
Vzácné anémie ze skupiny vrozených syndromů selhání kostní dřeně
Rare anemias from the group of congenital bone marrow failure syndromes
This review summarizes the pathophysiology, genetic background and clinical symptoms of anemias belonging to the group of inherited bone marrow failure syndromes with unilineage failure of erythropoiesis. It sums up the current knowledge of three diseases: Diamond-Blackfan anemia, congenital dyserythropoietic anemia and Fanconi anemia whose pathophysiology was elucidated in detail during the last decade, owing to the rapid development of new molecular-genetic techniques, especially next-generation sequencing. Fanconi anemia is included in this overview because of macrocytosis and/or anemia detected in the majority of the patients before they develop bone marrow failure. The paper also aims at pointing out typical associated anomalies in these diseases which might be overlooked and which can lead to early diagnosis. Unfortunately, the correct diagnosis is often established later in adulthood and, in some cases, as late as at the time of manifestation of malignant disease. Accurate and timely diagnosis of these conditions is extremely important for the determination of appropriate treatment approach, diagnosis of affected family members (especially in the process of bone marrow donor search), and genetic counselling, which can substantially influence the prognosis of these diseases.
Key words:
congenital dyserythropoietic anemia – Diamond-Blackfan anemia – DNA repair – dyserythropoiesis – Fanconi anemia – inherited bone marrow failure – ribosomopathies
Autoři:
Dagmar Pospíšilová
Působiště autorů:
Dětská klinika LF UP a FN Olomouc
Vyšlo v časopise:
Vnitř Lék 2018; 64(5): 488-500
Kategorie:
Přehledné referáty
Souhrn
Přehledný článek přibližuje patofyziologii, molekulárně-genetickou podstatu a klinické projevy anémií ze skupiny vrozeného selhání kostní dřeně s postižením erytroidní linie. Zahrnuje nejnovější poznatky o třech onemocněních, jejichž podstata byla odhalena až v posledních letech díky pokrokům v oblasti molekulární genetiky: Diamondově-Blackfanově anémii, kongenitální dyserytropoetické anémii a Fanconiho anémii. Fanconiho anémie je do přehledu zařazena pro častou přítomnost makrocytózy nebo anémie při manifestaci onemocnění, u většiny pacientů se však postupně vyvíjí selhání všech hematopoetických linií. Cílem přehledu je upozornit současně na méně nápadné příznaky uvedených nemocí, které vedou k jejich přehlédnutí a pozdní diagnostice. V řadě případů je správná diagnóza stanovena až v dospělém věku nebo dokonce až při rozvoji maligního onemocnění. Včasná diagnóza je nezbytná ke stanovení správného léčebného postupu, diagnostice nemoci u členů rodiny, především při vyhledávání dárců kostní dřeně, a ke genetickému poradenství. Cílem práce je rovněž přispět ke včasné diagnostice anémií doprovázených typickými somatickými anomáliemi.
Klíčová slova:
Diamondova-Blackfanova anémie – dyserytropoéza – Fanconiho anémie – kongenitální dyserytropoetická anémie – ribosomopatie – syndromy selhání kostní dřeně – systém oprav DNA – zvýšené riziko vzniku maligního onemocnění
Úvod
Do skupiny vrozených syndromů selhání kostní dřeně (Inherited Bone Marrow Failure Syndromes – IBMFS) jsou řazeny vzácné poruchy krvetvorby charakterizované:
- geneticky podmíněným selháním funkce jedné nebo více hematopoetických linií (erytrocytární, granulocytární nebo trombocytární)
- vrozenými anomáliemi nebo funkčními poruchami postihujícími různé tkáně a orgány: kůži, skelet, srdce, plíce, ledviny, centrální nervový systém, často doprovázené i malým vzrůstem
- zvýšeným rizikem vzniku maligních onemocnění: nejčastěji akutní myeloidní leukemie (AML), myelodysplastického syndromu (MDS) nebo některých typů solidních nádorů
Společným rysem těchto onemocnění je výrazná fenotypová variabilita jak hematologických, tak i somatických změn. Závažnější formy onemocnění se zřetelnými vrozenými anomáliemi, časnou manifestací cytopenií a rozvojem selhání kostní dřeně jsou obvykle diagnostikovány již v časném dětství v kojeneckém, batolecím nebo předškolním věku. Lehčí formy s nenápadnými anomáliemi a pozdní manifestací cytopenií mohou na druhé straně dlouho unikat pozornosti a jsou potom diagnostikovány až v dospělosti, nebo dokonce až při vypuknutí maligního onemocnění. Tuto skupinu onemocnění dělíme obvykle podle nejvýrazněji postižené linie krvetvorby a rozsahu jejich postižení na izolované cytopenie a pancytopenie.
U izolovaných cytopenií (Diamondova-Blackfanova anémie, kongenitální dyserytropoetická anémie, těžká vrozená neutropenie, trombocytopenie s chyběním radia) nedochází obvykle k rozvoji pancytopenie, u ostatních onemocnění se postupně rozvíjí pancytopenie s aplazií kostní dřeně (Fanconiho anémie, dyskeratosis congenita, Shwachmanův-Diamondův syndrom, kongenitální amegakaryocytární trombocytopenie).
Do skupiny vrozených syndromů setkání kostní dřeně jsou dnes řazena následující onemocnění:
- Fanconiho anémie (FA)
- dyskeratosis congenita (DC)
- syndrom hypoplastických chrupavek a vlasů (Cartilage-Hair Hypoplasia – CHH)
- Diamondova-Blackfanova anémie (DBA)
- Shwachmanův-Diamondův syndrom (SDS)
- těžká vrozená neutropenie (Severe Congenital Neutropenia – SCN)
- retikulární dysgeneze (RD)
- amegakaryocytární trombocytopenie (Congenital Amegakaryocytic Thrombocytopenia – CAMT)
- trombocytopenie s chyběním radia (thrombocytopenia with absent radius – TAR)
- Pearsonův syndrom (PS)
- kongenitální dyserytropoetická anémie (Congenital Dyserythropoietic Anemia – CDA) – tato byla zařazena do skupiny IBMFS teprve v posledních letech
Epidemiologie
Všechna tato onemocnění jsou vzácná, incidence některých z nich není přesně známa. V řadě případů je regionálně odlišná a závislá na lokální frekvenci nosičství mutací příslušných genů. V některých uzavřených populačních skupinách je vyšší frekvence výskytu v důsledku tzv. efektu zakladatele (founder effect), což znamená ztrátu genetické variability, která nastává při vytvoření uzavřené nové populace s malým počtem jedinců. Typ dědičnosti je u jednotlivých chorob variabilní a závisí na charakteru vyvolávajícího genetického defektu. Při významném zlepšení diagnostických možností se v posledním desetiletí zvyšuje i počet diagnostikovaných případů. Předpokládá se tedy, že tato onemocnění jsou nedostatečně diagnostikována, a to především jejich mírnější formy. Včasné stanovení diagnózy a určení genetického defektu je přitom velmi důležité pro stanovení adekvátního plánu dlouhodobého sledování pacienta, léčebného postupu a prognózy onemocnění. Má velký význam i pro genetické poradenství a vyhledávání dalších postižených členů rodiny [1].
Vrozené syndromy kostní dřeně s postižením erytroidní linie
Diamondova-Blackfanova anémie
Charakteristika
Diamondova-Blackfanova anémie (DBA) je vzácná vrozená hypoplazie erytropoézy, charakterizovaná normochromní makrocytární anémií, těžkou retikulocytopenií, normocelulární kostní dření se selektivním nedostatkem erytroidních prekurzorových buněk, normálním počtem leukocytů a normálním nebo lehce vyšším počtem trombocytů [2]. U 40–50 % pacientů jsou přítomny přídatné anomálie (tab. 1), asi u 30 % pacientů růstová retardace. Riziko rozvoje hematologických maligních onemocnění (MDS a AML) a solidních nádorů je 5–6krát vyšší než u zdravé populace [3].

DBA patří mezi nejčastěji se vyskytující onemocnění ve skupině syndromů selhání kostní dřeně. Poprvé byla popsána jako klinická jednotka v roce 1938 americkými pediatry Louisem K. Diamondem, považovaným za zakladatele pediatrické hematologie, a Kennethem Blackfanem.
Diagnóza je v 90 % stanovena v novorozeneckém nebo kojeneckém věku. Méně často bývá onemocnění zjištěno později, v posledních letech není výjimkou stanovení diagnózy u pacientů s mírnější formou onemocnění až v dospělosti [2]. Výjimečně může být nemoc odhalena až při diagnostice maligního onemocnění v dospělém věku [2], což potvrzuje i vlastní zkušenost autorky. Rozvoj selhání kostní dřeně je vzácný.
Epidemiologie a dědičnost
Incidence je udávána v rozmezí 5–8 případů na 1 milion živě narozených. DBA se vyskytuje u mužského a ženského pohlaví v poměru 1,1 : 1. Většinu pacientů tvoří příslušníci indoevropské populace, nemoc však byla popsána i u africké negroidní populace, Japonců a Arabů.
Ve 20–40 % případů se jedná o hereditární onemocnění s autosomálně dominantním typem dědičnosti. U nově popsaných mutací v extraribosomálních genech kódujících GATA1 transkripční faktor a TSR2 protein je dědičnost vázaná na chromosom X. Ve zbývajících případech se jedná o sporadicky se vyskytující onemocnění, v jejichž etiologii se uplatňují mutace vznikající de novo.
Klinické a laboratorní nálezy
U většiny pacientů je diagnóza stanovena v 1. roce života, u části pacientů je anémie vyžadující podání transfuze diagnostikována již v novorozeneckém věku. Prvními klinickými příznaky DBA jsou bledost, dušnost, u kojenců je často popisováno neprospívání. Vzácně byl popsán i fetální hydrops. U 30–50 % pacientů s DBA jsou přítomny vrozené anomálie, postihující převážně oblast lbi a obličeje (kraniofaciální dysmorfie, mikrocefalie, rozštěp patra, hypertelorizmus a gotické patro). K dalším anomáliím patří vývojové vady palce, horní končetiny (tříčlánkový palec, duplikace článku palce nebo hypoplazie palcového valu (thenar), srdce, ledvin, urogenitálního traktu a kostí (tab. 1, obr. 1, obr. 2).
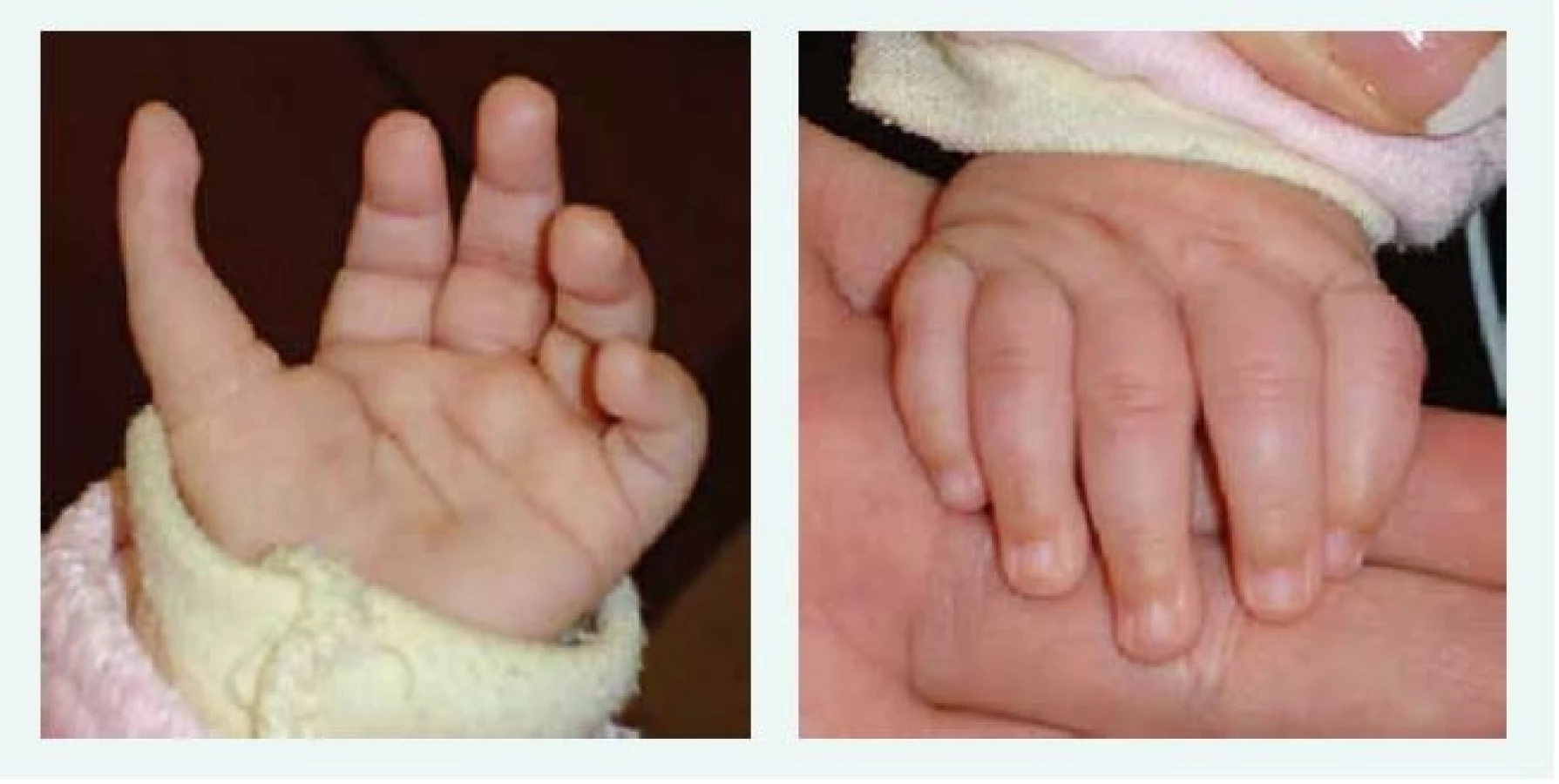

U 30–40 % pacientů je popisován malý vzrůst. Na růstové retardaci u starších pacientů se může podílet dlouhodobá léčba kortikosteroidy i přetížení organizmu železem.
Při laboratorním vyšetření je dominujícím nálezem makrocytární anémie s retikulocytopenií. Může být zvýšená hladina fetálního hemoglobinu (HbF) jako známka stresové erytropoézy. Důležitým znakem je zvýšení hladiny erytrocytární adenozindeaminázy (e-ADA), které může být jedním z prvních diagnostických vodítek. I přes specificitu tohoto nálezu pro DBA nebyla dosud příčina zvýšení e-ADA vysvětlena. V kostní dřeni je v typických případech výrazně snížen počet erytroidních prekurzorových buněk, často na 1–5 %.
Podle analýzy údajů z registru DBA v USA (DBAR) je riziko rozvoje solidních nádorů nebo leukemie u DBA 5,4krát vyšší než v populaci [3]. Zvýšená incidence byla popsána specificky pro některé typy maligních onemocnění: MDS, AML, karcinom plic, tlustého střeva, bazaliom, osteosarkom, nádory prsu a gynekologické karcinomy u žen. Dle DBAR je incidence solidních nádorů vyšší než výskyt leukemií. Kumulativní incidence solidních nádorů a AML u DBA byla průměrně 20 % ve 4. decéniu ve srovnání se 30 % u FA a DC kdykoliv ve stejné věkové kategorii; riziko solidních nádorů u netransplantovaných pacientů začíná narůstat po 30. roce věku, tedy později než u FA a DC. Malý počet popsaných případů a značná diverzita typů maligních onemocnění však zatím nedovoluje definitivní závěry.
Molekulární podstata onemocnění
Po desetiletích snahy o nalezení příčiny DBA byla teprve v roce 1999 poprvé odhalena její genetická podstata: překvapivý nález heterozygotní mutace genu kódujícího ribosomální protein (RP) RPS19, součásti malé ribosomální podjednotky u švédského pacienta. Mutace genu kódujícího RPS19 byly postupně prokázány u 25 % pacientů s DBA. RPS19 je komponentou malé 40S ribosomální podjednotky a je lokalizován v nukleolu, hlavním buněčném místě transkripce při biogenezi ribosomů. Role některých RP není dosud u vyšších eukaryont přesně známa. Delece jedné alely RP vede k poruše růstu a snížené tvorbě 40S ribosomální podjednotky, tedy k tzv. „ribosomálnímu stresu“. Buňka reaguje aktivací p53 proteinu a indukcí apoptózy. Dalším důsledkem je snížení translace [5], a tím i proteosyntézy, což může ovlivnit procesy s velkým nárokem na přísun proteinů – tedy s rychlým obratem produkce buněk, jako je hematopoéza, obnova kožních buněk a buněk GIT.
V dalších letech byly postupně nalezeny mutace genů kódujících dalších 20 RP: RPS7, RPS10, RPS15A, RPS17, RPS19, RPS24, RPS26, RPS27, RPS28, RPS29; RPL5, RPL9, RPL11, RPL15, RPL18, RPL26, RPL27, RPL31, RPL35, and RPL35A [5]. Ojediněle byly nalezeny mutace dvou dalších genů, které nekódují RP: GATA1, důležitého transkripčního faktoru pro erytropoézu a TSR2, který kóduje protein ovlivňující apoptózu a maturaci ribosomů. DBA tedy vzniká v důsledku haploinsuficience ribosomálních proteinů. Aktivace p53 proteinu i snížení translace bylo prokázáno na buněčných [5] i zvířecích modelech DBA. Jsou dále intenzivně studovány extraribosomální funkce RP a jejich možná úloha při vzniku somatických anomálií.
Odhalení poruchy biogeneze ribosomů jako příčiny hematologického onemocnění patří mezi nejzajímavější objevy moderní hematologie a otevřelo mnoho otázek týkajících se souvislosti poruch ribosomální biogeneze, somatických anomálií a zvýšené incidence vzniku maligního onemocnění. Diamondova-Blackfanova anémie tak dala vznik nové skupině onemocnění nazvaných ribosomopatie.
Diagnostika
Pro diagnózu DBA svědčí makrocytární anémie s retikulocytopenií při normálním počtu trombocytů a leukocytů. V době stanovování diagnózy se výjimečně může objevit trombocytóza a/nebo neutropenie. Pro diagnostiku je stěžejní vyšetření kostní dřeně, v níž je klasickým nálezem nízký počet erytroidních prekurzorových buněk v jinak normocelulární kostní dřeni. U pacientů, kteří nejsou závislí na transfuzní terapii ovlivňující výsledky vyšetření, je přínosné vyšetření erytrocytární adenozindeaminázy (eADA), jejíž hodnota je zvýšena. Normální hladina e-ADA nevylučuje onemocnění. Jedinou metodou, která jednoznačně potvrzuje diagnózu, je nález mutací genů pro ribosomální proteiny, případně genu kódujícího TSR2 protein a GATA1 transkripční faktor.
Při diagnostice je vždy nutné vyloučit některá jiná onemocnění, o kterých lze diferenciálně diagnosticky uvažovat. Především se jedná o: Fanconiho anémii, deficit G-6PD, erytroblastopenii vzniklou po infekci parvovirem B19, tranzientní erytroblastopenii dětského věku (Transient Erythroblastopenia of Childhood – TEC) a další získané erytroblastopenie, u pacientů s DBA v remisi s normálním počtem erytroidních prekurzorů, vzácně i kongenitální dyserytropoetickou anémii vzhledem k dysplastickým změnám erytropoézy.
Léčba
Léčebně je v 1. roce života obvykle nezbytné podávání transfuzí erytrocytů. Po 1. roce je indikováno podání kortikoidů, na které až 60–80 % pacientů odpoví akcelerací erytropoézy. Kortikoidy se podávají nejprve po dobu 4 týdnů v dávce 2 mg/kg/den, při dobré odpovědi je potom dávka prednisonu postupně pomalu snižována s cílem dosažení obdenní dávky nepřesahující 0,5 mg/kg. Dlouhodobé podávání denních dávek vyšších než 0,5 mg/kg se nedoporučuje vzhledem k riziku významných vedlejších účinků v dětském věku: rozvoj osteoporózy, přírůstek na váze, cushingoidní vzhled, rozvoj hypertenze a diabetu, růstová retardace, patologické fraktury, vznik žaludečních vředů, katarakty nebo glaukomu a zvýšenou náchylnost k infekcím.
U pacientů, kteří neodpovídají příznivě na léčbu kortikoidy, je nutno podávat dlouhodobě transfuze erytrocytární masy. Cílem transfuzního programu je udržení koncentrace hemoglobinu mezi 80–100 g/l, což je hladina adekvátní pro udržení normálního růstu a vývoje dítěte [2].
Z celkového počtu DBA pacientů je přibližně 40 % pacientů steroid-dependentních, asi 40 % je závislých na transfuzích a 20 % dosáhne remise.
U pacientů, kterým jsou podávány pravidelně transfuze erytrocytární masy, je nezbytné podávání chelátorů železa (desferioxamin, deferasirox) k prevenci orgánového postižení při přetížení železem. Léčbu chelátory je vhodné zahájit již po podání prvních 12 transfuzí, po nichž dosáhne sérový feritin obvykle hladin 1 000–1 500 µg/l, nebo když koncentrace železa v jaterní tkáni dosáhne 6–7 mg/g sušiny jaterní tkáně. Podávání deferasiroxu je vhodné až od 2 let věku. Deferipron běžně používaný k léčbě přetížení železem u pacientů s talasemií major není pro dlouhodobou léčbu u DBA doporučován pro zvýšené riziko vzniku agranulocytózy [6]. Je nutno sledovat pečlivě růst pacienta a monitorovat možné nežádoucí účinky přetížení železem i chelatační léčby. Nedílnou součástí týmu pečujícího o pacienty s DBA je proto i endokrinolog, jehož úkolem je včas podchytit a léčit poruchy růstu, kostní denzity, fertility, onemocnění štítné žlázy a hypofýzy vznikající v důsledku léčby kortikoidy a/nebo přetížení železem. U části pacientů (asi 20–30 %) může nastat remise onemocnění (spontánně nebo při opakované léčbě kortikoidy), která nastupuje obvykle do 8.–10. roku věku, velmi vzácně i později.
Zkoumá se i efekt nových látek s pozitivním efektem na erytropoézu s výhledem léčebného využití: aminokyseliny leucinu [7,8] a sotaterceptu [9].
Pokud lze nalézt v rodině pacienta příbuzenského dárce, případně shodného dárce v rámci registrů, je u pacientů trvale závislých na podávání transfuzí indikována transplantace kmenových buněk (Hematopoietic Stem Cell Transplantation – HSCT), která je zatím jedinou kurativní léčbou pro DBA. Pacienti trvale závislí na transfuzích nebo ti, u kterých se vyvinuly další cytopenie, jsou indikováni k HSCT. Otázka vhodných režimů a optimálního věku transplantace je předmětem diskusí, zatím se její provedení doporučuje do 10 let věku. HSCT od nepříbuzenských dárců má horší výsledky, i když v posledních letech došlo k jejich významnému zlepšení.
V průběhu těhotenství žen s Diamondovou-Blackfanovou anémií byly popsány četné komplikace preeklampsie, intrauterinní růstová retardace a úmrtí plodu, zhoršení anémie, předčasné porody [10].
Kongenitální dyserytropoetické anémie
Charakteristika
Kongenitální dyserytropoetické anémie (Congenital Dyserythropoietic Anemias – CDA) zahrnují skupinu vzácných vrozených poruch erytropoézy charakterizovaných chronickou hyporegenerativní anémií s neefektivní erytropoézou, relativní retikulocytopenií, hemolytickou složkou s mírnou hyperbilirubinemií a splenomegalií, typickými morfologickými změnami erytroblastů v kostní dřeni a v některých případech i přetížením železem. Většinou se jedná o poruchy vyzrávání erytroidních buněk v pozdních stadiích erytropoézy. V dnešní době je CDA řazena většinou hematologů do skupiny syndromů selhání kostní dřeně vzhledem k poruše diferenciace a proliferace buněk erytroidní linie [11].
Termín dyserytropoéza označuje kvalitativní defekt erytroidních prekurzorů nebo erytrocytů vedoucí k abnormální (neefektivní) erytropoéze s postižením diferenciace a proliferace erytroidních buněk, které v kostní dřeni nevyzrávají nebo částečně zanikají před vyplavením do periferní krve. V některých případech je doprovázena i zkráceným přežíváním erytrocytů. Dyserytropoéza provází řadu jak vrozených (talasemie a jiné hemoglobinopatie, hereditární sférocytóza, sideroblastická anémie, DBA, FA), tak získaných hematologických onemocnění (deficit železa, vitaminů, MDS), což může být zdrojem diferenciálně diagnostických rozpaků a obtížné diagnostiky.
Epidemiologie a dědičnost
CDA patří mezi vzácné anémie. Prevalence a geografická distribuce se liší mezi jednotlivými oblastmi. Nejnižší výskyt je popisován v severských zemích (0,04 případů na 1 milion obyvatel), nejvyšší výskyt v Itálii (2,4 případu na 1 milion obyvatel) [12]. Některé mutace se vyskytují s vysokou frekvencí v určitých populacích (u beduínské populace R1042W v genu pro codanin – CDAN1, v populaci zemí kolem Středozemního moře – Maroko, Izrael, Itálie – R14W v genu SEC23B). Výskyt jednotlivých alel však svědčí o vlivu mnohočetného efektu zakladatele (founder effect) v jednotlivých geografických oblastech.
Dědičnost jednotlivých typů CDA je většinou autosomálně recesivní, v některých případech dominantní.
Klinické a laboratorní nálezy
Klinicky se CDA projevují celoživotní středně těžkou až těžkou makrocytární nebo normocytární anémií s časně vznikajícím ikterem, splenomegalií a hepatomegalií. Často se u pacientů tvoří žlučové kameny. V některých případech může být prvním projevem onemocnění fetální hydrops nebo intrauterinní růstová retardace. U 4–14 % pacientů jsou přítomny vrozené anomálie, především v oblasti skeletu končetin. V důsledku opakovaných transfuzí a zvýšené resorpce železa se u polytransfundovaných pacientů, ale i části pacientů bez nutnosti transfuzí se rozvíjí přetížení železem se sekundární hemochromatózou. Jednou z příčin je i nízká hladina hepcidinu.
Při laboratorním vyšetření nacházíme v krevním obrazu normocytární nebo makrocytární anémii spojenou s anizocytózou, poikilocytózou, nálezem bazofilního tečkování erytrocytů, Howellových-Jollyových tělísek a relativní retikulocytopenií. V kostní dřeni je typická hyperplazie erytropoézy s výraznými dysplastickými změnami: dvoujaderné a mnohojaderné erytroblasty, internukleární můstky, vakuolizace cytoplazmy, nepravidelná kontura jader (obr. 3).

Typické změny jsou popisovány při vyšetření elektronovou mikroskopií (obr. 4).

Podezření na CDA tedy vychází z klinického obrazu a hematologických nálezů v periferní krvi. Vždy je nutno provést vyšetření kostní dřeně se zhodnocením morfologie erytroblastů. Významnou roli v diagnostice hraje i elektronová mikroskopie. Diagnóza je definitivně potvrzena nálezem patogenních variant pomocí molekulárně-genetického vyšetření, které jsou typické pro jednotlivé subtypy onemocnění (tab. 2).

Diagnóza
Pracovní klasifikace navržená poprvé Heimpelem a Wendtem (CDA I, II, a III) je stále v klinické praxi používána [13]. V některých případech anémie splňuje obecnou definici CDA, nelze ji však zařadit do žádného ze základních 3 subtypů, proto byly tyto označeny jako tzv. CDA varianty. Molekulárně genetický podklad obou nejčastějších typů: CDA Ia a II je znám již delší dobu, kauzální genetické změny u typu III, IV a u ostatních CDA variant však byly popsány teprve v posledních letech [6,7,9], což výrazně zpřesnilo diagnostiku této skupiny anémií.
Molekulární podstata onemocnění
Vzhledem k významným pokrokům v oblasti molekulární biologie, které přináší především sekvenování nové generace, byly dnes již u jednotlivých typů CDA identifikovány kauzální molekulárně-genetické změny, což významně přispělo k upřesnění klasifikace u této skupiny nemocí, a především k postupnému porozumění jejich patogenezi. Podle genetických změn je dnes rozlišováno 5 základních podtypů CDA (Ia, Ib, II, III a IV), tab. 3.
![Vrozené vývojové anomálie u pacientů s Fanconiho anémií. Upraveno podle [31]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/e133b864cbf217f4d2cdf7efcdf458a8.jpeg)
Některé další varianty dyserytropoetické anémie jsou součástí komplexních onemocnění či syndromů a jsou některými autory označované jako typ V a VI. S objevem kauzálních genetických variant se otevřela nová éra genetického a klinického výzkumu s cílem detailního odhalení funkce proteinů, v jejichž kódujících genech jsou popisovány mutace. Stanovení molekulární podstaty dnes umožňuje diagnostiku nosičství genu i prenatální diagnostiku, může být vodítkem při hledání nových alternativ léčby. Molekulárně-genetické charakteristiky jsou uvedeny u jednotlivých podtypů CDA.
Kongenitální dyserytropoetická anémie typu I
Kongenitální dyserytropoetická anémie typu I (CDA I) je autosomálně recesivně dědičná anémie charakterizovaná makrocytární anémií MCV (100–120 fL) a u některých pacientů i anomáliemi distálních částí končetin. Při vyšetření kostní dřeně je přítomna erytroidní hyperplazie s přítomností méně než 10 % dvojjaderných erytroblastů. Typickou změnou jsou internukleární můstky přítomné v 5–8 % erytroblastů. Elektronová mikroskopie ukazuje houbovitý vzhled heterochromatin označovaný jako „vzhled švýcarského sýra“ („Swiss cheese appearance“).
Většina pacientů s CDA I má makrocytární anémii s variabilní hladinou hemoglobinu, výjimečně může být anémie v dětském věku normocytární. Relativní retikulocytopenie je doprovázena zvýšeným rozpadem erytrocytů se zvýšenou hladinou nepřímého bilirubinu, vysokou hladinou laktátdehydrogenázy (LDH), snížením hladiny haptoglobinu a hyperbilirubinemií. Všichni pacienti mají od adolescentního věku splenomegalii, 20 % dětí má vrozené anomálie, nejčastěji syndaktylie na rukou nebo nohou, chybění nehtů nebo nadpočetné palce, deformity hrudníku a malý vzrůst. Byly popsány i deformity lebky v důsledku výrazné expanze kostní dřeně. Výjimečně byl popsán fetální hydrops a anémie vyžadující intrauterinní transfuzi. Asi 1 ze 3 plodů je menší než odpovídá jeho gestačnímu věku. Po porodu může být přítomna hepatomegalie a ikterus, v některých případech je nutná i transfuze erytrocytů.
Příčinou vzniku jsou mutace CDAN1 genu kódujícího codanin, které byly popsány v roce 2003 [14]. Dle dosavadních poznatků hraje tento protein důležitou roli v organizaci jaderného heterochromatinu v průběhu replikace DNA. Codanin 1 je rovněž součástí cytosolického Asf1-H3-H4-importin-4 komplexu, který má důležitou funkci při sestavování a degradaci nukleosomu. Pravděpodobně hraje roli i v časné fázi vývoje embrya.
Nález kauzálních mutací dalšího genu, C15ORF41, u pacientů s CDA I vedl k rozdělení CDA I na 2 podtypy: CDA 1a a 1b (tab. 2). Tento gen kóduje restrikční endonukleázu s dosud neznámou funkcí. Bylo však prokázáno, že C15ORF41 kooperuje s Asf1b, což podporu hypotézu, že primárním defektem u CDA 1b je rovněž patologie DNA replikace a sestavování chromatinu [15].
Kongenitální dyserytropoetická anémie typu II
CDA II je nejčastější variantou CDA. V kostní dřeni je více než 10 % binukleárních erytroblastů, elektronová mikroskopie ukazuje vezikuly s obsahem proteinů endoplazmatického retikula lokalizované pod plazmatickou membránou. U CDA II je pozitivní Hamův test (lýza erytrocytů s acidifikovaným alogenním sérem), který byl dříve používán k diagnostickým účelům. Vzhledem k potřebě několika kontrolních vzorků se dnes již běžně nepoužívá.
Analýza proteinů erytrocytární membrány SDS PAGE elektroforézou na polyakrylamidovém gelu, která identifikuje abnormality glykosylace rychle migrujícího pruhu 3 (the anion exchanger 1) a pruhu 4,5, je vysoce senzitivní a specifická. K potvrzení diagnózy je používána molekulárně-genetická analýza. V roce 2012 byla poprvé popsána molekulární příčina onemocnění: mutace genu SEC23B kódujícího protein COPII, jenž je součástí proteinového komplexu podílejícího se na tvorbě a transportu vezikul v buňce [16]. Teprve po několika letech však byl objasněn patogenetický mechanizmus vzniku patologických změn erytroblastů u CDA II.
Fenotyp multinuklearity vzniká při aberantní glykosylaci specifických proteinů nezbytných pro dělení buněk, což vede k defektům v tomto procesu. Specificita efektu na erytropoézu u takto ubikvitního genu je pravděpodobně způsobena tkáňově specifickou expresí SEC23B během erytroidní diferenciace.
Kongenitální dyserytropoetická anémie typu III
CDA III je nejvzácnějším typem CDA. Poprvé byla popsána v rodině z USA, většina pacientů jsou členové jedné velké rodiny ze Švédska. Dědičnost je v obou rodinách dominantní. Bylo však popsáno i několik případů s autosomálně recesivní dědičností. Splenomegalie většinou není přítomna. Nález v kostní dřeni je charakterizován erytroidní hyperplazií a velkými typickými multinukleárními erytroblasty. U mnoha členů švédské rodiny byla diagnostikována monoklonální gamapatie a u některých došlo k rozvoji mnohočetného myelomu.
Příčinou onemocnění je mutace genu KIF23 kódujícího MKLP1 protein, což je mitotický protein důležitý pro cytokinezi [17,18]. Porucha funkce tohoto proteinu vede ke vzniku multinuklearity erytroblastů.
Kongenitální dyserytropoetická anémie typu IV
CDA IV je velmi vzácným typem této skupiny. Kauzální jsou mutace genu KLF1 kódujícího transkripční faktor [19,20]. V roce 2010 byla u pacienta s CDA poprvé popsána mutace genu KLF1 lokalizovaná v exonu 3 (jedná se o mutaci měnící smysl: c.973 G>A, p.E325K). Dosud byly popsány pouze 4 případy onemocnění, 5. dosud nepublikovaný případ stejné mutace byl popsán v České republice.
Krüppel-like factor 1 (KLF1) je důležitý transkripční faktor v procesu erytropoézy. Účastní se regulace vyzrávání multipotentních kmenových buněk do erytroidní linie, hemoglobinového přepínání z fetálního na adultní hemoglobin a pozdní diferenciaci erytroidních buněk. Je důležitý pro udržování buněčné stěny v klidovém období života erytrocytů. Poslední studie ukázaly, že mutace v některých lokusech KLF1 genu se vyskytují poměrně často a jsou většinou asymptomatické, vedou však ke zvýšení hladin fetálního hemoglobinu (α2γ2) a adultního HbA2 (α2δ2).
Kongenitální dyserytropoetické anémie variantní
Do dnešního dne bylo popsáno několik dalších CDA variant. Tato onemocnění mají buďto izolovaný CDA fenotyp nebo se jedná o definované syndromy, u kterých je přítomen různý rozsah dyserytropoézy. Do této skupiny jsou řazeny následující jednotky:
- Makrocytární dyserytropoetická anémie s dominantní dědičností vázanou na chromosom X: charakterizovaná zvýšenou hladinou feritinu (800 ng/ml) – byla popsána u několika osob ženského pohlaví v jedné rodině – v kostní dřeni je výrazná dyserytropoéza, bez známek kumulace železa nebo sideroblastů; příčinou je překvapivě mutace genu pro ALAS2 (Y365C), která se podílí na syntéze hemu, recesívní mutace ALAS2 byly doposud asociovány se sideroblastickou anémií s dědičností vázanou na X chromosom (tzv. XLSA)
- Trombocytopenie s, nebo bez dyserytropoetické anémie s recesivní dědičností vázanou na chromosom X (XLTDA), charakterizovaná anémií různé závažnosti od hydrops fetalis a závislosti na transfuzích k projevům dyserytropoézy bez anémie, makrotrombocytopenií s hypogranulovanými trombocyty a tendencí ke krvácení, až absenci demarkace trombocytární membrány – příčinou jsou mutace genu kódujícího transkripční faktor GATA1 lokalizovaného na chromosomu X. GATA1 transkripční faktor hraje důležitou roli ve vývoji a udržování buněk erytroidní a megakaryocytární linie, o rozdílném efektu různých typů mutací svědčí různé fenotypy onemocnění, které dosud byly popsány podobně jako např. v případě KLF1 transkripčního faktoru – různé typy popsaných mutací GATA1 se přitom vyznačují různým fenotypem onemocnění, což je způsobeno rozdílným efektem jednotlivých mutací na funkci GATA1 transkripčního faktoru
- Kongenitální dyserytropoetická anémie byla popsána i jako součást klinického obrazu Majeed syndromu, vzácného autosomálně recesivního onemocnění charakterizovaného časným rozvojem chronické rekurentní multifokální osteomyelitidy, dermatitidami a hypochromní mikrocytární anémií s dyserytropoézou, erytroidní hyperplazií s binukleárními a trinukleárními erytroblasty – onemocnění je způsobené mutacemi genu LPIN2, kódujícího lipin 2, fosfatázu důležitou pro metabolizmus lipidů
- Dyserytropoetické anémie spojené s exokrinní pankreatickou dysfunkcí a hyperostózami lebky, způsobené mutacemi genu COX4I2 kódujícího podjednotku cytochrom C oxidázy (COX) podílející se na dýchacím řetězci v mitochondriích
- Těžká dyserytropoetická anémie spojená s recesívně dědičným deficitem mevalonát kinázy (MVK) v důsledku mutace genu pro MVK
Diferenciální diagnóza
Dyserytropoéza je často přítomna u anémií při deficitu železa, folátu, vitaminu B6 a E v podmínkách s relativní hypoxií jako je neonatální období, vrozené srdeční vady, chronická onemocnění bronchů, kdy se může objevit sekundární dyserytropoéza. Dyserytropoetické změny jsou přítomny i v kostní dřeni pacientů s vrozenými syndromy selhání kostní dřeně, především u DBA a Fanconiho anémie. Jsou jedním z diagnostických znaků u myelodysplastického syndromu. Všechna tato onemocnění musí být před stanovením diagnózy vyloučena.
Léčba
Při těžké anémii je nutné pravidelné podávání transfuzí, intramuskulární nebo subkutánní podání interferonu (IFN), a to INFα2a nebo INFα2b podávaného 2–4krát týdně. To vede u většiny pacientů s CDA I. typu ke zvýšení hladiny Hb a snížení přetížení železem [21,22]. U několika pacientů s těžkou formou CDA byla provedena úspěšná alogenní transplantace kmenových buněk, která by však měla být zvažována pouze u dětí závislých na transfuzích, které nereagovaly na podávání interferonu [23,24].
U pacientů v pravidelném transfuzním programu je nutná prevence sekundárních komplikací, především důsledné monitorování přetížení železem (každé 3 měsíce) včetně ročního vyšetření myokardiálního T2* MRI a jaterního R2* MRI od 10 let věku. Nezbytná je důsledná medikamentózní léčba přetížení železem za použití dostupných chelátorů železa (desferioxamin, deferipron, deferasirox). Pravidelné flebotomie jsou vzhledem k anémii nevhodné.
Závěr
Vzhledem k obrovskému pokroku v odhalování genetické příčiny CDA byly analyzovány kauzální mutace vedoucí ke vzniku CDA a vysvětlena jejich patogenetická role při vzniku tohoto onemocnění. To s sebou přináší i možnosti genetického poradenství, a to jak vyhledávání patologických mutací u členů rodiny, tak možnost prenatální diagnostiky a částečně i některé možnosti léčby jako je použití interferonu α u CDA.
V České republice bylo diagnostikováno dosud okolo 15 pacientů s CDA.
Fanconiho anémie
Charakteristika
Fanconiho anémie (FA) je klinicky i geneticky výrazně heterogenní onemocnění charakterizované postupně progredujícím selháním kostní dřeně, přítomností různých kombinací somatických anomálií a zvýšeným výskytem maligních onemocnění: MDS, AML a solidních nádorů rezistentních na léčbu. Prvním příznakem bývá trombocytopenie nebo anémie.
Poprvé byla popsána v roce 1927 švýcarským pediatrem Guidem Fanconim [25], molekulární podstata onemocnění byla postupně odhalena v 90. letech minulého století.
Epidemiologie a dědičnost
Incidence onemocnění je 1–5 na 1 milion živě narozených, přičemž jsou popsány výrazné regionální rozdíly. Frekvence nosičů genu je v populaci USA odhadována na 1 : 181, vyšší je popisována např. v populaci aškenázských (středo - a východoevropských) Židů (1 : 93) [26] a u černošské populace v malarických oblastech subsaharské Afriky, v nichž je incidence FA udávána asi u 1 na 40 000 živě narozených, což je významně vyšší než se původně předpokládalo [27]. Kauzální delece FANCG je zde zřejmě „founder mutation“ u populace kmene Bantu.
Onemocnění je autosomálně recesivně dědičné kromě jednoho z genetických defektů (FANCB) u kterého je dědičnost gonosomální vázaná na chromosom X.
Klinické a laboratorní nálezy
Na onemocnění může již v útlém věku upozornit přítomnost pestré palety vrozených anomálií. Jsou popisovány u 80 % pacientů, patří k nim: malý vzrůst spojený často s intrauterinní růstovou retardací, hyperpigmentace kůže, malformace palce, skeletu, ledvin, srdce, poruchy sluchu, a hypogonadizmus (tab. 3).
Selhání KD se klinicky manifestuje obvykle kolem 7. roku věku trombocytopenií, anémií, postupně se rozvíjí pancytopenie. Asi ve 4 % případů se aplazie kostní dřeně rozvíjí již během kojeneckého věku. Vzácně se naopak může cytopenie objevit až v dospělosti, někdy až současně s rozvojem maligního onemocnění. U 50 % pacientů, kteří se prezentují trombocytopenií, dochází k rozvoji pancytopenie v průběhu následujících 4 let, kumulativní incidence rozvoje selhání kostní dřeně je 50–90 % ve věku 40 let.
Některé literární zdroje uvádějí, že u 25–40 % jedinců s FA jsou vrozené anomálie nenápadné, nebo dokonce nejsou přítomny vůbec. Totéž platí pro známky selhání kostní dřeně, které mohou být pouze diskrétní – mírná anémie nebo trombocytopenie, a mohou tak dlouho unikat pozornosti [28,29].
U pacientů s FA je 50krát vyšší riziko vzniku maligních onemocnění ve srovnání se zdravou populací: kumulativní incidence MDS, AML a solidních nádorů ve věku 40 let je 30,10 a 30 % [7]. Nejčastějšími solidními tumory jsou spinocelulární karcinomy hlavy, krku, jazyka, jícnu a vulvy.
Molekulární podstata onemocnění
Charakteristickou vlastností buněk pacientů s FA je chromosomální nestabilita, kterou je možno in vitro dále akcentovat přidáním látek navozujících klastogenní stres (mitomycin C, diepoxybutan). Působení těchto látek indukuje vznik chromosomálních zlomů a fúzí s charakteristickými radiálními formami. Popisuje se i zvýšená tendence k apoptóze a zvýšená náchylnost k toxickému působení volných kyslíkových radikálů. Pestrému a variabilnímu klinickému obrazu odpovídá i výrazná heterogenita vyvolávajících genetických defektů, z nichž většina byla postupně analyzována až v posledních letech. Jednotlivé geny kódují proteiny, které jsou složkami systému reparace DNA. Geny, jejichž mutace byly popsány u pacientů s FA, se nazývají FANC geny – tzv. FANC komplementační skupiny Jejich genové produkty regulují 3 důležité procesy v průběhu oprav meziřetězcových křížových vazeb DNA: nukleolytické incize, translézní opravu DNA a homologní rekombinaci. Buňky pacientů s FA mají v důsledku poruchy funkce uvedených genů nízkou toleranci vůči poškození DNA jak in vitro, tak in vivo.
Od prvního popisu molekulárně-genetického defektu u FA v roce 1992 bylo dodnes identifikováno přes 1 000 různých mutací 21 genů (FANCA, FANCB, FANCC, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCM, PALB2 (FANCN), RAD51C (FANCO), BRCA2, BRIP1, a SLX4 (FANCP), FANCQ, FANCV, FANCD1, FANCJ, FANCR, FANCS, FANCU. Největší skupinu mutací tvoří FANCA mutace, z ostatních jsou nejčastější FANCB a FANCC [30]. Asi 84 % pacientů je řazeno do subtypu A, C nebo G [5].
Z FA proteinů se jich 8 sdružuje v tzv. jaderný komplex a společně s FAN1 proteinem jsou nezbytné pro monoubikvitinaci FANCD2 proteinu. Porucha funkce každého z proteinů tohoto komplexu přerušují FANCD2 monoubikvitinaci, klíčový krok, který ovlivňuje translokaci jaderného komplexu k chromatinu a jeho asociaci s ostatními jadernými proteiny účastnící se buněčné odpovědi na poškození DNA (schéma).

FANCD1 gen je BCRA2 supresorový gen, poruchy jeho funkce vedou k narušení systému reparace DNA. FANCI gen kóduje helikázu, enzym který odvíjí DNA. FANCM gen ovlivňuje DNA translokaci, FANCL gen sdílí homologii s E3 ubikvitinovými ligázami.
FANCD2 gen je rovněž fosforylován ATM kinázou (ataxia-teleangiectasia protein) a ovlivňuje pak S-fázi buněčného cyklu se zástavou DNA syntézy při radiaci. Všechny tyto procesy hrají tedy důležitou roli v buněčné odpovědi na poškození DNA. Gen BRCA2 má souvislost s predispozicí ke vzniku karcinomu prsu a ovaria.
Diagnostika a léčba
U všech pacientů s anémií nebo trombocytopenií a typickými somatickými anomáliemi, u kterých vyslovíme podezření na FA, je indikováno provedení testů chromosomální fragility z lymfocytů periferní krve s použitím diepoxybutanu (DEB) nebo mitomycinu C (MMC). V případech chromosomálního mozaicizmu však tyto testy mohou prokázat pouze malé procento buněk s chromosomálními zlomy, průměrný počet zlomů potom může výsledně být normální. Proto u pacientů s podezřením na FA a negativním DEB testem se doporučuje doplnit vyšetření chromosomální nestability z fibroblastů periferní krve. Vzhledem ke znalosti kauzálních genů a k možnostem sekvenování nové generace je v dnešní době molekulárně-genetická diagnostika k jednoznačnému průkazu diagnózy lépe přístupná a snáze proveditelná.
Při plánování léčebné strategie je nezbytný komplexní multidisciplinární přístup, na kterém se kromě hematologů podílejí specialisté z oboru genetiky, onkologie, neurologie, kožního lékařství. Nedílnou součástí týmu by měl být i endokrinolog, protože u pacientů s FA se setkáváme s poruchami růstu, glukózového a lipidového metabolizmu, funkcí štítné žlázy, gonadálních funkcí a kostní denzity. Pacienti jsou hypersenzitivní k radiaci, kyslíkovým radikálům, a zejména k vlivu chemických mutagenů, proto musí být důsledně chráněni před jejich vlivem v běžném životě a zejména pak v případě nutnosti léčby maligních onemocnění.
Zcela nezbytné je pečlivé monitorování hematologických nálezů a periodické vyšetření kostní dřeně k vyloučení rozvoje MDS. K nepříznivému vývoji hematologického onemocnění může dojít velmi rychle. Proto je vhodné připravit rámcový plán transplantace kostní dřeně (HSCT). Vzhledem k radiosenzitivitě a chemosenzitivitě je nezbytné použít přípravné režimy s redukovanou intenzitou. Na funkci kostní dřeně, může mít u některých pacientů příznivý efekt léčba androgeny, na druhé straně však může mít nežádoucí účinky včetně zvýšeného rizika hepatoceluárního karcinomu.
K časnému odhalení karcinomů v oblasti hlavy, krku a genitálu je nezbytné periodické sledování ve specializovaných ambulancích, většinou 1krát za půl roku.
Lepším porozuměním genetické a molekulární podstatě onemocnění a zlepšením výsledků léčby se Fanconiho anémie v současné době mění na vzácné chronické onemocnění, které již zdaleka není doménou dětské hematologie, ale je diagnostikováno častěji i u dospělých a vyžaduje sofistikovaný multidisciplinární přístup. Je jedním z příkladů umožňujících hlubší porozumění interakcí genetické informace s prostředím v etiologii vrozených anomálií, hematopoézy a rozvoje maligního onemocnění.
Jsou vyvíjeny nové techniky prevence a léčby FA, např. HSCT od sourozence po výběru geneticky zdravého embrya, genová léčba, genomové editování s použitím genetické rekombinace nebo konstruovaných nukleáz. Tyto přístupy však jsou zatím v experimentální fázi.
prof. MUDr. Dagmar Pospíšilová, Ph.D.
Dětská klinika LF UP a FN Olomouc
Doručeno do redakce 12. 1. 2018
Přijato po recenzi 17. 3. 2018
Zdroje
- Pospíšilová D. Vrozené syndromy selhání kostní dřeně. Postgraduální medicína 2015; 17(6): 589–601.
- Vlachos A, Ball S, Dahl N et al. Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. Br J Haematol 2008; 142(6): 859–876. Dostupné z DOI: <http://dx.doi.org/10.1111/j.1365–2141.2008.07269.x>.
- Vlachos A, Rosenberg PS, Atsidaftos E et al. Incidence of neoplasia in Diamond Blackfan anemia: a report from the Diamond Blackfan Anemia Registry. Blood 2012; 119(16): 3815–3819. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2011–08–375972>.
- Da Costa L, O’Donohue MF, van Dooijeweert B et al. Molecular approaches to diagnose Diamond-Blackfan anemia: The EuroDBA experience. Eur J Med Genet 2017; pii: S1769–7212(17)30505–0. Dostupné z DOI: <http://dx.doi.org/10.1016/j.ejmg.2017.10.017>.
- Cmejlova J, Dolezalova L, Pospisilova D et al. Translational efficiency in patients with Diamond-Blackfan anemia. Haematologica 2006; 91(11): 1456–1464.
- Henter JI, Karlen J. Fatal agranulocytosis after deferiprone therapy in a child with Diamond-Blackfan anemia. Blood 2007; 109(12): 5157–5159. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2007–02–065805>.
- Pospisilova D, Cmejlova J, Hak J et al. Successful treatment of a Diamond-Blackfan anemia patient with amino acid leucine. Haematologica 2007; 92(5): e66-e67.
- Narla A, Payne EM, Abayasekara N et al. L-Leucine improves the anaemia in models of Diamond Blackfan anaemia and the 5q - syndrome in a TP53-independent way. Br J Haematol 2014; 167(4): 524–528. Dostupné z DOI: <http://dx.doi.org/10.1111/bjh.13069>.
- Ear J, Huang H, Wilson T et al. RAP-011 improves erythropoiesis in zebrafish model of Diamond-Blackfan anemia through antagonizing lefty1. Blood 2015; 126(7): 880–890. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2015–01–622522>.
- Faivre L, Meerpohl J, Da Costa L et al. High-risk pregnancies in Diamond-Blackfan anemia: a survey of 64 pregnancies from the French and German registries. Haematologica 2006; 91(4): 530–533.
- Gambale A, Iolascon A, Andolfo I et al. Diagnosis and management of congenital dyserythropoietic anemias. Expert Rev Hematol 2016; 9(3): 283–296. Dostupné z DOI: <http://dx.doi.org/10.1586/17474086.2016.1131608>.
- Russo R, Gambale A, Langella C et al. Retrospective cohort study of 205 cases with congenital dyserythropoietic anemia type II: definition of clinical and molecular spectrum and identification of new diagnostic scores. Am J Hematol 2014; 89(14): E169-E175. Dostupné z DOI: <http://dx.doi.org/10.1002/ajh.23800>.
- Heimpel H, Kellermann K, Neuschwander N et al. The morphological diagnosis of congenital dyserythropoietic anemia: results of a quantitative analysis of peripheral blood and bone marrow cells. Haematologica 2010; 95(6): 1034–1036. Dostupné z DOI: <http://dx.doi.org/10.3324/haematol.2009.014563>.
- Dgany O, Avidan N, Delaunay J et al. Congenital dyserythropoietic anemia type I is caused by mutations in codanin-1. Am J Hum Genet 2002; 71(6): 1467–1474. Dostupné z DOI: <http://dx.doi.org/10.1086/344781>.
- Babbs C, Roberts NA, Sanchez-Pulido L et al. Homozygous mutations in a predicted endonuclease are a novel cause of congenital dyserythropoietic anemia type I. Haematologica 2013; 98(9): 1383–1387. Dostupné z DOI: <http://dx.doi.org/10.3324/haematol.2013.089490>.
- Schwarz K, Iolascon A, Verissimo F et al. Mutations affecting the secretory COPII coat component SEC23B cause congenital dyserythropoietic anemia type II. Nat Genet 2009; 41(8): 936–940.Dostupné z DOI: <http://dx.doi.org/10.1038/ng.405>.
- Irvine AF, Vikberg AL et al. Congenital dyserythropoietic anemia type III (CDA III) is caused by a mutation in kinesin family member, KIF23. Blood 2013; 121(23): 4791–4799. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2012–10–461392>.
- Sandström H, Wahlin A, Eriksson M et al. Intravascular haemolysis and increased prevalence of myeloma and monoclonal gammapathy in congenital dyserythropoietic anaemia, type III. Eur J Haematol 1994; 52(1): 42–46.
- Arnaud L, Saison C, Helias V et al. A dominant mutation in the gene encoding the erythroid transcription factor KLF1 causes a congenital dyserythropoietic anemia. Am J Hum Genet 2010; 87(5): 721–727. Dostupné z DOI: <http://dx.doi.org/10.1016/j.ajhg.2010.10.010>.
- Jaffray JA, Mitchell WB, Gnanapragasam MN et al. Erythroid transcription factor EKLF/KLF1 mutation causing congenital dyserythropoietic anemia type IV in a patient of Taiwanese origin: review of all reported cases and development of a clinical diagnostic paradigm. Blood Cells Mol Dis 2013; 51(2): 71–75. Dostupné z DOI: <http://dx.doi.org/10.1016/j.bcmd.2013.02.006>.
- Parez N, Dommergues M, Zupan V et al. Severe congenital dyserythropoietic anaemia type I: prenatal management, transfusion support and alpha-interferon therapy. Br J Haematol 2000; 110(2): 420–423.
- Lavabre-Bertrand T, Ramos J, Delfour C et al. Long-term alpha interferon treatment is effective on anaemia and significantly reduces iron overload in congenital dyserythropoiesis type I. Eur J Haematol 2004; 73(5): 380–383. Dostupné z DOI: <http://dx.doi.org/10.1111/j.1600–0609.2004.00310.x>.
- Unal S, Russo R, Gumruk F et al. Successful hematopoietic stem cell transplantation in a patient with congenital dyserythropoietic anemia type II. Pediatr Transplant 2014; 18(4): E130-E133. Dostupné z DOI: <http://dx.doi.org/10.1111/petr.12254>.
- Ayas M, Al-Jefri A, Baothman A et al. Transfusion-dependent congenital dyserythropoietic anemia type I successfully treated with allogeneic stem cell transplantation. Bone Marrow Transplant 2002; 29(8): 681–682. Dostupné z DOI: <http://dx.doi.org/10.1038/sj.bmt.1703526>.
- Fanconi G. Familiäre, infantile, perniziosaartige Anämie (perniziöses Blutbiod and Konstitution). Jahrb Kinderheilk 1927; 117(2): 257–280.
- Verlander PC, Kaporis A, Liu Q et al. Carrier frequency of the IVS4 + 4 A-->T mutation of the Fanconi anemia gene FAC in the Ashkenazi Jewish population. Blood 1995; 86(11): 4034–4038.
- Neil VM, Fahmida E, Demuth I et al. A common Fanconi anemia mutation in black populations of sub-Saharan Africa. Blood 2005; 105(9): 3542–3544. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2004–10–3968>.
- Giampietro PF, Verlander PC, Davis JG et al. Diagnosis of Fanconi anemia in patients without congenital malformations: an international Fanconi Anemia Registry Study. Am J Med Genet 1997; 68(1): 58–61.
- Bogliolo M, Surrallés J. Fanconi anemia: a model disease for studies on human genetics and advanced therapeutics. Curr Opin Genet Dev 2015; 33 : 32–40. Dostupné z DOI: <http://dx.doi.org/10.1016/j.gde.2015.07.002>.
- Cheung RS, Taniguchi. T Recent insights into the molecular basis of Fanconi anemia: genes, modifiers, and drivers. Int J Hematol 2017; 106(3): 335–344. Dostupné z DOI: <http://dx.doi.org/10.1007/s12185–017–2283–4>.
- Alter BP. Inherited bone marrow failure syndromes. In: Nathan DG, Orkin SH, Ginsburg D, Look AT (eds). Nathan and Oski‘s Hematology of Infancy and Childhood. Vol 1, 6th ed. WB Saunders Philadelphia 2003 : 281–365. ISBN 978–1-4160–3430–8.
Štítky
Diabetologie Endokrinologie Interní lékařstvíČlánek vyšel v časopise
Vnitřní lékařství

2018 Číslo 5
- Inovace v hojení ran: krytí Zetuvit Plus Silicone Border pro optimální management exsudátu z ran
- Prognostický význam hladiny natriuretických peptidů při léčbě empagliflozinem
- Ivabradin zlepšuje kvalitu života starších pacientů se srdečním selháním
- Fixní kombinace kandesartan/amlodipin v terapii arteriální hypertenze
Nejčtenější v tomto čísle
- Autoimunitní hemolytická anémie
- Diferenciální diagnostika anémií
- Vrozené a získané krvácivé stavy
- Hemoglobinopatie
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy