
Autoimunitní hemolytická anémie
Autoimmune hemolytic anemia
Autoimmune hemolytic anemia (AIHA) is caused by auto-antibodies directed against self red blood cell (RBC) surface antigens. A consequence may be an intravascular hemolysis mediated by activated complement or extravascular hemolysis caused by destruction of complex of RBC with autoantibody in spleen and liver. The basic classification subdivides AIHA in primary/idiopathic and secondary with known underlying disease. A classification according to the thermal range of antibody recognizes warm AIHA, cold aglutinin disease (CAD), mixed AIHA and paroxysmal cold hemoglobinuria. Pathogenesis of AIHA consists of a defective antigen presentation to immunocompetent cells, insufficient process of T-lymphocyte tolerance to autoantigens and induction of autoantibody production by B-lymphocytes. For the diagnosis of AIHA are essential direct and indirect antiglobulin tests. The first-line therapy for warm AIHA is still administration of corticosteroids. For non-responding patients, second-line treatment includes rituximab or splenectomy. Combination of other immunosuppressive drugs represents a third-line treatment for resistant/relapsing patients. Rituximab is a treatment of choice for patients with CAD.
Key words:
anemia hemolytic – autoimmunity – corticosteroids – diagnosis – pathogenesis – rituximab – splenectomy – treatment
Autoři:
Jaroslav Čermák 1,2; Martin Písačka 1,2
Působiště autorů:
Ústav klinické a experimentální hematologie 1. LF UK, Praha
1; Ústav hematologie a krevní transfuze, Praha
2
Vyšlo v časopise:
Vnitř Lék 2018; 64(5): 514-519
Kategorie:
Přehledné referáty
Souhrn
Autoimunitní hemolytická anemie (AIHA) vzniká v důsledku tvorby autoprotilátek namířených proti některému z membránových antigenů erytrocytu, což vede k intravaskulární hemolýze aktivací komplementu, či k zániku erytrocytů s navázanou protilátkou v monocyto-makrofágovém systému sleziny. Základní klasifikace rozděluje autoimunitní hemolytické anémie na primární (idiopatickou) a sekundární, způsobenou přítomností známé vyvolávající příčiny. Podle charakteru přítomných protilátek dělíme onemocnění na AIHA s tepelnými protilátkami, AIHA s chladovými protilátkami, AIHA se smíšeným typem protilátek a paroxyzmální chladovou hemoglobinurii. V patogenezi AIHA se uplatňuje porucha prezentace antigenů imunokompetentním buňkám, porucha indukce tolerance T-lymfocytů vůči autoantigenům a indukce tvorby protilátek B-lymfocyty Základním diagnostickým testem u AIHA je přímý a nepřímý Coombsův antiglobulinový test. V léčbě AIHA s tepelnými protilátkami se v I. linii stále uplatňuje podávání kortikosteroidů. U nemocných neodpovídajících na I. linii léčby je v současnosti ve II. linii léčby doporučována splenektomie či podávání rituximabu, kombinovaná imunosuprese se uplatňuje jako další léčba linie při relapsech či malém efektu I. a II. linie léčby. U choroby z chladových aglutininů je lékem volby rituximab.
Klíčová slova:
anémie hemolytická – autoimunita – diagnostika – kortikosteroidy – léčba – patogeneze – rituximab – splenektomie
Úvod
Autoimunitní hemolytická anémie (AIHA) patří mezi tzv. extrakorpuskulární hemolytické anémie, u nichž jsou příčinou destrukce krvinek mechanizmy vznikající mimo červenou krvinku. Autoimunitní mechanizmy vedou k tvorbě autoprotilátek proti erytrocytům s různými vlastnostmi a různou cílovou specifitou. Vazba protilátek namířených proti některému z membránových antigenů erytrocytu na povrch červené krvinky vede k intravaskulární hemolýze aktivací komplementu, či k zániku erytrocytů s navázanou protilátkou v monocyto-makrofágovém systému sleziny. Incidence onemocnění je asi 0,6/100 000 obyvatel.
Základní klasifikace rozděluje autoimunitní hemolytické anémie na primární (idiopatickou) a sekundární, způsobenou přítomností známé vyvolávající příčiny (autoimunitní onemocnění, infekce, nádory, léky aj). Podle charakteru přítomných protilátek dělíme onemocnění na AIHA s tepelnými protilátkami, AIHA s chladovými protilátkami, AIHA se smíšeným typem protilátek, paroxyzmální chladovou hemoglobinurii a hemolytické anémie vyvolané léky.
Základní rozdělení autoimunitních hemolytických anémií
- AIHA s tepelnými protilátkami
- AIHA s chladovými protilátkami
- AIHA se smíšeným typem protilátek
- paroxyzmální chladová hemoglobinurie
- hemolytická anémie způsobená léky
Etiologie a patogeneze
I když v etiopatogenezi autoimunní hemolytické anémie zůstává stále řada nejasností, ukazuje se, že důležitým momentem je ztráta imunologické tolerance vůči antigenům exprimovaným na povrchu erytrocytů. Na tomto procesu se podílejí 3 základní činitelé: porucha prezentace autoantigenů imunokompetentním buňkám, porucha indukce tolerance T-lymfocytů vůči autoantigenům a indukce tvorby protilátek B-lymfocyty (obr. 1) [1].

Porucha prezentace autoantigenů imunokompetentním buňkám
V této fázi se předpokládá určitá defektní funkce tzv. antigen prezentujících buněk (makrofágy, dendritické buňky), porucha maturace těchto buněk může být spojena s poruchou funkce, jež se projevuje defektním zpracováním a abnormální prezentací antigenního podnětu [2]. Důsledkem je chybění apoptotického stimulu pro autoreaktivní T-lymfocyty, jenž autoantigeny za normálních okolností indukují, a naopak přetrvávání imunitního aktivačního podnětu. V poslední době se diskutuje i role polymorfizmu receptorů pro Fcγ řetězec IgG v indukci imunitní odpovědi [3].
Porucha indukce tolerance T-lymfocytů vůči autoantigenům
Na poruše tolerance T-lymfocytů vůči autoantigenům se kromě přetrvávání imunitního aktivačního podnětu vyvolaného autoantigeny podílí i určitá nerovnováha mezi prozánětlivými a protizánětlivými mechanizmy. Recentně byl u AIHA popsán zvýšený počet Th17-lymfocytů produkujících IL17, jenž se uplatňuje v zánětlivé stimulaci a aktivaci autoreaktivních T-lymfocytů. Naopak, byl zjištěn snížený počet CD4+ CD25hiFox3+ T-regulačních lymfocytů uplatňujících se v inhibici právě Th17 lymfocytů a v udržování autotolerance [4].
Indukce tvorby autoprotilátek B-lymfocyty
Aktivace autoreaktivních T-lymfocytů vede k indukci přeměny Th0 na Th1-lymfocyty a k sekreci cytokinů (IFNγ, IL2, TNFβ), jež indukují tvorbu protilátek B-lymfocyty. Oxidativní stres a zvýšená hladina CRP mohou ovlivňovat aktivitu hemolýzy cestou aktivace komplementu a Fcγ receptorů [5].
Charakteristika protilátek a principy hemolýzy
Charakteristika protilátek
V tab je uvedena charakteristika protilátek, jež jsou přítomny u AIHA s tepelnými protilátkami, AIHA s chladovými protilátkami a paroxyzmální chladové hemoglobinurie. Tepelné protilátky patří do skupiny IgG, mají optimum aktivity při 37 ºC a jsou schopny vázat receptor pro Fcγ či C3b na povrchu makrofágů, vázat komplement díky přítomnosti receptoru pro C1q složkou komplementu a při dostatečně vysokém titru vyvolat jeho aktivaci. Chladové protilátky patří do skupiny IgM, optimum jejich aktivity je při 4 ºC, jsou rovněž schopny aktivovat komplement a vázat se na povrchu makrofágů. U paroxyzmální chladové hemoglobinurie mají protilátky IgG charakter, avšak maximum jejich aktivity je při 4 ºC.

Principy hemolýzy
Schéma 1 znázorňuje základní principy protilátkami vyvolané hemolýzy.

Vazba protilátky na antigen na povrchu erytrocytu může vést přímo k vazbě Fcγ řetězce protilátky s receptorem pro Fcγ na povrchu makrofágu a k fagocytóze erytrocytu s navázanou protilátkou ve slezině.
Vazba antigen-protilátka vede k fixaci C1q složky komplementu a k aktivaci C1-C3b složek komplementu. Při vysokém titru protilátek a dostatečně pevné vazbě může pokračovat aktivace komplementu až k aktivaci jeho termálního komplexu a k intravaskulární hemolýze způsobené aktivovaným komplementem. Pokud je titr protilátek nízký a vazba není pevná, dojde k převaze inhibičních mechanizmů komplementu (CD55 a CD59 antigeny) a konverzi C3b složky komplementu na C3d. Erytrocyt s navázanou C3b složkou komplementu pak podléhá extravaskulární hemolýze díky vazbě s C3b receptorem makrofágů zejména v játrech.
IgG zprostředkovaná hemolýza
Základní mechanizmy hemolýzy zprostředkované IgG protilátkami jsou uvedeny na obr. 2. Nejčastějším mechanizmem u IgG zprostředkované hemolýzy je zánik erytrocytů s navázanou protilátkou ve slezině vazbou Fcγ IgG s receptorem na povrchu makrofágů sleziny. Při vysokém titru protilátek je možná aktivace komplementu s následnou intravaskulární hemolýzou. IgG protilátky jsou většinou polyklonální, přičemž je třeba si uvědomit, že protilátky podtřídy IgG1 a IgG3 jsou účinnější v indukci fagocytózy a vazbě komplementu.

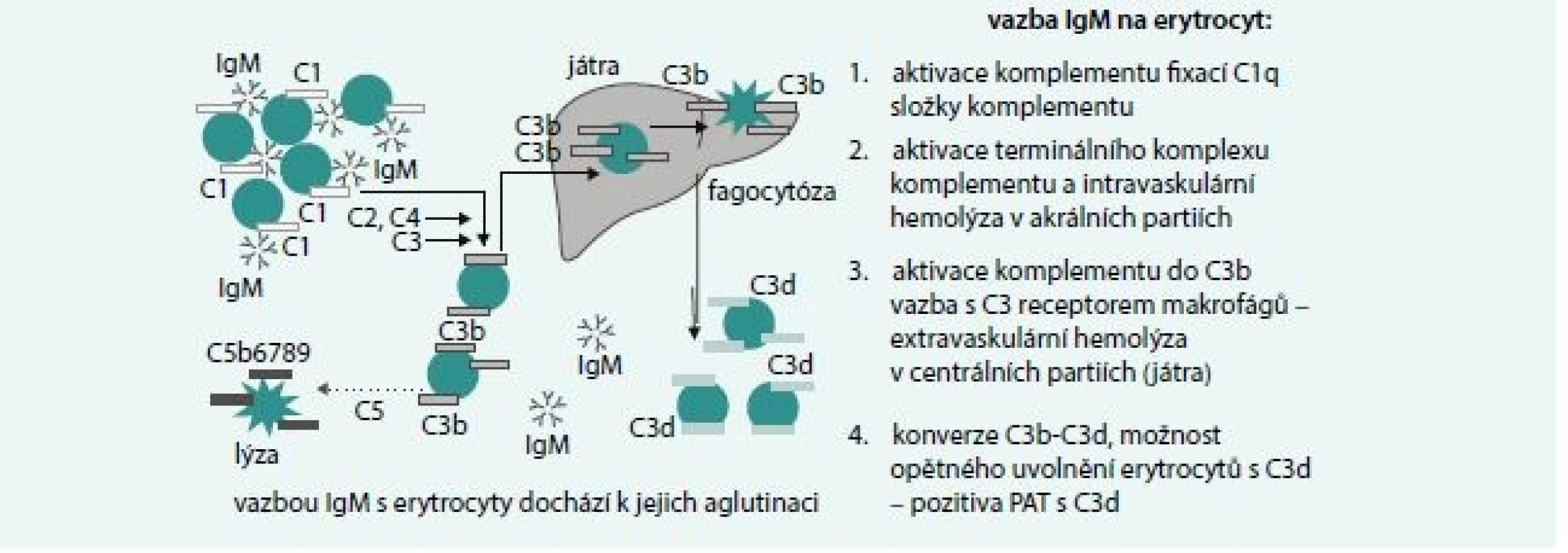
IgM zprostředkovaná hemolýza
Základní mechanizmy hemolýzy jsou uvedeny na obr. 3. IgM je pentamer, takže aktivace komplementu je snazší než u IgG a může dojít k aktivaci terminálního komplexu komplementu a intravaskulární hemolýze zejména v chladnějších akrálních partiích organizmu. Vazba je však často labilní a v centrálních partiích dochází k uvolnění navázaných protilátek, inhibici aktivace komplementu a vazbě C3b složky komplementu fixované na erytrocytech s receptorem na jaterních makrofázích a extravaskulární hemolýze.
Diagnostika a diferenciální diagnostika
Základními diagnostickými testy jsou přímý a nepřímý antiglobulinový (Coombsův) test. U AIHA s tepelnými protilátkami je pozitivní přímý Coombsův test (PAT) většinou jak při použití anti-IgG, tak při užití anti C3d, při nízkém titru protilátek však může být pozitivní pouze reakce s anti C3d či může být PAT negativní a je třeba při detekci protilátek užít citlivější metody (např. průtokovou cytometrii). U AIHA s chladovými protilátkami je pozitivní PAT s anti C3d. Při diagnostice AIHA postupujeme od testů s polyspecifikým sérem k monospecifickým sérům pro IgG , IgA, IgM, C3c, C3d a následně při pozitivitě PAT pro IgG určujeme titr anti-IgG a poměr anti-IgG1/anti Ig-G3 pomocí ID karet.
V diferenciální diagnostice je třeba odlišit korpuskulární hemolytické anémie, u nichž je příčina hemolýzy v defektním erytrocytu, k čemuž slouží speciální testy (autohemolýza, stanovení aktivity erytrocytárních enzymů, ELFO hemoglobinu (Hb), detekce mutací Hb pomocí molekulárně genetických metod). U paroxyzmální noční hemoglobinurie je intravaskulární hemolýza doprovázena určitým stupněm cytopenie v dalších řadách, PAT je negativní a je přítomen deficit inhibičních systémů komplementů (CD55 a CD59 antigenů) na povrchu erytrocytů, granulocytů a monocytů detekovatelný pomocí průtokové cytometrie. Pro neimunitní hemolytické anémie (mikroagiopatická hemolytická anémie, hemolyticko-uremický syndrom) je typická kromě intravaskulární hemolýzy přítomnost schistocytů v periferní krvi a kombinace s postižením dalších orgánů, zejména ledvin a CNS. U Gilbertovy choroby jsou negativní testy na hemolýzu a je možno prokázat mutaci UGT 1A1 genu. U některých forem myelodysplastického syndromu (MDS) s vysokým podílem inefektivní erytropoézy může být přítomna výrazná hemolytická složka, zejména pokud je přítomna přídatná získaná membránová porucha erytrocytů, ale i samotný MDS se může komplikovat hemolýzou na autoimunitním podkladě.
Léčba
Léčba AIHA s IgG protilátkami
Léčba první linie
Lékem první linie je stále podávání kortikosteroidů. Dávka a způsob podávání závisí na iniciální hodnotě hemoglobinu (Hb). U nemocných s lehčí formou hemolýzy s Hb > 80 g/l je podáván prednison v dávce 1,0–1,5 mg/kg/den po dobu 3–4 týdnů. Nemocní s těžkou formou hemolýzy by měli být hospitalizováni na specializovaném hematologickém oddělení či na jednotce intenzivní péče k podání i.v. kortikoidů (metylprednisolon – SoluMedrol® v dávce 250–1 000 mg denně po dobu 3–5 dnů) v monoterapii či v kombinaci s i.v. imunoglobuliny (0,5 g/kg/den 3–5 dnů) k vyvázání Fcγ receptorů makrofágů či s plazmaferézou při vysokém titru volných cirkulujících protilátek. Jako efektivní léčbu hodnotíme dosažení stabilní hodnoty Hb > 100 g/l bez potřeby transfuzí erytrocytů [6]. Následně je možno dávku kortikosteroidů redukovat o 10–15 mg týdně do dávky 20–30 mg/den a poté dávku snižovat jen pozvolna o 5 mg za 1–2 týdny do dávky 15 mg/den a o 2,5 mg po 2 týdnech při trvajícím efektu až do vysazení léčby při opakované negativitě PAT.
Odpověď na léčbu hodnocená trvalým vzestupem hodnoty Hb > 110 g/l, dosažením kompletní remise či stabilizace klinického a laboratorního nálezu je pozorována u 70–85 % nemocných. Nicméně, trvalého vyléčení dosáhne méně než 20 % nemocných [7]. U poloviny nemocných je třeba podávat kortikosteroidy dlouhodobě v udržovací dávce, u 20–30 % nemocných pak v kombinaci s jinými imunosupresivy. Celková doba podávání kortikosteroidů má být nejméně 3–4 měsíce (u nemocných léčených kratší dobu bývá vyšší incidence relapsů choroby), všichni nemocní by měli být vyšetřeni na možnou přítomnost vyvolávající choroby (zánět, nádor).
Léčba druhé linie
Léčba druhé linie je indikována u nemocných refrakterních na léčbu I. linie (nedostatečná odpověď do 3 týdnů po zahájení léčby s trvající hodnotou Hb < 100 g/l), u nemocných, u nichž je nutno podávat vysokou udržovací dávku kortikosteroidů (prednison v dávce > 15 mg/den po 6 měsících léčby) a při opakovaných relapsech choroby zejména v 1. roce léčby [6]. U nemocných mladších 65 let v dobrém klinickém stavu je možno ve 2. linii léčby indikovat splenektomii, zejména pokud je možno ji provést laparoskopicky. Odpověď na splenektomii je pozorována přibližně u 65 % nemocných, u 30 % trvá více než 3 roky a asi 20 % nemocných nepotřebuje žádnou další léčbu [7]. V současné době je splenektomie provázena poměrně nízkou mortalitou (2–3 %), příčinou mohou být infekce či trombotické komplikace. Nemocní vyžadující po splenektomii další léčbu mohou odpovědět na nízké dávky kortikosteroidů, nebo jsou indikováni k podání rituximabu.
Rituximab (Mabthera®) je indikován při selhání I. linie léčby zejména u nemocných, u nichž není indikována splenektomie. Je podávána dávka 375 mg/m2 ve 4 dávkách v týdenních intervalech, efekt u IgG zprostředkované hemolýzy je asi u 50–60 % nemocných [8]. Při selhání léčby rituximabem je možno provést splenektomii (pokud je indikovaná), podat druhý cyklus rituximabu či některý z léků III. linie. V poslední době je zkoušeno podání nízkých dávek rituximabu (100 mg/m2 4krát) v primoléčbě, efekt byl pozorován téměř u 90 % nemocných [9].
Léčba třetí a další linie
Cyklofosfamid je používán v dávce 100 mg/den, azatioprin (Imuran®) je podáván v dávce 2–3 mg/kg/den, cyklosporin A je podáván v iniciální dávce 3 mg/kg/den a dávka je upravována dle jeho plazmatické hladiny. Z dalších léků byl určitý efekt pozorován u danazolu, a mykofenolát mofetilu. U rezistentních forem je v tzv. záchranné léčbě užíván alemtuzumab (Mabcampath®) nebo cyklofosfamid ve vysokých dávkách (50 mg/kg/den 4krát).
Indikace k podání transfuzí erytrocytů závisí na věku, hloubce anémie a rychlosti jejího vzniku a na stavu kardiovaskulárního systému. Vždy je třeba nemocného monitorovat na lůžku a podávat transfuze pod clonou kortikoidů, k vyloučení skrytých aloprotilátek je třeba provést extenzivní genotypizaci.
Léčba AIHA s IgM protilátkami
Léčba je indikována u zhruba poloviny nemocných, kteří mají příznaky choroby (symptomy hemolýzy, pokles Hb pod 90–100 g/l). Steroidy mají většinou malý efekt, stejně tak není efektivní splenektomie vzhledem k tomu, že erytrocyty s navázaným komplexem C3-IgM jsou přednostně vychytávány jaterními makrofágy. Lékem I. linie je u IgM zprostředkované hemolýzy rituximab, efektivní u asi 85 % nemocných a navozující kompletní remisi u 55–60 % nemocných [10]. Při užití kombinace rituximabu s fludarabinem bylo dosaženo kompletní remise až u 74 % nemocných [11].

Paroxyzmální chladová hemoglobinurie
Paroxyzmální chladová hemoglobinurie je charakterizována intravaskulární hemolýzou s následnou hemoglobinurií po vystavení chladu. Příčinou je přítomnost tzv. bifázického Donathova-Landsteinerova IgG hemolyzinu, jenž se váže na erytrocyty za chladu s následnou aktivací C1q složky komplementu, jejímž důsledkem je intravaskulární lýza erytrocytů aktivovaným komplementem. Při teplotě > 10 ºC dochází k uvolnění vazby hemolyzinu s erytrocyty a menší část hemolýzy může být způsobena i fagocytózou erytrocytů s navázanou C3b složkou komplementu jaterními makrofágy. Onemocnění se dříve hojně vyskytovalo při lues, dnes je nejčastěji pozorováno u dětí při spalničkách nebo infekci parvovirem B19. U nemocných je pozitivní PAT s C3d a tzv. Donathův-Landsteinerův test, při němž po inkubaci erytrocytů se sérem nemocného za chladu dojde po přidání komplementu k hemolýze při 37 ºC.
Sekundární hemolytické anémie
Hemolytická anémie může být jedním z příznaků jiného probíhajícího onemocnění a často může jít dokonce o první příznak. Nejčastěji se jedná o 3 skupiny onemocnění:
- autoimunitní choroby (primární progresivní polyartritida – až 20 % nemocných, antifosfolipidový syndrom – 10 %, systémový lupus erytematodes – 7,5 %), většinou mají protilátky charakter IgG a může být přítomen Evansův syndrom
- zánětlivá onemocnění (Crohnova choroba a ulcerózní kolitida – 2 %), infekce (CMV, mykoplazma, parvovirus B19)
- nádory – nejčastěji lymfoproliferativní onemocnění (T angioimunoblastický lymfom – 15 %, splenický lymfom z marginálních lymfocytů – 10 %, chronická lymfatická leukemie – 5–10 %, lymfoplazmocytární lymfom – 3–5 %, maligní lymfogranulom – 1–2 %), mohou být přítomny jak IgG, tak IgM protilátky (tzv. syndrom chladových aglutininů bývá nejčastěji přítomen u angioimunoplastického lymfomu, lymfoplazmocytárního lymfomu a splenických lymfomů)
Výše uvedené nálezy znovu podtrhují důležitost celkového vyšetření všech nemocných s autoimunní hemolytickou anémií.
doc. MUDr. Jaroslav Čermák, CSc.
Ústav hematologie a krevní transfuze, Praha
Doručeno do redakce 3. 12. 2017
Přijato po recenzi 15. 3. 2018
Zdroje
- Marcus N, Attas D, Tamary H. Autoimmune hemolytic anemia: current understanding of pathophysiology. Hematology Education: educational program of the annual congress of EHA 2014; 8 : 321–328.
- Hawiger D, Inaba K, Dorsett Y et al. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J Exp Med 2001; 194(6): 769–779.
- Vidarsson G. Factors contributing to the pathogenesis of IgG-mediated autoimmune disease. Hematology Education: educational program of the annual congress of EHA 2015; 9 : 249–256.
- Noack M, Miossec P. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun Rev 2014; 13(6): 668–677. Dostupné z DOI: <http://dx.doi.org/10.1016/j.autrev.2013.12.004>.
- Kurien BT, Scofield RH. Autoimmunity and oxidatively modified autoantigens. Autoimmun Rev 2008; 7(7): 567–573. Dostupné z DOI: <http://dx.doi.org/10.1016/j.autrev.2008.04.019>.
- Zanella A, Barcellini W. Treatment of autoimmune hemolytic anemias. Haematologica 2014; 99(10): 1547–1554. Dostupné z DOI: <http://dx.doi.org/10.3324/haematol.2014.114561>.
- Lechner K, Jager U. How I treat autoimmune hemolytic anemias in adults. Blood 2010; 116(11): 1831–1838. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2010–03–259325>.
- Barcellini W, Zanella A. Rituximab therapy for autoimmune haematological diseases. Eur J Intern Med 2011; 22(3): 220–229. Dostupné z DOI: <http://dx.doi.org/10.1016/j.ejim.2010.12.016>.
- Barcellini W, Zaja F, Zaninoni A et al. Low-dose rituximab in adult patients with idiopathic autoimmune hemolytic anemia: clinical efficacy and biological studies. Blood 2012; 119(16): 3691–3697. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2011–06–363556>.
- Berentsen S, Ulvestad E, Gjertsen BT et al. Rituximab for primary chronic cold agglutinin disease: a prospective study of 37 courses of therapy in 27 patients. Blood 2004; 103(8): 2925–2928. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2003–10–3597>.
- Berentsen S, Randen U, Vagan AM et al. High response rate and durable remissions following fludarabine and rituximab combination therapy for chronic cold agglutinin disease. Blood 2010; 116(17): 3180–3184. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2010–06–288647>.
Štítky
Diabetologie Endokrinologie Interní lékařstvíČlánek vyšel v časopise
Vnitřní lékařství

2018 Číslo 5
- Inovace v hojení ran: krytí Zetuvit Plus Silicone Border pro optimální management exsudátu z ran
- Ivabradin zlepšuje kvalitu života starších pacientů se srdečním selháním
- Fixní kombinace kandesartan/amlodipin v terapii arteriální hypertenze
- Clopidogrel je v prevenci kardiovaskulárních příhod přínosnější než kyselina acetylsalicylová
Nejčtenější v tomto čísle
- Autoimunitní hemolytická anémie
- Diferenciální diagnostika anémií
- Vrozené a získané krvácivé stavy
- Hemoglobinopatie
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy