotypovými projevy jako brachydaktylie, polydaktylie či syndaktylie, nikoliv však typická synpolydaktylie (31–33). V práci Nakano et al. (33) bylo u 100 testovaných pacientů s různými malformacemi distální části končetiny identifikováno sedm nových mutací kódující sekvence HOXD13 a 2 nové mutace v nekódující sekvenci směrem 5’ od tohoto genu.
Tato i jiné recentní práce dobře dokumentují dnešní možnosti stanovení přesné mutace genu zodpovědné za příslušnou malformaci končetiny.
ZÁVĚR
Znalost mutace podmiňující vrozenou vadu sice dnes ještě neusnadňuje její léčbu (34, 35), kumulace dílčích údajů z jednotlivých případů však bezpochyby přispívá k hlubšímu poznání genové regulace prenatálního vývoje a má svůj význam pro genetické poradenství. A to je i výzva klinikům, kteří se s těmito pacienty setkávají, aby využili možnosti genetické diagnostiky, která je dnes k dispozici.
Zkratky
bp – páry bazí (base pairs)
FGF – růstový faktor fibroblastů (fibroblast growth factor)
HGF – růstový faktor hepatocytů (hepatocyte growth factor)
Lx – luxate, název mutace způsobující polydaktylii u potkana s mutací v genu Plzf. Není homologická myší mutaci Lx.
OMIM – Onlline Mendelian Inheritance in Men (viz http://www.ncbi.nlm.nih.gov/omim)
Plzf – promyelocytic leukemia zinc finger
SF – scatter factor
SHH – sonic hedgehog
TBX – T-box
WT – wild type, fyziologický jedinec
Tato práce vznikla v průběhu řešení projektů MSM 0021620806, LC06061 a GA ČR 301/07/P178.
Autoři děkují akad. malíři Ivanu Helekalovi, autorovi obrázků.
Adresa pro korespondenci:
MUDr. Pavel Šnajdr
Anatomický ústav 1. LF UK
U Nemocnice 3, 128 00 Praha 2
fax.: +420 224 965 770,
e-mail: pavel.snajdr@lf1.cuni.cz
Zdroje
1. Národní registr vrozených vad České republiky – ÚZIS ČR, http://www.vrozene-vady.cz/vrozene-vady/kvartaly/KVART_ 2008_4.pdf
2. Kozin SH. Upper-extremity congenital anomalies. J Bone Joint Surg Am 2003; 85-A: 1564–1576.
3. Sedmera D, Novotna B, Bila V, et al. The role of cell death in limb development of rats manifesting Lx allele on different genetic backgrounds. Eur J Morphol 1998; 36 : 173–181.
4. Hinrichsen KV, Jacob HJ, Jacob M, et al. Principles of ontogenesis of leg and foot in man. Ann Anat 1994; 176 : 121–130.
5. Franz T, Kothary R, Surani MA, et al. The Splotch mutation interferes with muscle development in the limbs. Anat Embryol (Berl) 1993; 187 : 153–160.
6. Brand-Saberi B, Seifert R, Grim M, et al. Blood vessel formation in the avian limb bud involves angioblastic and angiotrophic growth. Dev Dyn 1995; 202 : 181–194.
7. Szeder V, Grim M, Halata Z, et al. Neural crest origin of mammalian Merkel cells. Dev Biol 2003; 253 : 258–263.
8. Grier DG, Thompson A, Kwasniewska A, et al. The pathophysiology of HOX genes and their role in cancer. J Pathol 2005; 205 : 154–171.
9. Lewis EB. A gene complex controlling segmentation in Drosophila. Nature 1978; 276 : 565–570.
10. Soshnikova N, Duboule D. Epigenetic regulation of HOX gene activation: the waltz of methyls. Bioessays 2008; 30 : 199–202.
11. Deschamps J. Ancestral and recently recruited global control of the HOX genes in development. Curr Opin Genet Dev 2007; 17 : 422–427.
12. Tarchini B, Duboule D. Control of HOXd genes‘ collinearity during early limb development. Dev Cell 2006; 10 : 93–103.
13. Yekta S, Tabin CJ, Bartel DP. MicroRNAs in the HOX network: an apparent link to posterior prevalence. Nat Rev Genet 2008; 9 : 789–796.
14. Apiou F, Flagiello D, Cillo C, et al. Fine mapping of human HOX gene clusters. Cytogenet Cell Genet 1996; 73 : 114–115.
15. Horan GS, Ramirez-Solis R, Featherstone MS, et al. Compound mutants for the paralogous hoxa-4, hoxb-4, and hoxd-4 genes show more complete homeotic transformations and a dose-dependent increase in the number of vertebrae transformed. Genes Dev 1995; 9 : 1667–1677.
16. Wellik DM. HOX patterning of the vertebrate axial skeleton. Dev Dyn 2007; 236 : 2454–2463.
17. Zakany J, Duboule D. The role of HOX genes during vertebrate limb development. Curr Opin Genet Dev 2007; 17 : 359–366.
18. Barna M, Hawe N, Niswander L, et al. Plzf regulates limb and axial skeletal patterning. Nat Genet 2000; 25 : 166–172.
19. Liska F, Snajdr P, Sedova L, et al. Deletion of a conserved noncoding sequence in Plzf intron leads to Plzf down-regulation in limb bud and polydactyly in the rat. Dev Dyn 2009; 238 : 673–684.
20. Kren V. Genetics of the polydactyly-luxate syndrome in the Norway rat, Rattus norvegicus. Acta Univ Carol Med Monogr 1975; 1–103.
21. Thompson AA, Nguyen LT. Amegakaryocytic thrombocytopenia and radio-ulnar synostosis are associated with HOXA11 mutation. Nat Genet 2000; 26 : 397–398.
22. Mortlock DP, Innis JW. Mutation of HOXA13 in hand-foot-genital syndrome. Nat Genet 1997; 15 : 179–180.
23. Goodman FR, Bacchelli C, Brady AF, et al. Novel HOXA13 mutations and the phenotypic spectrum of hand-foot-genital syndrome. Am J Hum Genet 2000; 67 : 197–202.
24. Innis JW, Goodman FR, Bacchelli C, et al. A HOXA13 allele with a missense mutation in the homeobox and a dinucleotide deletion in the promoter underlies Guttmacher syndrome. Hum Mutat 2002; 19 : 573–574.
25. Shrimpton AE, Levinsohn EM, Yozawitz JM, et al. A HOX gene mutation in a family with isolated congenital vertical talus and Charcot-Marie-Tooth disease. Am J Hum Genet 2004; 75 : 92–96.
26. Goodman FR, Mundlos S, Muragaki Y, et al. Synpolydactyly phenotypes correlate with size of expansions in HOXD13 polyalanine tract. Proc Natl Acad Sci U S A 1997; 94 : 7458–7463.
27. Amiel J, Trochet D, Clement-Ziza M, et al. Polyalanine expansions in human. Hum Mol Genet 2004; 13 (Spec No 2): R235–243.
28. Utsch B, McCabe CD, Galbraith K, et al. Molecular characterization of HOXA13 polyalanine expansion proteins in hand-foot-genital syndrome. Am J Med Genet A 2007; 143A: 3161–3168.
29. Zakany J, Duboule D. Synpolydactyly in mice with a targeted deficiency in the HOXD complex. Nature 1996; 384 : 69–71.
30. Goodman FR, Majewski F, Collins AL, et al. A 117-kb microdeletion removing HOXD9-HOXD13 and EVX2 causes synpolydactyly. Am J Hum Genet 2002; 70 : 547–555.
31. Johnson D, Kan SH, Oldridge M, et al. Missense mutations in the homeodomain of HOXD13 are associated with brachydactyly types D and E. Am J Hum Genet 2003; 72 : 984–997.
32. Zhao X, Sun M, Zhao J, et al. Mutations in HOXD13 underlie syndactyly type V and a novel brachydactyly-syndactyly syndrome. Am J Hum Genet 2007; 80 : 361–371.
33. Nakano K, Sakai N, Yamazaki Y, et al. Novel mutations of the HOXD13 gene in hand and foot malformations. Int Surg 2007; 92 : 287–295.
34. Chomiak J. Vrozené vady končetin a systémové vady skeletu. In: P. Dungl. Ortopedie. Praha: Grada 2005; 245–314.
35. Smrčka V. Vrozené a získané vady horní končetiny. In: J. Měšťák. Úvod do plastické chirurgie. Praha: Karolinum 2005; 66–70.
Štítky
Adiktologie Alergologie a imunologie Angiologie Audiologie a foniatrie Biochemie Dermatologie Dětská gastroenterologie Dětská chirurgie Dětská kardiologie Dětská neurologie Dětská otorinolaryngologie Dětská psychiatrie Dětská revmatologie Diabetologie Farmacie Chirurgie cévní Algeziologie Dentální hygienistkaČlánek vyšel v časopise
Časopis lékařů českých

- Jak a kdy u celiakie začíná reakce na lepek? Možnou odpověď poodkryla čerstvá kanadská studie
- I mozek má svou krizi středního věku. Jak tyto změny souvisejí s rizikem demence ve stáří?
- Efektivita kartáčku Sonicare For Kids u dětí předškolního věku
- Nová generika s fixní kombinací salmeterol/flutikason pro terapii astmatu
- Nástroje k hodnocení závažnosti psoriázy v klinických studiích
Nejčtenější v tomto čísle
- Mužská plodnost a onkologická léčba
- Edukace u schizofrenie: Jak pacienti a příbuzní hodnotí program prevence relapsu PREDUKA
- Posuzování zdravotního stavu a pracovní schopnosti u duševních poruch a poruch chování
- HOX geny a vývoj končetin v klinické medicíně i v experimentu
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy