
Od chloromu k akutní promyelocytární leukemii – pohledy do historie
From chloroma to acute promyelocytic leukemia – a historical perspective
The progressing knowledge on chloroma and chloroleukemia is reviewed. However, it is uneasy to identify with certainty the cases of acute promyelocytic leukemia (APL) among the historical descriptions of chloroma since 1823. In part, this is due to confusion produced by the historical cytological nomenclature – in practice, the leukemic promyelocytes were regarded, quite inaccurately, as a subtype of paramyeloblasts until early 1960’s. The term promyelocytic leukemia first appeared in Naegeli’s text-book in 1931. However, no clinical associations of the morphological description were then given. As a clinico-pathological entity, APL was defined by Hillestad in Norway in 1957 and on a more detailed level, by Bernard et al in France in 1959. However, the descriptions of chloroma by Butterfield in 1909 and of leukemia with panmyelophthisis and defibrination by Risak in 1935 in the German-written literature and the case of chloroma described by Libánský in Czech in 1939 were cases of APL with high probability. Further on, experimental studies leading to the postulation of possible differentiation-inducing therapy of leukemia are reviewed – the importance of introducing of clonal culture of hematopoietic cells in the 1960’s and the proof of the possibility of bypassing the maturation block in the works of Sachs’s group in Rehovot, Israel, is stressed, as well as the later experiments with differentiation inducers in the cell line models in the 1970’s and 1980’s. The first trials with retinoids in therapy of APL are mentioned, along with the interesting background of the French-Chinese collaboration (under the auspices of professors Degos and Wang) in the introduction of retinoids into practice in the second half of the 1980’s. It is discussed how the success of the clinical trials led to enormous progression in the field of molecular genetics of APL, which, in turn, led to development of a new generation of remedies, i.e. to the renaissance of arsenic in the treatment of APL in the 1990’s, among others thanks to the current Chinese minister of health, Chen Zhu.
Key words:
history – chloroma – chloroleukemia – acute promyelocytic leukemia – morphology – differentiation induction – retinoids – ATRA – fusion genes – arsenic
Autoři:
J. Schwarz
Působiště autorů:
Klinický úsek Ústavu hematologi e a krevní transfuze Praha, přednosta doc. MUDr. Petr Cetkovský, Ph. D.
Vyšlo v časopise:
Vnitř Lék 2008; 54(7-8): 686-700
Kategorie:
Přehledné referáty
Souhrn
V práci je podán přehled vývoje poznání o chloromu a chloroleukemiích. Mezi historickými líčeními od roku 1823 se však nedaří s jistotou identifikovat případy akutní promyelocytární leukemie (APL). Je to zčásti na vrub poněkud málo přesné historické cytologické nomenklatury – leukemické promyelocyty byly v praxi považovány za subtyp paramyeloblastů až do začátku 60. let. 20. století. Prvně se objevil termín promyelocytární leukemie v Naegeliho učebnici z roku 1931, ale nebyly tehdy vylíčeny žádné klinické souvislosti popsané morfologie. APL byla definována jako klinicko-patologická jednotka teprve Hillestadem v Norsku v roce 1957 a podrobněji Bernardem et al ve Francii v roce 1959. Nicméně již popisy chloromu Butterfielda z roku 1909 a leukemie s panmyeloftízou a defibrinací Risaka z roku 1935 v německy psané literatuře a Libánského případ chloromu z roku 1939 v českém písemnictví byly patrně popisem APL s vysokou pravděpodobností. Dále je podán přehled experimentálních prací, které vedly k postulaci možné diferenciační terapie leukemií. Je zdůrazněn význam zavedení klonální kultivace hematopoetických buněk v 60. letech a prověření možnosti překonání maturačního bloku induktory diferenciace v pracích Sachsovy skupiny v izraelském Rehovotu, jakož i význam pozdějších experimentů s induktory diferenciace v modelu buněčných linií v 70. a 80. letech minulého století. Jsou zmíněny první klinické pokusy s retinoidy v léčbě APL i zajímavé pozadí francouzsko-čínské spolupráce (zastřešené profesory Degosem a Wangem) při zavádění retinoidů do praxe ve 2. polovině 80. let 20. století. Je zde popsáno, jak úspěch v klinické léčbě vedl k ohromnému rozvoji poznání molekulární genetiky APL, která obratem vedla k vývoji další generace léků, a to k renezanci arzeniku v léčbě APL v 90. letech minulého století, mj. i velkou zásluhou současného čínského ministra zdravotnictví Chena Zhu.
Klíčová slova:
historie – chlorom – chloroleukemie – akutní promyleocytární leukemie – morfologie – indukce diferenciace – retinoidy – ATRA – fúzní geny – arzenik
Věnování a úvod
Tento přehled obsahuje řadu (dnes z větší části) obsoletních, pro praxi většinou ne zcela potřebných informací a citací. Záměrně je však zařazuji jako projev úcty k nesčetným badatelům a badatelkám, ve své době slavným, ale také zcela neznámým, bez nichž bychom nebyli ve výzkumu a léčbě akutní promyelocytární leukemie (APL) tak daleko. Mají zásluhu na tom, že se z nejmalignějšího typu leukemie stalo onemocnění, při kterém dokážeme vrátit do plně kvalitního života většinu postižených. Cesty, po nichž se ubíralo poznávání nemoci, kterou dnes označujeme APL, byly značně klikaté. Nicméně z dnešního pohledu jsou takřka učebnicovým příkladem plodné spolupráce klinické medicíny a základního výzkumu, která, jak věřím, by mohla inspirovat i lékaře a výzkumníky stojící mimo obor hematologie.
Klinicko-patologická jednotka APL byla popsána až koncem 50. let 20. století. Avšak definice APL jistě v té době „nespadla z nebe“ – předcházelo jí 130leté bádání. Vydejme se po jeho stopách i v širších souvislostech a sledujme, jakým směrem se věda kolem APL ubírala dál.
Případ dr. Libánského
Začnu zcela nedidakticky od prostředku, líčením případu dr. Josefa Libánského (později profesora, přednosty Klinického oddělení ÚHKT v Praze) z roku 1939, kdy pracoval jako externí lékař pod vedením prof. Hynka na I. české klinice chorob vnitřních na Karlově náměstí (současná I. interní klinika 1. LF UK a VFN) v Praze. Jde o jeden z prvních podrobných popisů chloromu v česky psané literatuře (vůbec první zmínkou je krátký abstrakt Hynka a Kadlického v Časopise lékařů českých z roku 1911 [1] o případu, který pak byl in extenso publikován německy ve Vídni). Libánský popisuje případ „4letého děvčátka z rolnické rodiny od Českých Budějovic“ [2]. Po „angině těžšího průběhu s horečkou“ se u ní v květnu roku 1939 objevila malá bulka pod pravým horním víčkem, která byla vyoperována 7. 6. 1939 na Oční klinice prof. Dienstbiera. Dle „zelenavého zbarvení, jakož i lokalizace nádoru učinil prof. Kadlický, který nemocnou operoval, diagnózu chloromu“. Klobouk dolů před vzdělaným očařem. Histologický nález nebyl příliš reprezentativní: „…tkáň byla následkem pozdní nebo nedokonalé fixace silně autolyzována. Mikroskopický nález nevylučoval ev. leukeamický infiltrát.“ O týden později, při přijetí na I. internu, popisuje dr. Libánský bledost pacientky a v krajině spánkové vpravo ploché zduření velikosti holubího vejce a před ním menší, velikosti lískového oříšku. V té době měla pacientka hemoglobin 65 g/l, 252 000 trombocytů a počet leukocytů byl 17,7. V diferenciálním rozpočtu popisuje hiatus leukaemicus, s převahou myeloblastů a paramyeloblastů (celkem 53 % atypických buněk, včetně 3 % „chloromových“; o tom, co to jsou paramyeloblasty, viz níže). Asi v 5–10 % myeloblastů se nacházely tyčinkovité nebo vřetenovité útvary, jež pak dále autor ztotožňuje s „Auerovými tyčinkami“. Libánský připojil 2 mikrofotografie, obě ukazovaly po jedné buňce: obě buňky silně připomínají promyelocyty u APL. První je poměrně malý promyelocyt s excentricky uloženým bilobárním jádrem, cytoplazma je přeplněná tmavou hrubou granulací. Druhá buňka obsahuje v poměrně sporé, ale také granulární cytoplazmě 2 paralelně uložené Auerovy tyče. Libánský píše, že byly nalezeny mitózy a také „buňky s dvojdílným jádrem“. Pacientka však měla normální krvácivost dle Duka, srážlivost dle Bürkera a negativní Rumpel-Leedův fenomén. Nebyla popsána jakákoli hemoragická diatéza. Pacientka byla léčena malými dávkami arzenu, ale „než mohlo býti přikročeno k ozařování RTG, byla dne 24. 6. 1939 na žádost svých rodičů propuštěna domů“. Popis osudu děvčátka dále zmiňuje návštěvu u ní doma ze 6. 7. 1939. Byla kachektická, subfebrilní, mrzutá a plačtivá, s protruzí levého bulbu, po jehož laterální straně vyrůstal z orbity tumor velikosti holubího vejce. Na konci kazuistiky Libánský zmiňuje, že „podle zprávy došlé při 2. korektuře pacientka zemřela“. Celkové přežití děvčete tedy nemohlo být delší než 6 měsíců.
Otázka nyní zní: Jedná se o jeden z prvních popisů promyelocytární leukemie s extramedulárním postižením v české literatuře, nebo ne? Ve světle současných poznatků bychom spíše hádali, že patrně ano. Mikrofotografie buněk jsou pro APL typické a vysoce sugestivní. Jediný problém je v tom, proč by u dívky se 17,7 × 109/l leukocyty a tumoriformním postižením při diagnóze APL nebyly Libánským popsány krvácivé projevy, proč nebyla trombocytopenická, a proč navíc byly normální výsledky vyšetření hemostázy. Přežití blížící se zhruba polovině roku je rovněž překvapivě dlouhé – že by to bylo vlivem terapie arzenem? Bez léčby by pacientka s takto rozsáhlým postižením (leukocytózou a extramedulárními projevy) sotva žila déle než několik týdnů (viz dále Bernardovy případy). Další stín pochybnosti pramení z toho, že výskyt extramedulárního onemocnění při první prezentaci APL je vzácný – mnohem častěji se s ním setkáváme až v relapsu, zatímco u akutní myeloidní leukemie (AML) M2 dle FAB klasifikace je chlorom (čili zelenavý myelosarkom) či jiné formy extramedulárního postižení při první prezentaci onemocnění přece jen častější.
V diskuzi zmíněného článku [2] se uvádí, že myeloické chloromy s chloroleukemií jsou onemocněním převážně mladých lidí, manifestujícím se často nejprve hemoragickou diatézou – Libánský cituje další případ (patrně nepublikovaný prof. Hynkem): 8leté děvčátko mělo neztišitelné krvácení po extrakci stoličky, poruchy zraku a sluchu následkem hemoragické diatézy. U té šlo o leukopenickou formu myeloického chloromu. V tomto případě bychom si mohli být ještě jistější, že šlo o APL: nebyla zde leukocytóza, a navíc byla přítomna hemoragická diatéza.
Od chloromu k akutní promyelocytární leukemii (APL)
První případ chloromu, jako onemocnění sui generis, popsal Allan Burns v roce 1823 [3]. Celkově čtvrtý případ byl vylíčen pražským patologem Dittrichem: při sekci si povšiml špinavě zelených tumorů na periostu obratlů, žeber a lebky. Byly přítomny i v ledvinách a ovariích. Dittrichův případ byl publikován v časopise Pragere Vierteljahrsschrift für die praktische Heilkunde (sv. II, s. 104) v roce 1846 a je citován v článku Docka z Ann Arbor [4], který pak zmiňuje ještě další 2 případy z Prahy: jeden Dresslerův z roku 1866 a další Chiariho z roku 1883. Při publikaci celkově pátého případu v literatuře se onemocnění dostalo jména – termín chlorom pochází od Kinga z roku 1853 [5]. V té době byl také poznán vztah nádorů k lebečním kostem. V roce 1854 popsal Aran [6] chlorom jako „cancer vert“, pro jeho histologicky atypickou stavbu jej zařadil mezi maligní nádory a zároveň zdůraznil spojitost nádoru s lebečními kostmi. Popsal rovněž nádorové masy na tvrdé pleně (citace dle [7,8]). První zmínka o vztahu chloromu a leukemie pochází od Waldsteina z roku 1883 [9] (citace dle [4,8]): v jím popsaném případě se leukemie vyvinula až později v průběhu onemocnění chloromem. O definitivní charakteristiku vztahu mezi chloromem a chloroleukemií se zasloužil Dock [4,10]. Sám ve své práci [4] cituje, že myšlenka o souvislosti chloromu a leukemie byla ještě před ním vyslovena v roce 1885 von Reclinghausenem. Donner et al to v našem písemnictví líčili následovně: „...vyslovili dnes obecně uznávaný názor, že jde jen o zvláštní formu tumoriformní leukemie, při níž leukemické infiltráty, složené z nezralých krevních buněk, jeví nazelenalou barvu různé sytosti a označují se názvem chlorom“ [11]. Podobně již dřívější Libánského diskuze [2] končila konstatováním, že „dnes není pochyby o velmi blízkém vztahu chloromu k leukaemiím, za jejichž vzácnou varietu se pokládá“.
Dlouhá léta se však mělo za to, že jde o lymfoidní nádor [9]; teprve Türk v roce 1903 [12] (citace dle [8]) poprvé popsal myeloidní chlorom, o rok později ve výše citované práci Dock s Warthinem popisují rovněž případ nádoru z myeloidní řady. Již v roce 1909 publikoval Butterfield [13] pozorování 2 mnichovských případů myeloidních tumorů, které pečlivě popsal jak cytologicky, tak histologicky. Z jeho líčení bychom dnes neuměli jeho případ I zařadit, ale případ II (24letý truhlář) s četnými ekchymózami silně připomíná APL (ovšem z hlediska diagnózy APL s extrémní leukocytózou 60 000 × 109/l, terminálně až 200 000 × 109/l, a navíc překvapivě pacient neměl hyperplastickou dřeň – alespoň ne ve femoru a žebrech), nejvíce byly postiženy zelenavými tumory mezenteriální uzliny. Butterfield uváděl, že téměř všechny krevní buňky (80 %) obsahovaly tzv. „Kernkonvolut“. Takto označil mnohonásobně stočená jádra. (V dalším článku v tomto čísle časopisu Vnitřní lékařství o diagnostice APL je uvedeno, že buňky s takto „přeloženým“ jádrem jsou typické především pro APL s morfologií M3v dle klasifikace FAB.) Nález „Kernkonvolutu“ se pak relativně dlouho považoval za poměrně typický pro chlorom. Nicméně dle Libánského [2] již Naegeli ve své učebnici klinické hematologie (v citovaném vydání z roku 1919) uváděl, že „Kernkonvolut“ někdy u chloromu chybí, a naopak že bývá nalézán u akutních myelóz bez chloromu. Tatáž učebnice ve svém pozdějším vydání z roku 1931 [14] již ani tento termín nezmiňuje. Jaderný konvolut je vyobrazen na mikrofotografii v Libánského článku [2]. Rozsáhlý článek Askanazyho [7] je provázen kresbou buněk, u nichž se barví jádra, ale také (ve zjevně dezintegrující se buňce) mnohočetné Auerovy tyče [7], dle Libánského [2] prvně popsané Auerem v roce 1906. V letech 1909–1926 začal převažovat názor, že všechny chloromy jsou ve své podstatě myeloidní [2,8]. Dorsey Brennan poukazuje na nedostatečnou dokumentaci u dříve publikovaných „lymfoidních“ typů a všímá si, že v žádném z těchto případů nebyla provedena oxidázová reakce [8]. Chloromy se léčily arzenem (sic!) a RTG zářením, podávaly se transfuze, ale do konce 50. let minulého století byly takřka vždy fatální.
Chloromy a chloroleukemie dostaly své jméno podle zelenavého zbarvení tumoru a kostní dřeně, např. při sekci anebo při histologickém vyšetření tumoru. Dnes tento jev častěji pozorujeme při laboratorním vyšetření prstence mononukleárních leukocytů (tzv. „buffy-coatu“) po gradientní centrifugaci krve nebo dřeně pacientů s APL: bývá zelenavý. O podstatě zelenavé barvy, která mizí během několika hodin expozicí tkáně či buněk atmosférickému kyslíku (to popsal v roce 1909 Butterfield [13]), se vedly zhruba 75 let spory. Problém byl definitivně vyřešen až v roce 1959 [15]: primární granula promyelocytů (leukemických i normálních) obsahují zelenavou krystalickou myeloperoxidázu a dávají červenou fluorescenci v ultrafialovém světle. Tento červeně fluoreskující materiál byl identifikován jako porfyriny – bylo postulováno, že z porfyrin-proteinových komplexů vzniká myeloperoxidáza, která katalyzuje oxidaci aminokyselin peroxidem vodíku [15–17]. Jak poznamenávají Filip s Bednářem, zelenavou barvu lze zpět vyvolat redukčními činidly, např. hydrosulfitem sodným [16].
Kámen úrazu historických bádání o chloromech spočíval v tom, že pojem chlorom označoval tumoriformní projevy nejrůznějších subtypů leukemií. Připouštělo se, že v některých případech nemusí být chlorom nazelenalý. (Dnes bychom řekli, že chloromy jsou nazelenalým podtypem myelosarkomů – v angloamerické literatuře ještě méně přesně nazývaných granulocytární sarkomy.) Že se nejedná o lymfoidní tumory, bylo osvětleno na počátku 20. století [8,10,12]. Avšak i myeloidní subtyp chloromů zahrnoval nejen onemocnění, kterým bychom ve světle současných poznatků přiznali „nárok“ na zelené zbarvení nádorových buněk, tj. leukemické tumory z vyzrávajících buněk, jaké pozorujeme např. při APL, leukemii s translokací t(8;21), při některých peroxidáza-pozitivních myelomonocytárních leukemiích, popř. chronické myeloidní leukemii. Zahrnoval však i nádory u akutních leukemií, které per se nemají „nárok“ na zelené zbarvení, jako např. na peroxidázu negativní akutní leukemie monocytoidního subtypu, ev. vzácné myelosarkomy u myelodysplastického syndromu. Lze si představit, že příležitostné zelené zbarvení některého ze zjištěných nalezených extramedulárních projevů mohlo také souviset s putridními infekcemi, při kterých je přítomný hnis, nazelenalý vlivem příměsi polymorfonukleárních granulocytů (neutrofilní granula obsahují také myeloperoxidázu). Nutno si proto uvědomit, že jen výraznou menšinu z historických popisů chloromů a chloroleukemií bychom v současnosti klasifikovali jako suspektní APL s extramedulárním postižením.
Pátrání po původních popisech APL poněkud ztěžují i historické morfologické pojmy. Naegeli v roce 1900 popsal pojem myeloblast (jako základní kmenovou buňku pro neutrofilní, eozinofilní i bazofilní řadu) [14,18]. Myeloblast byl dosti striktně agranulární buňka, připouštěl se maximálně pouze náznak granulace. (Toto pojetí zůstalo víceméně platné dodnes.) Pro leukemie z buněk podobných myeloblastům se již užívalo pojmu akutní myeloblastová leukemie. Naegeli správně usuzoval, že leukemické buňky se liší od normálních, proto leukemií myeloblastového typu byla jen menšina z těch, které bychom nazvali AML. Většina z nich byla nazývána leukemiemi paramyeloblastovými. Termínu paramyeloblasty [14,19] se používalo pro leukemické buňky, které byly jakkoli odlišné ve srovnání s typickými myeloblasty; v písemnictví tento termín nacházíme až do začátku 60. let minulého století. Velmi pravděpodobně by proto byla APL označena jako leukemie paramyeloblastová, což však neříká nic konkrétního o její morfologii. Zároveň platil názor, že nejspolehlivějším znakem paramyeloblastu je přítomnost Auerových tyček, které bývaly v učebnicích či atlasech vykresleny zásadně v agranulárních myeloblastech, a opomíjel se jejich popis na úrovni promyelocytu [14,19,20] (obr. 1). Tady je další kámen úrazu pro naše pátrání, jelikož APL v moderním pojetí nejsou, až na raritní výjimky, agranulární. Prof. Netoušek ve své učebnici [19] rozlišuje paramyeloblasty promyelocytoidní, monocytoidní a retikuloidní. V pátrání po historických popisech APL by tedy značně ulehčilo situaci, kdyby chloromové či chloroleukemické buňky byly označeny jako promyelocytoidní paramyeloblasty. Žel, nic podobného jsem v literatuře nenašel. Přestože i Naegeli (po vzoru Pappenheima) používal termínu promyelocyt [14], vžitější označení pro tuto buňku bylo minimálně do 2. světové války „nezralý myelocyt“. V typickém diferenciálu té doby však byl uváděn jen myelocyt. Proto leukemické promyelocyty musely být ve starších studiích schovány mezi paramyeloblasty a myelocyty. Možná i proto trvalo relativně dlouho, než byla definována APL.

Na tomto místě jen dodejme, že již Naegeli v roce 1931 užil termín promyelocytová leukemie, aniž by jej však spojoval s buňkami obsahujícími v cytoplazmě Auerovy tyče [14]. Tento termín se však tak či onak nevžil. Proto spolehlivě nemůžeme (až do roku 1935 – viz níže) žádný z publikovaných případů zařadit k APL. Přitom vzhledem k tomu, že incidence APL v západní a střední Evropě představuje zhruba 7–10 % všech AML, i vzhledem k jisté tendenci APL k tvorbě extramedulárních tumorů a infiltrátů si můžeme být jisti, že četné z historických popisů chloromů musely být popisem chloroleukemie typu APL. Žel, jak řečeno, nedokážeme to již ex post dešifrovat. (Koneckonců ještě v literatuře o chloromech z 80. a 90. let 20. století často, ne‑li většinou, chybí cytogenetické rozbory.)
Velký význam pro pozdější definici APL měly práce o klinicko-laboratorních souvislostech. Vídeňský internista Erwin Risak publikoval v roce 1935 obsáhlou práci o fibrinopeniích [21]. Z celkem 17 případů se 3 kazuistiky týkaly hematoonkologických onemocnění. Jeden případ („Fall 6“) se týkal akutní leukemie 62letého muže se septickými horečkami, počtem leukocytů 2 000 a trombocytů 2 000 (jednotky se tehdy ve středoevropské literatuře neuváděly). V diferenciálu převažovaly lymfocyty (83 %), ale bylo popsáno vyplavování 6 % myeloblastů, 1 % myelocytů a 1 % metamyelocytů, a navíc mezi jednojadernými lymfocyty byl slovně popsán výskyt atypických buněk, nazvaných „mikromyeloblasty“. Hladina fibrinogenu byla 0,05 % (patrně 0,5 g/l). Onemocnění mělo podle Risakova popisu typický rychlý průběh, s hemoragickou diatézou. V posledním krevním obraze před smrtí pacienta bylo již 60 % myeloblastů při 15 000 leukocytech, hladina fibrinogenu 0,04 %. Risak uzavíral, že šlo o akutní myeloblastovou leukemii, s typickou panmyeloftízou (nyní bychom řekli pancytopenií), s rychlým průběhem a s krvácením, zapříčiněným nejen trombopenií s projevy kapilárního krvácení, ale také fibrinopenií [21]. Zdá se, že šlo o první zcela exaktní popis APL s její typickou koagulopatií. Doba od diagnózy k poslednímu krevnímu obrazu však nebyla přesně udána, bylo jen řečeno, že vývoj byl prudký, s progredující leukocytózou u neléčeného onemocnění. Na tuto Risakovu práci navazovala práce Croizata a Favre-Gillyho o hemoragickém syndromu u akutních leukemií [22]. Tito autoři postavili proti sobě běžně vídané případy hemoragické diatézy u 6 myeloblastových a 6 lymfoblastových leukemií, kde za hlavní příčinu hemoragie považovali trombocytopenii, proti případu 28letého pacienta s krvácivou diatézou při hodnotě destiček relativně vyšší (35 000) než u jiných případů akutní leukemie, zato však s těžkou hypofibrinogenemií 0,35 g/l a s ní spojeným prodloužením trombinového času. Nemocný měl v době diagnózy 7 000 bílých krvinek, v kostní dřeni převládaly myeloblasty a promyelocyty. Zemřel během 8 dní na generalizovanou krvácivou diatézu. Autoři v diskuzi poznamenávají, že podobné případy umírají začasto následkem krvácení, a navíc zmiňují možnost „meningeální“ hemoragie. Popisují dále vlastní zkušenost, že transfuze čerstvé krve bohaté na destičky pomáhají více než transfuze konzervované krve [22]. Některými autory (viz pěkný přehled amerických autorů [23]) je právě tato práce považována za první popis APL. Je však pozoruhodné, že velmi citovaný americký článek o chloromu a chloroleukemiích z 50. let minulého století [24], uvádějící jednu kazuistiku spolu s rozsáhlým literárním přehledem, sice detailně zpracoval historii, ale naprosto opominul v tomto odstavci zmíněné, na svou dobu moderní klinicko-patologické souvislosti.
Nejpodstatnějším přínosem na poli bádání o leukemických promyelocytech od konce 2. světové války do poloviny 60. let minulého století byly práce francouzských hematologů. Soustavně se promyelocytům a leukemiím z těchto buněk věnoval především Jean Bernard (v souladu s některými meziválečnými pracemi nazýval tyto buňky někdy také „premyelocyty“). Bernard publikoval sice hojně, ale zpočátku bez většího „impactu“ výhradně ve francouzštině. Patrně proto přišel o primát sdělení o APL v anglicky psané literatuře. Vzal mu jej Leif Hillestad z Rikshospitalet v Oslu. Ten v roce 1957 publikoval práci o 3 nemocných s akutní pro-myelocytární leukemií. Břitký postřeh internisty (publikujícího jinak o rozmanitých interních onemocněních) jej dovedl k poznání, že existuje vztah mezi rychlým a fatálním průběhem APL s nitrolebním krvácením a typickou hypergranulární morfologií promyelocytů a lapidárně popsal APL jako novou klinicko-patologickou entitu [25]. O jednom z těchto 3 pacientů bylo pojednáno Stormorkenem v norštině již o rok dříve [26]. Podobné závěry jako Hillestad, ale na základě studie mnohem většího souboru pacientů z roku 1959 (celkem 20 nemocných) pak publikovali Bernard et al [27], opět francouzsky, v časopise Schweizerische medizinische Wochenschrift. Krvácivou diatézu jako první připisovali diseminované intravaskulární koagulaci (DIC) a hyperfibrinolýze. Zmiňovali dosažení kompletní remise u jednoho pacienta a přechodné zlepšení u dalšího nemocného. Rosenthal et al [28] popsali ve shodě s předešlou prací, že nejobvyklejší příčina úmrtí u APL je intracerebrální krvácení (pozorované u 17/24 pacientů).
Bernard et al zanedlouho (v roce 1963) v tabulce o pitevních nálezech APL pacientů uvedli (ale nijak nediskutovali) nález trombózy u jednoho z pacientů zemřelých na APL [29]. V již citovaném článku Rosenthala et al je uveden jeden případ mnohočetné trombózy [28]. Fakt, že k přirozenému průběhu APL patří nejen krvácení, ale zdánlivě paradoxně i trombóza, popsala práce Bakera et al z New Yorku [30]. Jde o kazuistiku s podrobným rozborem klinického průběhu i pitevního nálezu. Zabývala se příčinami krvácení i mnohočetných trombóz. I tato práce viděla podstatu koagulopatie při APL a příčinu časných úmrtí v hypofibrinogenemii spojené s DIC. Referovali také o použití heparinu v léčbě těchto komplikací. Vzhledem k rovněž popisované hyperfibrinolýze bylo u popisovaného pacienta vyzkoušeno i podávání antifibrinolytika, a sice kyseliny ε-aminokapronové (EAC), jakož i substituce fibrinogenu [30]. Otázka, zda je výhodnější podávat heparin, antifibrinolytika, jejich kombinace, nebo ev. nepodávat žádnou prevenci, nebyla do současnosti vyřešena (ačkoli, alespoň odhadem, se na většině pracovišť podává heparin). Podávání heparinu však bylo v pozdějších letech chápáno spíše jako prevence a léčba krvácivé diatézy, která je důsledkem DIC; méně se bralo na vědomí, že podstatnou měrou se na úmrtích následkem koagulopatie mohou uplatňovat i trombózy. Na tento fakt mnohem později upozornila i naše skupina. Při soustavném pátrání po nich jednak klinicky a jednak v pitevním materiálu, jsme alespoň bílé tromby odhalili ve většině případů APL (ve skupině pacientů s leukocytózou u 6/7 a u pacientů bez leukocytózy u 2/8 případů) [31]. Výskyt trombózy nebyl spojen s podáváním kyseliny transretinové (ATRA), nýbrž s leukocytózou při diagnóze.
V 50. letech minulého století se prováděly první studie léčby leukemií jednak pomocí kortikoidů, které se používaly v kombinaci s novými látkami – cytostatiky (antimetabolity, např. 6‑merkaptopurinem) anebo s antagonisty kyseliny listové (aminopterinem nebo metopterinem). Byly popisovány případy kompletních remisí. V sestavě 20 pacientů Bernarda et al byla remise v krvi i dřeni dosažena v letech 1957–1959 pouze u 1 pacienta, navíc netrvala dlouho [27]. Průběh onemocnění byl hyperakutní, 2 týdny přežilo 8/20 pacientů a 6 týdnů nepřežil žádný z 19 pacientů, kteří nedosáhli remise. Zemřeli na hemoragické a infekční komplikace. Práce též popsala infiltraci mening při pitvě [27]. Revolučním počinem v léčbě APL bylo až zavedení antracyklinových cytostatik. První, kdo publikoval výsledky o monoterapii APL daunorubicinem, byl (kdo jiný než) prof. Bernard se svými spolupracovníky [32]. Byla popsána do té doby nevídaná míra dosažení kompletní remise (CR): 55 %. V 80. letech minulého století se pak standardem léčby APL (jakož i ostatních akutních myeloidních leukemií) stala kombinace založená na antracyklinu a cytarabinu. U APL bylo takto možné dosáhnout kolem 70 % CR s tím, že řada pacientů byla patrně vyléčena, neboť žila minimálně 5 let bez projevů onemocnění. Výsledky u APL byly v té době dosti podobné jako u stejně léčených ostatních subtypů AML [33,34].
V 60. a 70. letech minulého století také výrazněji pokročily morfologické studie. Bessis a Bretonová-Goriusová shrnuli své poznatky o elektronopticky vyšetřované ultrastruktuře buněk u akutních myeloidních leukemií v přehledném článku – relevantní směrem k APL jsou četná vyobrazení struktury Auerových tělísek [35] (dnes je nazýváme spíše tyčky). Tan et al publikovali elektronoptickou studii buněk osmi pacientů s APL [36]. Popsali různé typy azurofilní granulace a rozdělovali Auerova tělíska, odvozená z lyzozómů, na tyčkovitá („rod forms“) a třískovitá („splinter forms“). Byla rovněž popsána základní cytochemie APL buněk, jakož i základní ultrastrukturální odlišnosti mezi normálními a leukemickými promyelocyty [36]. Jelikož 70. léta 20. století přinesla již značně strukturované znalosti morfologie AML, nebylo překvapením, že se hematologové pokusili AML klasifikovat. V klasifikaci Galtona a Dacieho z roku 1975 bylo odlišeno 7 základních typů AML (M0 až M6), kde M0 byly nediferencované leukemie a M3 byly APL [37]. V podstatě velmi podobná klasifikace francouzsko-britsko-americké (FAB) skupiny, publikovaná v roce 1976, pak byla po dlouhá desetiletí základem diagnostiky i vzájemného odlišení podtypů AML [38]. V tomto klasifikačním systému figurovala APL jako M3 leukemie (z minulého návrhu vypustili pouze subtyp M0, aby jej znovu adoptovali v roce 1990). Podrobně se jí bude zabývat další článek v tomto čísle časopisu Vnitřní lékařství. Zde jen uveďme, že díky FAB klasifikaci se do obecného povědomí dostal pojem „faggot cells“, popisující APL buňky s mnohočetnými Auerovými tyčkami, často tvořícími snopce či otýpky. Ke konci 70. let minulého století však byla objevena i cytogenetická podstata APL a brzy se zjistilo, že řada případů s t(15;17) má odlišnou morfologii než FAB M3 (viz odstavec níže). Na tato zjištění celkem promptně reagovala FAB skupina, která 4 roky po své první publikaci [38] definovala variantní (hypo - nebo mikro-granulární) formu APL, kterou označila M3v [39]. Postupem času byly popsány nejrůznější morfologické varianty promyelocytárních leukemií – v tomto ohledu odkazuji opět na článek o diagnostice v tomto čísle časopisu Vnitřní lékařství a na základní citace [40–44].
Konečně v posledních 25 letech minulého století se ve výzkumu leukemií naplno uplatnily nové biotechnologie a pokroky v genetice. Skutečným milníkem ve výzkumu APL bylo odhalení společné podstaty naprosté většiny případů APL, a to balancované translokace t(15;17)(q22–23;q12–21). Původní popis této translokace pochází od Rowleyové et al z roku 1977 [45]. [Jde o stejnou badatelku, která předtím se svými spolupracovníky v Chicagu objasnila, že tzv. filadelfský chromozom, specifický pro chronickou myelózu (původně objevený Nowellem a Hungerfordem v roce 1960), je rovněž výsledkem reciproké translokace, a sice t(9;22). Translokace t(15;17) je naprosto patognomonická pro APL, a naopak valná většina případů APL nese tuto aberaci. Ačkoli je cytogenetický průkaz t(15;17) při standardním G-proužkování (tj. v preparátech obarvených dle Giemsy) někdy značně obtížný a translokace bývala v praxi prokazována jen v relativně nízkém procentu případů APL, Larson et al v polovině 80. let 20. století prokázali t(15;17)(q22;q21.1) ve všech z 27 vyšetřených vzorků od pacientů s APL [46]. I u pacientů se subtypem M3v je aberace běžně přítomna [47]. Nicméně i přes Larsonovo optimistické sdělení se u řady typických případů průkaz t(15;17) v praxi nedaří. Standardní cytogenetické vyšetření se však stalo běžným konfirmačním testem při suspekci na APL až do počátku 90. let minulého století, kdy bylo z větší části nahrazeno molekulárními testy, především polymerázou řetězovou reakcí (PCR). Samozřejmě úspěšné standardní cytogenetické vyšetření rozpozná, na rozdíl od níže uvedených molekulárních technik, i komplexní a variantní translokace a také přídatné aberace.
Dalším významným předělem byly pokroky v molekulární biologii. Vše začalo velmi akademickým výzkumem retinoidů a jejich regulačního uplatnění v morfogenezi a diferenciaci buněk. Ve Francii a zároveň v USA byl v roce 1987 izolován a klonován gen pro nukleární receptor pro kyselinu retinovou-α (RARα) [48,49]. Za-krátko byly objeveny i další nukleární receptory pro retinoidy (RARβ, RARγ a RARα‑γ – viz přehledný článek [50] a článek MUDr. Kořístka et al v tomto čísle časopisu Vnitřní lékařství). Nukleární receptory retinoidů patří podle své struktury do stejné rodiny nukleárních receptorů jako receptory pro ty-roidní hormony a steroidy [51]. Přestože je člověk závislý na pří-sunu retinolu z potravy (na rozdíl od zmíněných hormonů), není překvapivé, že retinoidy mají ve svých mechanizmech účinku leccos společného jak s tyroidními, tak i se steroidními hormony – signalizují cestou stejných „response“ elementů [52]. V roce 1990 vyšlo několik publikací z různých zemí o přestavbě genu RARα u APL a ještě týž rok byl zveřejněn objev fúzního (tehdy se spíše užívalo termínu „hybridního“) genu PML/RARα [53]. Dvě nezávislé skupiny v roce 1992 publikovaly téměř zároveň zjištění, že mohou existovat 3 různé zlomy v genu PML, zatímco zlomové místo v genu RARα je konstantní [54,55]. Zlomy v DNA genu PML jsou označovány bcr1, bcr2 a bcr3; nacházejí se v intronu 3, exonu 6, resp. intronu 6. Na úrovni mRNA jsou pak rozlišovány 3 různé transkripty: dlouhá (L-), variabilní (V‑) a krátká (S‑) forma, vzniklé fúzí celého exonu 6, nebo různě zkráceného exonu 6, anebo exonu 3 genu PML s exonem 3 genu RARα. Chenová et al vyvinuli nested (tedy dvoukrokovou) RT-PCR metodiku pro záchyt všech 3 izoforem PML/RARα [54]. RT-PCR metodika byla natolik zdokonalena („hot-start“ amplifikace při jednokrokové metodě), že je možné dojít k výsledku během 5–6 hod [56]. Klinický obraz APL s různými izoformami PML/RARα se poněkud liší (např. APL se zlomem bcr3 mají v průměru více leukocytů, častěji se mezi nimi vyskytne morfologicky variantní forma; APL se vzácnou izoformou bcr2 jsou zatím jen podezírány z celkově horšího klinického průběhu se zkráceným přežitím) [57,58]. Fúzi PML/RARα mají až na vzácné výjimky všechny případy APL. Naopak reciproký gen PML/RARα nese jen 67–81 % případů APL, které se klinicky neliší od těch, které jej neexprimují [59]. Exprese RARα/PML tedy patrně nemá, na rozdíl od PML/RARα, zásadní význam pro leukemogenezi APL, ale ukazuje na následující: t(15;17) není v případech bez exprese RARα/PML balancovaná a reciproká. Může se jednat i o kryptické translokace či inzerce [60,61]. Tyto případy mají horší nebo nemají vůbec žádnou šanci být zachyceny standardní cytogenetikou, jejíž úspěšnost v praxi bývá zhruba 41–97 % [59,62–64]. Molekulární vyšetření (RT-PCR) jako první v Česku zavedl ve své laboratoři v ÚHKT dr. Cedrik Haškovec v roce 1994.
PCR metodiky přispěly ke zlepšení péče o pacienty s APL i jinak: RT-PCR se hodí nejen k diagnostice v době záchytu onemocnění, ale i ke sledování minimálního reziduálního onemocnění (MRO), ať již standardní dvoukrokovou metodou s pomocí „nested“ primerů, anebo kvantitativní metodou RT-PCR v reálném čase [65,66]. APL se z hlediska onkologie stala prvním onemocněním, u kterého výsledky molekulárního stanovení MRO rozhodují o dalším terapeutickém postupu [67]. Předběžné výsledky z ÚHKT ukazují, že by monitorování fúze PML/RARα z kostní dřeně mohlo být nahrazeno sledováním exprese WT1 genu v periferní krvi [68].
Stanovení fúzního genu PML/RARα se stalo v posledních 15 letech nejsnadnějším a zároveň nejvalidnějším průkazem APL se 100% specificitou. WHO klasifikace zavedla onemocnění AML s t(15;17) nebo s jinými, velmi vzácnými translokacemi jako samostatnou jednotku, přičemž již nezáleží na tom, jakou mají buňky morfologii [69,70]. Jelikož byly popsány AML s morfologií APL, které neexprimují fuzní gen PML/RARα, zabývala se jimi mezinárodní expertní skupina [61]. Z 18 pacientů mělo alternativní fúzní gen 13 případů, 11 z nich mělo fúzi PLZF/RARα, u 9 z nich s prokazatelnou t(11;17), a 2 pacienti měli t(5;17), přičemž jeden z nich měl fúzi NPM/RARα, zatímco druhý nikoli. U 5 pacientů se nezdařilo vysvětlit molekulární etiologii APL [61], takže stále zbývá prostor pro další bádání.
Od experimentální hematologie v Izraeli k cílené terapii APL a k čínskému ministru zdravotnictví
Na počátku moderní, cílené léčby APL stál řetězec experimentálních prací. Byly umožněny pokroky v tkáňové kultivaci, ať již v suspenzní kultuře, anebo o pár let později zavedením klonální kultivace v polotuhých médiích. (V ÚHKT Praha existovalo Oddělení tkáňových kultur již od počátku 60. let minulého století pod vedením dr. Jaroslava Činátla, který napsal krásnou monografii o buněčné kultivaci a byl prvním autorem 2 článků v prestižním časopise Nature – viz např. [71].) Peter Nowell (ten, který téhož roku spolu s Hungerfordem objevil Ph chromozóm) byl v roce 1960 prvním, kdo popsal fenomén spontánní diferenciace leukemických buněk, pěstovaných in vitro s přidáním séra v suspenzní kultuře [72]. Skupiny kolem Lea Sachse v Rehovotu v Izraeli a kolem Donalda Metcalfa v australském Melbourne zvládly v polovině 60. let 20. století klonální kultivaci hematopoetických buněk. Klonálně vzniklé kolonie buněk rostly na polotuhém médiu – agaru [73–75]. Publikace Parana et al z roku 1970 [76] ukázala na možnost diferenciace leukemických buněk pacientů s leukemií v polotuhém agaru účinkem kondiciovaného média ze slezinných buněk (obsahovalo směs různých, v té době nedefinovaných cytokinů). O rok později se objevila práce o možnosti indukovat diferenciaci (syntézu hemoglobinu) polárním dimetylsulfoxidem (DMSO) v modelu myší virem indukované erytroleukemické linie Friendové [77]. Skupiny v Rehovotu a v Melbourne pak ukázaly, že diferenciační blok je za určitých okolností překonatelný i u myeloidních leukemických buněk pěstovaných in vitro [78,79]. U permanentních linií myších leukemických buněk docházelo působením některých fyziologicky se vyskytujících induktorů diferenciace k diferenciaci a maturaci myeloblastů do zralejších forem granulocyto-makrofágové řady. (Jak ukázali Činátl et al, zralé granulocyty, na rozdíl od makrofágů, již nemohou dále proliferovat [71].) V době prvních pokusů v Izraeli a Austrálii nebylo ani zdaleka jasné, které působky tehdy indukovaly růst a diferenciaci v klonech jejich myších leukemických buněk M1 a WEHI-3. Teprve značně později (až když byly k dispozici jasně definované rekombinantní růstové faktory) bylo objasněno, že hlavními hráči ve zmíněných in vitro experimentálních systémech byly kolonie-stimulující faktory GM-CSF, M-CSF a G-CSF a interleukiny-3 a -6 [80,81]. Uvedené výzkumné skupiny tedy přišly nejen se zajímavým konceptem diferenciační léčby, ale zároveň se značnou měrou podílely na definování regulátorů normální hematopoezy.
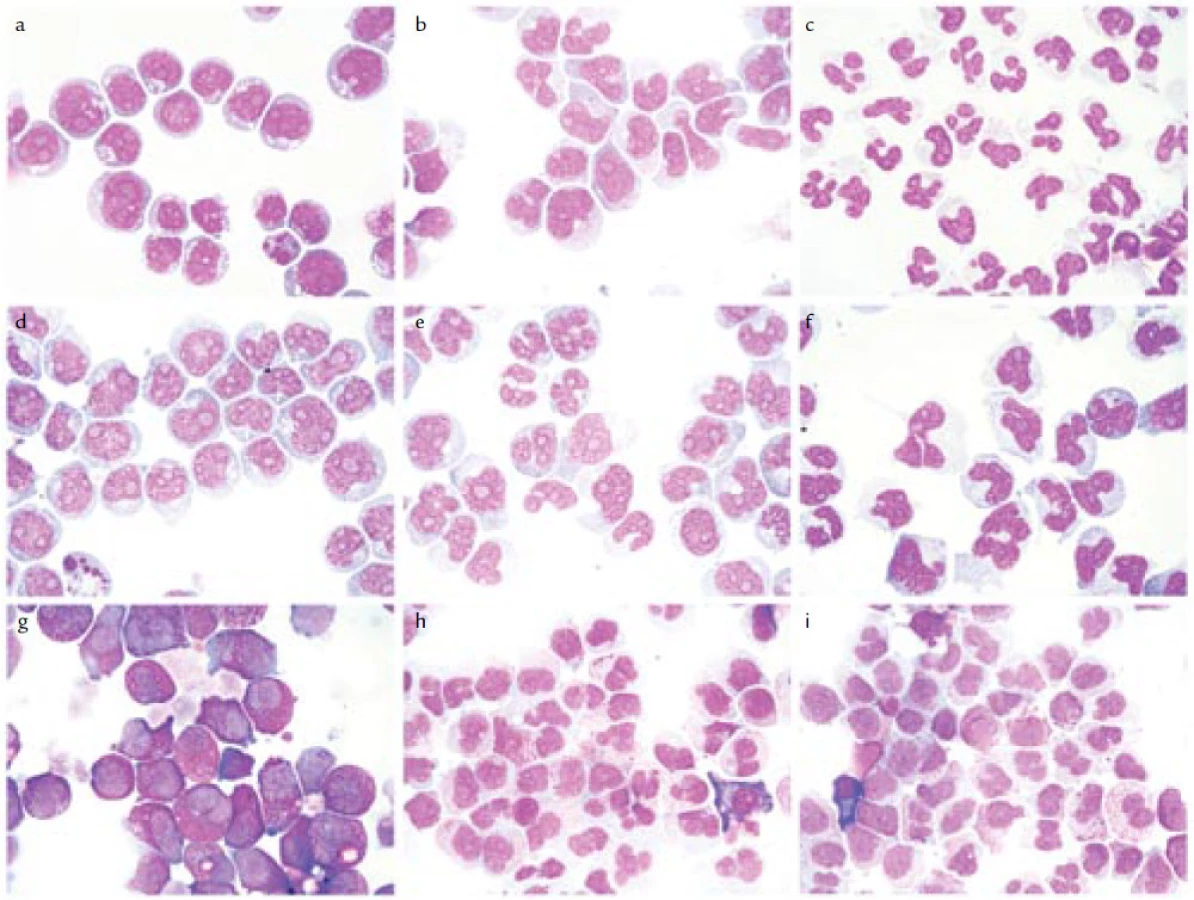
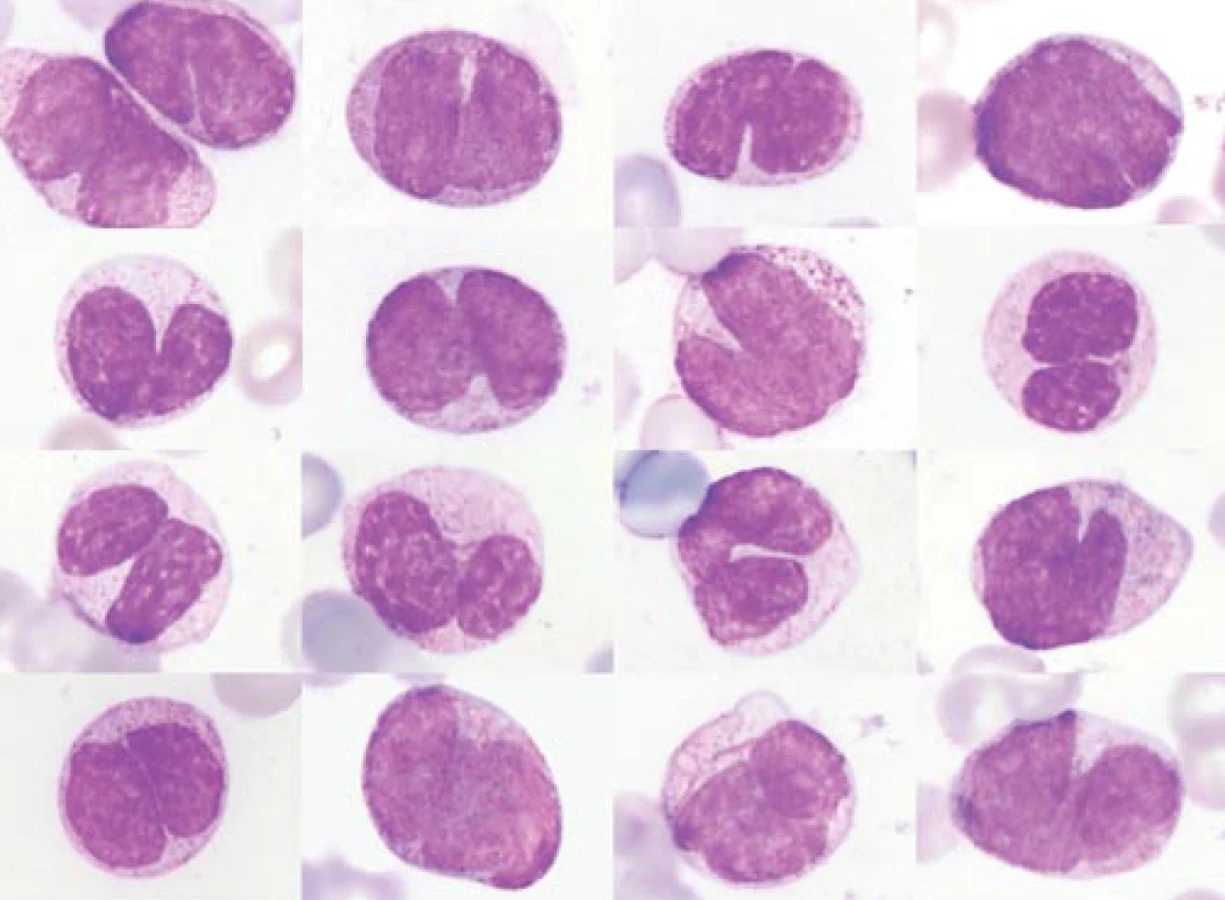
Jako první kliničtí hematologové si povšimli výše uvedených zajímavých experimentálních prací Gunz s Vincentem, a to ve své anotaci v prvním čísle nového časopisu Leukemia Research v roce 1977 [82]. Uvedli užití induktorů diferenciace jako jednu z možných léčeb budoucnosti. O rok později pak jasně formuloval program léčebného využití těchto látek ve své anotaci také Sachs [83]. Výzkum se pak přesunul k lidským leukemickým buňkám, neboť v roce 1977 byla ustanovena první permanentní myeloidní leukemická linie HL-60 [84] a o něco později řada dalších, např. linie ML-1 až ML-3 [85]. Původně se předpokládalo, že linie HL-60 pochází od pacienta s APL [84]; teprve následný detailnější klinický rozbor ukázal, že u nemocného šlo o případ M2 subtypu dle FAB, bez t(15;17) [86]. Na linii HL-60 provedli v National Cancer Institutu v Bethesdě v USA řadu experimentů s induktory diferenciace. V roce 1978 zjistili, že různé polární látky, jako např. DMSO anebo butyrát, jsou v tomto ohledu mimořádně účinné [87]. Jako další induktor diferenciace, tentokrát „fyziologický“, byla v systému buněk HL-60 a o málo později i v primokulturách buněk od pacientů s APL popsána kyselina transretinová (tretinoin; „all-trans retinoic acid“ – ATRA) (obr. 2). Byla takto účinná v evidentně suprafyziologické 10–6 M koncentraci [88–90]. (Normální hladina ATRA se pohybuje v oblasti o 3 řády nižší [91].) Leukemické myeloblasty řady dalších lidských leukemických linií diferencovaly v dosti podivné buňky se segmentovaným jádrem (obr. 3), které si však zachovávaly jisté známky nezralosti, resp. maturační asynchronie. V segmentovaném jádře byla např. stále přítomna jadérka (na rozdíl od normálních, panopticky obarvených polymorfonukleárů). Značná maturační asynchronie byla pozorována jak ve smyslu morfologickém (asynchronie mezi tvarem jádra a strukturou chromatinu, ale také mezi tvarem jádra a relativně méně zralou cytoplazmou) (obr. 3), tak i ve smyslu imunofenotypu [92,93]. V modelu těchto linií tedy nešlo o zcela terminální diferenciaci a nebylo zdaleka jisté, že by léčba ATRA in vivo mohla vést k ireverzibilní zástavě proliferace a klonální extinkci terminálně diferencovaných buněk (jakožto logickému předpokladu možného vyléčení pacientů) [93]. Podobné „ATRA-buňky“ byly později pozorovány také in vivo [94] (obr. 4). V západním světě v době objevu diferenciačních účinků ATRA nebyla tato látka k dispozici ke klinickým účelům. Proto byl v několika klinických experimentech u jednotlivých pacientů s rozvinutým onemocněním APL vyzkoušen alespoň dostupný 13-cis stereoizomer kyseliny retinové (13cRA; isotretinoin) (obr. 2), který byl předtím v in vitro pokusech poněkud méně účinný než ATRA. Přesto byly u těchto prvních jednotlivých případů pozorovány odpovědi na léčbu, včetně kompletní remise [95,96]. Historie vedoucí k objevu vitaminu A je sama o sobě nesmírně poutavá. Zvídavé čtenáře, kteří by se chtěli dovědět o historii vitaminu A a retinoidů od empirické léčby očních projevů hypovitaminózy A ve starém Egyptě v dobách kolem roku 3500 př. n. l. až k moderní medicíně, onkologii a dermatologii, odkazuji na přehledné články [97–102].
![Chemická struktura retinoidů. Chemické vzorce retinolu (provitaminu A, pro člověka zdroje vitaminu A v potravě), aktivního vitaminu A, tj. kyseliny retinové (tretinoinu, kyseliny transretinové – ATRA) a jejích stereoizomerů, kyseliny 13-cis retinové (isotretinoinu, 13cRA) a 9- cis retinové (alitretinoinu, 9cRA) [102].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/1a72068476586621f0a24451e4963056.jpg)
Milníky v léčbě APL se v posledních 20 letech daří překračovat hlavně Číňanům. První výsledky léčby APL pomocí monoterapie ATRA publikoval Huang Meng-Er pod vedením prof. Wanga Zhen-Yi [103] ze Šanghajského ústavu hematologie v nemocnici Rui-Jin – kompletní remise dosáhlo 23 z 24 pacientů (z toho bylo 8 rezistentních k předchozí terapii a 16 z 24 nemocných mělo de novo APL). Zbývající pacient dosáhl kompletní remise až po přidání nízkých dávek cytarabinu k ATRA [103]. Nedávný přehledný článek v Bloodu dává nahlédnout i do životopisu hlavních protagonistů tohoto vývoje [104]. Pozadí, jak došlo k tomuto a dalším objevům v Číně, by mohlo být námětem i k beletristickému zpracování. Prof. Wang Zhen-Yi promoval v roce 1948 (ještě před komunistickou revolucí) na francouzské jezuitské univerzitě v Šanghaji – proto ovládal plynně francouzštinu. Později pracoval tamtéž jako hematolog (tehdy v Číně zcela nový obor). V období Kulturní revoluce byl na dlouhých 11 let maoistickým režimem zavržen a nesměl jako reakcionářský živel působit ve výzkumu. Po svém návratu k vědecké hematologii v roce 1978 byl zaujat výše zmíněnými pokusy s indukcí diferenciace. Byly k tomu patrně 2 důvody. Jednak v Číně té doby byl naprostý nedostatek léků běžně užívaných na západě, jednak tento způsob „opravy“ leukemické buňky velmi vyhovoval čínskému myšlení v duchu Konfucia. Laurent Degos, vedoucí hematolog v Hôpital Saint-Louis v Paříži, psal v roce 1990 v referátu ze sympózia o retinoidech o tom, jak se v roce 1985 s Wangem setkal – tzn. nedlouho předtím, než Wang zahájil klinickou studii v Šanghaji [105]. V tehdejší Číně nebyla běžně dostupná cytostatika, podobně tomu bylo i s podáváním transfuzí. Možná i proto se Wangovi tolik líbila myšlenka, že možnost opravy (tj. indukce diferenciace) je lepší než zabíjení (rozuměj: buněk). Prof. Wang měl štěstí, že v Šanghaji (na rozdíl od západních zemí) byla dostupná ATRA pro dermatologické potřeby. Proto začal v roce 1980 s in vitro pokusy s ATRA a po setkání s prof. Degosem v Paříži v roce 1985 se rozhodl ji v nemocnici Rui-Jin podávat nemocným s APL. Vůbec první pacientkou bylo 5leté děvče v těžkém stavu se sepsí, rezistentní na předchozí chemoterapii včetně antracyklinů. Děvčátko bylo léčeno v šanghajské dětské nemocnici dávkou ATRA 45 mg/m2/den (ostatní nemocní dostali dávky od již zmíněné až po 100 mg/m2/den). Po jednom týdnu u ní došlo k ústupu horečky a po dalších 3 týdnech se „zázračně“ dostala do kompletní remise. Nyní 28letá žena stále žije a vede dobrou kariéru [104]. (Poznámka autora: Dávkování použité u 1. pacientky se používá dosud, jen u dětí a mladistvých se aplikují nižší dávky. Je velmi pravděpodobné, že Číňané dosahují o něco nižších plazmatických koncentrací, možná následkem rozdílů v metabolizmu retinoidů, možná také proto, že vstřebávají ATRA méně kvůli nižšímu obsahu tuků ve stravě, takže výskyt toxicity ve formě akutní hypervitaminózy A – pseudotumoru cerebri [106] je u nich patrně podstatně nižší.) Jak již bylo uvedeno výše, své mimořádné výsledky pak Wangova skupina publikovala v Bloodu [103].
Odbočíme zpět do západního civilizace. Článek Huanga Meng-er et al [103] představoval pro většinu světa dosti šokující informaci a jak jinak, účinnost ATRA se začala verifikovat v různých zemích. Postgraduální studentka Christine Chomienne v Hôpital Saint-Louis (dnes profesorka tamtéž) publikovala se svými kolegy první vlastní výsledky terapie APL pomocí 13-cRA, resp. ATRA: zatímco 2 pacienti léčení 13-cRA nedosáhli remise, 2 pacienti v relapsu APL léčení ATRA jí dosáhli [107]. Sám prof. Degos pak se svými spolupracovníky zveřejnil studii, ve které bylo docíleno monoterapií ATRA kompletní remise u 19 z 20 pacientů s relapsem APL [108]. ATRA pro francouzské pacienty byla zprvu dodávána prof. Wangem (čínští postdoktorandi ji vozili do Paříže díky podpoře společnosti Air France – prvním takovým cestovatelem byl právě Huang Meng-er [109]). Po neblahých událostech na náměstí Tchien-An-Men v Pekingu v roce 1989 však francouzské úřady vyvinuly tlak na ukončení vědecké spolupráce s lidovou Čínou a ve Francii vznikl problém s dodávkou ATRA. Prof. Laurent Degos (v autorovi jediném známém článku týkajícím se explicitně historie APL ve světovém písemnictví) pak popisuje ve 3. osobě svou vlastní roli coby katalyzátora toho, aby se ATRA dostala do výroby v západních zemích [109]. Nejdříve přesvědčil zástupce firmy Roche ve Francii, aby vyrobila ATRA v „chemicky čistší“ formě. Francouzská divize však omezila použití přípravku, v souladu s firemní politikou, pouze na území Francie. V roce 1989 se proto prof. Degos sešel s dr. Lorettou Itri, viceprezidentkou americké divize firmy Roche v Nutley, která následně převzala výrobu. Její manžel, Raymond Warrell jr., se v krátké době postavil do čela klinického výzkumu ATRA u APL v USA (jak píše Degos, Warrell byl přítomen jeho setkání s dr. Itri a vylíčené účinky ATRA jej nesmírně překvapily) [109]. Firma Hoffman LaRoche (nyní Roche) vyrábí ATRA dodnes pod názvem Vesanoid®. Degos však nepíše o další významné skutečnosti, a sice o tom, že firma Hoffman LaRoche měla skvělé předpoklady pro to, aby se věnovala výrobě retinoidů, a to díky spolupráci s dr. Wernerem Bollagem, expertem na retinoidy, pod jehož vedením bylo ve firmě v Basileji nasyntetizováno přes 1 000 různých retinoidů. Nicméně švýcarské vedení původně Degosův návrh na zavedení výroby ATRA odmítlo a Degos uspěl až cestou spolupráce s americkou divizí firmy.
Remise při monoterapii ATRA trvají obvykle 2–6 měsíců. Postremisní podávání samotné ATRA (ev. v kombinaci s nízkodávkovou chemoterapií) vedlo ve všech případech k relapsu [103,104,108]. Později bylo vysvětleno, že relapsy jsou patrně důsledkem snižující se plazmatické hladiny ATRA, která je fyziologicky katabolizována enzymy podobnými cytochromu P-450. Ke značnému snížení její hladiny dochází již během 1. týdne podávání [91,110]. Postulát, že se na snižování koncentračních špiček při jejím dlouhodobém podávání podstatnou měrou podílí její katabolizmus, je založen na pozorování stoupání močové exkrece jejího katabolitu 4-oxo-ATRA ve formě glukuronidu [91]. Úbytku hladiny lze předejít jednak intermitentním podáváním léku (týden ano, týden ne) [111] anebo současným podáváním inhibitorů cytochromu P-450, např. antimykotika ketokonazolu [112] nebo flukonazolu (který v kombinaci s ATRA může naopak vyvolat příznak hypervitaminózy A – pseudotumor cerebri [106]). Čínské výsledky s ATRA u APL byly brzy ověřeny také v USA a Japonsku [94,113]. Studium specifických přestaveb 2. intronu genu RARα v Číně a také klonální analýzy provedené v Austrálii ověřily, že zralé granulocyty v remisi po léčbě ATRA skutečně pocházejí z leukemického APL klonu [114,115] a že dochází ke klonální extinkci leukemických buněk [113]. V Česku byla první, tehdy 30letá pacientka s relapsem APL léčena ATRA v ÚHKT v roce 1992. Pacientka byla vegetariánka na beztukové dietě (což jsme zpočátku vítali vzhledem k obavám z hyperlipidemie, uváděné jako možný vedlejší účinek ATRA). Léčba u ní měla po 4 týdnech jen malý účinek (v té době by se normálně už mohla blížit ke klinické remisi). Tehdy jsme si uvědomili, že pacientka následkem svých dietních zvyklostí patrně ATRA vstřebává naprosto minimálně. V té době však pacientka zemřela následkem krvácivých komplikací. Teprve druhá pacientka s APL, v roce 1993 v relapsu po autologní transplantaci, dosáhla kompletní remise a po další chemoterapii (mj. s antracykliny) žije bez leukemie dodnes. Vzhledem k tomu, že po podávání ATRA v monoterapii docházelo k relapsům, začala se zkoumat udržovací anebo konsolidační chemoterapie včetně transplantací [103,108]. První randomizované srovnání mezi monoterapií ATRA a chemoterapií (daunorubicin + cytarabin ve standardní dávce) v indukční léčbě APL provedla studie APL-91, původně koncipovaná ve Francii. Vedli ji spolu prof. Degos z Paříže a Pierre Fenaux z Lille. Zásluhou druhého z nich byla studie rozšířena na evropskou, pod záštitou v Bruselu sídlící EORTC se jí účastnila řada zemí včetně Česka. Studie ukázala o něco vyšší procento kompletních remisí ve větvi ATRA (91 vs 81 %; výsledek nebyl statisticky signifikantní). Co se však příliš nepředpokládalo, ale nakonec mělo zásadní význam, bylo to, že se signifikantně zlepšilo přežití pacientů po ATRA a konsolidační chemoterapii ve srovnání s pacienty ve větvi s indukční chemoterapií, kteří pak dostali stejnou konsolidaci. Ve větvi s ATRA tedy došlo k signifikantnímu úbytku relapsů [116]. Extrémní úspěch klinického použití ATRA pak zaznamenala při užití kombinace s chemoterapií (idarubicinem) v indukci rozsáhlá společná studie italských skupin GIMEMA a pediatrické AIEOP [67]. Mezi 240 hodnotitelnými pacienty z celkem 71 institucí bylo dosaženo 95 % kompletních remisí. Dle užitého protokolu s poetickým názvem AIDA (odvozeným od ATRA plus IDArubicin) se podávaly ještě tři konsolidační chemoterapie, sestávající z různých dávek idarubicinu nebo mitoxantronu, a 2leté udržovací terapie pomocí ATRA (jen zpočátku se v 1 ze 4 větví udržovacích schémat po randomizaci nepodávala žádná léčba). Přežití bez události ve dvou letech bylo z hlediska léčby leukemií rekordní – 79 % – a vedlo na svou dobu k nebývale vysoké míře vyléčení pacientů. Podobně úspěšná byla i španělská studie skupiny PETHEMA, která užívala jen mírně modifikovaný protokol ve srovnání s AIDA – pouze v konsolidačních léčbách zrušila podávání méně potřebných cytostatik [117]. Naopak zklamání přinesl klinický pokus s použitím 9-cis izomeru kyseliny retinové (alitretinoinu; 9cRA) (obr. 2). Ačkoli in vitro indukovala diferenciaci ještě výraznější měrou než ATRA (viz také obr. 3; 9cRA působí podobně jako ATRA na nukleární receptor RARα, ale navíc i na receptory RXR), v klinickém pokusu 9cRA indukovala kompletní remisi pouze u 1 ze 7 pacientů s relapsem APL. U de novo APL však má 9cRA patrně účinnost podobnou jako ATRA [118,119].
A ještě zpět do Číny. Neobvyklý vývoj kariéry měl i šanghajský žák prof. Wanga, a sice Dr. Chen Zhu, pocházející z lékařské rodiny. Toho zastihla Kulturní revoluce na střední škole, kterou musel opustit a živit se na venkově zemědělskou prací. Po samostudiu tam pak působil coby „bosonohý“ lékař. Po návratu do Šanghaje dostudoval a začal pracovat v týmu prof. Wanga. Ten jej jako nadaného pracovníka poslal do pařížské Hôpital Saint-Louis, kde na oddělení prof. Degose mj. studoval i in vitro aspekty indukované diferenciace pomocí ATRA, zatímco doma v Šanghaji probíhal první klinický pokus. Pobyt v Paříži zakončil v roce 1989 doktorskou prací na téma přestaveb genů T-receptoru‑γ. Své nabyté znalosti z molekulární biologie pak uplatnil doma v Číně [104]. Ve spolupráci s francouzskými molekulárními biology a cytogenetiky popsal jeho tým prioritně alternativní translokaci u APL, a sice t(11;17), na jejímž podkladě se tvoří fúzní gen PLZF/RARα [104,120]. Tento vzácný typ APL se vyznačuje poněkud jinou morfologií buněk a relativní neodpovídavostí na terapii ATRA. [Francouzi – de Thé et al, ale i jiné skupiny, mezitím zjistili molekulární podstatu APL s t(15;17) – tvorbou fúzního genu PML/RARα – viz výše, podrobněji pak v článku Kořístka et al v tomto čísle časopisu Vnitřní lékařství.] Ale zpět k Chenovi: dále pak vedl tým, který vrátil do exaktní medicíny starou látku, známou svými účinky již Hippokratovi, a později užívanou v tradiční čínské medicíně, a sice bílý arzenik (As2O3) [104]. Lze jím rovněž mimořádně úspěšně léčit APL. Jelikož působí jiným mechanizmem než ATRA (má méně vyjádřený diferenciační účinek in vitro, ale působí výrazně proapoptoticky), je účinný in vivo i u ATRA-rezistentních relapsů [121,122]. Mimořádnou účinnost v léčbě APL pak má kombinace ATRA s oxidem arzenitým [104]. Charizmatický vědec Chen Zhu se posléze stal místopředsedou Čínské akademie věd a v roce 2007 ministrem zdravotnictví Čínské lidové republiky [104]. Poznamenejme jen, že za zády mu stála jeho manželka, Chen Sai-Juan, která byla začasto první autorkou prioritních prací skupiny [54,115,120].
Moderní historie výzkumu APL je pozoruhodná v mnoha směrech. Je to příběh demonstrující úžasně plodné sepětí klinické vědy a teoretického výzkumu: jak praxi vzdálené experimenty na myších buňkách v Izraeli a Austrálii vedly k formulaci hypotetické možnosti léčby, která byla za velmi krátkou dobu skutečně přivedena do klinických studií v Číně a krátce po té i do běžné klinické praxe v celém světě, a to navíc s mimořádným úspěchem. Ale nejen to: tyto klinické úspěchy v další fázi zase probudily zájem o výzkum APL, který přinesl obratem další vědecká poznání velkého dosahu, jež byla následně znovu zúročena renezancí dalšího léčiva, konkrétně arzeniku, v klinické praxi. Zároveň jde o příběh, ve kterém se do klinické praxe dostal principálně nový způsob léčby, jenž nemá obdoby v žádném jiném podoboru onkologie, a sice diferenciační terapie, překonávající maturační blok.
Poděkování
Doc. Petru Stöckbauerovi, který ve mně hned po nástupu do ÚHKT v roce 1983 vzbudil zájem o diferenciační induktory, které pak byly tématem mé kandidátské práce. Prof. Karlu Smetanovi sr. za pomoc před 20 lety při interpretaci mých nálezů „nepatřičných jadérek“ v segmentovaných buňkách vznikajících diferenciací leukemických myeloblastů působením ATRA a za zapůjčení krásných „obsoletních“ knížek z historie hematologie v roce 2008. A konečně Ing. Janě Markové za připomínky k rukopisu.
Doručeno do redakce: 9. 6. 2008
MUDr. Jiří Schwarz, CSc.
www.uhkt.cz
e‑mail: jiri.schwarz@uhkt.cz
Zdroje
1. Hynek K, Kadlický R. Zprávy z lékařských spolkův a sjezdů. První odborná schůze českých očních lékařů. Čas Lék Čes 1911 : 931–937.
2. Libánský J. Chlorom. Popis případu. Čas Lék Čes 1939; 78 : 996–1007.
3. Burns A. Observations on the surgical anatomy of the head and neck. Baltimore: Lucas Publishers 1823.
4. Dock G. Chloroma and its relation to leukæmia. Am J Med Sci 1893; 106 : 152–185.
5. King A. A case of chloroma. Monthly J Med Sci 1853; 17 : 91.
6. Aran FA. Note sur une forme particulière et encore peu connue de cancer de la dure-mère et des os du crâne (cancer vert, chloroma). Arch Gén Méd 1854; 2 : 385–412.
7. Askanazy M. Einiges zum Verständnis der Chlorome. Beitr Pathol Anat 1916; 63 : 22–59.
8. Brennan D. Chloroma. The recent literature and a case report. Bull Johns Hopkins Hosp 1926; 3 : 189–215.
9. Waldstein L. Ein Fall von progressiver Anämie und darauffolgender Leukocythemie mit Knochenmarkerkrankung und einem sogenannten Chlorom (Chlorolymphom). Virchows Arch Pathol Anat 1883; 91 : 12.
10. Dock G, Warthin AS. A new case of chloroma with leukemia with a study of cases reported since 1893. Med News 1904; 85 : 971.
11. Donner L, Středa A, Vaněk J. Chloroleukemie. Čas Lék Čes 1951; 90 : 278–284.
12. Türk W. Akute myeloide Leukämie mit Grünfärbung des Knochenmarks. Mitt Gesellschft Innere Med Kinderhk 1903; 2 : 39.
13. Butterfield EE. Beitrag zum Morphologie der Chloromzellen. Folia Haematol 1909; VIII: 179–184.
14. Naegeli O. Blutkrankheiten und Blutdiagnostik. 5. vyd. Berlin: Springer 1931.
15. Schultz J, Rosenthal S. Iron (II) inactivation of myeloperoxidase. J Biol Chem 1959; 234 : 2486–2490.
16. Filip O, Bednár B. Chloroma and chloroleukemia. Neoplasma 1966; 13 : 525–530.
17. Wiernik PH, Serpick AA. Granulocytic sarcoma (chloroma). Blood 1970; 35 : 361–369.
18. Türk W. Vorlesungen über klinische Hämatologie. Wien: Braumüller 1904.
19. Netoušek M. Klinická hematologie. Praha: Státní zdravotnické nakladatelství 1962.
20. Ławkowicz W, Krzemińska-Ławkowicz I. Hematologický atlas. Směrnice pro rozpoznání nemocí krvetvorné soustavy. Praha: Státní zdravotnické nakladatelství 1953.
21. Risak E. Die Fibrinopenie. Z Klin Med 1935; 128 : 605–629.
22. Croizat P, Favre-Gilly J. Les aspects du syndrome hémorragique des leucémies; a propos de 12 cas de thrombocytopénie et d‘un cas de fibrinopénie. Sang 1949; 20 : 417–421.
23. Jones ME, Saleem A. Acute promyelocytic leukemia. A review of literature. Am J Med 1978; 65 : 673–677.
24. Ross RR. Chloroma and chloroleukemia. Am J Med 1955; 18 : 671–676.
25. Hillestad LK. Acute promyelocytic leukemia. Acta Med Scand 1957; 159 : 189–194.
26. Stormorken H. Fibrinolytic purpura associated with myelogenous leukemia; case. Tidsskr Nor Lægeforen 1956; 76 : 754.
27. Bernard J, Mathé G, Boulay J et al. La leucose aiguë à promyélocytes. Etude portant sur vingt observations. Schweiz Med Wochenschr 1959; 89 : 604–608.
28. Rosenthal RL. Acute promyelocytic leukemia associated with hypofibrinogenemia. Blood 1963; 21 : 495–508.
29. Bernard J, Lasneret J, Chome J et al. A cytological and histological study of aute premyelocytic leukaemia. J Clin Pathol 1963; 16 : 319–324.
30. Baker WG, Bang NU, Nachman RL et al. Hypofibrinogenemic hemorrhage in acute myelogenous leukemia treated with heparin. With autopsy findings of widespread intravascular clotting. Ann Intern Med 1964; 61 : 116–123.
31. Schwarz J, Lemez P, Lukasova M et al. High incidence of thromboembolic events in leukemic patients with acute promyelocytic leukemia (APL). Blood 1996; 88 (Suppl 1, Part 2): 169b. Abstract 3404.
32. Bernard J, Weil M, Boiron M et al. Acute promyelocytic leukemia: results of treatment by daunorubicin. Blood 1973; 41 : 489–496.
33. Cunningham I, Gee TS, Reich LM et al. Acute promyelocytic leukemia: treatment results during a decade at Memorial Hospital. Blood 1989; 73 : 1116–1122.
34. Sanz MA, Jarque I, Martín G et al. Acute promyelocytic leukemia. Therapy results and prognostic factors. Cancer 1988; 61 : 7–13.
35. Bessis M, Breton-Gorius J. Pathologie et asychronisme de dévelopment des organelles cellulaires au cours des leucémies aiguës granulocytaires. Etude au microscope électronique. Nouv Rev Fr Hématol 1969; 9 : 245–277.
36. Tan HK, Wages B, Gralnick HR. Ultrastructural studies in acute promyelocytic leukemia. Blood 1972; 39 : 628–636.
37. Galton DAG, Dacie JV. Classification of acute leukaemias. Blood Cells 1975; 1 : 17–24.
38. Bennett JM, Catovsky D, Daniel MT et al. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol 1976; 33 : 451–458.
39. Bennett JM, Catovsky D, Daniel MT et al. A variant form of hypergranular promyelocytic leukaemia (M3). Br J Haematol 1980; 44 : 169–170.
40. Liso V, Troccoli G, Grande M. Cytochemical study of acute promyelocytic leukaemia. Blut 1975; 30 : 261–268.
41. Das Gupta A, Sapre RS, Shah AS et al. Cytochemical and immunophenotypic heterogeneity in acute promyelocytic leukemia. Acta Haematol 1989; 81 : 5–9.
42. Neame PB, Soamboonsrup P, Leber B et al. Morphology of acute promyelocytic leukemia with cytogenetic or molecular evidence for the diagnosis: characterization of additional microgranular variants. Am J Hematol 1997; 56 : 131–142.
43. Sainty D, Liso V, Cantù-Rajnoldi A et al. A new morphologic classification system for acute promyelocytic leukemia distinguishes cases with underlying PLZF/RARA gene rearrangements. Blood 2000; 96 : 1287–1296.
44. Liso V, Bennett J. Morphological and cytochemical characteristics of leukaemic promyelocytes. Best Pract Res Clin Haematol 2003; 16 : 349–355.
45. Rowley JD, Golomb HM, Dougherty C. 15/17 translocation, a consistent chromosomal change in acute promyelocytic leukaemia. Lancet 1977; 1 : 549–550.
46. Larson RA, Kondo K, Vardiman JW et al. Evidence for a 15;17 translocation in every patient with acute promyelocytic leukemia. Am J Med 1984; 76 : 827–841.
47. Golomb HM, Rowley JD, Vardiman JW et al. “Microgranular” acute promyelocytic leukemia: a distinct clinical, ultrastructural, and cytogenetic entity. Blood 1980; 55 : 253–259.
48. Petkovich M, Brand NJ, Krust A et al. A human retinoic acid receptor which belongs to the family of nuclear receptors. Nature 1987; 330 : 444–450.
49. Giguere V, Ong ES, Segui P et al. Identification of a receptor for the morphogen retinoic acid. Nature 1987; 330 : 624–629.
50. Chambon P. A decade of molecular biology of retinoic acid receptors. FASEB J 1996; 10 : 940–954.
51. Evans RM. The steroid and thyroid hormone receptor superfamily. Science 1988; 240 : 889–895.
52. Umesono K, Giguere V, Glass CK et al. Retinoic acid and thyroid hormone induce gene expression through a common responsive element. Nature 1988; 336 : 262–265.
53. de Thé H, Chomienne C, Lanotte M et al. The t(15;17) translocation of acute promyelocytic leukaemia fuses the retinoic acid receptor alpha gene to a novel transcribed locus. Nature 1990; 347 : 558–561.
54. Chen SJ, Chen Z, Chen A et al. Occurrence of distinct PML‑RARα fusion gene isoforms in patients with acute promyelocytic leukemia detected by reverse transcriptase/polymerase chain reaction. Oncogene 1992; 7 : 1223–1232.
55. Pandolfi PP, Alcalay M, Fagioli M et al. Genomic variability and alternative splicing generate multiple PML/RARα transcripts that encode aberrant PML proteins and PML/RARα isoforms in acute promyelocytic leukaemia. EMBO J 1992; 11 : 1397–1407.
56. Diverio D, Riccioni R, Pistilli A et al. Improved rapid detection of the PML/RARα fusion gene in acute promyelocytic leukemia. Leukemia 1996; 10 : 1214–1216.
57. Gallagher RE, Willman CL, Slack JL et al. Association of PML‑RARα fusion mRNA type with pretreatment hematologic characteristics but not treatment outcome in acute promyelocytic leukemia: an Intergroup molecular study. Blood 1997; 90 : 1656–1663.
58. Slack JL, Willman CL, Andersen JW et al. Molecular analysis and clinical outcome of adult APL patients with the type V PML‑RARα isoform: results from Intergroup protocol 0129. Blood 2000; 95 : 398–403.
59. O‘Connor SJ, Evans PA, Morgan GJ. Diagnostic approaches to acute promyelocytic leukaemia. Leuk Lymphoma 1999; 33 : 53–63.
60. Grimwade D, Howe K, Langabeer S et al. Establishing the presence of the t(15;17) in suspected acute promyelocytic leukaemia: cytogenetic, molecular and PML immunofluorescence assessment of patients entered into the M.R.C. ATRA trial. Br J Haematol 1996; 94 : 557–573.
61. Grimwade D, Biondi A, Mozziconacci MJ et al. Characterization of acute promyelocytic leukemia cases lacking the classic t(15;17): results of the European Working Party. Blood 2000; 96 : 1297–1308.
62. Borrow J, Solomon E. Molecular analysis of the t(15;17) translocation in acute promyelocytic leukaemia. Baillière’s Clin Haematol 1992; 5 : 833–856.
63. Frankel SR. Acute promyelocytic leukemia. New insights into diagnosis and therapy. Hematol Oncol Clin North Am 1993; 7 : 109–138.
64. Mancini M, Nanni M, Cedrone M et al. Combined cytogenetic, FISH and molecular analysis in acute promyelocytic leukaemia at diagnosis and in complete remission. Br J Haematol 1995; 91 : 878–884.
65. Miller WH Jr, Kakizuka A, Frankel SR et al. Reverse transcription polymerase chain reaction for the rearranged retinoic acid receptor alpha clarifies diagnosis and detects minimal residual disease in acute promyelocytic leukemia. Proc Natl Acad Sci USA 1992; 89 : 2694–2698.
66. Cassinat B, Zassadowski F, Balitrand N et al. Quantitation of minimal residual disease in acute promyelocytic leukemia patients with t(15;17) translocation using real-time RT‑PCR. Leukemia 2000; 14 : 324–328.
67. Mandelli F, Diverio D, Avvisati G et al. Molecular remission in PML/RARα-positive acute promyelocytic leukemia by combined all‑trans retinoic acid and idarubicin (AIDA) therapy. Blood 1997; 90 : 1014–1021.
68. Polák J, Marková J, Schwarz J et al. Užití kvantitativního stanovení exprese genu Wilmsova tumoru 1 pro monitorování reziduální nemoci pacientů s akutní myeloidní leukemií. Čas Lék Čes 2006; 145 : 36–42.
69. Brunning RD, Bennett J, Matutes E et al. Acute myeloid leukaemia with recurrent genetic abnormalities. In: Jaffe ES, Harris NL, Stein H et al (eds).World Health Organization classification of tumours. Pathology and genetics. Tumours of haematopoietic and lymphoid tissues. Lyon: IARC 2001 : 81–87.
70. Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood 2002; 100 : 2292–2302.
71. Cinátl J, Paluska E, Chudomel V et al. Culture of macrophage cell lines from normal mouse bone marrow. Nature 1982; 298 : 388–389.
72. Nowell PC. Differentiation of human leukemic leukocytes in tissue culture. Exp Cell Res 1960; 19 : 267–277.
73. Pluznik DH, Sachs L. The cloning of normal “mast” cells in tissue culture. J Cell Physiol 1965; 66 : 319–324.
74. Pluznik DH, Sachs L. The induction of clones of normal mast cells by a substance from conditioned medium. Exp Cell Res 1966; 43 : 553–563.
75. Bradley TR, Metcalf D. The growth of mouse bone marrow cells in vitro. Aust J Exp Biol Med Sci 1966; 44 : 287–299.
76. Paran M, Sachs L, Barak Y et al. In vitro induction of granulocyte differentiation in hematopoietic cells from leukemic and non-leukemic patients. Proc Natl Acad Sci USA 1970; 67 : 1542–1549.
77. Friend C, Scher W, Holland JG et al. Hemoglobin synthesis in murine virus‑induced leukemic cells in vitro: stimulation of erythroid differentiation by dimethyl sulfoxide. Proc Natl Acad Sci USA 1971; 68 : 378–382.
78. Lotem J, Sachs L. Different blocks in the differentiation of myeloid leukemic cells. Proc Natl Acad Sci USA 1974; 71 : 3507–3511.
79. Metcalf D. Humoral regulators in the development and progression of leukemia. Adv Cancer Res 1971; 14 : 181–230.
80. Lotem J, Sachs L. Independent regulation of myeloid cell growth and differentiation inducing proteins: in vivo regulation by compounds that induce inflammation. Int J Cancer 1985; 35 : 93–100.
81. Sachs L, Lotem J, Shabo Y. The molecular regulators of macrophage and granulocyte development. Role of MGI-2/IL‑6. Ann N Y Acad Sci 1989; 557 : 417–435.
82. Gunz FW, Vincent PC. Towards a cure of acute granulocytic leukemia? Leuk Res 1977; 1 : 51–66.
83. Sachs L. The differentiation of myeloid leukaemia cells: new possibilities for therapy. Br J Haematol 1978; 40 : 509–517.
84. Collins SJ, Gallo RC, Gallagher RE. Continuous growth and differentiation of human myeloid leukaemic cells in suspension culture. Nature 1977; 270 : 347–349.
85. Minowada J. Immunology of leukemic cells. In: Gunz W, Henderson ES (eds). Leukemia. 1st ed. New York: Grune and Stratton 1978 : 119–139.
86. Dalton WT Jr, Ahearn MJ, McCredie KB et al. HL‑60 cell line was derived from a patient with FAB-M2 and not FAB-M3. Blood 1988; 71 : 242–247.
87. Collins SJ, Ruscetti FW, Gallagher RE et al. Terminal differentiation of human promyelocytic leukemia cells induced by dimethyl sulfoxide and other polar compounds. Proc Natl Acad Sci USA 1978; 75 : 2458–2462.
88. Breitman TR, Selonick SE, Collins SJ. Induction of differentiation of the human promyelocytic leukemia cell line (HL‑60) by retinoic acid. Proc Natl Acad Sci USA 1980; 77 : 2936–2940.
89. Honma Y, Takenaga K, Kasukabe T et al. Induction of differentiation of cultured human promyelocytic leukemia cells by retinoids. Biochem Biophys Res Commun 1980; 95 : 507–512.
90. Breitman TR, Collins SJ, Keene BR. Terminal differentiation of human promyelocytic leukemic cells in primary culture in response to retinoic acid. Blood 1981; 57 : 1000–1004.
91. Muindi JR, Frankel SR, Huselton C et al. Clinical pharmacology of oral all‑trans retinoic acid in patients with acute promyelocytic leukemia. Cancer Res 1992; 52 : 2138–2142.
92. Stöckbauer P, Gahmberg CG, Andersson LC. Changes in cell surface glycoproteins and antigens during differentiation of the human myeloid leukemia cell lines ML‑1, ML‑2, and HL‑60. Cancer Res 1985; 45 : 2821–2826.
93. Schwarz J, Stöckbauer P, Soucek J et al. Features of immaturity in cells derived from granulocytic differentiation inducer treated human myeloid leukaemia (ML‑1) cells. Leuk Res 1987; 11 : 869–876.
94. Warrell RP Jr, Frankel SR, Miller WH Jr et al. Differentiation therapy of acute promyelocytic leukemia with tretinoin (all‑trans‑retinoic acid). N Engl J Med 1991; 324 : 1385–1393.
95. Flynn PJ, Miller WJ, Weisdorf DJ et al. Retinoic acid treatment of acute promyelocytic leukemia: in vitro and in vivo observations. Blood 1983; 62 : 1211–1217.
96. Daenen S, Vellenga E, van Dobbenburgh OA et al. Retinoic acid as antileukemic therapy in a patient with acute promyelocytic leukemia and Aspergillus pneumonia. Blood 1986; 67 : 559–561.
97. Wolf G. A history of vitamin A and retinoids. FASEB J 1996; 10 : 1102–1107.
98. Smith MA, Parkinson DR, Cheson BD et al. Retinoids in cancer therapy. J Clin Oncol 1992; 10 : 839–864.
99. Lippman SM, Kessler JF, Meyskens FL Jr Retinoids as preventive and therapeutic anticancer agents (Part I). Cancer Treat Rep 1987; 71 : 391–405.
100. Lippman SM, Kessler JF, Meyskens FL Jr. Retinoids as preventive and therapeutic anticancer agents (Part II). Cancer Treat Rep 1987; 71 : 493–515.
101. Pokorný M, Pavcová Š, Voltr J. Retinoidy a jejich použití v dermatologii. Prakt Lék 1983; 63 : 685–686.
102. Warrell RP Jr. Applications for retinoids in cancer therapy. Semin Hematol 1994; 31 : 1–13.
103. Huang ME, Ye YC, Chen SR et al. Use of all‑trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood 1988; 72 : 567–572.
104. Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood 2008; 111 : 2505–2515.
105. Degos L. Introduction to the round table on retinoids and differentiation therapy. Nouv Rev Fr Hématol 1990; 32 : 25–26.
106. Visani G, Manfroi S, Tosi P et al. All‑trans‑retinoic acid and pseudotumor cerebri. Leuk Lymphoma 1996; 23 : 437–442.
107. Chomienne C, Ballerini P, Balitrand N et al. Retinoic acid therapy for promyelocytic leukaemia. Lancet 1989; 2 : 746–747.
108. Degos L, Chomienne C, Daniel MT et al. Treatment of first relapse in acute promyelocytic leukaemia with all‑trans retinoic acid. Lancet 1990; 336 : 1440–1441.
109. Degos L. The history of acute promyelocytic leukaemia. Br J Haematol 2003; 122 : 539–553.
110. Smith MA, Adamson PC, Balis FM et al. Phase I and pharmacokinetic evaluation of all‑trans‑retinoic acid in pediatric patients with cancer. J Clin Oncol 1992; 10 : 1666–1673.
111. Adamson PC, Bailey J, Pluda J et al. Pharmacokinetics of all‑trans‑retinoic acid administered on an intermittent schedule. J Clin Oncol 1995; 13 : 1238–1241.
112. Adamson PC. Pharmacokinetics of all‑trans‑retinoic acid: clinical implications in acute promyelocytic leukemia. Semin Hematol 1994; 31 : 14–17.
113. Ohashi H, Ichikawa A, Takagi N et al. Remission induction of acute promyelocytic leukemia by all‑trans‑retinoic acid: molecular evidence of restoration of normal hematopoiesis after differentiation and subsequent extinction of leukemic clone. Leukemia 1992; 6 : 859–862.
114. Elliott S, Taylor K, White S et al. Proof of differentiative mode of action of all‑trans retinoic acid in acute promyelocytic leukemia using X‑linked clonal analysis. Blood 1992; 79 : 1916–1919.
115. Chen SJ, Zhu YJ, Tong JH et al. Rearrangements in the second intron of the RARA gene are present in a large majority of patients with acute promyelocytic leu-kemia and are used as molecular marker for retinoic acid‑induced leukemic cell differentiation. Blood 1991; 78 : 2696–2701.
116. Fenaux P, Le Deley MC, Castaigne S et al. Effect of all transretinoic acid in newly diagnosed acute promyelocytic leukemia. Results of a multicenter randomized trial. Blood 1993; 82 : 3241–3249.
117. Sanz MA, Martín G, Rayón C et al. A modified AIDA protocol with an-thracycline‑based consolidation results in high antileukemic efficacy and reduced toxicity in newly diagnosed PML/RARα-positive acute promyelocytic leukemia. PETHEMA group. Blood 1999; 94 : 3015–3021.
118. Miller WH Jr, Jakubowski A, Tong WP et al. 9-cis retinoic acid induces complete remission but does not reverse clinically acquired retinoid resistance in acute promyelocytic leukemia. Blood 1995; 85 : 3021–3027.
119. Soignet SL, Benedetti F, Fleischauer A et al. Clinical study of 9-cis retinoic acid (LGD1057) in acute promyelocytic leukemia. Leukemia 1998; 12 : 1518–1521.
120. Chen SJ, Zelent A, Tong JH et al. Rearrangements of the retinoic acid receptor alpha and promyelocytic leukemia zinc finger genes resulting from t(11;17)(q23;q21) in a patient with acute promyelocytic leukemia. J Clin Invest 1993; 91 : 2260–2267.
121. Chen GQ, Shi XG, Tang W et al. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): I. As2O3 exerts dose‑dependent dual effects on APL cells. Blood 1997; 89 : 3345–3353.
122. Shen ZX, Chen GQ, Ni JH et al. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood 1997; 89 : 3354–3360.
Štítky
Diabetologie Endokrinologie Interní lékařstvíČlánek vyšel v časopise
Vnitřní lékařství

2008 Číslo 7-8
- Diosmin a hesperidin: Co ukazuje farmakokinetika?
- Omeprazol a mechanismus hojení krvácení z peptického vředu
- Porovnání farmakologických vlastností mikronizovaného diosminu a hesperidinu v terapii chronické žilní insuficience a hemoroidů
- Srovnání vlivu omeprazolu a pantoprazolu na antiagregační účinek klopidogrelu
Nejčtenější v tomto čísle
- Urgentní stav v hematologii: akutní promyelocytární leukemie – principy diagnostiky
- Koagulopatie a diferenciační syndrom: hlavní komplikace úvodní léčby akutní promyelocytární leukemie
- Stručné kazuistiky ilustrující úvodní průběh u akutní promyelocytární leukemie
- Akutní promyelocytární leukemi e: cesta k nejlépe léčitelné akutní leukemii dospělých
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy