
Diferenciální diagnostika anémií
Differential diagnosis of anemia
Anemia, defined as hemoglobin level under lower normal limit, is a symptom of different pathologic conditions and the accurate differential diagnosis is necessary to determine the cause of anemia. The article uses the morphological classification of anemia to distinguish macrocytic, normocytic and microcytic types of anemias and divides anemias with increased amount of peripheral blood reticulocytes as a special group. It describes commonly known clinical units as iron deficiency anemia or anemia of chronic disease, as so as rare clinical units, which are always need to think about in a differential diagnosis of an anemic patient. There is an increasing incidence of rare blood disorders due to introduction of molecular genetics methods into diagnostics, prolonged overall survival of patients and increasing migration from areas with endemic occurrence of these diseases. Etiology, basic pathophysiological mechanisms, main clinical features together with important diagnostic examinations are described by each clinical unit. Due to the differential diagnostic focus of the article only basic knowledge about therapy is mentioned. The authors are members of the IHBT Center for Rare Disorders of Hematopoiesis, which is focused mainly on congenital and acquired disorders of red blood cell.
Key words:
anemia – differential diagnosis – enzymopathies – hemoglobinopathies – iron – macrocytosis – microcytosis – rare blood disorders – reticulocytosis
Autoři:
Jan Válka; Jaroslav Čermák
Působiště autorů:
Ústav hematologie a krevní transfuze, Praha
Vyšlo v časopise:
Vnitř Lék 2018; 64(5): 468-475
Kategorie:
Přehledné referáty
Souhrn
Anémie, definovaná jako pokles koncentrace hemoglobinu pod dolní hranici normálních hodnot, je projevem mnoha chorobných stavů a pro adekvátní terapii pacienta je vždy nutné diferenciálně diagnosticky upřesnit její příčinu. Článek využívá morfologického dělení anémií na makrocytární, normocytární a mikrocytární a jako zvláštní skupinu vyčleňuje anémie se zvýšením počtu retikulocytů v periferní krvi. Popsány jsou jednak klinické jednotky známé a obecně rozšířené jako anémie sideropenická, či anémie chronických chorob, ale také jednotky raritní, na které je však třeba v diagnostice anemického pacienta vždy pomýšlet. Incidence těchto vzácných onemocnění se navíc zvyšuje, a to jednak zavedením molekulárně-genetických metod do diagnostiky, jednak prodlužující se délkou přežití nemocných a rapidně se zvyšující migrací z oblastí s endemickým výskytem těchto onemocnění. U každé klinické jednotky je popsána etiologie, základní patofyziologické mechanizmy a klinické příznaky spolu s diagnostickými vyšetřeními. Terapie je, z důvodu diferenciálně diagnostického zaměření článku, zmíněna jen okrajově. Autoři jsou členy Centra pro vzácné choroby krvetvorby Ústavu hematologie a krevní transfuze (ÚHKT), které se zaměřuje zejména na vrozené a získané choroby červené krevní řady.
Klíčová slova:
anémie – diferenciální diagnostika – enzymopatie – hemoglobinopatie –makrocytóza – mikrocytóza – retikulocytóza – vzácná onemocnění krve – železo
Úvod
Anémie je definována jako pokles koncentrace hemoglobinu (Hb) pod dolní hranici normálních hodnot, tedy < 130 g/l u mužů a < 120 g/l u žen, nebo pokles hematokritu pod 40 % u mužů a pod 35 % u žen. Anémie je projevem mnoha chorobných stavů a vždy je nutno diferenciálně diagnosticky upřesnit její příčinu. Anémii lze klasifikovat podle různých hledisek. Z patofyziologického hlediska, které lépe objasňuje příčinu anémie, lze anémie rozdělit na
- anémie z poruchy tvorby erytrocytů
- anémie ze zvýšených ztrát erytrocytů
- posthemoragické anémie [1]
Pro základní diagnostickou rozvahu je praktičtější rozdělení anémií podle dvou základních parametrů krevního obrazu: středního objemu červené krvinky (mean cell volume – MCV) a počtu retikulocytů. Střední objem erytrocytu je přesným parametrem vypovídajícím o morfologii červené krvinky a jeho normální hodnota se pohybuje v rozmezí 82–98 fl. Počet retikulocytů je důležitým údajem vypovídajícím o produkci erytrocytů v kostní dřeni, obvykle se vyjadřuje jako procentuální zastoupení retikulocytů v erytrocytech periferní krve s normální hodnotou od 0,5–2,5 %. Anémie spojená s retikulocytózou svědčí o zachovalé schopnosti adekvátní produkce erytrocytů v kostní dřeni, nedostatečná retikulocytární odpověď při anémii je naopak obrazem poruchy produkce v kostní dřeni nebo neefektivní erytropoézy [2]. Při vyšetření pacienta je anémie obecně spojena s anemickým syndromem s bledostí kůže a sliznic, slabostí, únavností, nevýkonností, poruchami soustředění, závratěmi, palpitacemi, dušností námahovou i klidovou a hučením v uších.
Mikrocytární anémie s nezvýšeným počtem retikulocytů
Anémie z nedostatku železa neboli anémie sideropenická je celosvětově nejčastější anémií. Typickým znakem je malý objemem červené krvinky, tzv. mikrocytóza, spojená navíc s nedostatečnou hemoglobinizací erytrocytu – hypochromií [3]. Železo je nepostradatelným prvkem pro tvorbu a funkci erytrocytů, tvoří centrální část hemu, který je obsažen v každém ze 4 globinových řetězců molekuly hemoglobinu. Zásoby železa v organizmu jsou komplexně regulovány tak, aby bylo k dispozici pro tvorbu hemoglobinu a dalších proteinů a současně aby nedocházelo k přetížení organizmu železem s následným poškozením zejména jater a myokardu. Na deficitu železa v organizmu se často podílí více faktorů, které můžeme shrnout do následujících skupin.
- zvýšená potřeba železa, zejména v růstu, v těhotenství a při laktaci
- snížený příjem železa, který může představovat nedostatek železa v potravě (ohroženou skupinou jsou zejména vegetariáni a vegani) nebo nedostatečná resorpce železa z gastrointestinálního traktu/GIT, typicky při celiakii, achlorhydrii nebo po resekci žaludku
- zvýšené ztráty železa, typicky při chronických krevních ztrátách u maligních i nemaligních chorob GIT, při gynekologických a urologických malignitách, při nadměrném menstruačním krvácení a dalších méně častých příčinách krevních ztrát
- vzácné poruchy transportu železa spojené s deficitem železo-transportujícího proteinu transferinu, nebo přítomností protilátek proti tomuto proteinu
Při klinickém vyšetření můžeme kromě typických příznaků anemického syndromu nalézt také atrofii kůže a sliznic, zvýšenou lomivost nehtů, orální ragády, či atrofii sliznice jazyka. Základním diagnostickým ukazatelem nedostatku železa je snížená sérová koncentrace zásobního proteinu feritinu, která daleko přesněji odráží stav zásob železa v organizmu než samotná snížená hladina sérového železa. Na základě těchto 2 parametrů a hodnoty hemoglobinu můžeme rozlišit 3 stadia deficitu železa. Stadium prelatentní, charakterizované vyčerpáním zásob železa v organizmu, s normální hodnotou hemoglobinu a sérového železa a nízkou koncentrací feritinu, stadium latentní, v němž již dochází ke snížení hladiny železa v séru, ale stále se ještě neobjevuje anémie, a stadium manifestní anémie s poklesem hodnoty hemoglobinu [2]. K odlišení sideropenické anémie od dalších chorobných stavů je nutné podrobnější vyšetření metabolizmu železa v organizmu včetně snížené saturace transferinu, parametr daný procentuálním podílem transferinu, na který je navázáno železo k celkovému množství transferinu. Terapie sideropenické anémie spočívá jednak v léčbě onemocnění, které vedlo ke krevním ztrátám, a dále v substituci železa perorální cestou. Parenterální podávání železa je vyhrazeno pro pacienty netolerující perorální substituci nebo nemocné s malabsorpcí.
Diferenciálně diagnosticky je třeba nejčastěji odlišit anémii chronických chorob provázející chronická zánětlivá a nádorová onemocnění. Tato anémie může být až u čtvrtiny nemocných mikrocytární. Podkladem anémie v těchto případech není deficit železa, ale snížení jeho sérové hladiny způsobené poruchou metabolizmu železa. V důsledku zvýšené sekrece některých cytokinů (IL6, TNFα, INFγ) dochází ke sníženému vstřebávání železa ze střeva a k retenci zásobního železa v monocyto-makrofágovém systému [4]. Typickým biochemickým nálezem u anémie chronických chorob je snížená hladina železa v séru spolu s normální saturací transferinu a normální nebo zvýšenou koncentrací feritinu. Jako pomocný parametr muže sloužit vyšetření hladiny cirkulujících transferinových receptorů v séru, hladina je zvýšena u sideropenie a bývá v normě u anémie při chronickém onemocnění. Léčba spočívá v terapii základního onemocnění, u symptomatických nemocných v substituci erytropoetinu nebo v substituci železa při jeho současném deficitu.
Nápadná mikrocytóza a hypochromie jsou přítomny u nemocných s talasemií. B-talasemie je autosomálně recesivní onemocnění vznikající v důsledku různého stupně snížení tvorby β-globinového řetězce [5]. Z důvodu rozdílné závažnosti mutace, která může buď zcela potlačit tvorbu β-globinu (β0-talasemie), či jí do určité míry zachovat (β+-talasemie) a velmi variabilní expresivitě mutovaných genů vzniká široké spektrum klinických projevů od klinicky němých forem s nálezem hypochromních mikrocytů v krevním obraze až po těžkou anémii s hepatosplenomegalií, ikterem, deformitami skeletu a závislostí na podávání transfuzí erytrocytů s rozvojem přetížení železem těžkého stupně již v dětství. V naší populaci se vyskytují téměř výlučně nemocní s heterozygotní formou β+-talasemie, kteří mají klinický obraz thalasemia minima s pouhými laboratorními změnami v krevním obraze nebo formu thalasemia minor s mírnou anémií nevyžadující podávání transfuzí [6]. Mimo mikrocytózy a hypochromie je pro β-talasemii charakteristické zvýšení hladiny fetálního hemoglobinu HbF a hemoglobinu HbA2 při elektroforetickém vyšetření hemoglobinu. Počet retikulocytů nebývá zvýšen. Diagnózu potvrdí molekulárně genetické vyšetření se záchytem mutace. A-talasemie je rovněž autosomálně dědičné onemocnění charakterizované poruchou tvorby α-globinu v důsledku mutace postihující jeden či více α-globinových genů [7]. Anémie má obdobný charakter jako u β-talasemie (hypochromie s mikrocytózou a nezvýšeným počtem retikulocytů), na rozvoji anémie se podílí i výraznější hemolýza daná nestabilitou vznikajících tetramerů β řetězců (HbH). Mutace jednoho α-globinového genu vede většinou pouze k laboratornímu nálezu mírné hypochromie a mikrocytózy bez klinických příznaků. Postižení 2 α-globinových genů vede k obrazu thalassemia minor s lehce sníženými hodnotami hemoglobinu, hypochromií a makrocytózou. (tzv. α thalassemia trait). Diagnosticky se opět uplatňuje elektroforéza hemoglobinu s průkazem HbH a molekulárně genetické vyšetření s průkazem mutace α-globinového genu. U obou typů talasemií je terapie založena na substituci erytrocytů, u těžkých forem potom lze přistoupit ke splenektomii nebo indikovat nemocného k alogenní transplantaci krvetvorby v dětském věku.
Mikrocytóza muže být někdy přítomna i u sideroblastické anémie charakterizované ukládáním železa v mitochondriích sideroblastů s následnou tvorbou prstenčitých sideroblastů v kostní dřeni. Příčinou tohoto typu anémie mohou být vrozené poruchy syntézy hemu nebo metabolizmu železa v mitochondriích, dále také alkohol nebo některé léky. Nález prstenčitých sideroblastů je charakteristický také pro některá nádorová onemocnění krvetvorby, např. v rámci myelodysplastického syndromu (refrakterní anémie se zmnožením prstenčitých sideroblastů).
Makrocytární anémie s nezvýšeným počtem retikulocytů
Tuto skupinu je možné rozdělit na anémie s přítomností megaloblastů a makrocytární anémie nemegaloblastové [8]. Nejčastější příčinou megaloblastových anémií je nedostatek vitaminu B12 či kyseliny listové, jenž ve svém důsledku vede k poruše tvorby tymidinových bází a syntézy DNA. Výskyt těchto anémií se zvyšuje po 60. roce života. Vitamin B12 z potravy se v žaludku váže na intrinsic (vnitřní) faktor produkovaný parietálními buňkami žaludku, komplex vnitřního faktoru s navázaným vitaminem B12 je pak resorbován v terminálním ileu, v plazmě je pak vitamin transportován ve vazbě na transkobalamin. Deficit vitaminu B12 je spojen s řadou stavů porušujících tuto fyziologickou dráhu.
Perniciózní anémie je termín vyhrazený pro anémii z deficitu B12 způsobenou autoimunitní destrukcí parietálních buněk žaludku s následnou atrofií žaludeční sliznice s achlorhydrií, můžeme nalézt také protilátky proti vnitřnímu faktoru [2,9]. Dalšími příčinami deficitu vitaminu B12 jsou stavy po resekci žaludku či terminálního ilea, malabsorpční syndromy, alkoholizmus, vzácněji např. gastrinom, střevní parazitární infekce nebo deficit transkobalaminu.
Kyselina listová (folát) je absorbována v proximálním jejunu a v plazmě přenášena ve vazbě na albumin. Etiologie deficitu folátu souvisí zejména s malnutricí (alkoholizmus, anorexie) nebo malabsorpcí (celiakie, střevní zánětlivá onemocnění). Potřeba folátu je zvýšena v období těhotenství a růstu, ale také při malignitách a chronické hemolýze. Klinickými projevy megaloblastových anémií jsou mimo anemického syndromu také neurologické projevy, syndrom zadních provazců s poruchou chůze, čití a taxe u deficitu vitaminu B12 a dráždivost, porucha paměti a neuropatie u deficitu kyseliny listové. Může být také porušena trofika kůže a sliznic (Hunterova glositida jazyka, gastritida). Perniciózní anémie je spojena s rizikem vzniku adenokarcinomu žaludku. V diagnostice se uplatňuje nález snížené sérové hladiny vitaminu B12 nebo kyseliny listové. V nátěru z periferní krve bývá současně s anémií s přítomností megalocytu, resp. megaloblastu mírná leukopenie s hypersegmentací neutrofilů a trombocytopenie. V důsledku inefektivní erytropoézy s destrukcí prekurzorů erytrocytů v kostní dřeni (obr. 1) je zvýšena hladina volného i přímého bilirubinu, zásoby železa jsou v normě. Diagnosticky cenným nálezem je průkaz protilátek proti vnitřnímu faktoru a přítomnost atrofické gastritidy s achlorhydrií u perniciózní anémie. Terapie spočívá v substituci vitaminu B12 nebo kyseliny listové, přitom je nutno sledovat hladinu železa pro riziko jeho vyčerpání při obnově krvetvorby. Pacienti s perniciózní anémií by měli být dispenzarizování a pravidelně gastroskopicky vyšetřováni.

V diferenciální diagnóze je nutné vyloučit zejména anémii v rámci myelodysplastického syndromu (MDS), při němž je zvýšený objem erytrocytu výrazem poruchy erytropoézy v důsledku mutace kmenové krvetvorné buňky, jež dává vznik patologickému klonu [10]. Pro odlišení MDS je třeba provést sternální punkci, při nedostatku vitaminu B12 či kyseliny listové převažuje hyperplastická erytropoéza s přítomností megaloblastů, nálezem makrocytózy v bílé řadě a s polyploidií megakaryocytu, u MDS bývají výraznější morfologické dysplastické změny krvetvorby, u pokročilých forem MDS dochází současně k nárůstu počtu myeloblastů. K odlišení MDS muže přispět i nález aberací v karyotypu při cytogenetickém vyšetření, nález některých abnormálních enzymatických reakcí při cytochemickém vyšetření a přítomnost nadbytku nezralých CD34+ prekurzorů u pokročilých forem choroby při vyšetření aspirátu pomocí průtokové cytometrie. Diagnózu MDS je třeba ověřit trepanobioptickým vyšetřením kostní dřeně. Hladina vitaminu B12 muže být snížena u forem s výrazným stupněm inefektivní erytropoézy, ale odpověď na podání vitaminu B12 je u nemocných s MDS jen částečná a přechodná na rozdíl od nemocných s perniciózní anémií [8].
Makrocytární anémie bez přítomnosti megaloblastů bývá rovněž nalézána u nemocných s jaterní chorobou a alkoholiků, makrocytóza je zde způsobena poruchou struktury erytrocytární membrány při změnách v metabolizmu lipidů. U nemocných s hypotyreózou vzniká makrocytární anémie v důsledku poruchy metabolizmu cholesterolu, často se však kombinuje s perniciózní anémií při současné přítomnosti protilátek proti vnitřnímu faktoru.
Diamondova-Blackfanova anémie (DBA) s aplazií červené krevní řady patří mezi vrozené syndromy selhání kostní dřeně. Onemocnění je řazeno mezi tzv. ribosomopatie, neboť základním patogenetických momentem je mutace některého z genů pro ribosomální proteiny, v jejímž důsledku dochází k poškození tvorby ribosomů. Onemocnění je dominantně dědičné a má poměrně velkou fenotypovou rozmanitost od forem zcela klinicky němých, po těžké anémie spojené s vrozenými orgánovými malformacemi. Základním klinickým projevem je makrocytární anémie, asi u poloviny nemocných mohou být přítomny malformace skeletu (palce, horní končetiny), a orgánů (srdce, urogenitální systém), diagnostika se opírá o nález v krevním obraze (anémie, makrocytóza, retikulocytopenie) a chybění erytropoézy v kostní dřeni [11]. Je zvýšena hladina HbF a ADA (adenozindeaminázy), mutace některého z genů pro ribosomální proteiny bývá přítomna u 60–65 % nemocných. Krom substituce transfuzemi erytrocytů se v léčbě DBA uplatňuje podávání kortikosteroidů, jež mohou zmírnit závislost na podávání transfuzí. U nemocných s nutností transfuzní léčby je nutná monitorace zásob železa a chelatační terapie k zabránění orgánového poškození. Alogenní transplantace krvetvorných buněk je možnou léčebnou metodou, je však zatížena poměrně velkým rizikem komplikací. Na DBA je třeba myslet u každé nově vzniklé makrocytární anémie v období dospívání, kdy může dojít k pozdní manifestaci lehčích forem řady vrozených poruch krvetvorby [12].
Normocytární anémie s nezvýšeným počtem retikulocytů
Normocytární anémie bez retikulocytózy tvoří heterogenní skupinu anémií, jejichž příčina muže být v kostní dřeni či mimo ni. Můžeme sem řadit některé anémie chronických chorob, anémie při jaterních chorobách, endokrinopatiích, anémie vznikající z důvodu nedostatku erytropoetinu při renální insuficienci a také anémii při otravě olovem.
Z definovaných hematologických chorob se normocytární anémií bez retikulocytózy manifestuje aplastická anémie definovaná jako selhání krvetvorby s aplazií nebo těžkou hypocelularitou kostní dřeně a pancytopenií v periferním krevním obraze. U těžké aplastické anémie klesá počet retikulocytů < 0,1 %, počet neutrofilů < 0,5 × 109/l a počet trombocytů < 20 × 109/l, u velmi těžké aplastické anémie klesá počet neutrofilních granulocytů až < 0,2 × 109/l [13]. Aplastické anémie dělíme na vrozenou a sekundární. Sekundární aplastickou anémii můžeme vidět např. po expozici ionizujícímu záření, po podání určitých léků (sulfonamidy, chloramfenikol, tyreostatika, antidiabetika, antirevmatika), po infekci (hepatitidy, EBV, HIV), po expozici chemikáliím (benzen, insekticidy) nebo na autoimunitním podkladě u pacientů s tymomem či protilátkami proti hematopoetickým kmenovým buňkám. Pro diagnózu je rozhodující sternální punkce a trepanobiopsie kostní dřeně. Kostní dřeň bývá tuková, její buněčnost nepřesahuje 25 % normy. Diferenciálně diagnosticky je nutno odlišit zejména hypoplastickou formu MDS, u aplastické anémie nejsou přítomny morfologické známky dysplazie ani změny při cytochemickém vyšetření a vyšetření karyotypu, ani nejsou zmnoženy blasty [14]. Terapií volby je u mladších pacientů alogenní transplantace od shodného příbuzného dárce, u ostatních pacientů pak transplantace ve druhé linii po selhání imunosupresivní terapie.
Mezi vrozené aplastické anémie patří řada vzácných, nicméně závažných chorob. Fanconiho anémie je charakterizována hypoplazií kostní dřeně s pancytopenií v krevním obraze. Příčinou je mutace některého ze skupiny genů podílejících se na reparaci poškozené DNA v buňce tzv. homologní rekombinací [15]. Onemocnění je autosomálně recesivní. Anémie může být spojena s malformacemi skeletu (mikrocefalie, defekty paprsku radia), poškozením ledvin a kožními skvrnami. Mimo makrocytózu je při vyšetření přítomna zvýšená hodnota HbF a zvýšená fragilita chromosomů. K potvrzení diagnózy slouží molekulárně genetické vyšetření. Onemocnění je spojeno s rizikem přechodu do hematologické malignity (MDS nebo akutní leukemie) nebo vznikem solidních nádorů. Terapie spočívá v podpůrné hematologické léčbě a onkologické dispenzarizaci, u těžších forem onemocnění je indikována alogenní transplantace krvetvorby. Shwachmanův-Diamondův syndrom se projevuje těžkou granulocytopenií a různě závažným stupněm anémie a trombocytopenie. Onemocnění může být doprovázeno insuficienci exokrinní části pankreatu, kostními malformacemi a anomáliemi jater. Příčinou je mutace SBDS genu. Dyskeratosis congenita je onemocnění spojené se selháním kostní dřeně a zvýšeným rizikem vzniku malignit. Při fyzikálním vyšetření můžeme vidět retikulární pigmentaci kůže, leukoplakii nebo dystrofii nehtů.
Normocytární anémie spojená s různým stupněm leukopenie a trombocytopenie bývá přítomna u akutní leukemie a také při infiltraci kostní dřeně u chronických lymfoproliferativních chorob spojených s fibrózou kostní dřeně, zejména u tzv. leukemie z vlasatých lymfocytů, nebo u metastatického postižení kostní dřeně buňkami solidních tumorů. Pro diagnózu je rozhodující nález ve sternálním punktátu a v bioptickém vzorku kostní dřeně.
Anémie se zvýšeným počtem retikulocytů
Na rozdíl od předešlých skupin je u těchto anémií přítomna vystupňovaná tvorba erytrocytů, která kompenzuje jejich zvýšený zánik, ať již v důsledku inefektivní erytropoézy, či periferní hemolýzy. Diferenciálně diagnosticky rozlišujeme hemolytické anémie vrozené (korpuskulární) s poruchou erytrocytu a získané (extrakorpuskulární), jejichž příčina leží mimo červenou krvinku. Korpuskulární hemolytické anémie jsou v našich podmínkách poměrně vzácné, můžeme je dále dělit na
- anémie vznikající v důsledku poruchy erytrocytární membrány
- anémie vznikající důsledku porušeného enzymatického vybavení erytrocytu
- hemoglobinopatie s poruchou tvorby molekuly hemoglobinu [8]
Nejčastěji se vyskytující vrozenou hemolytickou anémií je v našich podmínkách dědičná (hereditární) sférocytóza (HS). Je charakterizována defektem různých genů, které se podílejí na tvorbě skeletu erytrocytární membrány a vedou k deficitu ankyrinu, spektrinu či dalších proteinů membrány červené krvinky [16]. Důsledkem je zvýšená propustnost membrány pro ionty Na+, s nutností zvýšené aktivity Na+/K+ pumpy, to vede k postupnému energetickému vyčerpání krvinky, změně jejího tvaru a ke snížené deformovatelnosti (obr. 2). Důsledkem je porucha průchodu erytrocytu slezinnými kapilárami a destrukce erytrocytu makrofágy sleziny – extravaskulární hemolýza. Onemocnění je většinou autosomálně dominantní a značná část pacientů je diagnostikována již v dětském věku. Klinickému nálezu dominuje splenomegalie. V diagnostice se uplatní průkaz snížené osmotické rezistence erytrocytů, elektroforéza proteinů erytrocytární membrány s deficitem spektrinu a ankyrinu a průtoková cytometrie. Není prokázána přítomnost antierytrocytárních protilátek. U těžších forem onemocnění je indikována splenektomie.
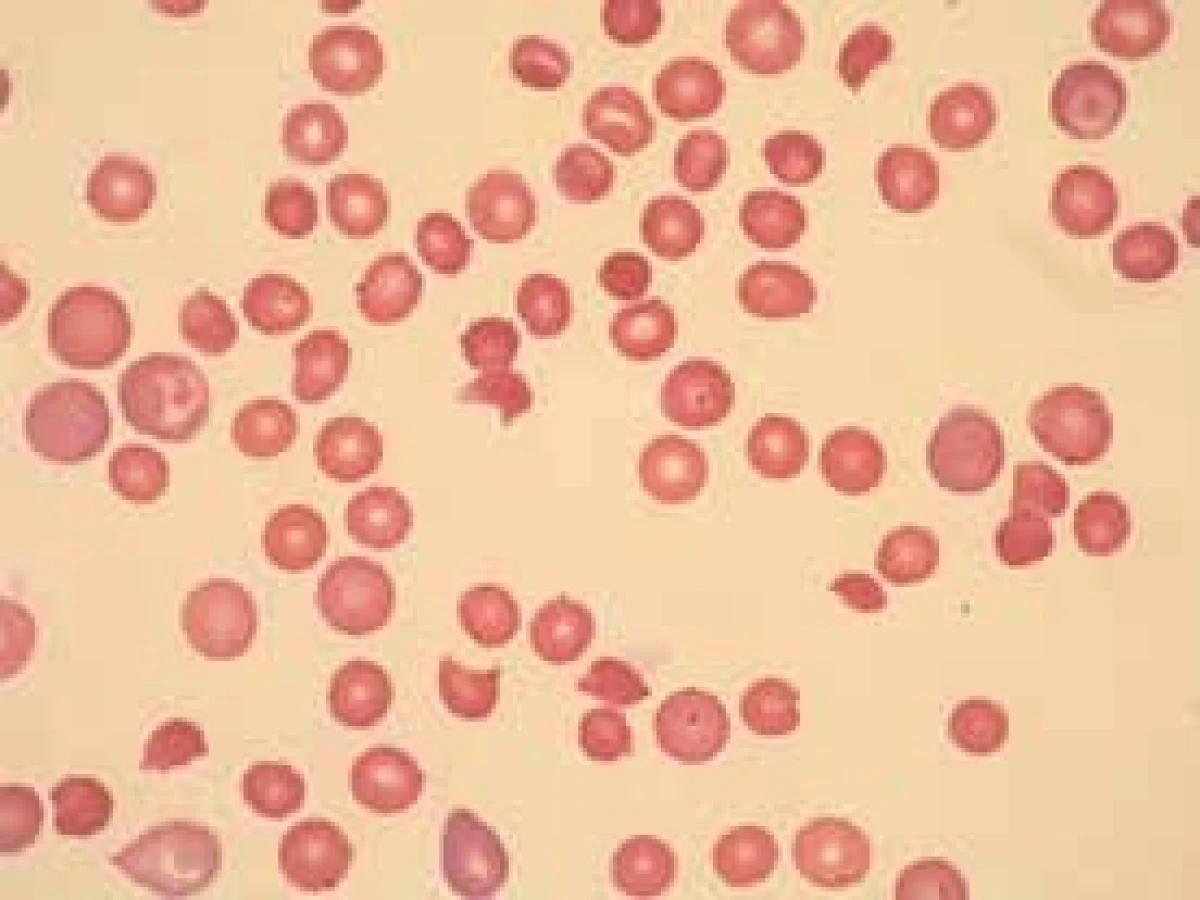
Do skupiny nesférocytárních hemolytických anémií jsou řazeni nemocní, u nichž je přítomna korpuskulární hemolýza, ale laboratorní nálezy nesvědčí ani pro HS, ani pro některou z erytrocytárních enzymopatií. Hereditární xerocytóza je způsobena abnormální propustnosti membrány erytrocytu s neschopností zadržet K+ a následně H2O v buňce. Hereditární stomatocytóza vzniká v důsledku retence kationtů a vody uvnitř erytrocytu. Nemocní mají různý stupeň anémie s nálezem stomatocytů v periferní krvi.
Anémie vznikající v důsledku poruchy enzymatických systémů červené krvinky jsou u nás poměrně vzácné, nejčastěji se lze setkat s deficitem pyruvátkinázy a glukóza-6-fosfátdehydrogenázy, k odlišení od jiných typů korpuskulárních anémií je většinou nutné stanovit hladinu příslušného enzymu. Deficit pyruvátkinázy je spojen s výrazným energetickým deficitem buňky z důvodu poruchy glykolytické dráhy v cytoplazmě, jenž se na rozdíl od HS neupravuje in vitro po přidání glukózy [17]. Klinicky se choroba projevuje různě těžkým stupněm hemolytické anémie, jež může být spojena se splenomegalií a komplikována cholelitiázou, nebo rozvojem přetížení železem při vysokém obratu erytropoézy a v důsledku opakovaných transfuzí erytrocytů. Deficit glukóza-6-fosfátdehydrogenázy se projevuje poruchou pentózofosfátového cyklu, jehož produkty zabraňují oxidaci hemoglobinu a umožňují jeho udržení v redukovaném stavu [18]. Onemocnění vede k variabilnímu stupni hemolýzy po požití oxidačních činidel (potraviny, léky). Může se projevovat až těžkou záchvatovitou hemolýzou (favizmus).
Hemoglobinopatie jsou reprezentovány zejména srpkovitou anémií. Bodová mutace β-globinového genu vede ke vzniku hemoglobinu HbS se sníženou rozpustností v redukovaném stavu, tím je snížena deformovatelnost krvinky. Příznaky se vyskytují zejména u homozygotů, z nichž většina pochází ze subsaharské nebo rovníkové Afriky. Hemolýza může být jak extravaskulární ve slezině, tak intravaskulární při tzv. hemolytických krizích [19]. Hemolytické krize mohou být vyprovokovány infekcí, dehydratací, prochladnutím. Tzv. srpkovitá krize vzniká obstrukcí kapilár rigidními srpky s obrazem akutní respirační insuficience, bolestí břicha nebo ischemickým postižením CNS. Diagnostiku srpkovité anémie lze provést z kapilární elektroforézy hemoglobinu, u homozygotů nejdeme > 50 % HbS, heterozygoti mají < 50 % HbS. Terapie spočívá v substituci erytrocytů s cílem snížení procenta HbS. Při krizích se uplatní zejména erytrocytaferéza, transfuze erytrocytů a symptomatická terapie bolesti analgetiky.
Extrakorpuskulární hemolytické anémie se dělí podle příčiny hemolýzy na autoimunitní a neimunitní. Autoimunitní hemolytická anémie je způsobena tvorbou protilátek proti povrchovým antigenům erytrocytu. Tyto protilátky dělíme na tepelné s vazbou na erytrocyty při 37 °C, většinou třídy IgG, a chladové s aktivací při nízké teplotě patřící většinou do třídy IgM. Tepelné protilátky způsobují převážně extravaskulární hemolýzu, protilátky chladové mohou díky aktivaci komplementového systému způsobovat i hemolýzu intravaskulární. Spouštěčem tvorby protilátek může být prodělaná infekce (např. mykoplazmata, mononukleóza, hepatitida B a C, human immunodeficiency virus – HIV), lymfoproliferativní onemocnění krve (chronická lymfocytární leukemie – CLL), malignity, autoimunitní choroby (revmatoidní artritida, lupus, tyreoididita) a některé léky (antibiotika, metyldopa) [20]. Spouštěcí mechanizmus nemusí být ovšem vždy identifikován. Přítomnost protilátek prokazujeme přímým a nepřímým antiglobulinovým testem (Coombsův test), je přítomna retikulocytóza v periferní krvi. V séru prokazujeme zvýšenou hladinu nekonjugovaného bilirubinu a laktátdehydrogenázy (LDH). Při těžké hemolýze muže být přítomna zvýšená hladina volného hemoglobinu v plazmě spolu s poklesem haptoglobinu a hemoglobinurií.
Z neimunitních extrakorpuskulárních hemolytických anémií je nejzávažnější mikroangiopatická hemolytická anémie vznikající v důsledku mechanického poškození erytrocytu fibrinem ukládajícím se v drobných cévách. Nejčastější příčinou je trombotická trombocytopenická purpura (TTP) a diseminovaná intravaskulární koagulace (DIC). Diagnosticky důležitým nálezem je přítomnost schistocytů, fragmentovaných krvinek v periferní krvi, často je současně přítomna trombocytopenie. Vzácně muže dojít k přímému poškození erytrocytu mechanicky (umělé chlopně), toxicky (acetylfenylhydrazin, toxin Clostridium perfringens) nebo parazitem (Plasmodium falciparum).
Paroxyzmální noční hemoglobinurie (PNH) je vzácné získané onemocnění kmenové krvetvorné buňky vzniklé na podkladě mutace PIG-A genu, což vede k chybění proteinů CD55 a CD59 na povrchu buněk [21]. V důsledku ztráty těchto inhibitorů komplementu dochází k aktivaci komplementu a komplementem zprostředkované intravaskulární hemolýze erytrocytů. Mimo klasické příznaky anemického syndromu a příznaků hemolýzy se PNH manifestuje také arteriálními nebo venózními trombózami, často v neobvyklých lokalizacích (GIT, CNS), progredující renální insuficiencí a plicní hypertenzí. Dalším typickým projevem PNH jsou známky selhávání kostní dřeně s cytopeniemi v periferní krvi. Často nacházíme určité procento PNH buněk také u nemocných s MDS a aplastickou anémií a tyto stavy mohou volně přecházet mezi sebou. Diagnostika PNH spočívá v průkazu CD55 a CD59 deficitních erytrocytů, granulocytů a monocytů v periferní krvi metodou průtokové cytometrie, molekulárně geneticky může být prokázána mutace v PIG-A genu. V diferenciální diagnostice je nutno pomýšlet na přítomnost MDS, v anamnéze pacientů se často setkáváme s údajem o terapii aplastické anémie. Určitý stupeň aplazie krvetvorby může přetrvávat. Mimo podpůrné terapie spočívající v substituci erytrocytů a udržování zásob železa (substituce nebo chelatace) se u pacientů s převažující hemolýzou uplatňuje léčba inhibitorem C5 složky komplementu ekulizumabem, u nemocných s progredující aplazií dřeně nebo známkami dysplazie je nutno uvažovat o provedení transplantace krvetvorby.
Závěr
Z předchozího textu vidíme, že anémie může být spojena s mnoha chorobnými stavy a může doprovázet nejen postižení erytrocytu, kostní dřeně, ale i celé řady jiných orgánů. Anémie může být projevem řady maligních onemocnění a naopak některé vrozené syndromy, manifestující se zpočátku anémií, mohou být spojeny s vyšším rizikem vzniku malignity. Těžká anémie, zejména rychle vzniklá, vede často k dekompenzaci stavu pacientů s kardiovaskulárními chorobami. Stanovení příčiny anémie je důležité jednak pro správnou a cílenou terapii nemocného a současně ke snížení možných rizik a komplikací, které mohou být s daným typem anémie spojeny. Diferenciální diagnostiku anémií shrnuje tab. a schéma.


MUDr. Jan Válka
Ústav hematologie a krevní transfuze, Praha
Doručeno do redakce 18. 12. 2017
Přijato po recenzi 15. 3. 2018
Zdroje
- Buliková A. Anémie. In: Penka M, Buliková A et al. Neonkologická hematologie. 2. ed. Grada: Praha 2009 : 39–95. ISBN 978–80–247–2299–3.
- Vydra J, Čermák J. Anémie. In: Vydra J, Cetkovský P (eds) et al. Hematologie v kostce. Mladá fronta: Praha 2015 : 11–68. ISBN 978–80–204–3698–6.
- Camaschella C. Iron-deficiency anemia. N Engl J Med 2015; 372(19): 1832–1843. Dostupné z DOI: <http://dx.doi.org/10.1056/NEJMra1401038>.
- Nemeth E, Ganz T. Anemia of Inflammation. Hematol Oncol Clin N Am 2014; 28(4): 671–681, vi. Dostupné z DOI: <http://dx.doi.org/10.1016/j.hoc.2014.04.005>.
- Rund D, Rachmilewitz E. β thalassemia. N Engl J Med 2005; 353(11): 1135–1146. Dostupné z DOI: <http://dx.doi.org/10.1056/NEJMra050436>.
- Divoka M, Partschova M, Kucerova J et al. Molecular characteristics of β-thalassemia in the Czech and Slovak populations: Mediterranean, Asian and unique mutations. Hemoglobin 2016; 40(3): 156–162. Dostupné z DOI: <http://dx.doi.org/10.3109/03630269.2016.1152581>.
- Harteveld CR, Higgs CR. α-thalassemia. Orphanet J Rare Dis 2010; 5 : 13. Dostupné z DOI: <http://dx.doi.org/10.1186/1750–1172–5-13>.
- Čermák J. K diferenciální diagnostice a léčbě anémií. Remedia 2003; 13(4): 258–265.
- Bizzaro N, Antico A. Diagnosis and classification of pernicious anemia. Autoimmun Rev 2014; 13(4–5): 565–568. Dostupné z DOI: <http://dx.doi.org/10.1016/j.autrev.2014.01.042>.
- Papaemmanuil E, Gerstung M, Malcovati L et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013; 122(22): 3616–3627. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2013–08–518886>.
- Vlachos A, Ball S, Dahl N et al. Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. Br J Haematol 2008; 142(6): 859–876. Dostupné z DOI: <http://dx.doi.org/10.1111/j.1365–2141.2008.07269.x>.
- Shimamura A, Alter BP. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev 2010; 24(3): 101–122. Dostupné z DOI: <http://dx.doi.org/10.1016/j.blre.2010.03.002>. Erratum in Blood Rev 2010; 24(4–5): 201.
- Young NS, Maciejewski J. The pathophysiology of acquired aplastic anemia. N Engl J Med 1997; 336(19): 1365–1372. Dostupné z DOI: <http://dx.doi.org/10.1056/NEJM199705083361906>.
- Čermák J. Myelodysplastický syndrom. In: Starý J, Mayer J (eds). Leukémie. Grada: Praha 2002 : 221–234. ISBN 80–7169–991–8.
- D´Andrea AD, Grompe M. Molecular Biology of Fanconi Anemia: Implications for Diagnosis and Therapy. Blood 1997; 90(5): 1725–1736.
- Bolton-Maggs PHB, Langer JC, Iolascon A et al. Guidelines for the diagnosis and management of hereditary spherocytosis – 2011 update. Br J Haematol 2004; 156(1): 37–49. Dostupné z DOI: <http://dx.doi.org/10.1111/j.1365–2141.2011.08921.x>.
- Grace RF, Zanella A, Neufeld EJ et al. Erythrocyte pyruvate kinase deficiency: 2015 status report. Am J Hematol 2015; 90(9): 825–830. Dostupné z DOI: <http://dx.doi.org/10.1002/ajh.24088>.
- Beutler E. Glucose-6-phosphate dehydrogenase deficiency: a historical perspective. Blood 2008; 111(1): 16–24. <http://dx.doi.org/10.1182/blood-2007–04–077412>.
- Stuart MJ, Nagel RL. Sickle cell disease. Lancet 2004; 364(9442): 1343–1360. Dostupné z DOI: <http://dx.doi.org/10.1016/S0140–6736(04)17192–4>.
- Zanella A, Barcellini W. Treatment of autoimmune hemolytic anemias. Haematologica 2014; 99(10): 1547–1554. Dostupné z DOI: <http://dx.doi.org/10.3324/haematol.2014.114561>.
- Válka J. Paroxysmální noční hemoglobinurie pro praktické lékaře. XIV. kongres praktických lékařů v Praze – abstract. Med Praxi 2017; 14(Suppl E): 11.
Štítky
Diabetologie Endokrinologie Interní lékařstvíČlánek vyšel v časopise
Vnitřní lékařství

2018 Číslo 5
- Inovace v hojení ran: krytí Zetuvit Plus Silicone Border pro optimální management exsudátu z ran
- Prognostický význam hladiny natriuretických peptidů při léčbě empagliflozinem
- Srovnání vlivu omeprazolu a pantoprazolu na antiagregační účinek klopidogrelu
- Ivabradin zlepšuje kvalitu života starších pacientů se srdečním selháním
- Fixní kombinace kandesartan/amlodipin v terapii arteriální hypertenze
Nejčtenější v tomto čísle
- Autoimunitní hemolytická anémie
- Diferenciální diagnostika anémií
- Vrozené a získané krvácivé stavy
- Hemoglobinopatie
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy