
Získaná uniparentální disomie v buňkách kostní dřeně nemocných s myelodysplastickými syndromy a komplexním karyotypem
Acquired uniparental disomy in bone-marrow cells of patients with myelodysplastic syndrome and complex karyotype
Complex karyotypes are seen in approximately 20% of patients with myelodysplastic syndromes (MDS) and are associated with a high risk of transformation into acute myeloid leukaemia (AML) and poor prognosis. Acquired uniparental disomy (aUPD, i.e. both copies of a chromosome pair or its part originate from one parent) may contribute to increased genomic instability in bone-marrow cells of patients with MDS. The pathological potential of aUPD, which arises as a clonal aberration in a proportion of somatic cells, involves tumour suppressor gene and oncogene homozygous mutations.
The aim of this study was to evaluate the frequency and implications of uniparental disomy (UPD) in a cohort of 57 patients with MDS and complex karyotype using array comparative genomic hybridization with detection of single nucleotide polymorphisms (aCGH/SNP).
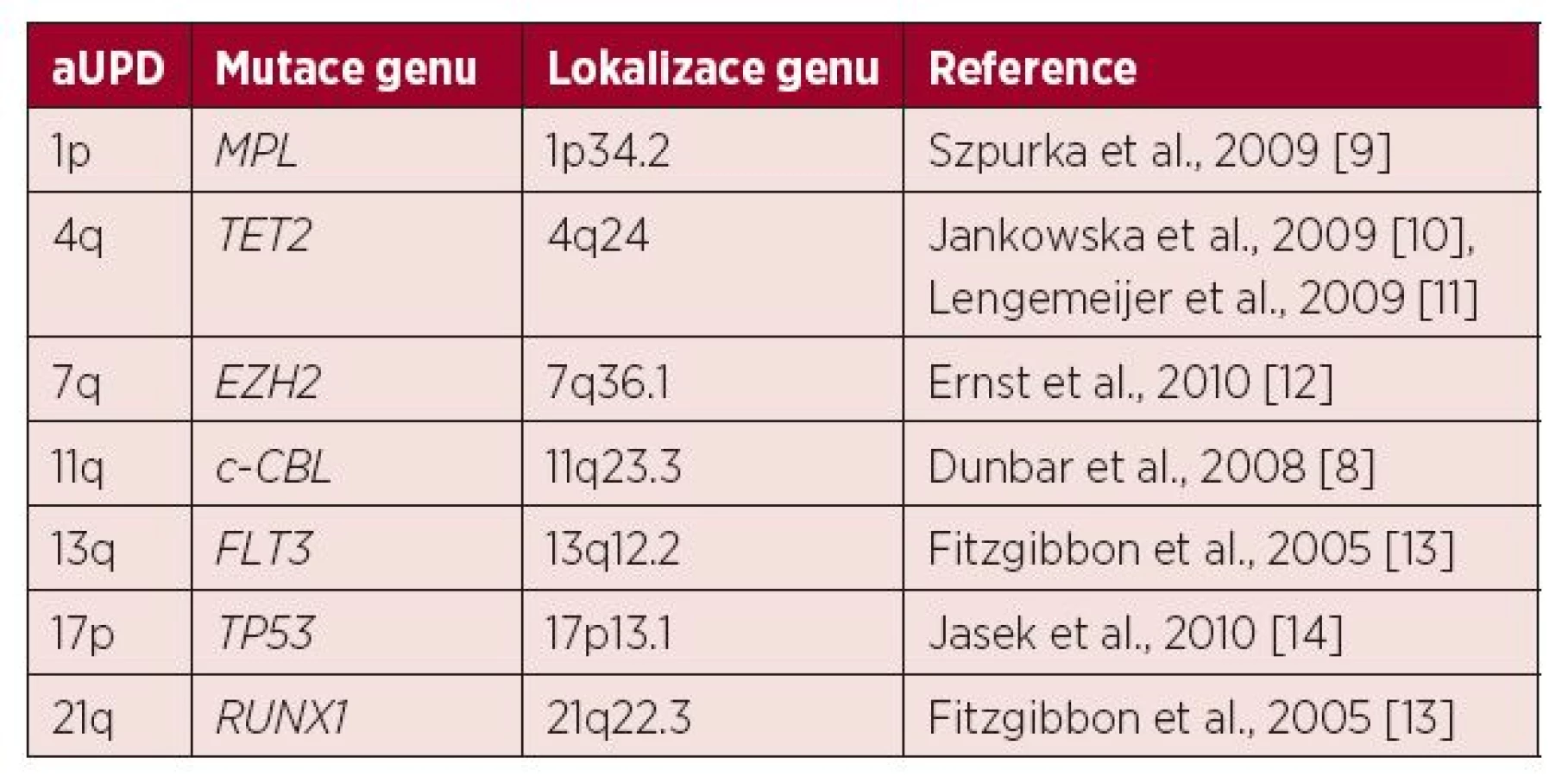
UPD was found to be present in 40 regions in 21 of 57 patients (36.8%). Almost half of these involved non-recurrent changes (19/40). Chromosome X UPD was detected in six patients (Xp22.11–Xp22.2 in four and Xq13.3–Xq21.1 in two, respectively). UPD of 17q with a variable extent from 17q22 to 17q24.2 was observed in two patients. The most common finding was aUPD of the short arm of chromosome 17, which was detected in 13 of 57 patients (22.8%). Mutational analysis confirmed a homozygous mutation of the TP53 gene in all samples with this finding, including two frameshift mutations that are not registered in the IARC (International Agency for Research on Cancer TP53) database. This finding correlated with very poor prognosis (median OS 4 months). aUPD 17p was strongly associated with complex karyotype in the studied cohort. However, other previously published aUPDs in MDS (1p, 4q, 7q, 11q, 13q, 21q) were not found in our study.
UPD regions appear to be balanced (i.e. without change of DNA copies) by conventional and molecular cytogenetic methods. Therefore, aCGH/SNP represents an ideal method for the identification and further characterization of UPDs and affected genes significant for disease development and progression.
Key words:
uniparental disomy, myelodysplastic syndromes, complex karyotype, mutation, TP53 gene
Autoři:
K. Svobodová 1; Z. Zemanová 1; H. Lhotská 1; M. Nováková 1; J. Březinová 2; M. Beličková 2; A. Berková 1; I. Šárová 2; L. Lizcová 1; S. Izáková 1; A. Jonášová 3; J. Čermák 2; K. Michalová 1,2
Působiště autorů:
Centrum nádorové cytogenetiky, Ústav lékařské biochemie a laboratorní diagnostiky, VFN a 1. LF UK, Praha
1; Ústav hematologie a krevní transfuze, Praha
2; I. interní klinika, VFN a 1. LF UK, Praha
3
Vyšlo v časopise:
Transfuze Hematol. dnes,21, 2015, No. 3, p. 126-134.
Kategorie:
Souhrnné práce, původní práce, kazuistiky
Souhrn
U 10–20 % nemocných s myelodysplastickými syndromy (MDS) je nalézán komplexní karyotyp, který je spojen s vysokým rizikem transformace do akutní myeloidní leukemie (AML) a špatnou prognózou. Jedním z jevů, které se mohou podílet na zvýšené genomové nestabilitě buněk kostní dřeně u nemocných s MDS, je získaná uniparentální disomie (aUPD), tj. stav, kdy obě kopie určitého chromozomu nebo jeho části pochází od jednoho rodiče. Patologický potenciál této změny, která vzniká jako klonální aberace v některých somatických buňkách, spočívá v přítomnosti homozygotních mutací tumor supresorových genů a onkogenů.
Cílem studie bylo zjistit frekvenci a význam uniparentální disomie (UPD) v souboru 57 nemocných s MDS a komplexním karyotypem za použití array komparativní genomové hybridizace kombinované s detekcí jednonukleotidových polymorfismů (aCGH/SNP).
V souboru 57 nemocných bylo nalezeno celkem 40 oblastí UPD u 21 pacientů (36,8 %). U zhruba poloviny nálezů šlo o sporadický výskyt (19/40). U šesti pacientů byly detekovány UPD na chromozomu X lokalizované v oblastech Xp22.11–Xp22.2 (4/6) a Xq13.3–Xq21.1 (2/6) a u dvou pacientů byla pozorována UPD 17q s proměnlivým rozsahem v pruzích 17q22 až 17q24.2. Nejčastějším nálezem byla aUPD krátkých ramen chromozomu 17, která byla detekována celkem u 13/57 pacientů (22,8 %). U všech 13 pacientů mutační analýza potvrdila homozygotní mutaci genu TP53, z toho dvě nalezené posunové (frameshift) mutace nebyly dosud popsány v databázi IARC (International Agency for Research on Cancer TP53 database). Nález byl spojen s velmi špatnou prognózou (medián přežití 4 měsíce). Vzhledem k četnosti výskytu ve studovaném souboru byla aUPD 17p silně asociována s komplexním karyotypem na rozdíl od jiných v literatuře dosud popsaných oblastí aUPD (1p, 4q, 7q, 11q, 13q, 21q), které nebyly v souboru detekovány vůbec.
Oblasti UPD se jeví při analýzách metodami klasické a molekulární cytogenetiky jako balancované, tedy bez změny počtu kopií DNA. Čipové technologie s detekcí jednonukleotidových polymorfismů jsou ideální metodou pro identifikaci a bližší charakterizaci oblastí UPD a s nimi souvisejících genů významných pro vznik a progresi onemocnění.
Klíčová slova:
uniparentální disomie, myelodysplastické syndromy, komplexní karyotyp, mutace, gen TP53
ÚVOD
Myelodysplastické syndromy (MDS) představují heterogenní skupinu klonálních hematologických onemocnění, která se projevují neefektivní krvetvorbou, morfologickou dysplazií krvetvorných buněk a periferní cytopenií postihující jednu či více krvetvorných linií. Incidence tohoto onemocnění se pohybuje okolo 4–5 nemocných na 100 000 obyvatel ročně, převážně jde o pacienty starší 65 let. Chromozomové aberace jsou v době diagnózy popisovány přibližně u 50 % nemocných s primárními MDS a až u 80 % pacientů se sekundárními MDS.
U 10–20 % nemocných je nalézán komplexní karyotyp zahrnující 3 a více chromozomové aberace [1]. Komplexní karyotyp je spojen s vysokým rizikem transformace do akutní myeloidní leukemie (AML), špatnou odpovědí na léčbu a vyšším rizikem relapsu. Vznik komplexních aberací je odrazem zvýšené genomové nestability nádorových buněk, její příčiny však dosud nebyly spolehlivě objasněny. Významnou úlohu mohou hrát např. kryptické genové mutace nebo tzv. získané uniparentální disomie (aUPD, acquired UPD) některých chromozomových oblastí nebo celých chromozomů, v jejichž důsledku dochází k inaktivaci tumor supresorových genů nebo naopak ke zvýšení exprese genů ovlivňující proliferaci patologického klonu.
Uniparentální disomie (UPD) je stav, kdy obě kopie určitého chromozomu nebo jeho části pochází od jednoho rodiče. Jedná se tedy o balancovanou změnu, při které dochází ke ztrátě heterozygozity, ale nikoliv ke změně počtu kopií DNA sekvencí. Vrozená UPD vzniká chybou v meióze během zárodečného vývoje a všechny buňky vyvíjejícího se organismu následně nesou tutéž změnu. Tento stav může mít u člověka za následek klinické projevy vrozených vývojových poruch, a to na základě vytvoření homozygotního stavu dané oblasti nebo odlišného maternálního či paternálního imprintingu.
V posledních letech je odbornou veřejností diskutován pojem získané uniparentální disomie. Objevuje se u části somatických buněk během mitotického dělení a je součástí patologického klonu odpovědného za vznik a progresi choroby, zejména u onkologických onemocnění.
aUPD může vzniknout několika různými mechanismy (obr. 1). Chybnou segregací v mitóze dojde k trisomii nebo monosomii jednoho chromozomu. Během dalšího dělení dochází ke ztrátě nadbytečného, případně ke zdvojení zbývajícího chromozomu, mitotickou nondisjunkcí, a tak ke vzniku aUPD celého chromozomu. Prostřednictvím jedné či více mitotických rekombinací, případně vlivem opravy dvouřetězcového zlomu, vzniká segmentální aUPD.

Patologický potenciál aUPD spočívá v přítomnosti mutací tumor supresorových genů, onkogenů, genů pro miRNA, imprintovaných genů, hyper - nebo hypometylovaných genů, deacetylovaných genů, případně ve ztrátě malých úseků obsahujících geny důležité pro tumorigenezi [2]. Duplikace heterozygotní somatické či vrozené mutace vede ke ztrátě funkce tumor supresorových genů, případně ke zvýšené expresi mutovaných onkogenů kvůli přítomnosti jejich dvou kopií.
Jedním z prvních objevů upozorňujících na výskyt uniparentální disomie u hematoonkologických onemocnění byl nález aUPD krátkých ramen chromozomu 9 u nemocných s diagnózou polycythaemia vera [3]. aUPD byla popsána u 33 % pacientů s tímto onemocněním a posléze spojena s výskytem homozygotní aktivující mutace genu JAK2 [4, 5].
U MDS a AML se aUPD vyskytuje zhruba u 20 % nemocných a mezi nejčastěji nalézané a potenciálně prognosticky významné patří aUPD 1p, 4q, 7q, 11q, 13q, 17p a 21q (tab. 1) [6, 7, 8]. Pacienti, u nichž jsou nalezeny tyto změny, mají obecně horší prognózu (15).
Cílem naší studie bylo zjistit frekvenci a klinický význam aUPD u nově diagnostikovaných nemocných s MDS a komplexním karyotypem s použitím array komparativní genomové hybridizace kombinované s detekcí jednonukleotidových polymorfismů (aCGH/SNP, array comparative genomic hybridization/single nucleotide polymorphism).
MATERIÁL A METODIKA
V období let 2011–2014 bylo retrospektivně vyšetřeno 57 vzorků kostní dřeně pacientů s MDS, u kterých byl v době stanovení diagnózy konvenčním G-pruhováním a/nebo mFISH nalezen komplexní karyotyp (≥ 3 chromozomové aberace). Do souboru bylo zařazeno 32 mužů a 25 žen, medián věku nemocných v době diagnózy byl 70 let a medián přežití 2 měsíce. Skupina byla rozdělena podle WHO klasifikace následovně: refrakterní anémie (RA) – 2 pacienti, refrakterní anémie s přebytkem blastů v rozmezí 5–9 % myeloblastů (RAEB I) – 7 nemocných, refrakterní anémie s přebytkem blastů v rozmezí 10–19 % myeloblastů (RAEB II) – 16 nemocných, MDS neklasifikovatelný (MDS NS) – 3 pacienti, refrakterní cytopenie s multilineární dysplazií (RCMD) – 5 nemocných, AML s myelodysplastickými změnami (MDS-AML) – 24 pacientů [16]. Všichni pa-cienti podepsali informovaný souhlas s použitím vzorků pro výzkumné účely.
Nestimulované buňky kostní dřeně byly kultivovány 24 hodin v médiu RPMI 1640 s 10% fetálním telecím sérem, dále zpracovány standardním cytogenetickým postupem za použití kolcemidu, hypotonického roztoku a fixace (metanol/kyselina octová 3 : 1). Chromozomové preparáty byly pro potřeby konvenční cytogenetické analýzy barveny Wrightovým barvivem (G-pruhy) a následně bylo hodnoceno alespoň 20 mitos (pokud byly na preparátu přítomny). Karyotypy byly popsány podle mezinárodní ISCN nomenklatury 2013 [17]. K ověření chromozomových aberací nalezených konvenčními technikami byly použity metody interfázní fluorescenční in situ hybridizace (I-FISH), mnohobarevná FISH (mFISH), mnohobarevné pruhování (mBAND) a aCGH/SNP. Metody byly prováděny podle standardních postupů doporučených výrobci.
K průkazu nebalancovaných změn karyotypu, upřesnění zlomových míst a detekci UPD byly použity čipy Cytochip Cancer SNP 4x180K v 2.0 (BlueGnome, UK; Illumina, USA). U každého vzorku bylo hybridizováno 1000 ng izolované DNA (QIAamp DNA Blood Mini kit, Qiagen Inc., Německo) podle návodu popsaného výrobcem. Koncentrace a kvalita DNA byla kontrolována spektrofotometrem NanoDrop 2000 (Wilmington, USA). Čip byl naskenován pomocí systému Agilent G2565CA Microarray Scanner (Agilent, USA). Vyhodnocení oligonukleotidových sond v programu BlueFuse Multi (verze 3.1–4.1) proběhlo na základě porovnání intenzit fluorescenčního signálu značeného vzorku a referenční DNA, přičemž každý byl značen jiným fluorescenčním barvivem. Analýza SNP sond byla vyhodnocena na základě intenzity fluorescenčního signálu navázané DNA, která závisí na jednonukleotidových polymorfismech v místě štěpení restriktázami AluI a RsaI. Podle informací výrobce je tento typ čipu schopen rozlišit UPD o velikosti minimálně 5 Mb.
K ověření počtu kopií genu TP53 jsme použili I-FISH se sondou LSI TP53/CEP 17 (Abbott Vysis, USA).
Mutace genu TP53 (exony 4–11) byly analyzovány za využití sekvenačních metod nové generace (454 GS Junior, Roche, Švýcarsko). Průměrná hloubka čtení byla 800 pro sekvenované exony a umožnila nastavit detekční limit pro záchyt mutace na 2 % nádorových buněk ve vzorku. Primery použité pro analýzu byly ověřené v rámci mezinárodní studie IRON-II (Interlaboratory Robustness Of Next generation sequencing) a sekvenační protokol byl dodržen podle doporučení výrobce. Všechny mutace s alelickou frekvencí ≥ 20 % byly ověřeny Sangerovým sekvenováním (Applied Biosystems 3500, USA).
VÝSLEDKY
V souboru 57 nemocných s MDS a komplexními karyotypy jsme nalezli 40 oblastí UPD celkem u 21 pacientů (36,8 %). U necelé poloviny případů šlo o sporadické nálezy (19/40) pozorované na chromozomech 1, 2, 3, 5, 7, 9, 13, 14, 16, 17, 19, 20, 22 a X. Opakovaně jsme pozorovali UPD na krátkých i dlouhých ramenech chromozomů X (6krát) a 17 (15krát). Přehled těchto rekurentních nálezů, věk nemocných, klinické subtypy MDS a celkové přežití jednotlivých pacientů v této skupině uvádí tabulka 2.

Nejčastějším nálezem byla aUPD krátkých ramen chromozomu 17 (13/57), celkem 22,8 % případů. Medián věku pacientů s aUPD 17p v době diagnózy byl 64 let a medián doby přežití 4 měsíce. Velikost aUPD 17p se pohybovala od 8 do 20 Mb.
U 11 z 13 pacientů s aUPD 17p jsme současně prokázali deleci dlouhých ramen chromozomu 5, dva nemocní měli komplexní karyotyp bez delece 5q (obr. 2). K dalším rekurentním strukturním/početním změnám patřila delece dlouhých ramen nebo monosomie chromozomu 7 (8/13), delece krátkých ramen chromozomu 3 (8/13) a delece krátkých ramen chromozomu 12 zahrnující gen ETV6 (8/13).

U většiny nemocných (10/13) s aUPD 17p jsme nenalezli žádné další strukturní nebo numerické aberace chromozomu 17. U dvou pacientů jsme kombinací použitých metod prokázali balancovanou translokaci zahrnující chromozom 17 (KD6, KD9). U vzorku KD6 jsme metodou mFISH nalezli komplexní přestavbu chromozomu 17, při které byla část dlouhých ramen chromozomu 17 translokována na dlouhá ramena chromozomu 5 a zároveň byla do oblasti 17p přemístěna část dlouhých ramen chromozomu 19. U pacienta KD9 došlo k rozdělení chromozomu 17 na dvě části, přičemž dlouhá ramena spolu s centromerou vytvořila dicentrický chromozom s chromozomem 20 - dic(17;20)(p11;q11) a krátká ramena byla přemístěna na marker chromozom tvořený materiálem z částí dlouhých ramen chromozomů 10, 17 a 21. aCGH/SNP v obou případech prokázala, že jde o balancovanou translokaci bez změny počtu kopií DNA chromozomu 17. U pacienta KD8 jsme nalezli nebalancovanou translokaci t(7;17) se dvěma delecemi dlouhých ramen chromozomu 17 v pruzích 17q11.2 a 17q21.31–17q21.33 o velikosti 2 a 5 Mb.
U pacientů s prokázanou aUPD 17p jsme provedli mutační analýzu genu TP53 (tab. 3). U všech 13 nemocných jsme prokázali přítomnost homozygotní mutace genu TP53. Velikost patologického klonu se pohybovala od 70 do 97 %. Ve vzorku KD12 jsme detekovali dvě různé mutace s velikostí patologického klonu 60,6 % a 13,4 %. Mutace byly nalezeny v exonech 5, 6, 7 a 8. Většinou se jednalo o mutace měnící smysl (missense mutace) v DNA vazebné doméně (9/14), u dvou pacientů jsme prokázali nesmyslné (nonsense) mutace a u dvou posunové (frameshift) mutace. U pacienta KD10 jsme nalezli homozygotní mutaci genu TP53 ve 4. intronu. Nalezené mutace byly porovnávány s databází IARC (International Agency for Research on Cancer TP53 database, http://www-p53.iarc.fr/). jedenáct z 13 prokázaných mutací bylo již v databázi zaznamenáno. Dvě nalezené posunové mutace (c.421_442del22, c.686_687insA) nebyly dosud popsány. Dvě bodové mutace (c.733G>T, c.434T > C) jsou již známy, ale nebyly nalezeny u hematoonkologických onemocnění a zbývající uvedené mutace byly již v nízkém procentu u těchto onemocnění pozorovány (0,11–4,79 %).
Dále jsme u dvou pacientů pozorovali UPD 17q s proměnlivým rozsahem v pruzích 17q22 a 17q24.2 a velikostí 6,4–7,7 Mb.
U šesti pacientů jsme detekovali UPD na chromozomu X lokalizované v oblastech Xp22.11–Xp22.2 (4/6) a Xq13.3–Xq21.1 (2/6). Rozsah UPD Xp se pohyboval mezi 5,2–6,1 Mb a velikost UPD Xq byla 6,3 a 8,4 Mb.
DISKUSE
Získaná uniparentální disomie je popisována u nemocných s různými typy nádorových onemocnění a předpokládá se, že souvisí s jejich progresí. Při vzniku aUPD dochází ke ztrátě heterozygozity, ale ne ke změně počtu kopií DNA příslušné oblasti. Tato oblast může obsahovat mutované tumor supresorové geny, případně onkogeny, u nichž dochází ke ztrátě funkce nebo naopak ke zvýšení exprese genu. Chromozomové oblasti s aUPD se jeví při analýzách metodami klasické a molekulární cytogenetiky jako normální (bez aberací). Ideální metodou pro jejich identifikaci jsou čipové technologie se schopností detekce SNP, případně analýza mutací konkrétních genů.
Současné studie uvádí, že aUPD bývá obvykle větší než 25 Mb, zahrnuje centromerickou nebo telomerickou oblast a většinou obsahuje homozygotní mutace genů, imprintované geny, případně různě metylované a acetylované geny ovlivňující vznik a vývoj choroby [2, 15]. Naopak vrozená UPD zasahuje výrazně menší oblasti chromozomů, bývá intersticiální a obvykle neovlivňuje fenotyp nositele.
Zhruba polovina nalezených UPD v našem souboru (19/40 oblastí UPD) charakterem odpovídala spíše vrozené formě UPD. Usuzujeme tak na základě velikosti těchto oblastí pohybující se mezi 5–10 Mb, úseky neobsahují žádné známé kauzální geny zodpovědné za tumorigenezi a také jde o sporadické výskyty. Nicméně u některých nemocných jsme detekovali více malých UPD na různých chromozomech (10/21 pacientů). Zvýšený počet oblastí aUPD u jednoho pacienta může být ukazatelem chromozomové nestability patologického klonu a známkou klonálního vývoje, během něhož dochází ke vzniku nových balancovaných i nebalancovaných aberací. Tento jev tak představuje zvýšené riziko progrese choroby.
Ve čtyřech případech jsme pozorovali UPD v oblasti Xp22.11–Xp22.2 (viz tab. 2). V této oblasti se nalézá gen REPS2/POB1 (Xp22.2). Jeho mutace byly popsány u karcinomu prostaty, kde potlačují apoptózu nádorových buněk, a zajišťují tak přežívání patologického klonu [18, 19].
Kromě toho jsme na chromozomu X dvakrát nalezli UPD v oblasti Xq13.3–Xq21.1, která zahrnovala gen ABCB7 (Xq13.3) – viz tabulka 2. Snížená exprese tohoto genu je asociována se zvýšeným počtem prstenčitých sideroblastů v kostní dřeni u nemocných s diagnózou RA s prstenčitými sideroblasty (20). Mutace ABCB7 byly nalezeny u čtyř rodin s diagnózou vrozené X-vázané sideroblastické anémie a ataxie [21]. U získané formy této choroby dosud nebyla potvrzena žádná mutace ani metylace genu ABCB7 a není zcela objasněno, čím je způsobena jeho snížená exprese [22].
Dále jsme u dvou pacientů detekovali UPD dlouhých ramen chromozomu 17 s proměnlivým rozsahem v pruzích 17q22 a 17q24.2 (viz tab. 2). Tato oblast obsahuje několik kandidátních genů, které jsou spojeny obecně s tumorigenezí. Mezi nimi jsou například geny BRIP1 (17q23.2) a AXIN2 (17q24.1). BRIP1 je gen asociovaný zejména s rakovinou prsu, zárodečné mutace genu byly popsány u Fanconiho anémie a ve spojitosti s fa-miliárním výskytem nádoru prsu [23, 24, 25]. Mutace genu AXIN2 a zvýšená exprese byla pozorována např. u kolorektálního karcinomu a celé řady další typů zhoubných nádorů [26].
U 22,8 % pacientů v našem souboru jsme metodou aCGH/SNP potvrdili aUPD krátkých ramen chromozomu 17, proto jsme se na tuto oblast detailněji zaměřili (viz tab. 2). aUPD 17p je popisována zhruba u 16 % pa-cientů s MDS a komplexním karyotypem a podle údajů v literatuře se její velikost pohybuje od 5 do 12 Mb [14]. Velikost aUPD 17p v našem souboru se pohybovala v rozmezí 8–20 Mb.
Obecně jsou ztráty heterozygozity (delece, mutace, UPD) krátkých ramen chromozomu 17 asociovány s komplexním karyotypem a delecemi dlouhých ramen chromozomů 5 a 7 [14]. Shodný trend jsme pozorovali i ve skupině našich pacientů, kde u většiny byly nalezeny delece dlouhých ramen, chromozomu 5 (11/13) a delece dlouhých ramen případně monosomie chromozomu 7 (8/13) – viz obrázek 2. Kromě toho byla u osmi ze 13 pacientů potvrzena delece krátkých ramen chromozomu 3 a delece krátkých ramen chromozomu 12 ve všech případech zahrnující gen ETV6 (12p13). Tento gen je znám jako translokační partner tvořící celou řadu fúzních genů negativně ovlivňujících prognózu pacienta.
S výskytem aUPD 17p je úzce asociována homozygotní mutace genu TP53 (14). Při srovnání pacientů s normálním chromozomem 17 (mutace genu TP53 nalézána v cca 5 % případů) a nemocných s delecí 17p (mutace druhé alely genu TP53 popisována až v 78 % případů), je výskyt mutace genu TP53 u pacientů s aUPD 17p nejvyšší, bývá prokázána až v 83 % případů (obr. 3). Tomu odpovídají výsledky mutační analýzy genu TP53 u pacientů s aUPD 17p v naší studii. U všech jsme nalezli homozygotní mutaci genu TP53 (tab. 3). Mutace byly detekovány v exonech 5, 6, 7 a 8. V souladu s literaturou byla většina nalezených mutací charakteru mutací měnících smysl (9/14), tedy bodové mutace v DNA vazebné doméně [14]. U těchto mutací dochází k záměně nukleotidu a tím ke změně smyslu kodonu. Tato změna ovlivňuje stabilitu proteinu nebo jeho schopnost vazby k DNA. U dvou pacientů jsme dále prokázali nesmyslné mutace, při kterých dochází ke vzniku předčasného stop kodonu, a tím ke zkrácení a ztrátě funkce vznikajícího proteinu. Dva ze 13 nemocných měli posunové mutace, u kterých dochází k posunu čtecího rámce delecí nebo insercí několika nukleotidů, jejichž počet není dělitelný třemi, a tak ke vzniku nefunkčního proteinu. V našem případě šlo o deleci 22 nukleotidů a inserci adeninu. Všechny nálezy kromě námi pozorovaných posunových mutací (c.421_442del22, c.686_687insA) byly již zaznamenány v databázi IARC. Dvě bodové mutace jsou již známy (c.733G > T, c.434T > C), ale nebyly popsány u hematoonkologických onemocnění a zbývající mutace byly již v nízkém procentu u těchto onemocnění pozorovány (0,11–4,79 %). U pacienta KD10 jsme nalezli homozygotní mutaci v nekódující části genu (4. intron), kde došlo k záměně guaninu za adenin. Vzhledem k umístění mutace není jisté, zda má vliv na funkci vznikajícího proteinu p53. Přesto vysoké procento patologického klonu ve vzorku (70,9 %) a fakt, že podle databáze IARC byla tato mutace v jednom případě popsána u vzorku tumoru centrální nervové soustavy, naznačuje možnou souvislost s nádorovým onemocněním.
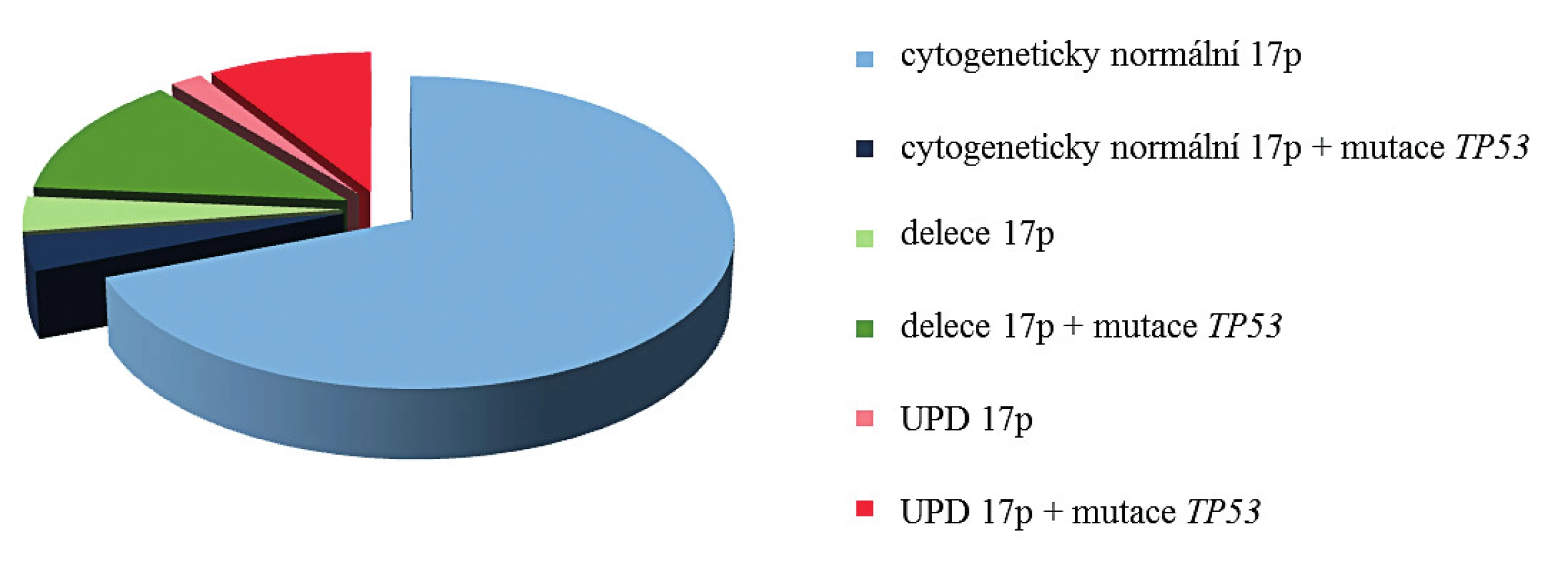

Prognóza pacientů s aUPD 17p je stejná nebo horší než u pacientů s delecí 17p [27]. Důvodem je právě výskyt homozygotní mutace genu TP53. Tento známý, tumor supresorový gen je zodpovědný za regulaci genové exprese na transkripční úrovni a také zajišťuje přechod buněk do apoptózy, pokud dojde k rozsáhlému poškození DNA.
Jak ukazuje tabulka 1, v literatuře bylo popsáno několik genomových oblastí, v nichž poměrně často dochází ke vzniku aUPD u nemocných s MDS a AML. Tyto oblasti jsou spojeny se změnami exprese konkrétních tumor supresorových genů a jsou příčinou horší prognózy. Dosavadní studie byly zaměřeny obecně na pacienty s těmito diagnózami bez ohledu na cytogenetický nález. V naší studii jsme se naopak zaměřili na relativně homogenní skupinu nemocných s MDS a komplexním karyotypem. Nejčastějším a potenciálně prognosticky významným nálezem v této skupině byla aUPD 17p asociovaná s homozygotní mutací genu TP53, kterou jsme detekovali celkem u 13/57 nemocných. Lze tedy říci, že tato změna silně koreluje s komplexním karyotypem a je spojena s velmi špatnou prognózou. Naopak jiné aUPD dosud popsané u MDS a AML uvedené v tabulce 1 nebyly v tomto souboru vůbec detekovány a zřejmě nejsou asociovány se vznikem komplexních aberací.
ZÁVĚR
S využitím metody aCGH/SNP jsme u 22,8 % (13/57) pacientů s MDS a komplexními karyotypy nalezli aUPD 17p. U všech nemocných s aUPD 17p jsme zároveň potvrdili homozygotní mutace genu TP53, přičemž dvě nalezené posunové mutace nebyly dosud popsány v databázi IARC. Medián přežití nemocných s prokázanou aUPD 17p byl 4 měsíce.
Kromě toho jsme detekovali UPD na krátkých a dlouhých ramenech chromozomu X (6 nemocných) a dlouhých ramenech chromozomu 17 (2 nemocní). Tyto oblasti dosud nebyly asociovány s MDS, přesto obsahují několik genů, které by mohly hrát významnou úlohu při rozvoji patologického klonu.
aCGH/ SNP a následná analýza mutací jsou ideálními metodami pro detekci nebalancovaných aberací a UPD. Bližší charakterizace oblastí UPD přináší možnost identifikace nových genů souvisejících se vznikem a progresí onemocnění, případně může přispět k hledání nových terapeutických cílů.
Poděkování
Práce byla podpořena granty RVO-VFN64165, GAČR P302/12/G157 a PRVOUK-P27/LF1/1, IGA MZ ČR NT14377, MZČR 00023736.
Podíl autorů na přípravě rukopisu
KS – vyhodnocení aCGH/SNP a I-FISH, zpracování podkladů a literatury, napsání rukopisu
ZZ – analýza komplexních karyotypů, mFISH/mBAND, připomínkování rukopisu, schválení finální verze
HL – vyhodnocení aCGH/SNP, úprava rukopisu
MN – vyhodnocení aCGH/SNP
JB – analýza komplexních karyotypů, mFISH/mBAND
MB – analýza mutací genu TP53
AB – vyhodnocení I-FISH
IŠ – vyhodnocení I-FISH a FISH s multicentromerickou sondou
LL – analýza komplexních karyotypů, vyhodnocení mFISH/mBAND
SI – analýza komplexních karyotypů, vyhodnocení mFISH/mBAND
AJ – diagnostika a léčba nemocných, sběr klinických dat
JČ – diagnostika a léčba nemocných, sběr klinických dat
KM – připomínkování rukopisu, schválení finální verze
Čestné prohlášení autora
Autor práce prohlašuje, že v souvislosti s tématem, vznikem a publikací tohoto článku není ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou. Toto prohlášení se týká i všech spoluautorů.
Doručeno do redakce: 17. 4. 2015
Přijato po recenzi: 16. 6. 2015
Mgr. Karla Svobodová
Centrum nádorové cytogenetiky
Ústav lékařské biochemie a laboratorní diagnostiky
VFN a 1. LF UK
U Nemocnice 499/2
12808 Praha 2
e-mail: karla.svobodova@vfn.cz
Zdroje
1. Schoch C, Haferlach T, Bursch S, et al. Loss of genetic material is more common than gain in acute myeloid leukemia with complex aberrant karyotype: a detailed analysis of 125 cases using conventional chromosome analysis and fluorescence in situ hybridization including 24-color FISH. Genes Chromosomes Cancer, 2002;35(1):20–29.
2. Tuna M, Knuutila S, Mills GB. Uniparental disomy in cancer. Trends Mol Med, 2009;15(3):120–128.
3. Kralovics R, Guan Y, Prchal JT. Acquired uniparental disomy of chromosome 9p is a frequent stem cell defect in polycythemia vera. Exp Hematol, 2002;30 : 229–236.
4. Kralovics R, Buser AS, Teo S, et al. Comparison of molecular markers in a cohort of patients with chronic myeloproliferative disorders. Blood, 2003;102(5):1869–1871.
5. Kralovics R, Passamonti F, Buser AS. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med, 2005;352 : 1779–1790.
6. Makishima H, Maciejewski JP. Pathogenesis and consequences of uniparental disomy in cancer. Clin Cancer Res, 2011;17 : 3913–3923.
7. O’Keefe C, McDevitt MA, Maciejewski JP. Copy neutral loss of heterozygosity: a novel chromosomal lesion in myeloid malignancies. Blood, 2010;115(14):2731–2739.
8. Dunbar AJ, Gondek LP, O’Keefe C, et al. 250K SNP array karyotyping identifies acquired uniparental disomy and homozygous mutations, including novel missense substitutions of c-Cbl, in myeloid malignancies. Cancer Res, 2008;68(24):10349–10357.
9. Szpurka H, Gondek LP, Mohan SR, Hsi ED, Theil KS, Maciejewski JP. UPD1p indicates the presence of MPL W515L mutation in RARS-T, a mechanism analogous to UPD9p and JAK2 V617F mutation. Leukemia, 2009;23 : 610–614.
10. Jankowska AM, Szpurka H, Tiu RV, et al. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood, 2009;113(25):6403–6410.
11. Langemeijer SMC, Kuiper RP, Berends M, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet, 2009;41(7):838–842.
12. Ernst T, Chase AJ, Score J, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet, 2010;42(8):722–726.
13. Fitzgibbon J, Smith L-L, Raghavan M. Association between acquired uniparental disomy and homozygous gene mutation in acute myeloid leukemias. Cancer Res, 2005;65 : 9152–9154.
14. Jasek M, Gondek LP, Bejanyan N, et al. TP53 mutations in myeloid malignancies are either homozygous or hemizygous due to copy number-neutral loss of heterozygosity or deletion of 17p. Leukemia, 2010;24(1):216–219.
15. Score J, Cross NCP. Acquired uniparental disomy in myeloproliferative neoplasms. Hematol Oncol Clin N Am, 2012;26 : 981–991.
16. Swerdlow S H, Campo E, Harris N L, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon, France. IARC Press 2008.
17. Shaffer LG, McGowan-Jordan J, Schmid M (eds). ISCN 2013: An International System for Human Cytogenetic Nomenclature. S. Karger, Basel 2013.
18. Ikeda M, Ishida O, Hinoi T, Kishida S, Kikuchi A. Identification and characterization of a novel protein interacting with Ral-binding protein 1, a putative effector protein of Ral. J Biol Chem, 1998;273 : 814–821.
19. Ooosterhoff JK, Penninkhof F, Brinkmann AO, Grootegoed JA, Blok LJ. REPS2/POB1 is downregulated during human prostate cancer progression and inhibits growth factor signalling in prostate cancer cells. Oncogene, 2003;22 : 2920–2925.
20. Boultwood J, Pellagatti A, Nikpour M, et al. The role of the iron transporter ABCB7 in refractory anemia with ring sideroblasts. PLoS ONE, 2008;3(4):1970.
21. D‘Hooghe M, Selleslag D, Mortier G, et al. X-linked sideroblastic anemia and ataxia: a new family with identification of a fourth ABCB7 gene mutation. Eur J Paediatr Neurol, 2012;16 : 730–735.
22. Nikpour M, Scharenberg C, Liu A, et al. The transporter ABCB7 is mediator of the phenotype of acquired refractory anemia with ring sideroblasts. Leukemia, 2013;27 : 889–896.
23. Levitus M, Waisfisz Q, Godthelp BC, et al. The DNA helicase BRIP1 is defective in Fanconi anemia complementation group. J Nat Genet, 2005;37 : 934–935.
24. Cantor SB, Bell DW, Ganesan S, et al. BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell, 2001;105 : 149–160.
25. Ren LP, Xian YS, Diao DM, Chen Y, Guo Q, Dang CX. Further evidence for the contribution of the BRCA1-interacting protein-terminal helicase 1 (BRIP1) gene in breast cancer suspectibility. Genet Mol Res, 2013;12(4):5793–5801.
26. Wu Z, Brabletz T, Fearon E. Canonical Wnt suppressor, Axin2, promotes colon carcinoma oncogenic aktivity. Proc Natl Acad Sci USA, 2012;109(28):11312–11317.
27. Tiu RV, Gondek LP, O’Keefe C, et al. Prognostic impact of SNP array karyotyping in myelodysplastic syndromes and related myeloid malignancies. Blood, 2011;117(17):4552–4560.
Štítky
Hematologie a transfuzní lékařství Interní lékařství OnkologieČlánek vyšel v časopise
Transfuze a hematologie dnes

2015 Číslo 3
- Diosmin a hesperidin: Co ukazuje farmakokinetika?
- Evaluace skóre m7-FLIPI u pacientů s folikulárním lymfomem odhalila slibný prediktivní biomarker k určení vhodné chemoterapie
- Neuropatie výrazně zpomaluje proces hojení u povrchových ran
- Diagnostika von Willebrandovy choroby krok za krokem
- Těhotenství a porod u ženy s VWD − kazuistika
Nejčtenější v tomto čísle
- Nové testy pro screening syfilis u dárců krve
- Protinádorové účinky klinicky používaných chelátorů železa – přehled literatury a vlastní zkušenosti
- Získaná uniparentální disomie v buňkách kostní dřeně nemocných s myelodysplastickými syndromy a komplexním karyotypem
- Kazuistika (ne)chelatovaného polytransfundovaného pacienta s 5q minus syndromem
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy