
Myopatie a smíšená hyperlipoproteinemie jako první projev systémové AL‑amyloidózy
Myopathy and mixed hyperlipoproteinemia as the first symptom of systemic AL-amyloidosis
Systemic AL-amyloidosis is a disorder brought about by the over-production and deposition of fragments of light immunoglobulin chains in tissues, the consequence of which is their functional impairment. The most often affected are the kidneys, the heart, the gastro-intestinal tract and the nervous system. We describe the case of a 57-year old patient in whom a rare disorder of the striated muscle (am yloid myopathy) was present, as the first clinical indication of systemic AL-amyloidosis. The main symptoms were muscle weakness and an increase in laboratory signs of muscle lesion. The patient was monitored for several years and treated with a diagnosis of primary idiopathic polymyositis (the likely diagnosis according to the diagnostic criteria of Bohan and Peter). Only after some years did hepatomegaly appear with elevated liver transaminases and a diagnosis of systemic AL-amyloidosis was established on the basis of a liver biopsy. By additional staining of striated muscle preparations with a dye for amyloid (in accordance with Maldyk) amyloid myopathy was confirmed. In addition to muscle affection, mixed hyperlipoproteinemia was present from the beginning. This was probably the first indication of amyloidosis. The case description points out the justified inclusion of systemic AL-amyloidosis in differential diagnosis of muscle weakness and signs of muscle lesion. Amyloidosis must be considered if in addition to myopathy there is also present a problem with any organ which can typically be affected by amyloidosis.
Key words:
Systemic AL-amyloidosis – myopathy – primary idiopathic polymyositis – mixed hyperlipoproteinemia – autologous peripheral stem cell transplantation
Autoři:
M. Tošovský 1; T. Soukup 1; P. Bradna 1; V. Maisnar 1; V. Tyčová 2; J. Tomš 1; M. Prixová 1
Působiště autorů:
II. interní klinika Lékařské fakulty UK a FN Hradec Králové, přednosta prof. MUDr. Jaroslav Malý, CSc.
1; Fingerlandův ústav patologie Lékařské fakulty UK a FN Hradec Králové, přednosta prof. MUDr. Aleš Ryška, Ph. D.
2
Vyšlo v časopise:
Vnitř Lék 2008; 54(10): 1014-1019
Kategorie:
Kazuistika
Souhrn
Systémová AL‑amyloidóza je onemocnění způsobené nadprodukcí a ukládáním fragmentů lehkých řetězců imunoglobulinů ve tkáních, které má za následek poruchu jejich funkce. Nejčastěji bývají postiženy ledviny, srdce, gastrointestinální trakt a nervový systém. Popisujeme případ 57letého pacienta, u kterého bylo přítomno vzácné postižení příčně pruhovaného svalu (amyloidová myopatie) jako první klinický projev systémové AL‑amyloidózy. Hlavními příznaky byla svalová slabost a zvýšení laboratorních známek svalové léze. Pacient byl několik let sledován a léčen s diagnózou primární idiopatické polymyozitidy (diagnóza pravděpodobná dle diagnostických kritérií dle Bohana a Petera). Teprve po letech se objevila hepatomegalie s elevací jaterních transamináz a na základě biopsie jater byla stanovena diagnóza systémové AL‑amyloidózy. Dodatečným obarvením preparátů příčně pruhovaného svalu barvením na amyloid (podle Maldyka) byla potvrzena amyloidová myopatie. Vedle postižení svalů byla navíc od počátku přítomna smíšená hyperlipoproteinemie. Pravděpodobně se jednalo o první projev amyloidózy. Popis případu má upozornit na oprávněné zařazení systémové AL‑amyloidózy v diferenciální diagnostice svalové slabosti a známek svalové léze. Na amyloidózu musíme pomýšlet, pokud je vedle myopatie přítomno postižení jakéhokoliv jiného orgánu typického pro postižení amyloidózou.
Klíčová slova:
systémová AL‑amyloidóza – myopatie – primární idiopatická polymyositida – smíšená hyperlipoproteinemie – autologní transplantace periferních kmenových buněk
Úvod
Systémová AL‑amyloidóza je vzácné onemocnění způsobené ukládáním fragmentů lehkých řetězců imunoglobulinů v tkáních. Tyto fragmenty se ukládají v tkáních a orgánech jako fibrilární protein zvaný amyloid, to má za následek poruchu jejich funkce, časté bývá též jejich zvětšení. Nejčastěji bývají postiženy ledviny (ledvinná amyloidóza), játra, srdce (amyloidová kardiomyopatie), trávicí trubice a nervový systém (periferní i centrální). Postižení příčně pruhovaného svalu (amyloidová myopatie) patří mezi postižení méně časté [1,2].
Popis případu
57letý pacient byl od roku 2002 sledován v revmatologické poradně. Důvodem byl subjektivní pocit svalové slabosti dolních končetin a celkové únavy provázený laboratorními známkami svalové léze (zvýšená aktivita sérové kreatinfosfokinázy a hladina myoglobinu – vstupní hodnoty 7,94 µkat/l, resp. 162 µg/l). V rodinné anamnéze pacienta nás zaujalo časné úmrtí otce, jehož příčina nebyla známa. V osobní anamnéze se kromě smíšené hyperlipoproteinemie (diagnóza v 53 letech) a arteriální hypertenze (diagnóza v 54 letech) nevyskytovala žádná jiná závažná onemocnění. Hodnoty lipidů v úvodu byly: celkový cholesterol 14,0 mmol/l, triacylglyceroly 20,1 mmol/l. Arteriální hypertenze byla úspěšně korigována beta‑blokátorem. Konzumace alkoholu byla minimální, pacient již několik let nekouřil. Fyzikální nález byl v úvodu kromě nápadněji muskulaturního typu postavy zcela v normě.
Při pátrání po příčině svalové slabosti a známkách svalové léze byla vyloučena endokrinopatie, infekce myotropními viry, byl proveden onkologický screening s negativním výsledkem. Nemocný nebyl do té doby léčen statiny či fibráty. Pro významnou hyperlipidemii byly později přechodně nasazeny, avšak pro další zhoršení laboratorních známek svalové léze byly pro další léčbu kontraindikovány. Pro podezření na polymyozitidu bylo vyšetřeno sérum pacienta na přítomnost antinukleárních protilátek a protilátek proti extrahovatelnému nukleárnímu proteinu, tato vyšetření však byla negativní. Elektromyografické vyšetření (EMG) dolních končetin prokázala lehkou demyelinizační periferní polyneuropatii motorických vláken, dále byla přítomna velmi lehká myogenní léze v m. tibialis anterior l. dx. Při magnetické rezonanci (MRI) dolních končetin byla nalezena suspektní ložiska myopatie (edém, tuková degenerace v oblasti obou m. gluteus maximus a m. quadriceps femoris), při cílené biopsii však zánětlivé změny nebyly zastiženy. I přes značné diagnostické rozpaky byl stav pacienta uzavřen jako primární idiopatická polymyozitida (dle diagnostických kritérií podle Bohana a Petera – diagnóza pravděpodobná) – tuto diagnózu do určité míry podporoval pozitivní terapeutický test s podáním 5krát 125 mg metylprednizolonu v květnu roku 2004, po kterém došlo k subjektivnímu i laboratornímu zlepšení (kreatinfosfokináza před léčbou 18,9 µkat/l, 1,8 µkat/l po léčbě), byla ponechána perorální kortikoterapie 32 mg metylprednizolonu denně.
V následující době zůstával stav pacienta stabilizovaný, přestože známky svalové léze v kolísavé míře neustále přetrvávaly. Problémem rovněž zůstávala hyperlipidemie, která byla navíc po nasazení kortikoterapie ještě výraznější. Pacientovi byla doporučena nízkocholesterolová dieta, léčba pryskyřicí (cholesyramin) byla bez efektu. Přechodně byla použita terapie pomocí extrakorporální plazmaferézy s dobrým, ale přechodným efektem. Koncentrace lipidů v séru zůstávaly vysoké (cholesterol nad 10 mmol/l, TAG nad 6 mmol/l).
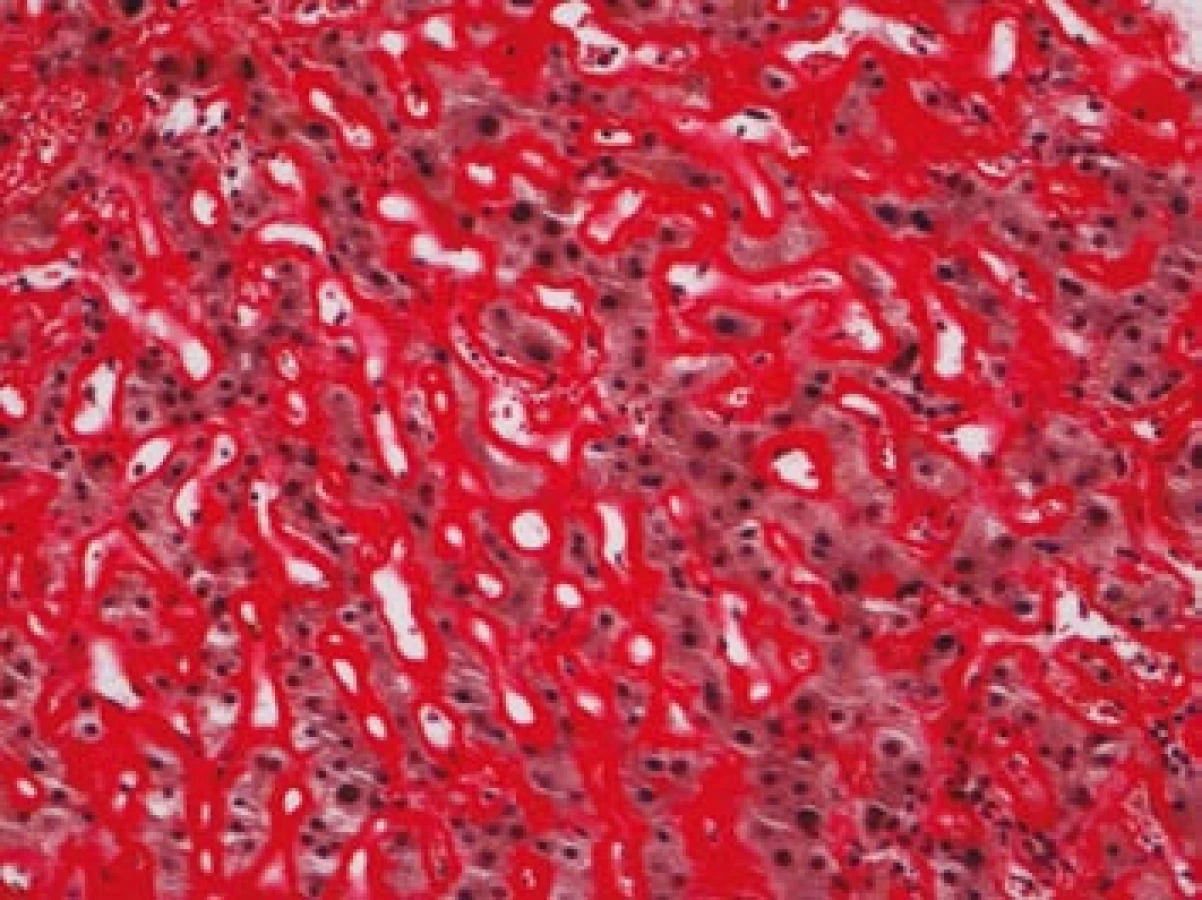
Na podzim roku 2006 byl zachycen vzestup jaterních transamináz – ALT 2,1 µkat/l, AST 1,71 µkat/l, GMT 10,21 µkat/l (do té doby jen mírně nadhraniční hodnoty), při fyzikálním vyšetření byla zjištěna hepatosplenomegalie (dle UZ 19,5 cm, resp. 14,5 cm, více než rok předtím byla velikost jater a sleziny dle UZ v normě). Z tohoto důvodu byla provedena necílená jaterní biopsie, která potvrdila podezření na společného jmenovatele pacientových obtíží – v cévách portobiliárních prostorů a v sinusech uvnitř Disseho prostorů byla přítomna eosinofilní homogenní hmota, která vykazovala pozitivní barvení na amyloid (obr. 1). Při dodatečném imunohistochemickém vyšetření vykazovala tkáň pozitivní průkaz κ lehkých řetězců (obr. 2), nález byl proto diagnosticky uzavřen jako systémová AL‑amyloidóza. Dodatečně byly na požádání obarveny na přítomnost amyloidu i preparáty biopsií z předchozích let. Vzorek příčně pruhovaného svalu z roku 2003 vykazoval přítomnost amyloidu drobných cév, v samotných svalových vláknech nebyl amyloid prokázán (obr. 3). V biopsii tračníku z roku 2004 (průjmy neurčené etiologie s normálním pankoloskopickým nálezem) byl po dodatečném obarvení amyloid prokázán rovněž ve stěnách drobných cév, ojedinělá depozita byla prokázána i ve vláknech hladké svaloviny (obr. 4). Následně bylo pátráno po známkách postižení dalších orgánů. Byla zjištěna koncentrická hypertrofie levé srdeční komory bez poruchy systolické funkce (možný orgánový projev amyloidózy), rovněž byla zjištěna nadhraniční proteinurie (0,34 g/den) jako možný projev postižení ledvin. Biopsie myokardu ani ledvin nebyla u nemocného provedena. Paraprotein v séru ani v moči pacienta nebyl metodou imunofixace detekován, sedimentace erytrocytů byla středně zvýšena (20/46 mm/hod, resp. 2 hod).



Po stanovení základní diagnózy byla zahájena systémová chemoterapie. Po 4 cyklech pulzní terapie AD (à 1 měsíc Doxorubicin 60 mg den 1, Dexamethason 40 mg p.o. den 1–4, 10–13, 20–23) a následné separaci periferních kmenových buněk (PKB) byla v květnu roku 2006 provedena autologní transplantace PKB s použitím přípravného režimu MEL200 (Melfalan 200 mg/m2). Již po zahájení indukční chemoterapie AD došlo u nemocného k poměrně výraznému snížení známek jaterní a svalové léze, současně došlo i ke zlepšení pacientova lipidogramu (tab. 1). K dalšímu zlepšení hodnot došlo po transplantaci PKB (tab. 1). Další trend ve vývoji pacientova zdravotního stavu včetně subjektivního vnímání svalové slabosti prokáže teprve následující čas.

Diskuze
U našeho pacienta jsme objevili vzácné onemocnění – systémovou AL‑amyloidózu. Incidence tohoto onemocnění je udávána mezi 5,1 a 12,8 na milion osob za rok [2,4]. Jedná se o onemocnění z početnější skupiny amyloidóz, v tomto případě jsou v tkáních ukládány fragmenty lehkých řetězců imunoglobulinů (častěji λ, méně často κ), které jsou produkovány klonem plazmatických buněk. AL‑amyloidóza se může vyskytnout samostatně, často bývá spojena s onemocněním mnohočetným myelomem. U našeho pacienta nebyl v séru ani moči paraprotein metodou imunofixace detekován. Jedná se tedy o méně častý případ, jelikož paraprotein bývá detekovatelný u pacientů s AL‑amyloidózou až v 90 % [5–7].
V našem případě jsme zjistili amyloidovou myopatii, která byla prvním projevem tohoto onemocnění. Postižení příčně pruhovaného svalu patří mezi méně časté příznaky systémové AL‑amyloidózy [8]. Myopatie jako první projev systémové AL‑amyloidozy je pak popisována výjimečně, ještě vzácněji se jedná o jediný orgánový projev systémové AL‑amyloidózy [9]. U pacienta byly přítomny typické příznaky amyloidové myopatie, jak jsou popisovány v literatuře. Nejčastěji bývá přítomna svalová slabost převážně pletencových svalů [10–12], ojediněle je popisována slabost distálních svalů [13]. Přítomny bývají rovněž laboratorní známky svalové léze a elektromyografický nález myopatie. V našem případě se nevyskytovala makroglosie s poruchou artikulace ani dysfagie, která bývá u pacientů s AL‑amyloidozou popisována. Byla však přítomna bolestivost a tzv. pseudohypertrofie svalů, které bývají u těchto pacientů rovněž přítomny [11,14]. Pacienti pak mohou budit zdání vypracované muskulaturní postavy. Histologickým korelátem bývá přítomnost hypertrofie svalových vláken, která však v našem případě popsána nebyla.
Při histologickém vyšetření (hematoxylin‑eosin) příčně pruhovaného svalu nebyla diagnóza vzhledem k diskrétnímu postižení zpočátku stanovena, teprve při dobarvení na amyloid byla depozita patrna ve stěnách drobných cév. Stejný histologický obraz bývá ve většině případů amyloidové myopatie uváděné v literatuře – maximum depozit bývá pouze ve stěnách cév (nejčastěji arteriol), postižení v oblasti intersticia svalových vláken je popisováno méně často [10,13,15–17]. Patogeneze myopatie není zcela objasněna. Jedním z uváděných mechanizmů je porucha prokrvení svalů kvůli depozitům amyloidu v cévních stěnách, což má za následek poruchu výživy svalu, váznutí odstraňování odpadních produktů a ischemii. Dalším uváděným mechanizmem je snížení svalové elasticity kvůli depositům amyloidu, ev. porucha šíření akčního potenciálu [8,15]. Včasná diagnóza amyloidové myopatie je důležitá, časné zahájení terapie amyloidové myopatie může vést dle dostupných literárních zdrojů ke zlepšení jejích příznaků [13,18,19].
U našeho pacienta jsme se zpočátku mylně domnívali, že se jedná o primární idiopatickou polymyozitidu, jelikož splňoval diagnostická kritéria dle Bohana a Petera na úrovni pravděpodobné diagnózy. Případy, kdy byl pacient v úvodu léčen jako primární idiopatická polymyozitida a později byla diagnostikována amyloidová myopatie, jsou v literatuře popisovány [17,20]. Pro doplnění uvádíme stručný přehled možných příčin myopatie (tab. 2).
![Příčiny myopatií [29].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/55c424a85d0656cc3c1f0ce0c921c332.png)
Domníváme se, že hyperlipoproteinemie byla způsobena amyloidovou he-pa-topatií. Částečně se pravděpodobně podílela pozdější medikace kortikoidy, jelikož po zahájení terapie kortikoidy došlo ke zhoršení lipidogramu. Postižení jater při amyloidóze je časté. Popisovány bývají hepatomegalie, elevace hladiny bilirubinu, zvýšená aktivita alkalické fosfatázy, γ-glutamyltransferázy a prodloužení protrombinového času (25 %, 11 %, 26 %, 34 %, resp. 16 %) [21]. Přítomnost hypercholesterolemie (nad 7,8 mmol/l) a hypertriglyceridemie (nad 3,4 mmol/l) je popisována v 27 %, resp. v 13 % případů [21]. Hyperlipidemie je často považována za sekundární v rámci nefrotického syndromu způsobeného amyloidózou. O souvislosti hyperlipidemie s postižením jater při amyloidóze přinášejí důkazy publikace, ve kterých jsou popisovány poruchy lipidového spektra bez současné přítomnosti nefrotického syndromu [22,23]. O hyperlipidemii jako první manifestaci primární amyloidózy pak hovoří pouze jediná publikace [21]. V literatuře je popisováno několik možných patogenetických mechanizmů, které mají za následek vznik hyperlipidemie – cholestáza, deficit jaterní lipázy a LCAT (lecitin‑cholesterol-acyl-transferáza), porucha odbourávání IDL (intermediate density lipoproteins) [22]. Významná úprava do té doby obtížně korigovatelného lipidogramu, která následovala během pulzní terapie AD a po autologní transplantaci periferních kmenových buněk, svědčí v našem případě pro možnou souvislost hyperlipoproteinemie a amyloidózy. Dlouhodobá dávka kortikoidů od zjištění diagnózy do transplantace zůstávala stejná, naopak byla navýšena o dexametason podávaný v rámci pulzní terapie AD, přesto docházelo k poklesu hladiny lipidů.
V našem případě se tedy jednalo o amyloidovou myopatii, která byla od počátku doprovázena hyperlipoproteinemií. Oba typy postižení jsou v případě prvního projevu onemocnění popisovány vzácně (na úrovni kazuistik), v našem případě se jedná o prvně popsaný společný výskyt těchto dvou příznaků [9,21]. Současná lehká neuropatie (diagnóza pomocí EMG) pak rovněž spadá do obrazu AL‑amyloidózy. Dodatečně bylo stanoveno postižení trávicí trubice a myokardu. Přestože na diagnózu amyloidózy můžeme mnohdy pomýšlet již na základě klinického obrazu, její definitivní stanovení je možné pouze na základě biopsie. Depozita amyloidu se ve světelném mikroskopu jeví jako amorfní hyalinní hmota, k ozřejmení je nutné barvení např. dle Maldyka, typ amyloidu pak určí imunohistochemické vyšetření [22–24].
Náš pacient byl léčen pomocí transplantace periferních kmenových bu-něk, která umožňuje použití vysokodávkové chemoterapie. Tato metoda je používána od poloviny 90. let minulého století a přináší zatím nejlepší výsledky [25–27]. Cílem přitom není jen zastavení další progrese onemocnění, ale po odstranění primárního činitele (nadprodukce fragmentů imunoglobulinů) je popisováno postupné vstřebání deponovaného amyloidu z postižených tkání a zlepšení zdravotního stavu nemocných. U ně-kte-rých pacientů je popisováno i kompletní uzdravení [28].
Závěr
Cílem našeho článku je upozornit na oprávněné zařazení systémové AL‑amyloidózy do diferenciální diagnostiky svalové slabosti s laboratorními známkami svalové léze. Do vyšetřovacího algoritmu těchto stavů bývá běžně zařazena svalová biopsie, k objasnění amyloidové myopatie pak postačí doplnit obarvení preparátu příčně pruhovaného svalu na amyloid, které může odhalit jeho případná depozita. K tomuto kroku by nás pak měla vést přítomnost jakéhokoliv dalšího příznaku budícího podezření na možnou amyloidózu (v našem případě hepatopatie a neuropatie). V případě postižení jiného orgánu vhodného k bioptickému vyšetření je vhodné provést biopsii k průkazu amyloidózy.
MUDr. Marian Tošovský
www.fnhk.cz
e‑mail: tosmar@post.cz
Doručeno do redakce: 4. 9. 2007
Přijato po recenzi: 30. 6. 2008
Zdroje
1. Falk RH, Comenzo RL, Skinner M. The systemic amyloidoses. N Engl J Med 1997; 337 : 898–909.
2. Gertz MA, Lacy MO, Dispenzieri A et al. Amyloidosis. Best Pract Res Clin Haematol 2005; 18 : 709–727.
3. Adam Z, Ščudla V. Clinical manifestation of AL‑amyloidosis and some other type of amyloidosis. Vnitř Lék 2001; 47 : 36–45.
4. Kyle RA, Linos A, Beard CM et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood 1992; 79 : 1817–1822.
5. Tichý M, Maisnar V. Laboratorní průkaz monoklonálních imunoglobulinů. Vnitř Lék 2006; 52 (Suppl 2): 41–45.
6. Kyle RA, Greipp PR. Amyloidosis (AL). Clinical and laboratory features in 229 cases. Mayo Clin Proc 1983; 58 : 665–683.
7. Sanders PW, Herrera GA, Kirk KA et al. Spectrum of glomerular and tubulointerstitial renal lesions associated with monotypical immunoglobulin light chain deposition. Lab Invest 1991; 64 : 527–537.
8. Roke ME, Brow WFE, Boughner D. Myo-pathy in primary systemic amyloidosis. Can J Neurol Sci 1988; 15 : 314–316.
9. Karacostas D, Soumpourou M, Mavromatis I et al. Isolated myopathy as the initial manifestation of primary systemic amyloidosis. J Neurol 2005; 252 : 853–854.
10. Chapin JE, Kornfeld M, Harris A. Amyloid myopathy: characteristic features of a still underdiagnosed disease. Muscle Nerve 2005; 31 : 266–272.
11. Roke ME, Brown WF, Bouhner D et al. Myopathy in primary systemic amyloidosis. Can J Neurol Sci 1988; 15 : 314–316.
12. Vaish AK, Mehrotra S, Kushwaha MR. Proximal muscle weakness due to amyloid deposition. J Neurol Neurosurg Psychiatry 1998; 64 : 409–410.
13. Smestad C, Monstad P, Lindboe CF et al. Amyloid myopathy presenting with distal atrophic weakness. Muscle Nerve 2004; 29 : 605–609.
14. Scola RH, Werneck LC, Ramos CS et al. Amyloiditic muscle pseudohypertrophy: case report. Arq Neuropsiquiatr 2001; 59 : 582–586.
15. Li K, Hizawa K, Numomura S et al. Systemic amyloid myopathy: light-microscopic and fine structural study of the skeletal muscles with histochemical and immunohistochemical study of amyloid. Acta Neuropathol (Berl) 1984; 64 : 114–121.
16. Yoshita M, Ishida C, Yanase D et al. Immunoglobulin light-chain (AL) amyloidosis with myasthenic symptoms and echocardiographic features of dilated cardiomyopathy. Intern Med 2006; 45 : 159–162.
17. Mandl LA, Folkerth RD, Pick MA et al. Amyloid myopathy masquerading as polymyositis. J Rheumatol 2000; 27 : 949–952.
18. Majolino I, Marcenò R, Pecoraro G. High‑dose therapy and autologous transplantation in amyloidosis – AL. Haematologica 1993; 78 : 68–71.
19. Merlini G. Treatment of primary amyloidosis. Semin Hematol 1995; 32 : 60–79.
20. Hull KM, Griffith L, Kuncl RW et al. A deceptive case of amyloid myopathy: clinical and magnetic resonance imaging features. Arthritis Rheum 2001; 44 : 1954–1961.
21. Couture P, Le Blanc F, Gagnon P et al. Hyperlipidemia as the first biochemical manifestation of primary amyloidosis. Am J Gastroenterol 1997; 92 : 1046–1047.
22. Levy Y, Magill PJ, Miller NE et al. Primary systemic amyloidosis presenting as extreme hyperlipidaemia with tendon xanthomas. Br Med J (Clin Res Ed) 1981; 283 : 699–700.
23. Buxbaum JN, Tagoe CE. The genetics of the amyloidoses. Annu Rev Med 2000; 51 : 543–569.
24. Serpell LC, Sunde M, Benson MD et al. The protofilament substructure of amyloid fibrils. J Mol Biol 2000; 300 : 1033–1039.
25. Comenzo RL, Gertz MA. Autologous stem cell transplantation for primary systemic amyloidosis. Blood 2002; 99 : 4276–482.
26. Gertz MA, Lacy MO, Gastineau DA et al. Blood stem cell transplantation as therapy for primary systemic amyloidosis (AL). Bone Marrow Transplant 2000; 26 : 963–969.
27. Adam Z, Ščudla V, Tomiska M. Treatment of AL‑amyloidosis and some other types of amyloidosis. Vnitř Lék 2001; 47 : 46–52.
28. Gillmore JD, Davies J, Iqbal A et al. Allogeneic bone marrow transplantation for systemic AL amyloidosis. Br J Haematol 1998; 100 : 226–228.
29. Miller ML. Approach to the patient with muscle weakness. UpToDate 16.1. [online]. 2008 [cit. 2008–06–24]. http://www.utdol.com/online/content/topic.do?topicKey =muscle/ 2406&selectedTitle=1~150&source=search_result.
Štítky
Diabetologie Endokrinologie Interní lékařstvíČlánek vyšel v časopise
Vnitřní lékařství

2008 Číslo 10
- Proces hojení ran krok za krokem a co ho může zkomplikovat
- Limity glykovaného hemoglobinu a význam dalších glykovaných proteinů
- Korelace dávky levothyroxinu s titrem autoimunitních protilátek u primární hypotyreózy
- Zeolit-jodový komplex pomáhá v péči o infikované rány
Nejčtenější v tomto čísle
- Duální protidestičková léčba
- Srdeční arytmie při obstruktivní spánkové apnoe
- Stanovení LDL‑cholesterolu – stále nevyřešený problém: vypočíst, nebo změřit? – editorial
- Hemoeliminační metody v léčbě sepse: současný stav
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy