
Urgentní stav v hematologii: akutní promyelocytární leukemie – principy diagnostiky
An urgency in hematology: acute promyelocytic leukemia – principles of diagnosis
A review of diagnosis of acute promyelocytic leukemia (APL) is presented. There are still many patients with progressive disease with leukocytosis at presentation. These are at greater risk of early death due to bleeding (often intracranial), or, less frequently, due to thrombotic complications. In Czechia, we have, in some instances, noted an unacceptably long time from the first symptoms to diagnosis and to administration of the highly specific differentiation therapy with tretinoin (ATRA) along with anthracycline chemotherapy. This combination is highly efficient – cures are seen in some 70% of patients. Therefore, we present a diagnostic minimum for each and every internist, and even better for every general practician, to get acquainted with. All cases of pancytopenia and consumption coagulopathy should be suspected of APL and referred to a specialized hematologist without any delay. In the following more detailed review of diagnostic measures, much attention is given to APL morphology, which is the first clue leading to diagnosis. The finding of the typical hypergranular FAB M3 morphology and of cells with bundles of Auer rods („faggot cells“), along with the HLA-DR–, CD33+ immunophenotype, is highly (but not absolutely) specific for APL. In cases of the micro-/hypo-granular variant FAB M3v form, and whenever APL cannot be ruled out with certainty, a test to prove the presence of the PML/RARα fusion gene is indicated, using either RT‑PCR or, eventually, immunological demonstration of the specific distribution of the PML protein in the cell nucleus. Given that morphology of APL cases, as defined according to WHO criteria (95% of which carry the PML/RARα fusion gene), admits extremely divergent morphological pictures of the variant forms, we recommend these investigations to be performed in every case of de novo acute myeloid leukemia. A review of the less frequent morphological, as well as genetic variants is given, and the principles of immunophenotypic, cytogenetic and molecular diagnostics are also reviewed.
Key words:
acute promyelocytic leukemia – diagnostics – morphology – FAB classification – WHO criteria – faggot cells – immunophenotype – cytogenetics – t(15;17) – molecular biology – fusion genes – PML/RARα
Autoři:
J. Schwarz 1; P. Kačírková 1; J. Marková 1; D. Mikulenková 1; I. Marinov 1; I. Ballingová 1; K. Michalová 1
Působiště autorů:
Klinický úsek Ústavu hematologie a krevní transfuze Praha, přednosta doc. MUDr. Petr Cetkovský, Ph. D.
1; Výzkumný úsek Ústavu hematologie a krevní transfuze Praha, přednosta prof. Ing. Jan E. Dyr, DrSc.
2
Vyšlo v časopise:
Vnitř Lék 2008; 54(7-8): 728-744
Kategorie:
Přehledný referát
Souhrn
Přehledný článek se zabývá diagnostikou akutních promyelocytárních leukemií (APL). Řada pacientů stále ještě přichází s rozvinutým onemocněním s leukocytózou. U těchto pacientů může častěji dojít k časné smrti následkem krvácení (často intrakraniálního) anebo vzácněji i trombotické komplikace. V Česku jsme zaznamenali v některých případech neúnosně dlouhou dobu od prvních symptomů k diagnóze a k nasazení léčby. Ta bývá v případě APL specifická při použití diferenciační terapie tretinoinem (ATRA) spolu s chemoterapií. Tato kombinace je zároveň vysoce účinná – lze vyléčit kolem 70 % nemocných. Proto uvádíme jisté diagnostické minimum, se kterým by měl být seznámen minimálně každý internista, a ještě lépe každý praktický lékař. Každé setkání s projevy pancytopenie a konzumpční koagulopatie by mělo vést k podezření na APL a pacient by měl být urychleně vyšetřen na specializovaném hematologickém pracovišti. V následujícím podrobném přehledu diagnostiky je věnována velká pozornost morfologii APL, která musí být prvním vodítkem k diagnóze. Nález typické hypergranulární FAB M3 morfologie a buněk s otýpkami Auerových tyčí („faggot cells“) je spolu s HLA-DR–, CD33+ imunofenotypem pro APL vysoce (avšak ne absolutně) specifický. V případech mikro-/hypogranulární variantní formy FAB M3v a vždy, když nelze s jistotou vyloučit, že se nejedná o APL, je indikován test k potvrzení přítomnosti fúzního genu PML/RARα pomocí RT‑PCR, ev. pomocí průkazu specifické distribuce PML proteinu v buněčném jádře imunologickými metodikami. Zhruba 95 % případů onemocnění definovaného podle WHO kritérií nese fúzní gen PML/RARα. Vzhledem k tomu, že morfologie onemocnění připouští extrémně různorodé morfologické obrazy variantních forem onemocnění, doporučujeme tato vyšetření provést u každé de novo akutní myeloidní leukemie. Je podán přehled vzácnějších morfologických obrazů i vzácnějších genetických variant, jsou rovněž shrnuty principy imunofenotypové, cytogenetické a molekulární diagnostiky.
Klíčová slova:
akutní promyelocytární leukemie – diagnostika – morfologie – klasifikace FAB – WHO kritéria – faggot cells – imunofenotyp – cytogenetika – t(15;17) – molekulární biologie – fúzní geny – PML/RARα
Úvod
Akutní promyelocytární leukemie (APL) je nejvyšší hematologickou urgencí především proto, že dokáže vést ke smrti pacienta během několika hodin, nejčastěji v důsledku koagulopatie, projevující se krvácením (často intrakraniálním), v některých případech naopak trombózou [1–4]. Sami jsme zaznamenali úmrtí např. v důsledku Budd-Chiariho syndromu, infarktu myokardu anebo venózní i arteriální trombózy s projevy phlegmasia alba dolens s gangrénami. V řadě případů nastalo úmrtí pacienta do několika hodin od přijetí na oddělení, někdy i dříve, než byla zahájena terapie (viz kazuistiky v tomto čísle časopisu Vnitřní lékařství). APL je v současnosti onemocněním s nejlepší prognózou mezi všemi typy akutních myeloidních leukemií (AML). Nejnebezpečnějším obdobím pro pacienta jsou první dny po diagnóze, většina z těchto časných úmrtí nastává zhruba do jednoho měsíce. Vždy jde o komplikace onemocnění – u APL s fúzním genem PML/RARα totiž neexistuje primární rezistence k indukční terapii kombinací antracyklinu a tretinoinu (kyseliny transretinové – ATRA). Relapsů je při správně vedené léčbě pod 10 %. Vzhledem k této mimořádně dobré prognóze je hned v okamžiku suspekce na APL třeba nasadit všechny síly k tomu, aby bylo onemocnění co nejrychleji diagnostikováno a specificky léčeno. V naší literatuře již bylo upozorněno na fakt, že diagnostika v Česku trvá neúnosně dlouho [5]. V případě pouhé suspekce na APL je třeba zahájit terapii ATRA, která významně zmírňuje průběh koagulopatie a snižuje riziko časného úmrtí, s tím, že bude co nejrychleji (do 24 hod) dokončena přesná diagnostika stavu. Je menší chybou indikovat ATRA u stavu, který se ukáže být jinou AML, než neindikovat ATRA u méně typické APL [4]. Současná situace v Česku je alarmující i z hlediska erudice na hematologických laboratořích – v nedávné studii kvality správně diagnostikovala vzorek s typickou APL pouze 1/3 z nich (nepublikováno).
Tento přehledný článek si klade za cíl vysvětlit principy diagnostiky APL coby urgentního stavu v hematologii – nejprve vysvětlíme, co by mělo vést k suspekci na APL, shrneme minimum pro rychlou diagnostiku onemocnění, dále se dotkneme otázky definice a také incidence APL, a konečně podáme podrobný přehled o diagnostice stavu.
Co musí vést k suspekci na APL
- Obraz pancytopenie v periferní krvi – je pro APL sice typický (vyskytuje se zhruba u 2/3 případů APL), ve zbývajících případech je však počet leukocytů normální anebo zvýšený. Diferenciální diagnostika pancytopenie je velmi široká. Přesto obraz snížení všech 3 základních krevních řad musí lékaře vždy vést k vážné úvaze, nejedná‑li se o APL. Od pancytopenie se rovněž odvíjí klinický obraz – nemocní mají v různém poměru vyjádřené symptomy infekční, anemické a krvácivé. U typických APL se vyplavuje do periferní krve minimum nezralých granulocytů (často nemusí jít o promyelocyty, mohou se objevit např. myelocyty a metamyelocyty), někdy nenajdeme nezralé granulocyty v diferenciálním obrazu vůbec. Pouze v případech s leukocytózou se do periferní krve vyplavují pravidelně i leukemické promyelocyty (mohou však mít jinou morfologii než ve dřeni). Výsledek krevního obrazu s mikroskopickým diferenciálním rozpočtem bývá k dispozici i s barvením do 40–60 min.
- Obraz konzumpční koagulopatie – ať již v koagulačních testech, anebo v klinickém nálezu. Klinicky může být významný i postřeh ošetřujícího lékaře, že míra krvácivých projevů neodpovídá zjištěné hloubce trombocytopenie. V koagulačních testech může být různá hodnota APTT (nejčastěji však zjistíme normální nebo lehce prodloužené časy), typické je snížení hodnot fibrinogenu, často prodloužený protrombinový čas (Quick) a vysoké hodnoty D‑dimerů. Je vhodné co nejvíce rozšířit koagulační vyšetření dle možností pracoviště – viz článek doc. Žáka et al v tomto čísle časopisu Vnitřní lékařství.
Minimum pro rychlou diagnostiku
- Morfologie aspirátu kostní dřeně – typický obraz APL [6] (obr. 1), při kterém v myelogramu dominují atypické promyelocyty, často s velmi intenzivní a hrubou granulací cytoplazmy; u morfologicky hypergranulární formy onemocnění představují tyto buňky většinu bílých krvinek v preparátu kostní dřeně, u hypogranulární formy [7] (obr. 2) jsou tyto buňky zastoupeny v menší míře. Ve většině případů APL je možné v nátěru najít tzv. „faggot cells“, tj. buňky s otýpkami či snopci Auerových tyčí, ev. s mnohočetnými Auerovými tyčemi (obr. 3). Přesná a obecně vžitá definice pro „faggot cell“ neexistuje [8]. Sainty et al definovali „faggot cell“ jako buňku obsahující alespoň 3 Auerovy tyče [9], avšak ani jejich definice se neujala a ani my ji neužíváme. Vždy je nutno po „faggot cells“ pátrat, někdy se snopce Auerových tyčí najdou spíše v rozpadajících se buňkách (obr. 3). Snáze než v panopticky obarvených preparátech je můžeme nalézt v preparátech barvených na průkaz myeloperoxidázy. Nález „faggot cells“ sice není zcela patognomonický (tzn. jednoznačně specifický pro diagnózu – ojediněle nalezneme podobné buňky i u AML M2 – viz obr. 3), avšak přítomnost většího počtu těchto buněk nás opravňuje učinit diagnózu APL se spolehlivostí zhruba 99 %. Avšak pozor: jak bylo uvedeno, zdaleka ne v každém případě APL lze „faggot cells“ nalézt. Naprostou většinu případů variantní formy FAB AML M3v [7] nutno považovat na podkladě pouhé morfologie za diagnosticky nejistou a je nutno je vždy co nejrychleji vyšetřit na přítomnost molekulární aberace. K výsledku morfologického vyšetření kostní dřeně dojdeme během 1 hod (tab. 1).
- Imunofenotyp buněk kostní dřeně je také poměrně specifický. APL je jediným typem myeloidní leukemie, která neexprimuje HLA‑DR. Proto nás kombinace znaků HLA-DR–, CD33+ přivede k diagnóze APL snadno (existují však výjimky – viz podrobná diagnostika). Spolehlivost imunofenotypové diagnózy se odhaduje zhruba na 95 %. K výsledku dojdeme během 2 hod (tab. 1).
- Průkaz t(15;17), definující ve WHO klasifikaci naprostou většinu případů [10,11]. Tato translokace je zcela patognomonická, nevyskytuje se u žádné jiné leukemie nebo malignity. K rychlému průkazu t(15;17) je nejvhodnější jednokroková RT‑PCR metoda k detekci specifického fúzního genu PML/RARα, produktu této translokace [12,13]. RT‑PCR prokáže tuto fúzi obvykle i u případů se zanedbatelnou měrou vyplavování APL promyelocytů v periferní krvi. Při rychlém postupu ve „STATIM“ režimu dojdeme k výsledku za 5–6 hod. Ještě rychlejší může být diagnóza pomocí hodnocení rozložení PML tělísek při užití anti-PML protilátky [13–17] (výsledek je k dispozici za 2–3 hod). Metoda fluorescenční in situ hybridizace (FISH) může být podobně rychlá jako RT‑PCR (za 6 hod – viz tab. 1), v praktickém životě to však trvá mnohdy podstatně déle.


![Srovnání specificity a rychlosti záchytu APL. Zčásti podle Lo Coca et al [18] a podle Sanze et al [4].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/775a1ddb9e6d02b073feaafea2b5564c.png)
Definice onemocnění
Francouzsko-americko-britská (FAB) klasifikace z let 1976–1980 ([6,7], podrobně viz též několik odstavců níže) definuje APL výlučně podle morfologických charakteristik buněk kostní dřeně a periferní krve.
Z hlediska cytogenetiky naprostá většina případů FAB M3 (APL) nese t(15;17), jejímž produktem je fúzní gen PML/RARα [23]. Zprvu se zdálo, že t(15;17) mají všichni nemocní s APL, teprve později bylo zjištěno, že raritně se mohou vyskytnout i případy s jinou translokací, např. s t(11;17), nebo s t(5;17) [9,24]. APL s t(11;17) s fúzním genem PLZF/RARα mívá sice morfologii velmi podobnou APL (AML FAB M3 nebo M3v), ale ve většině případů je morfologicky odlišitelná od typických případů APL s t(15;17) [9].
O syntézu morfologických a nověji definovaných genetických charakteristik se pokusila velice inteligentní, žel obecně málo užívaná klasifikace AML publikovaná Headem v roce 1996 [30]. Z jeho pohledu patří APL s t(15;17) do kategorie „true de novo AML“, charakterizovaných cytogenetickými translokacemi. Na rozdíl od AML staršího věku, pro které je charakteristická multilineární dysplazie a cytogenetické aberace podobné těm u myelodysplastického syndromu, je [až na výjimky, např. právě APL s t(11;17)] pro „true de novo AML“ typická relativně dobrá prognóza [30].
WHO klasifikace z roku 2001 rozeznává subtyp AML s rekurentními genetickými aberacemi, který zahrnuje APL, resp. AML s t(15;17)(q22;q12) či PML/RARα, anebo s variantními translokacemi [10,11]. Tato kategorie tedy zahrnuje nejen všechny APL, které mají t(15;17) anebo PML/RARα, ale i případy s variantními translokacemi, mezi kterými jsou v původní definici uvedeny tři, u nichž jsou fúzními partnery genu RARα následující geny: u t(11;17)(q23;q21) gen PLZF, u t(5;17)(q23;q12) gen NPM (kódující nukleofosmin) a u t(11;17)(q13;q21) gen NuMA [11]. Ve srovnání s FAB klasifikací [6,7] jsou tedy vyloučeny pouze případy AML, které sice mají morfologii kompatibilní s kategorií M3, či spíše M3v, ale nelze u nich prokázat jednu z výše uvedených genetických aberací (celkově zhruba 1 % všech pacientů s AML FAB M3 či M3v) [31]. Naopak v rámci WHO definice se vyskytují případy s morfologií kompatibilní s většinou ostatních morfologických FAB subtypů AML (za podmínky, že nesou uvedené translokace a/nebo příslušné fúzní geny) [10,11]. Níže bude vysvětleno, že zlom v genu RARα se u všech nyní správně lokalizuje v pruhu 17q21.
Je však třeba mít na paměti, že pro pacienta není tak důležité, jakou mají jeho buňky morfologii, nýbrž to, jak má být léčen. Rozdíl mezi APL s t(15;17) a s t(11;17) s fúzním genem PLZF/RARα spočívá hlavně v tom, že první z nich výborně odpovídá na léčbu ATRA (bez ohledu na morfologický nález), zatímco druhá jen málo (je však možné diferenciační účinek ATRA podpořit přidáním G‑CSF), navíc sníženou měrou odpovídá i na léčbu As2O3 [32]. S přihlédnutím k velmi nízké frekvenci výskytu atypických cytogenetických a molekulárních aberací u APL je ve velkých centrech (jako např. v ÚHKT Praha anebo na Hematoonkologické klinice v Brně-Bohunicích) pravděpodobnost záchytu těchto vzácných forem APL zhruba 1krát za 15–20 let. Praktický význam má tedy především co nejrychlejší záchyt aberace t(15;17), resp. jejího produktu, fúzního genu PML/RARα, u všech morfologicky sporných případů, ev. u těch vzácných případů AML, u kterých by ani zkušený hematolog nepojal podezření, že by se mohlo jednat o AML s t(15;17), resp. její produkt PML/RARα.
Prevalence a incidence
Přes svou relativní vzácnost je t(15;17) u APL nejčastěji se vyskytující translokací u AML – při metaanalýze 4 257 dospělých pacientů byla prokázána v 325 případech (7,6 %) [33]. V databázi ÚHKT je 500 pacientů s AML diagnostikovanou v letech 1991–2007. Mezi nimi je vedeno 58 nemocných (11,6 %) s APL s t(15;17) a/nebo fúzním genem PML/RARα. Na základě tohoto údaje odhadujeme četnost APL v Česku bez selekce dané institucí rovněž kolem 7–8 %. U románské populace v Evropě, ale i u „latinské“ populace v USA (los-angeleském okresu) a také v zemích Latinské Ameriky je zastoupení APL v rámci AML podstatně vyšší: 15–25 % (např. ve Španělsku 23 %) [34–38]. Zároveň se u případů ze středoamerických zemí relativně častěji zachytí zlom bcr1 v genu PML (viz níže), což však nevede k rozdílu v celkovém přežití [37,38]. Incidence onemocnění je poměrně konstantní ve všech věkových kategoriích [39], proto medián věku při záchytu bývá ve středním věku [11]. Jelikož s věkem dochází k nárůstu incidence jiných AML (s podobnými aberacemi jako u myelodysplastického syndromu a s multilineální dysplazií), činí zastoupení APL mezi ostatními AML postupně nižší procento [30]. U pediatrických pacientů např. v Itálii a na Kubě APL reprezentuje 30–31 % všech případů AML – incidence u dospělých Kubánců je přitom poloviční [36,40]. Existuje však jen minimum studií, které by přinesly údaje o incidenci APL. Jedna z mála těchto studií však naznačuje, že u „Hispaniků“ v USA není incidence APL v průběhu celého života vyšší než u „nehispánských“ bělochů [41]. (Pozn. autorů: Kdybychom však vztáhli toto tvrzení na studie ukazující vysoká procenta zastoupení APL v rámci AML, došli bychom k tomu, že incidence ostatních subtypů AML v těchto publikacích je v přepočtu na obyvatele nižší, což je dosti nepravděpodobné.) Jak však poukazují autoři ze zemí Latinské Ameriky, v uvedených studiích z USA jsou vnímána etnika především na základě jazyka (a pak autoři píší o „Hispáncích“ či o „Latinos“). Ve skutečnosti má např. průměrný obyvatel Mexika – mestic – 56 % genů pocházejících z indoevropské („kavkazské“) populace, 40 % genů původu indiánského (odvozeného od Asiatů, kteří překročili Behringovu úžinu před 12 000 lety) a 4 % z černé populace [42]. Zvýšené zastoupení zlomu bcr1 v Mexiku může být naopak dáno asijskými kořeny u mestické populace – také v Číně a v Japonsku je zastoupení tohoto zlomu vyšší [38]. Paradoxně však právě autoři z USA [34,37,41] mohou být blízko pravdy, neboť nelze vyloučit, že právě španělsky nebo portugalsky mluvící indoevropské národy (tj. jen vybraní jihoevropané) zanesli geny predisponující k APL do Latinské Ameriky. Vysvětlení, proč někteří Indoevropané (kavkazské národy) mají vyšší dispozici k APL a jiné menší, však neexistuje a stále nelze vyloučit vliv faktorů zevního prostředí.
Diagnostika podrobně
Morfologie
Jelikož je morfologické vyšetření stále standardním prvním krokem hematologické diagnostiky, je bezpodmínečně nutné, aby každý hematolog (nejen ve specializovaných centrech) bezpečně ovládal morfologickou diagnostiku APL a dokázal na základě svého nálezu indikovat molekulární vyšetření vedoucí ke správné diagnóze.
AML s fúzním genem PML/RARα může mít téměř jakýkoli morfologický obraz v rámci AML (jak ještě bude uvedeno níže), tj. nejen morfologii definovanou jako M3 a M3v dle francouzsko‑americko-britské (FAB) skupiny [6,7]. Proto je podle našeho názoru vhodné vyšetřovat přítomnost fúze PML/RARα rutinně u všech AML, a to co nejdříve od doby záchytu. Sice velmi vzácně, ale přece se i zkušený hematolog setká s velkým překvapením v podobě nečekané pozitivity tohoto vyšetření.
Definice FAB skupiny. APL ve své typické hypergranulární formě byla definována FAB skupinou v roce 1976 jako AML M3 a o 4 roky později byla stejnými autory definována variantní mikrogranulární forma onemocnění, označovaná M3v [6,7]. Rozpoznání typické hypergranulární formy APL většinou nečiní obtíže. Podle definice M3 FAB [6] (kurzivou je uvedeno přesné znění definice, zatímco komentáře autorů jsou uváděny standardním písmem) je velká většina buněk ve sternálním punktátu tvořena atypickými promyelocyty s charakteristickou výraznou granulací (obr. 1, 4). Buněčná jádra se značně liší jak velikostí, tak i tvarem. Mohou být ledvinovitá, často dvojlaločná, někdy jakoby přeložená (obr. 4), u některých buněk mohou být excentricky uložena. Chromatin bývá celkem jemný (přesto o něco více denzní než u méně zralých typů AML), jadérka bývají různých velikostí a v ně-kterých promyelocytech nemusí být jasně patrna [8,19,43,44]. U některých typických hypergranulárních promyelocytů však strukturu chromatinu nelze hodnotit, neboť jádro je překryto granulací (což je rys APL, který bývá pozorován u jiných subtypů AML velmi vzácně). Cytoplazma většiny leukemických promyelocytů nebývá příliš bazofilní, ale bývá objemná a je kompletně vyplněna těsně napěchovanými, či dokonce splývajícími velkými granuly barvícími se Romanowskiho barvivy světle růžově, červeně anebo purpurově (až fialovo-černě). V některých buňkách je cytoplazma vyplněna jemnými, prachovitými granuly. Charakteristické buňky obsahující otýpky Auerových tyčí („faggots“ – obr. 3) náhodně distribuované v cyto-plazmě jsou takřka vždy přítomny v kostní dřeni a někdy i v periferní krvi. Nález „faggot cells“ se v té době považoval za patognomonický (již jsme však uvedli, že to neplatí absolutně – viz též obr. 3). Cytoplazma „faggoty“ obsahujících buněk je často bez granul a je bledá, ale může obsahovat i azurofilní granula. Různě velký podíl hypergranulárních promyelocytů a „faggot cells“ se může rozpadnout a pak leží granula a Auerovy tyče volně mezi jinými buňkami, mohou ležet i přes ně. Je nutno odlišit případy M2 se zvýšeným procentem promyelocytů od M3. U M2 je cytoplazmatická granulace obvykle méně intenzivní a nezakrývá bazofilní cytoplazmu, jak je obvyklé u M3; nicméně příležitostně se u M2 zastihne buňka s cytoplazmou vyplněnou hrubými granuly. Na základě vlastní zkušenosti také doporučujeme srovnání velikosti leukemických promyelocytů s normálními protějšky: u APL jsou často (ale zdaleka ne vždy) o něco menší.


V roce 1980 byl publikován článek FAB skupiny s definicí variantní hypogranulární formy M3v [7]. Píše se v něm: Většina případů odpovídá popisu M3, avšak byla popsána atypická forma, charakterizovaná spíše minimální než excesivní granulací (4 citace, např. Galton et al [45]). Nejnápadnější cytomorfologický rys této „M3 varianty“ se vztahuje ke konfiguraci jádra a k relativně malému výskytu buněk s výraznou granulací a buněk obsahujících Auerovy tyče („faggot cells“). Jádro téměř každé buňky je dvojlaločné, multibilobulární, nebo ledvinovité, ale většina buněk je buď zcela prosta granulace, anebo obsahuje jen málo jemných azurofilních granul (obr. 2). Nicméně je přítomno několik buněk se všemi cytoplazmatickými rysy typické M3. Jsou‑li přehlédnuty, případ bude pravděpodobně chybně diagnostikován jako monocytární leukemie. Dodejme, že v průměru mohou mít M3v případy výraznější cytoplazmatickou bazofilii. A nyní pozor, důležitý dovětek autorů, na který stále řada hematologů zapomíná: Tato atypická morfologie je rysem buněk periferní krve (obr. 2). V kostní dřeni je morfologie bližší M3. Dále autoři uvádějí, že počet leukocytů bývá u případů M3v vyšší než u často leukopenické M3. Udávají 3 důvody, proč M3v považovat jen za variantu M3:
- byť v menším počtu, jsou přítomny morfologicky typické M3 buňky,
- oba typy leukemie jsou provázeny diseminovanou intravaskulární koa-gulací (DIC),
- t(15;17) se prokazuje u obou typů [7].




Udává se, že M3v morfologii má zhruba 15–27 % případů APL [9,46]. Vše ukazuje na to, že APL s morfologií M3, resp. M3v, nejsou různé subtypy, nýbrž různá stadia jednoho onemocnění. Byly pozorovány případy, kdy pacient s typickou APL v době diagnózy měl při relapsu morfologii „podobnou jiným AML“ [47]. Důvod, proč se zachytí M3v v USA častěji u nebílé rasy [46], může souviset i s otázkami sociálními – tito nemocní prostě mohou v průměru přijít k lékaři později než běloši. Z hlediska FAB klasifikace, založené na morfologii, hrozí u variantní formy M3v při relativně subjektivním hodnocení nátěrů mnohem větší nebezpečí záměny s jiným subtypem AML (především M5b, M5a, M4, M2, ev. M1). Nicméně lze říci, že i při variantní formě může být v typických případech (např. s průkazem „faggot cells“) diagnóza vyřčena s poměrně malým rizikem omylu. Je‑li naše pracovní diagnóza M3v, je její rozpoznání založeno na pozitivní identifikaci určitých morfologických charakteristik a riziko omylu je nízké. V opačném sledu, kdy morfolog nemá podezření na M3v, ale na jiný typ AML, je riziko nepoznání M3v formy podstatně vyšší.
Jen pro úplnost dodejme, že často můžeme v aspirátu kostní dřeně u APL nalézt nečetné zralejší elementy neutrofilní granulopoezy a ty mohou mít různé poruchy vyzrávání a nápadnou maturační asynchronii, jsou‑li již potomstvem leukemického klonu. Mohou mít např. Pelgerovu-Huëtovu anomálii, případné Auerovy tyče mohou doložit původ buňky z leukemického klonu. Naopak významnější dysplazie červené a megakaryocytární řady do obrazu nově diagnostikované APL nepatří.
Tan et al [48] v elektronoptické studii popsali typickou strukturu chromatinu APL buňky. Je více kondenzovaný u jaderné membrány a disperznější uprostřed jádra, s nukleoly (obsahujícími nukleolonemata a pars amorpha) typicky lokalizovanými spíše poblíž jaderné membrány. Při panoptickém barvení jsou jadérka někdy málo zřetelná, lépe jsou vidět v méně zralých buňkách M3 a obecně u varianty M3v, neboť chromatin těchto buněk je o něco jemnější. Pokud se týká azurofilní granulace, obsahuje FAB definice [6] informaci o tom, že granula mohou v některých buňkách splývat; neinformuje však, že granula mohou být co do velikosti i tvaru dosti bizarní, Sainty et al (viz níže [9]) ně-které z těchto atypií označují jako „Chédiak-Higashi‑like“ (viz též obr. 4). Auerovy tyče jsou u APL robustnější než u jiných subtypů AML [8].
Kromě výše uvedených morfologických subtypů APL M3 a M3v, mezi nimiž neexistuje ostrá hranice [8], byla popsána řada dalších morfologických nuancí, přičemž u všech z nich se prokazuje t(15;17) – nyní již tedy hovoříme o APL definované rekurentní chromozomální aberací, tzn. o onemocnění definovaném podle WHO klasifikace [11]. Avšak již v době, kdy ještě nebyla objevena souvislost APL s t(15;17), uváděli Liso et al [49] ve své cytochemické studii případ APL s granuly metachromaticky barvitelnými toluidinovou modří a diskutovali podobnost buněk s bazofilními promyelocyty (viz též obr. 4). Až o mnoho let později byla Kubonishim et al [50] popsána APL s bazofilní diferenciací, která nesla t(15;17). Koike et al a po nich Tallman et al ukázali, že tato další variantní forma může být spojena klinicky s hyperhistaminemií, šokovým stavem s ulceracemi v žaludku a duodenu, s rozšířením periferních cév a bolestmi hlavy, a cytogeneticky s přestavbou krátkého raménka chromozomu 12 (pruhu 12p13) [51,52]. Neměli bychom zaměnit popsané případy s bazofilními granuly s další variantou, a sice hyperbazofilní APL, prvně popsanou McKennou et al [53]. U té se bazofilie týká cytoplazmy, APL je mikrogranulární a má některé myelomonocytární rysy. Na monocytoidní rysy tohoto typu APL, jak v cytochemické, tak i imunofenotypové rovině, upozornila řada prací, včetně citované práce Lisa et al i cytochemické studie Lemeže [43,49,54,55]. Naeme et al v roce 1997 rozeznávali 5 morfologických subtypů APL – již zmíněné M3, M3v, McKennovu hyperbazofilní variantu a nově popsané případy podobné M1 a M2 [56]. Stejná skupina potom popsala pacienta s t(15;17), který byl odeslán k příjmu pod diagnózou akutní lymfoblastické leukemie. Buňky byly agranulární (viz též obr. 2), pouze v elektronovém mikroskopu byly nečetné buňky slabě pozitivní na peroxidázu, často byly přítomny cytoplazmatické výběžky [57] (viz též obr. 4). Ty jsou však často spojeny s hyperbazofilní variantou – připomínají někdy mikromegakaryocyty [43]. I imunofenotyp byl u uvedeného případu v souladu s nezralostí buněk a akutní leukemie byla považována za bifenotypovou – CD34+, DR+, CD2+, CD7+, CD13+, CD15+, ale CD33– [57] (ačkoli nebyla splněna kritéria pro bifenotypovou akutní leukemii [58]). Dopis editorovi o tomto případu byl v časopise zařazen hned za dalším (Aventín et al), popisujícím téměř stejný případ agranulární, HLA‑DR+ leukemie, s expresí PML/RARα se zlomem bcr3 [59]. Morgan et al [60] popsali i případ APL s megakaryoblastickými rysy jak na úrovni morfologické (buňky s cytoplazmatickými výběžky), tak i imunofenotypové – HLA‑DR– s expresí megakaryocytárního antigenu CD41. Rovněž již byl popsán případ AML s eozinofilní diferenciací, s průkazem fúze PML/RARα [61]. Byl publikován i případ imunologicky méně zralé (CD34+) APL s výraznou fibrózou dřeně, která ustoupila po standardní terapii (indukci daunorubicinem a ATRA, 3 konsolidacích) a autologní transplantaci [62]. Z uvedeného výčtu vidíme, že téměř jakýkoli morfologický nález akutní leukemie může být ve skutečnosti vzácnou formou AML s t(15;17) podle definice WHO. Onemocnění definované WHO má tedy podstatně širší diferenciální diagnostiku než APL definovaná skupinou FAB. Proto doporučujeme provádět vyšetření k vyloučení této translokace u všech případů AML.
Cytochemicky nacházíme u APL pozitivitu při průkazu myeloperoxidázy, naftol AS-D chloroacetát-esterázy a při barvení sudanovou černí B [8,19,43,49,63]. Velmi výraznou pozitivitu těchto reakcí nalezneme u typických M3 forem APL – u těch však obvykle nepotřebujeme cytochemická vyšetření, neboť obraz při standardním panoptickém barvení podle Maye-Grünwalda-Giemsy je dostatečně sugestivní, navíc výsledky cytochemie nejsou pro APL nijak specifické – podobné výsledky bývají pozorovány i u jiných subtypů AML s vyzráváním [43]. Na druhé straně zase často bývá u variantní formy M3v pozitivita v uvedených reakcích méně výrazná. Cytochemie však může být i moderní diagnostice platná dvěma způsoby:
- Pomocí peroxidázové reakce snáze odhalíme Auerovy tyče a „faggot cells“, které nemusejí být patrné při panoptickém barvení (vlastní zkušenost) – Liso s Bennettem za stejným účelem doporučují chloroacetát-esterázu, naopak myeloperoxidázu nedoporučují [19].
- Nápadně silná reakce myeloperoxidázy v hypogranulárních buňkách nás může upozornit, že daný případ AML by ve skutečnosti mohl být APL [8]. Je však nutno počítat s tím, že reakce myeloperoxidázy může být u M3v formy slabší; u hyperbazofilní formy naopak bývá reakce silná [19]. V případě, že laboratoř má k dispozici analyzátor krevních obrazů pracující na principu průkazu myeloperoxidázy (např. přístroj Advia 120 fy Bayer), je možné rychlé rozlišení mezi variantní formou APL (AML M3v) a AML M5.
Jak již bylo zmíněno v odstavci o definici onemocnění, APL s t(11;17) a fúzním genem PLZF/RARα má specifickou morfologii. Mezinárodní skupina na semináři v Monze v červnu roku 1997 probírala případy APL bez prokázané t(15;17). Vytvořila poměrně jednoduchou klasifikaci APL podle tvaru buněčného jádra (pravidelné kulaté nebo oválné versus nepravidelné, např. ledvinovité, dvojlaločné, přehnuté), stupně granularity (hyper – nebo hypo-) a přítomnosti Auerových tyčí. Takto bylo vytvořeno 12 základních kategorií buněk a k nim pak byly přidány ještě 3 speciální, a sice kategorie buněk s bazofilními granuly, dále kategorie buněk s granuly podobnými Chédiakově-Higashiho granulím (viz též obr. 4), a posledně kategorie zrajících pelgeroidních buněk. Morfologické nálezy pak byly porovnány s cytogenetickými a molekulárními údaji. Vyšlo najevo, že výrazně se odlišují APL s t(11;17) s fúzí PLZF/RARα. Na rozdíl od typické APL s t(15;17) mají především dosti pravidelná jádra (jinak typičtější pro M2 leukemie). Rovněž struktura jejich chromatinu bývá kondenzovanější (blíží se již k myelocytům). Výraznější než u APL s t(15;17) bývá zrající granulocytární komponenta, s četnými neutrofily s Pelgerovou-Huëtovou anomálií. Tento typ APL morfologicky často stojí na pomezí M2 a M3 ve FAB klasifikaci. Většina APL s t(11;17) je hypergranulární, jen v některých případech jsou přítomny i „faggot cells“ [9].
Existují i některé stavy, které by méně zkušený morfolog mohl mylně vydávat za APL. V první řadě je nutno odlišit promyelocytární leukemoidní reakce, často v souvislosti s reparač-ním procesem v kostní dřeni po útlumu krvetvorby, např. u revmato-logických pacientů léčených meto-trexátem nebo azathioprinem (obr. 5). Promyelocyty bývají u tohoto reaktivního stavu velké (jde o největší buňky kostní dřeně s výjimkou megakaryocytů), se silně bazofilní cytoplazmou a s výraznou toxickou azurofilní granulací. Na rozdíl od leukemických promyelocytů mívají projasnění v oblasti Golgiho zóny. Pochopitelně zde nenalezneme Auerovy tyče a extrémní míru atypie granulace, jako je běžné u APL. Jádra hyperproliferujících normálních promyelocytů jsou oválná nebo ledvinovitá podle míry jejich zralosti, chromatinová struktura je v souladu s maturačním stadiem buňky (oproti APL tedy chybí výraznější maturační asynchronie), mají středně zřetelná jadérka (obr. 5). Dalším stavem, který by mohl být ev. považován za APL, by mohl být ojedinělý případ myelodysplastického syndromu s maturačním blokem na úrovni promyelocytů. U takového stavu mohou (ale nemusí) být přítomny dysplastické změny ostatních řad krvetvorby, avšak k záměně s typickou APL by dojít nemělo.

Jaký je dopad znalosti morfologie u APL?
- V případě typické FAB M3 nás morfologie sama přivede k diagnóze. Pacient dostane ATRA a diagnózu pak bez většího spěchu verifikujeme molekulárně.
- Některé případy jsou ovšem morfologicky sporné. Platí to pro situace (a), ve kterých je diagnóza APL pravděpodobná, ale nikoli jistá, a pro situace (b), kdy s diagnózou APL počítáme pouze jako s diferenciálně-diagnostickou možností. V obou případech pak musíme velice spěchat s molekulární verifikací. V případě (a) musí pacient dostat léčbu ATRA (spěch je na místě proto, aby se nestalo, že pacient neexprimující PML/RARα bude dostávat neindikovaně terapii ATRA a zároveň nebude léčen dle protokolu vhodnějšího pro jiné typy AML). Případ (b) je nejtěžší k rozhodování, zda rovnou ordinovat ATRA – záleží na konkrétním posouzení klinika, morfologa a imunofenotypisty. Právě zde je místo pro zkušeného morfologa, aby uměl „vychytat“ podezřelé nálezy, a to obzvláště na pracovištích, která nedělají paušální screening na fúzní gen PML/RARα u všech pacientů s AML, aby zvážil indikaci pro jeho „STATIM“ vyšetření a dal popud k ev. nasazení léčby ATRA.
- Morfologie může vést k suspekci na t(11;17), kterou musíme verifikovat opět molekulárně a cytogeneticky. Tuto leukemii musíme léčit jinak než APL s t(15;17), neboť je relativně ATRA-rezistentní.
- Odlišení M3 a M3v má i své klinické a prognostické koreláty – M3v je spojena s leukocytózou i s tendencí k horší šanci docílit kompletní remise (po dosažení remise se však již vliv morfologického subtypu v době diagnózy neuplatní) [46].
Imunofenotyp
Pro diagnostiku APL znamenalo za-ve-dení monoklonálních protilátek významný posun. Jejich pomocí lze detekovat poměrně specifický imunofenotyp APL buněk – HLA‑DR–, CD33+, CD13+. Ačkoli tento fenotyp není zcela specifický, jeho vyšetření patří k základním diagnostickým opatřením. Navíc výsledek je k dispozici velmi rychle (tab. 1).

Jak jsme již uvedli, ve většině případů napomůže správné diagnóze průkaz HLA-DR– blastů a promyelocytů v kontextu myeloidního fenotypu, s pozitivitou znaku CD33 nebo CD13 [20,64] (v sestavě ÚHKT jsou oba tyto myeloidní znaky exprimovány u 98 % pacientů). Článek v knize WHO klasifikace připouští možnost proměnlivější (tj. méně konstantní) exprese CD13 ve srovnání s CD33 [11]. Chceme však zdůraznit, že průkaz DR negativity není nikterak patognomonický pro APL a diagnóza by měla být potvrzena dalším vyšetřením, především molekulárně biologickým, a měla by být v souladu s výsledky morfologie a cytochemie. Dále je nutno si uvědomit, že existují případy APL pozitivní na HLA-DR. Na konferenci o APL – A Curable Disease? – v Římě v roce 1993 referovaly dokonce 3 různé italské skupiny o incidenci DR+ u APL ve 4 %, resp. 14 %, resp. 22 % případů. V kapitole o morfologii byly zmíněny AML s t(15;17) s velice nezralým fenotypem, s pozitivitou HLA-DR. V ÚHKT jsme však viděli pouze jediného pacienta pozitivního na HLA-DR ze 41 hodnocených případů (2 %). Je však třeba počítat s tím, že reprodukovatelnost morfologických výsledků, alespoň pokud se týká typické hypergranulární APL, je poměrně vysoká, zatímco u imunofenotypových analýz může být naopak nižší, neboť může být zatížena použitím monoklonálních protilátek proti různým epitopům zkoumaného antigenu (velký pozor je nutno dávat především při volbě protilátky proti HLA‑DR), ev. i technickými chybami v méně zkušených rukách. Principy imunologické diagnostiky APL podle jednoho z nás (I. M.) jsou sumarizovány v tab. 2. Z ostatních znaků může být zhruba ve 30–40 % případů přítomen i antigen CD15, prokazatelný protilátkou proti sializovanému antigenu (klon VEP–9), avšak nikoli protilátkou proti nesializovanému antigenu (klon VIM–D5) [20]. Exprese CD15 může být pozorována pouze u případů CD34– [11]. Znak CD34 bývá exprimován především u morfologického subtypu M3v (v sestavě ÚHKT je pozitivní ve 23 % ze všech APL). Rovněž tak variantní forma M3v je o něco častěji spojena s expresí lymfoidních antigenů CD2, CD7 a CD19 (v ÚHKT jsme pouze ve 3 případech, tzn. u 6 % ze všech pacientů, našli koexpresi CD2, z ostatních uvedených lymfoidních antigenů jsme neprokázali žádný). Monocytoidní markery, resp. adhezivní molekuly CD14, CD11b a CD11c, se prokazují poměrně vzácně [20] (v naší sestavě 2 %, 21 %, resp. 11 %). CD117 (produkt genu C-KIT) jsme našli u 54 % nemocných v souladu s některými publikovanými výsledky [65], zatímco podle Paiettaové et al [66] je tento znak přítomen vždy. Poměrně hodně pozornosti bylo věnováno antigenu CD56, normálně exprimovanému NK buňkami (přirozenými zabíječi). Některé skupiny jej považují za marker nepříznivé prognózy při APL [67,68]. Nicméně žádný z uvedených znaků, kromě negativity HLA‑DR, nemá diskriminační hodnotu pro diferenciální diagnózu mezi APL a ostatními AML. Od APL je třeba odlišit jiné HLA-DR– akutní leukemie: malou část vyzrávajících AML FAB M2, akutní lymfoblastickou leukemii T řady a dále hypergranulární AML morfologicky silně připomínající APL s NK/myeloidním CD56+ fenotypem. Tato leukemie však nenese t(15;17), pouze 1/20 popsaných případů měl t(11;17) [69]. Podle Paiettaové et al by diskriminační hodnotu pro odlišení APL od jiných AML s relativně zralým imunofenotypem mohla mít negativita znaků CD11a a CD18 (leukocytárních integrinů) [22,66].
Cytogenetika a molekulární biologie
Pro diagnózu APL, resp. AML s t(15;17) [10,11], je nutno jakýmkoli způsobem tuto translokaci prokázat. Může být zachycena cytogeneticky na úrovni chromozomální analýzy, nebo imunologicky na úrovni proteinů anebo na úrovni nukleových kyselin metodami molekulární genetiky. Vzhledem k často vyžadované rychlosti průkazu (tab. 1) má v diagnostice zcela výsadní místo PCR metoda pro detekci fúzního genu PML/RARα [13,23,70,71]. O situacích, kdy by vyšetření mělo být požadováno ve „STATIM“ režimu, již bylo pojednáno výše (v odstavci Morfologie).
Metody konvenční cytogenetiky pro záchyt t(15;17) (G - a Q-pruhování, tj. identifikace pruhů na chromozómech po barvení podle Giemsy, resp. po fluorescenčním barvení chinakrinem), dovolily v roce 1976 Golombovi et al odhalit inzerci na chromozomu 17 u 2 pacientů s APL [72,73], o rok později stejná skupina z Chicaga (Rowleyová et al) zjistila při popisu 3. případu (vzorku z periferní krve!), že jde o translokaci mezi chromozomy 15 a 17 se zlomovými místy v oblastech t(15;17)(q22;q21), která je balancovaná [25], a navíc reciproká [74] (obr. 6). V roce 1984 pak stejná skupina publikovala, že tato translokace byla zachycena u každého z 27 vyšetřených pacientů s APL. V této práci jsou také přesně lokalizována zlomová místa, a sice 15q22 a 17q21.1 [24]. Pozdější molekulární analýzy potvrdily správné určení míst zlomů na obou chromozomech [75–77]. WHO klasifikace [11] tedy uvádí (podle dřívější cytogenetické literatury a projektu Human Gene Mapping 9 z roku 1987) patrně nesprávný lokus 17q11.2–12 pro zlom na chromozomu 17.

Praxe v jiných institucích se neukázala být tak dokonalá, jak naznačovala publikace se zjištěním translokace t(15;17) ve 100 % případů [24]: běžně se zachytí u 41–97 % morfologicky diagnostikovaných případů [76,78]. Může být problém získat dostatek metafází k vyšetření – APL buňky mají nízkou syntézu DNA (podle studií s inkorporací 3H-thymidinu nižší než normální promyelocyty) i nízký mitotický index [79,80], navíc problémem mohou být také kontaminující dělící se normální buňky, např. erytroblasty, navíc se stává, že je k dispozici málo materiálu k analýze, neboť se klinikovi odebírajícímu dřeň může vzorek ve zkumavce srazit následkem DIC. Pozorování, že fúzní gen PML/RARα byl přítomen u všech případů s APL, zatímco fúzní gen RARα/PML jen u 12/18 pacientů, ukázaly, že t(15;17) nemusí být vždy reciproká [76] a že může jít o kryptické, submikroskopické translokace [23,31,81].
Význam standardního cytogenetického vyšetření se dnes, v době molekulárních analýz, snížil především z hlediska diagnostiky typické t(15;17), avšak standardní analýza stále poskytuje důležitou možnost záchytu ně-kterých alternativních translokací. Dosud jich bylo popsáno 5: především t(11;17)(q23;q21) s fúzním genem PLZF/RARα a reciprokým transkriptem RARα/PLZF (vyskytuje se zhruba v 0,8 % případů APL), zhruba u 0,4 % případů APL lze prokázat t(5;17)(q35;q21) s transkriptem NPM/RARα (též i reciprokým). Zbylé 3 typy cytogenetických a molekulárních aberací byly popsány pouze v ojedinělých případech. Jedná se o t(11;17)(q13;q21) s fúzním genem NuMA/RARα (bez tvorby reciprokého transkriptu), o intersticiální deleci (3 Mb dlouhou), která vede k tvorbě fúzního genu STAT5b/RARα (logicky také bez reciprokého transkriptu) [31,32], a posledně o nedávno popsanou fúzi PRKAR1A/RARα, vzniklou patrně rovněž intersticiální delecí [82]. Jak již bylo uvedeno, stále existuje malý zlomek případů s APL (s FAB AML M3 morfologií), u nichž nelze prokázat žádnou z výše uvedených genetických aberací (celkově zhruba 1 % všech pacientů s AML FAB M3 či M3v) [31]. Význam možného záchytu komplexních translokací a ev. přídatných cytogenetických aberací dnes již není velký – pokud se prokážou, nemění prognózu onemocnění (nicméně nelze vyloučit, že z hlediska prognózy jednotlivého pacienta mohou mít některé komplexní přestavby negativní dopad). Zlomy na chromozomech 15 a 17 jsou při komplexních aberacích stejné jako u klasické t(15;17) a vždy lze prokázat fúzní gen PML/RARα [81,83]. Nejčastější sekundární cytogenetickou aberací je trizomie 8, vyskytuje se u 17 % nemocných [84]. V případě některých překvapivých výsledků cytogenetiky, jako byl např. náš záchyt t(14;22), je nutno pátrat po vrozené aberaci, nesouvisející s onemocněním. Výsledek konvenční cytogenetiky může být hotov nejdříve za 24 hod (tab. 1), obvykle to však trvá déle, mj. také podle zvolené délky kultivace, potřebné pro získání dostatečného počtu mitóz. Doporučuje se kultivovat buňky nejméně 48 hod, protože buňky s fúzním genem PML/RARα mají delší generační čas a při delší kultivaci snáze získáme dostatečný počet mitóz pro cytogenetickou analýzu.
Detekce t(15;17) pomocí fluorescenční in situ hybridizace (FISH) patří mezi metody molekulární cytogenetiky a při detekci fúze PML/RARα má oproti karyotypové analýze jader v metafázi k průkazu t(15;17) obecně tu výhodu, že může k vyšetření použít buňky v interfázi a nevyžaduje kultivaci. Pomocí FISH se zachytí fúze PML/RARα častěji než t(15;17) při standardní cytogenetické analýze. FISH dosahuje citlivosti 10–1 až 10‑2 a lze ji provést během několika hodin (tab. 1). Obvykle se v cytogenetické laboratoři provádí molekulární vyšetření jako doplňkové ke klasické analýze karyotypu.
Detekce PML/RARα pomocí polymerázové řetězové reakce po reverzní transkripci (RT‑PCR) je na většině pracovišť základní vyšetřovací metodou pro detekci fúze PML/RARα. Spočívá v amplifikaci genových úseků vymezených nukleotidovými primery specifickými pro jednotlivé zlomové oblasti fúzních genů (obr. 6). V ÚHKT používáme přední primery M2 a M4 a reverzní primery R5 a R8 podle jedné z původních prací Biondiho et al [27] o RT‑PCR metodice pro detekci PML/RARα (tab. 3). Místo hybridizace těchto primerů je znázorněno na obr. 7. Jiné primery jsou uvedeny v doporučeních skupiny BIOMED-1 [85]. RT‑PCR je jedinou dostupnou metodou rutinní diagnostiky, která je schopna definovat typ zlomového místa v genu PML (což má kruciální význam pro další sledování a detekci minimální reziduální nemoci). Ve většině laboratoří se rutinně detekují zlomy bcr1 (z hlediska výsledného transkriptu dlouhá L-forma) a bcr2 (variabilní V-forma) pomocí stejného „forward“ (předního) primeru v jedné PCR, zatímco případy se zlomem bcr3 (s výsledným krátkým transkriptem; S-formou) se zachytávají ve druhé, paralelně prováděné PCR (obr. 7) (13,70). Ve skupině 116 pacientů zachycených v ÚHKT mělo 43,1 % jedinců zlom bcr1 a 50,0 % zlom bcr3. Vzácné případy s variabilně umístěným zlomem bcr2 v 6. exonu genu PML (6,8 % záchytů na naší laboratoři) je nutné verifikovat sekvenací – na elektroforetickém gelu mohou být PCR produkty vzhledem k proměnlivé délce transkriptu u různých případů různě vysoko (obr. 8) a mohou být omylem (např. při slabší expresi PML/RARα) považovány i za nespecificitu. Původně se RT‑PCR prováděla ve dvou krocích, kdy ve druhém kroku byly užity jiné primery než v kroku prvním (tzv. „nested“ metoda) [27–29]. Jednokroková RT‑PCR [12] je stále ještě dostatečně citlivá (10–3 až 10–4), je však rychlejší – výsledek je k dispozici za 5–6 hod (tab. 1). Zásadní podmínkou úspěšného vyšetření přítomnosti genu PML/RARα metodou RT‑PCR a případně jeho další sledování ve formě minimálního reziduálního onemocnění (MRO) je kvalitní vzorek RNA a účinná reverzní transkripce [12]. Transkript PML/RARα je dosti fragilní a je třeba šetrně zacházet s odebraným vzorkem – již jsme viděli případ, kdy vlivem špatného zacházení (zahřátí v saku) nebyl amplifikovatelný gen PML/RARα, zatímco kontrolní gen ABL ještě amplifikovatelný byl. O indikacích molekulárního vyšetření, tj. o situacích, kdy by bylo vyslovenou chybou jej neučinit, již bylo referováno zevrubně výše (viz odstavec Morfologie). Zde ještě zmíníme, že někteří autoři (např. z Británie) nepovažují za nutné vyšetřovat každý případ záchytu AML na fúzní gen PML/RARα [86]. Ve studiích britského Medical Research Council našli pouze jednoho z 530 pacientů, u kterého byla ex post zjištěna tato fúze. Nutno si však uvědomit, že to byli zevrubně vyšetření pacienti zařazovaní do studií, nikoli běžný vzorek pacientů. Autoři citované práce nemohli vědět, kolik pacientů s nepoznaným fúzním genem PML/RARα jim mohlo „utéci“ mimo rámec studií. My se kloníme spíše k názoru jiných autorů (např. [57]), kteří z důvodu nemožnosti morfologického rozeznání zcela atypických případů molekulární detekci doporučují u všech AML. Mezi našimi 61 případy APL byli přinejmenším 3, které bychom na základě morfologického nálezu z APL vůbec nepodezírali.
![Sekvence primerů používaných v ÚHKT pro záchyt PML/RARa i detekci minimálního reziduálního onemocnění. Jsou velmi podobné (nikoli identické) s primery publikovanými Biondim et al [27], jsou i stejně označeny. M2 a M4 jsou přední primery, R5 a R8 reverzní. Místa jejich hybridizace s cDNA jsou znázorněna na obr. 7. Pro záchyt L a V transkriptů (se zlomy bcr1 a bcr2 v genu PML) užíváme dvojici primerů M2-R8, PCR produkt má 326 bp. Pro záchyt S transkriptů (se zlomem bcr3) požíváme primery M4‑R8, výsledný PCR produkt je 324 bp dlouhý.](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/1b435cd36083adc64e4ff16e22f7182d.png)
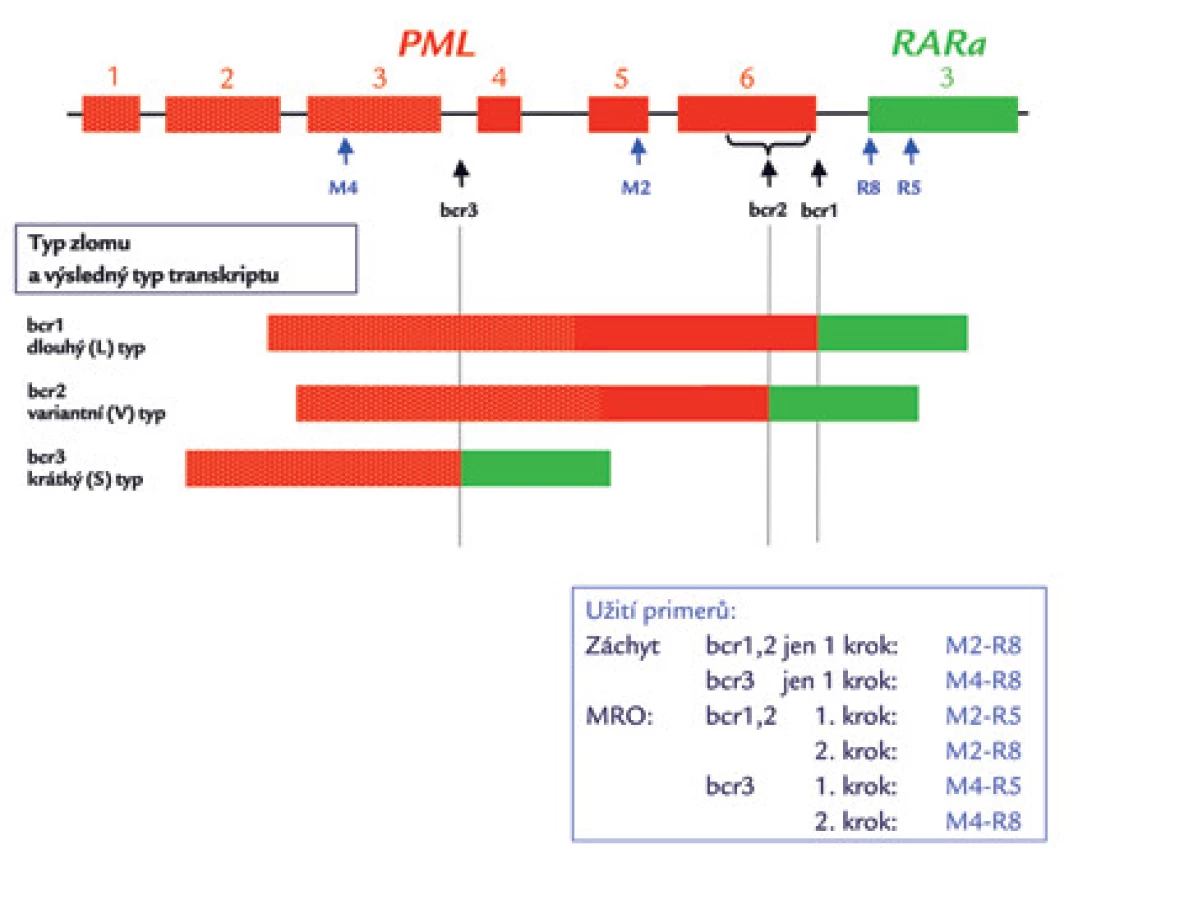

Záchyt PML/RARα pomocí RT‑PCR v reálném čase (RQ‑PCR) je možnou alternativou kvalitativní RT‑PCR (viz předchozí odstavec). Pro záchyt fúzních genů při zlomech genu PML bcr1, bcr2 a bcr3 se při RQ‑PCR používá společné sondy a reverzního primeru, zatímco přední primer je pro každý ze tří zlomů odlišný [77]. Tato metodika, vyvinutá v rámci programu Europe Against Cancer, má výhodu v tom, že již při záchytu odliší L od V formy transkriptu (tzn. PML/RARα se zlomy bcr1 a bcr2 v genu PML), při sledování minimálního reziduálního onemocnění je navíc ihned k dispozici výsledek dne 0 a odpadá potřeba provádět standardní, kvalitativní RT‑PCR při záchytu. Na této metodě je však problematické to, že reakce pro záchyt V formy dokáže zachytit pouze 80 % případů V formy PML/RARα s různými subtypy zlomů genu PML, souhrnně nazývanými bcr2 [77] (obr. 6 a obr. 7). To znamená, že při užívání této metody se diagnózu (na rozdíl od klasické RT‑PCR) nepodaří odhalit u zhruba 2 % pacientů s fúzí PML/RARα. V ÚHKT ji proto k záchytu onemocnění nepoužíváme.
Interní tandemová duplikace genu FLT3 (FLT3-ITD) je společně s mutací v kodónu D835 genu FLT3 nejčastější sekundární genetickou lézí u APL. Morfologicky variantní formy M3v jsou vůbec nejčastějším subtypem AML, u kterého je FLT3-ITD zjistitelná – vyskytuje se až v 65 % těchto případů a ve 23 % případů APL s klasickou morfologií M3 [87]. FLT3 je receptorová tyrozinová kináza a mutace jejího genu dává buňkám [nezávisle na ligandu, cytokinu FL (FLT3‑ligandu)] mocný proliferační impuls. FLT3-ITD je jednou z možných příčin, proč jsou v případech M3v v průměru vyšší počty leukocytů a pravděpodobně i horší šance na dosažení kompletní remise. Naopak je‑li remise dosaženo, je pak lhostejné, zda byla původně přítomna FLT3-ITD. Tato situace je naprosto odlišná od jiných subtypů AML, u kterých FLT3-ITD významně neovlivňuje dosažení kompletní remise, ale má dopad na incidenci relapsů [87–90].
Rozvržení proteinu PML v buněčném jádře – detekce monoklonální protilátkou
V chromatinu buněk pacientů s AML bez t(15;17) dochází ke shlukování PML proteinu v tzv. nukleárních tělískách, kterých bývá v jednom buněčném jádře zhruba 5–30. Někdy jsou popisována zkratkou POD (PML onkogenní domény). Označíme‑li PML v nukleárních tělískách anti-PML monoklonální protilátkou 5E10 nebo PG-M3, ev. polyklonálním antisérem, PML se pak v preparátu znázorní jako velmi hrubé skvrny. U AML s fúzním proteinem PML/RARα dochází působením tohoto proteinu k disrupci nukleárních tělísek a při označení PML protilátkou dostáváme typický mikrotečkovaný obrazec. K vizualizaci reakce s monoklonální protilátkou lze použít jak imunofluorescenční [13–17], tak i imunohistochemickou techniku (s použitím imuno-alkalické fosfatázy) [15]. Me-toda je vysoce specifická pro AML s t(15;17), avšak může být zatížena technickým problémem (jako v jednom případě v původní publikaci Dyckové et al [14,16]). Hlavní výhodou těchto metodik je jednak rychlost (výsledek je k dispozici do 2 hod, viz tab. 1), jednak se můžeme vyhnout potřebě drahých zařízení pro molekulární biologii. Hlavní nevýhodou je, že metoda neodliší různé zlomy v genu PML, které je nutné znát pro následné sledování minimálního reziduálního onemocnění, při kterém je doporučenou metodou RT‑PCR, nejlépe kvantitativní. V zahraničí byla metoda diagnostiky pomocí anti-PML protilátky relativně oblíbená, v Česku ji, pokud víme, nikdo neužívá.
Závěr
Shora jsme podali přehled diagnostického minima APL, se kterým by podle našich představ měl být seznámen každý internista, nejlépe však i každý praktický lékař. Věříme, že širší povědomí o tomto onemocnění pomůže urychlit diagnostický proces, který musí vést ke včasnému nasazení specifické léčby. Vzhledem k její vysoké úspěšnosti mohou být zachráněny lidské životy. Dále pak byly podrobně rozvedeny diagnostické a diferenciálně diagnostické otázky pro specialisty-hematology, kteří musí být v otázce APL vysoce erudováni.
Doručeno do redakce: 9. 6. 2008
MUDr. Jiří Schwarz, CSc.
www.uhkt.cz
e‑mail: jiri.schwarz@uhkt.cz
Zdroje
1. Bernard J, Mathé G, Boulay J et al. La leucose aiguë à promyélocytes. Etude portant sur vingt observations. Schweiz Med Wochenschr 1959; 89 : 604–608.
2. Baker WG, Bang NU, Nachman RL et al. Hypofibrinogenemic hemorrhage in acute myelogenous leukemia treated with heparin. With autopsy findings of widespread intravascular clotting. Ann Intern Med 1964; 61 : 116–123.
3. Schwarz J, Lemez P, Lukasova M et al. High incidence of thromboembolic events in leukemic patients with acute promyelocytic leukemia (APL). Blood 1996; 88 : 169b (Abstract 3404).
4. Sanz MA, Tallman MS, Lo-Coco F. Tricks of the trade for the appropriate management of newly diagnosed acute promyelocytic leukemia. Blood 2005; 105 : 3019–3025.
5. Lemež P, Schwarz J, Jelínek J et al. Pozdní a pomalá diagnostika akutních promyelocytárních leukemií – hlavní příčina časných smrtí. Vnitř Lék 1994; 40 : 654–659.
6. Bennett JM, Catovsky D, Daniel MT et al. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol 1976; 33 : 451–458.
7. Bennett JM, Catovsky D, Daniel MT et al. A variant form of hypergranular promyelocytic leukaemia (M3). Br J Haematol 1980; 44 : 169–170.
8. Kačírková P, Campr V, Karban J et al. Hematoonkologický atlas krve a kostní dřeně. Praha: Grada 2007.
9. Sainty D, Liso V, Cantù-Rajnoldi A et al. A new morphologic classification system for acute promyelocytic leukemia distinguishes cases with underlying PLZF/RARA gene rearrangements. Blood 2000; 96 : 1287–1296.
10. Harris NL, Jaffe ES, Diebold J et al. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues: report of the Clinical Advisory Committee meeting – Airlie House, Virginia, November, 1997. J Clin Oncol 1999; 17 : 3835–3849.
11. Brunning RD, Bennett J, Matutes E et al. Acute myeloid leukaemia with recurrent genetic abnormalities. In: Jaffe ES, Harris NL, Stein H et al (eds). World Health Organization classification of tumours. Pathology and genetics. Tumours of haematopoietic and lymphoid tissues. Lyon: IARC Press 2001 : 81–87.
12. Diverio D, Riccioni R, Pistilli A et al. Improved rapid detection of the PML/RARα fusion gene in acute promyelocytic leukemia. Leukemia 1996; 10 : 1214–1216.
13. Lo Coco F, Diverio D, Falini B et al. Genetic diagnosis and molecular monitoring in the management of acute promyelocytic leukemia. Blood 1999; 94 : 12–22.
14. Dyck JA, Warrell RP Jr, Evans RM et al. Rapid diagnosis of acute promyelocytic leukemia by immunohistochemical localization of PML/RARα protein. Blood 1995; 86 : 862–867.
15. Falini B, Flenghi L, Fagioli M et al. Immunocytochemical diagnosis of acute promyelocytic leukemia (M3) with the monoclonal antibody PG-M3 (anti‑PML). Blood 1997; 90 : 4046–4053.
16. Samoszuk MK, Tynan W, Sallash G et al. An immunofluorescent assay for acute promyelocytic leukemia cells. Am J Clin Pathol 1998; 109 : 205–210.
17. O‘Connor SJM, Evans PAS, Morgan GJ. Diagnostic approaches to acute promyelocytic leukaemia. Leuk Lymphoma 1999; 33 : 53–63.
18. Lo Coco F, Nervi C, Avvisati G et al. Acute promyelocytic leukemia: a curable disease. Leukemia 1998; 12 : 1866–1880.
19. Liso V, Bennett J. Morphological and cytochemical characteristics of leukaemic promyelocytes. Best Pract Res Clin Haematol 2003; 16 : 349–355.
20. Paietta E, Andersen J, Gallagher R et al. The immunophenotype of acute promyelocytic leukemia (APL): an ECOG study. Leukemia 1994; 8 : 1108–1112.
21. Paietta E, Andersen J, Racevskis J et al. Significantly lower P-glycoprotein expression in acute promyelocytic leukemia than in other types of acute myeloid leukemia: immunological, molecular and functional analyses. Leu kemia 1994; 8 : 968–973.
22. Paietta E. Expression of cell-surface antigens in acute promyelocytic leukaemia. Best Pract Res Clin Haematol 2003; 16 : 369–385.
23. Grimwade D, Howe K, Langabeer S et al. Establishing the presence of the t(15;17) in suspected acute promyelocytic leukaemia: cytogenetic, molecular and PML immuno-fluorescence assessment of patients entered into the M.R.C. ATRA trial. Adult leukaemia working party. Br J Haematol 1996; 94 : 557–573.
24. Larson RA, Kondo K, Vardiman JW et al. Evidence for a 15;17 translocation in every patient with acute promyelocytic leukemia. Am J Med 1984; 76 : 827–841.
25. Rowley JD, Golomb HM, Dougherty C. 15/17 translocation, a consistent chromosomal change in acute promyelocytic leukaemia. Lancet 1977; 1 : 549–550.
26. Mancini M, Nanni M, Cedrone M et al. Combined cytogenetic, FISH and molecular analysis in acute promyelocytic leukaemia at diagnosis and in complete remission. Br J Haematol 1995; 91 : 878–884.
27. Biondi A, Rambaldi A, Pandolfi PP et al. Molecular monitoring of the myl/retinoic acid receptor-alpha fusion gene in acute promyelocytic leukemia by polymerase chain reaction. Blood 1992; 80 : 492–497.
28. Chen SJ, Chen Z, Chen A et al. Occurrence of distinct PML‑RARα fusion gene isoforms in patients with acute promyelocytic leukemia detected by reverse transcriptase/polymerase chain reaction. Oncogene 1992; 7 : 1223–1232.
29. Miller WH Jr, Kakizuka A, Frankel SR et al. Reverse transcription polymerase chain reaction for the rearranged retinoic acid receptor alpha clarifies diagnosis and detects minimal residual disease in acute promyelocytic leukemia. Proc Natl Acad Sci USA 1992; 89 : 2694–2698.
30. Head DR. Revised classification of acute myeloid leukemia. Leukemia 1996; 10 : 1826–1831.
31. Grimwade D, Biondi A, Mozziconacci MJ et al. Characterization of acute promyelocytic leukemia cases lacking the classic t(15;17): results of the European Working Party. Blood 2000; 96 : 1297–1308.
32. Mistry AR, Pedersen EW, Solomon E et al. The molecular pathogenesis of acute promyelocytic leukaemia: implications for the clinical management of the disease. Blood Rev 2003; 17 : 71–97.
33. Mrózek K, Heerema NA, Bloomfield CD. Cytogenetics in acute leukemia. Blood Rev 2004; 18 : 115–136.
34. Douer D, Preston-Martin S, Chang E et al. High frequency of acute promyelocytic leukemia among Latinos with acute myeloid leukemia. Blood 1996; 87 : 308–313.
35. Tomás JF, Fernández-Rañada JM. About the increased frequency of acute promyelocytic leukemia among Latinos: the experience from a center in Spain. Blood 1996; 88 : 2357–2358.
36. Hernández P, Milanés MT, Svarch E et al. High relative proportion of acute promyelocytic leukemia in children: experience of a multicenter study in Cuba. Leuk Res 2000; 24 : 739–740.
37. Douer D, Santillana S, Ramezani L et al. Acute promyelocytic leukaemia in patients originating in Latin America is associated with an increased frequency of the bcr1 subtype of the PML/RARα fusion gene. Br J Haematol 2003; 122 : 563–570.
38. Ruiz-Argüelles GJ, Garcés-Eisele J, Reyes-Núñez V et al. More on geographic hematology: the breakpoint cluster regions of the PML/RARα fusion gene in Mexican Mestizo patients with promyelocytic leukemia are different from those in Caucasians. Leuk Lymphoma 2004; 45 : 1365–1368.
39. Vickers M, Jackson G, Taylor P. The incidence of acute promyelocytic leukemia appears constant over most of a human lifespan, implying only one rate limiting mutation. Leukemia 2000; 14 : 722–726.
40. Cantú-Rajnoldi A, Biondi A, Jankovic M et al. Diagnosis and incidence of acute promyelocytic leukemia (FAB M3 and M3 variant) in childhood. Blood 1993; 81 : 2209–2210.
41. Matasar MJ, Ritchie EK, Consedine N et al. Incidence rates of acute promyelocytic leukemia among Hispanics, blacks, Asians, and non-Hispanic whites in the United States. Eur J Cancer Prev 2006; 15 : 367–370.
42. Ruiz-Argüelles GJ. Promyelocytic leukemia in Mexican Mestizos. Blood 1997; 89 : 348–349.
43. Castoldi GL, Liso V, Specchia G et al. Acute promyelocytic leukemia: morphological aspects. Leukemia 1994; 8(Suppl 2): S27–S32.
44. Golomb HM, Rowley JD, Vardiman JW et al. “Microgranular” acute promyelocytic leukemia: a distinct clinical, ultrastructural, and cytogenetic entity. Blood 1980; 55 : 253–259.
45. Galton DAG, Dacie JV, Peto R. The relation between morphology and other features of acute myeloid leukaemia, and their prognostic significance. Report of the Medical Research Council‘s Working Party on Leukaemia in Adults. Br J Haematol 1975; 31 : 65–180.
46. Davey FR, Davis RB, MacCallum JM et al. Morphologic and cytochemical characteristics of acute promyelocytic leukemia. Am J Hematol 1989; 30 : 221–227.
47. Valdivieso M, Rodriguez V, Drewinko B et al. Clinical and morphological correlations in acute promyelocytic leukemia. Med Pediatr Oncol 1975; 1 : 37–50.
48. Tan HK, Wages B, Gralnick HR. Ultrastructural studies in acute promyelocytic leukemia. Blood 1972; 39 : 628–636.
49. Liso V, Troccoli G, Grande M. Cytochemical study of acute promyelocytic leukaemia. Blut 1975; 30 : 261–268.
50. Kubonishi I, Fujishita M, Niiya K et al. Basophilic differentiation in acute promyelocytic leukemia. Nippon Ketsueki Gakkai Zasshi 1985; 48 : 1390–1396.
51. Koike T, Tatewaki W, Aoki A et al. Brief report: severe symptoms of hyperhistaminemia after the treatment of acute promyelocytic leukemia with tretinoin (all‑trans‑retinoic acid). N Engl J Med 1992; 327 : 385–387.
52. Tallman MS, Hakimian D, Snower D et al. Basophilic differentiation in acute promyelocytic leukemia. Leukemia 1993; 7 : 521–526.
53. McKenna RW, Parkin J, Bloomfield CD et al. Acute promyelocytic leukaemia: a study of 39 cases with identification of a hyperbasophilic microgranular variant. Br J Haematol 1982; 50 : 201–214.
54. Das Gupta A, Sapre RS, Shah AS et al. Cytochemical and immunophenotypic heterogeneity in acute promyelocytic leukemia. Acta Haematol 1989; 81 : 5–9.
55. Lemež P. A case of acute promyelo-cytic leukaemia with “faggot cells” exhibiting strong alpha-naphthylbutyrate esterase activity. Br J Haematol 1988; 68 : 138–139.
56. Neame PB, Soamboonsrup P, Leber B et al. Morphology of acute promyelocytic leukemia with cytogenetic or molecular evidence for the diagnosis: characterization of additional microgranular variants. Am J Hematol 1997; 56 : 131–142.
57. Foley R, Soamboonsrup P, Kouroukis T et al. PML/RARα APL with undifferentiated morphology and stem cell immunophenotype. Leukemia 1998; 12 : 1492–1493.
58. Matutes E, Morilla R, Farahat N et al. Definition of acute biphenotypic leukemia. Haematologica 1997; 82 : 64–66.
59. Aventín A, Mateu R, Martino R et al. A case of cryptic acute promyelocytic leukemia. Leukemia 1998; 12 : 1490–1491.
60. Morgan DL, Dunn DM, Cobos E et al. Translocation t(15;17) in acute myelogenous leukemia with atypical megaka-ryoblastic features: diagnostic, clinical, and therapeutic implications. Cancer Genet Cytogenet 1996; 92 : 50–53.
61. Yu RQ, Huang W, Chen SJ et al. A case of acute eosinophilic granulocytic leukemia with PML‑RAR alpha fusion gene expression and response to all‑trans retinoic acid. Leukemia 1997; 11 : 609–611.
62. Fukuno K, Tsurumi H, Yoshikawa T et al. A variant form of acute promyelocytic leukemia with marked myelofibrosis. Int J Hematol 2001; 74 : 322–326.
63. Lemež P. Cytochemické nálezy u akutních promyelocytárních leukemií. Sb Lék 1988; 90 : 14–20.
64. Guglielmi C, Martelli MP, Diverio D et al. Immunophenotype of adult and childhood acute promyelocytic leukaemia: correlation with morphology, type of PML gene breakpoint and clinical outcome. A cooperative Italian study on 196 cases. Br J Haematol 1998; 102 : 1035–1041.
65. Di Noto R, Lo Pardo C, Schiavone EM et al. Stem cell factor receptor (c-kit, CD117) is expressed on blast cells from most immature types of acute myeloid mallignancies but is also a characteristic of a subset of acute promyelocytic leukaemia. Br J Haematol 1996; 92 : 562–564.
66. Paietta E, Goloubeva O, Neuberg D et al. A surrogate marker profile for PML/RARα expressing acute promyelocytic leukemia and the association of immunophenotypic markers with morphologic and molecular subtypes. Cytometry B Clin Cytom 2004; 59 : 1–9.
67. Murray CK, Estey E, Paietta E et al. CD56 expression in acute promyelocytic leukemia: a possible indicator of poor treatment outcome? J Clin Oncol 1999; 17 : 293–297.
68. Di Bona E, Sartori R, Zambello R et al. Prognostic significance of CD56 antigen expression in acute myeloid leukemia. Hae-matologica 2002; 87 : 250–256.
69. Scott AA, Head DR, Kopecky KJ et al. HLA‑DR–, CD33+, CD56+, CD16 – myeloid/natural killer cell acute leukemia: a previously unrecognized form of acute leukemia potentially misdiagnosed as French-American-British acute myeloid leukemia-M3. Blood 1994; 84 : 244–255.
70. Grignani F, Fagioli M, Ferrucci PF et al. The molecular genetics of acute promyelocytic leukemia. Blood Rev 1993; 7 : 87–93.
71. Schwarz J, Pechová R, Haškovec C. Místo PCR vyšetření hybridního genu PML/RARα v komplexní diagnostice AML. Scr Med (Brno) 1997; 70 : 291–297.
72. Golomb HM, Rowley J, Vardiman J et al. Partial deletion of long arm of chromosome 17: a specific abnormality in acute promyelocytic leukemia? Arch Intern Med 1976; 136 : 825–828.
73. Golomb HM, Vardiman J, Rowley JD. Acute nonlymphocytic leukemia in adults: correlations with Q-banded chromosomes. Blood 1976; 48 : 9–21.
74. Rowley JD, Golomb HM, Vardiman J et al. Further evidence for a non-random chromosomal abnormality in acute promyelocytic leukemia. Int J Cancer 1977; 20 : 869–872.
75. Mattei MG, Petkovich M, Mattei JF et al. Mapping of the human retinoic acid receptor to the q21 band of chromosome 17. Hum Genet 1988; 80 : 186–188.
76. Borrow J, Solomon E. Molecular analysis of the t(15;17) translocation in acute promyelocytic leukaemia. Baillière’s Clin Haematol 1992; 5 : 833–856.
77. Gabert J, Beillard E, van der Velden VHJ et al. Standardization and quality control studies of “real-time” quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia – a Europe Against Cancer program. Leukemia 2003; 17 : 2318–2357.
78. Frankel SR. Acute promyelocytic leukemia. New insights into diagnosis and therapy. Hematol Oncol Clin North Am 1993; 7 : 109–138.
79. Pileri A, Pegoraro L, Gavosto F. Cytogenetical and proliferative characteristics of acute promyelocytic leukaemia cells. Eur J Cancer 1966; 2 : 189–192.
80. Sjögren U. Mitotic activity in acute promyelocytic leukaemia and leukaemoid reactions. Acta Med Scand 1976; 199 : 181–183.
81. Brunel V, Lafage-Pochitaloff M, Alcalay M et al. Variant and masked translocations in acute promyelocytic leukemia. Leuk Lymphoma 1996; 22 : 221–228.
82. Catalano A, Dawson MA, Somana K et al. The PRKAR1A gene is fused to RARA in a new variant acute promyelocytic leukemia. Blood 2007; 110 : 4073–4076.
83. Slack JL, Arthur DC, Lawrence D et al. Secondary cytogenetic changes in acute promyelocytic leukemia – prognostic im-portance in patients treated with chemotherapy alone and association with the intron 3 breakpoint of the PML gene: a Cancer and Leukemia Group B study. J Clin Oncol 1997; 15 : 1786–1795.
84. Berger R, Le Coniat M, Derré J et al. Cytogenetic studies in acute promyelocytic leukemia: a survey of secondary chromosomal abnormalities. Genes Chromosomes Cancer 1991; 3 : 332–337.
85. van Dongen JJ, Macintyre EA, Gabert JA et al. Standardized RT‑PCR analysis of fusion gene transcripts from chromosome aberrations in acute leukemia for detection of minimal residual disease. Report of the BIOMED-1 concerted action: Investigation of minimal residual disease in acute leukemia. Leukemia 1999; 13 : 1901–1928.
86. Allford S, Grimwade D, Langabeer S et al. Identification of the t(15;17) in AML FAB types other than M3: evaluation of the role of molecular screening for the PML/RARα rearrangement in newly diagnosed AML. Br J Haematol 1999; 105 : 198–207.
87. Schnittger S, Schoch C, Dugas M et al. Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood 2002; 100 : 59–66.
88. Kiyoi H, Naoe T, Yokota S et al. Internal tandem duplication of FLT3 associated with leukocytosis in acute promyelocytic leukemia. Leukemia 1997; 11 : 1447–1452.
89. Schwarz J, Peková S, Cermák J et al. Prognosis in AML and APL: the role of FLT3 gene internal tandem duplications (ITDs) and other prognostic markers. Leuk Lymphoma 2003; 44 (Suppl): S64.
90. Au WY, Fung A, Chim CS et al. FLT-3 aberrations in acute promyelocytic leukaemia: clinicopathological associations and prognostic impact. Br J Haematol 2004; 125 : 463–469.
Štítky
Diabetologie Endokrinologie Interní lékařstvíČlánek vyšel v časopise
Vnitřní lékařství

2008 Číslo 7-8
- Limity glykovaného hemoglobinu a význam dalších glykovaných proteinů
- Inovace v hojení ran: krytí Zetuvit Plus Silicone Border pro optimální management exsudátu z ran
- Ivabradin zlepšuje kvalitu života starších pacientů se srdečním selháním
Nejčtenější v tomto čísle
- Urgentní stav v hematologii: akutní promyelocytární leukemie – principy diagnostiky
- Koagulopatie a diferenciační syndrom: hlavní komplikace úvodní léčby akutní promyelocytární leukemie
- Stručné kazuistiky ilustrující úvodní průběh u akutní promyelocytární leukemie
- Akutní promyelocytární leukemi e: cesta k nejlépe léčitelné akutní leukemii dospělých
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy