
Protinádorové účinky klinicky používaných chelátorů železa – přehled literatury a vlastní zkušenosti
Antitumor effects of clinically used iron chelators – review of literature and our own experience
Myelodysplastic syndrome represents a heterogeneous group of diseases, typically characterised by ineffective haematopoiesis and transfusion dependency. Iron chelators currently represent an important treatment modality in patients with myelodysplastic syndrome (MDS), who given the presence of anaemia, are dependent on repeated blood transfusions leading to the accumulation of toxic iron in organs. It has been shown that chelation therapy significantly improves overall survival and leukaemia-free survival in patients with MDS. Besides iron chelation, iron chelators also exhibit significant antiproliferative and pro-apoptotic effects on cancer cells. The suggested mechanisms of antitumor effects of iron chelators include inhibition of cell cycle progression, induction of endoplasmic reticulum stress, accumulation of DNA damage specifically in cancer cells and modulation of antitumor immune response. The exact mechanism of action of chelating agents on cancer cells is not yet fully understood and even our data suggest that it is likely to be very complex.
Key words:
deferoxamine mesylate, iron chelators, iron, myelodysplastic syndrome, cancer cells
Autoři:
L. Křupková 1,2; L. Rašková Kafková 1; Z. Somíková 1; M. Beličková 3; P. Lužná 1,4; J. Čermák 3; V. Divoký 1,2
Působiště autorů:
Ústav biologie, Lékařská fakulta Univerzity Palackého v Olomouci
1; Hemato-onkologická klinika, Lékařská fakulta Univerzity Palackého a Fakultní nemocnice Olomouc
2; Ústav hematologie a krevní transfuze, Praha
3; Ústav histologie a embryologie, Lékařská fakulta Univerzity Palackého v Olomouci
4
Vyšlo v časopise:
Transfuze Hematol. dnes,21, 2015, No. 3, p. 117-125.
Kategorie:
Souhrnné práce, původní práce, kazuistiky
Souhrn
Myelodysplastický syndrom je heterogenní skupina onemocnění, jejichž typickým projevem je neefektivní krvetvorba a závislost na krevních transfuzích. Chelátory železa v současné době představují důležitou léčebnou modalitu u těch pacientů s myelodysplastickým syndromem, u kterých v důsledku přítomnosti anémie a opakovaných krevních transfuzí dochází k hromadění toxického železa v orgánech. Bylo prokázáno, že chelatační terapie u pacientů s mye-lodysplastickým syndromem významně prodlužuje celkové přežití a oddaluje leukemickou transformaci. Kromě vlastní chelatace iontů železa bylo u těchto chelatačních látek prokázáno i výrazné antiproliferační a proapoptotické působení na nádorové buňky. Předpokládanými mechanismy protinádorových účinků chelátorů železa jsou inhibice progrese buněčného cyklu, vyvolání stresu endoplazmatického retikula, akumulace poškozené DNA specificky u nádorových buněk i modulace protinádorové imunitní odpovědi. Přesný mechanismus působení chelatačních látek na nádorové buňky však prozatím není zcela objasněn a i naše výsledky naznačují, že bude velmi komplexní.
Klíčová slova:
deferoxamin mesylát, chelátory železa, ionty železa, myelodysplastický syndrom, nádorové buňky
ÚVOD
Chelátory železa představují v současné době důležitou léčebnou modalitu u pacientů s myelodysplastickým syndromem (MDS). Jako MDS označujeme heterogenní skupinu onemocnění, která jsou charakterizována hromaděním nezralých krevních prekurzorů v kostní dřeni a inefektivní hematopoézou projevující se cytopenií v periferní krvi [1]. Klasifikace světové zdravotnické organizace (WHO) z roku 2008 dělí pacienty s MDS do několika podskupin: refrakterní cytopenie s dysplazií v jedné řadě (RCUD), refrakterní anémie s prstenčitými sideroblasty (RARS), MDS s izolovanou delecí 5q, refrakterní cytopenie s dysplazií ve více řadách (RCMD), refrakterní cytopenie s dysplazií ve více řadách s prstenčitými sideroblasty (RCMD-RS), refrakterní anémie s nadbytkem blastů (RAEB) a neklasifikovaný MDS (MDS-U). Mezinárodní prognostický skórovací systém IPSS (International Prognostic Scoring System) posuzuje onemocnění podle agresivity nemoci a prognózy pacienta. Pacienti jsou podle něj rozděleni do čtyř rizikových skupin: nízce riziková skupina, středně riziková skupina 1, středně riziková skupina 2 a vysoce riziková skupina. Tento prognostický systém byl roku 2012 revidován na tzv. IPSS‑R (IPSS‑Revised; [1]) a rozděluje nemocné do 5 rizikových skupin podle délky přežití a rizika přechodu do akutní myeloidní leukemie (AML).
MDS je označován jako tzv. „preleukemický stav“, který u 1/3 pacientů vede k transformaci do akutní myeloidní leukemie (AML). Úspěšnou léčbu MDS komplikuje zejména neznalost přesných mechanismů řídících iniciaci a progresi onemocnění [2]. Vedle neefektivní erytropoézy vedoucí k přetížení železem je velká část pacientů z důvodu přítomnosti anémie závislá na opakovaných transfuzích, jejichž hlavním nežádoucím účinkem je hromadění toxického železa v důležitých orgánech [3]. Toto přetížení železem je pak v klinické praxi eliminováno podáváním chelatačních přípravků u pacientů, kteří vykazují významnou akumulaci železa (laboratorně při hladině feritinu v séru více než 1 000 mg/l). Vedle známých účinků na regulaci zásob železa a prevenci kardiotoxicity železa účinkují chelátory také na metabolismus železa v nádorových buňkách a ovlivňují jejich proliferaci a přežití; mechanismům tohoto účinku je věnován tento přehledový článek.
PROTINÁDOROVÉ ÚČINKY CHELÁTORŮ ŽELEZA
Hlavním cílem chelatační terapie je snížit hladinu tzv. labilní hotovosti železa v buňce („labile iron pool“, LIP), a tím minimalizovat poškození buněk a orgánů. Kromě vlastní eliminace LIP byl u chelatačních látek rovněž prokázán antiproliferativní účinek na nádorové buňky [4, 5, 6], které mají díky své rychlé proliferaci vyšší metabolické požadavky na přísun železa [7]. Chelátory železa se tak jeví jako látky se slibným protinádorovým účinkem, nicméně přesný mechanismus jejich působení není prozatím zcela objasněn a pravděpodobně bude velmi komplexní. Předpokládá se, že jejich hlavní mechanismus působení spočívá v inhibici celulárního příjmu železa, čímž dochází:
- k inhibici ribonukleotidreduktázy (RR) jako obrat-limitujícího („rate limiting“) enzymu pro syntézu DNA závislého na železe [8, 9];
- k regulaci buněčného cyklu a jeho zastavení;
- ke zvýšené produkci reaktivních forem kyslíku („reactive oxygen species“, ROS) redox-aktivními chelátory vedoucí k peroxidaci lipidů, proteinů a poškození DNA (paradoxně i přes to, že železem indukovaný oxidační stres iniciuje tumorigenezi);
- k indukci nádorových supresorů (p53, NDRG1 – „N-myc downstream regulated gene-1, N-myc regulovaný gen-1) aj. [10] – obrázek 1.
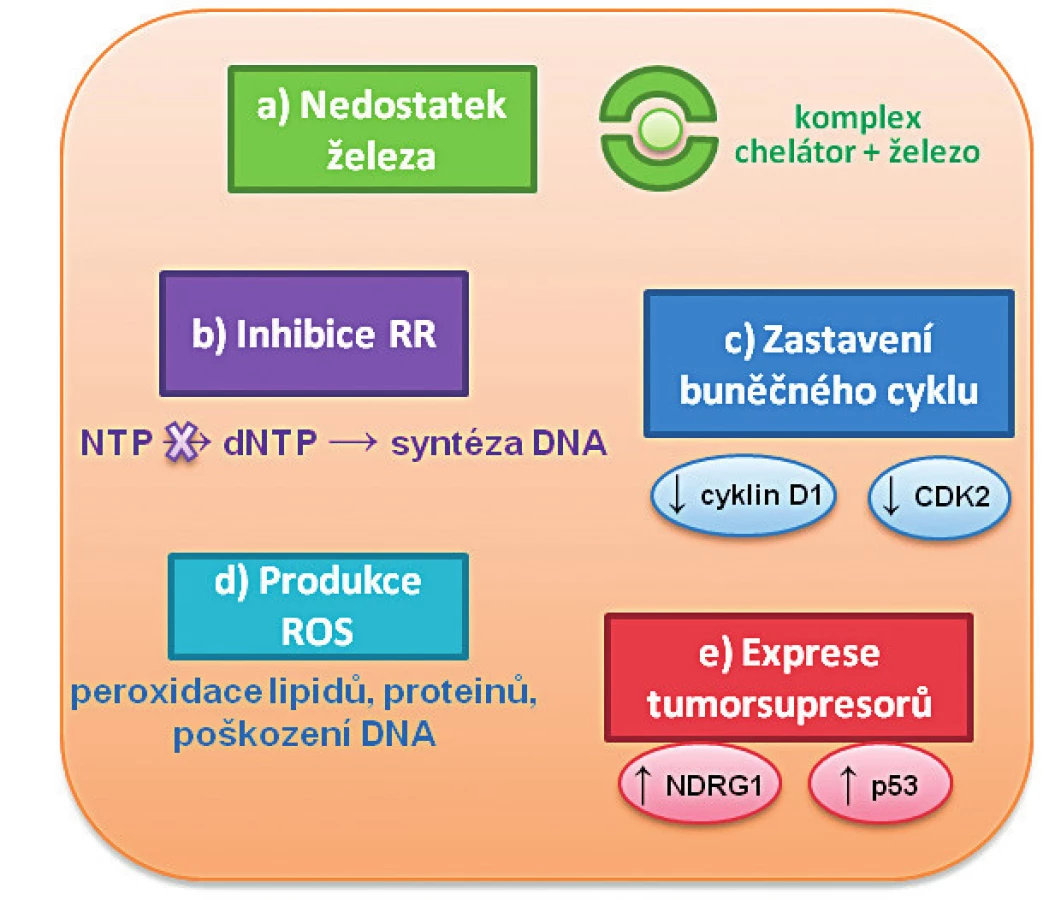
Chelátory železa mohou rovněž měnit nádorový fenotyp prostřednictvím inhibice epiteliálně-mezenchymálního přechodu, který je kritický pro metastázování (pravděpodobně prostřednictvím potlačení exprese transformujícího růstového faktoru‑β (TGF‑β), nebo regulací stresu endoplazmatického retikula (ER) [11, 12].
Chelatace železa u pacientů s MDS má pravděpodobně vliv na prodloužení jejich celkového přežití („overall survival“, OS). Medián přežití se zvýšil ze 49 měsíců u skupiny nechelatovaných pacientů na 79 měsíců u pacientů chelatovaných [13]. Publikované studie také ukazují oddálení doby do leukemické transformace („leukemia‑free survival“, LFS) u chelatovaných pacientů. Po dobu trvání studie (medián sledování 41 měsíců), zahrnující 146 pacientů na chelatační léčbě a 117 bez chelatační léčby, došlo k leukemické transformaci pouze u 4 sledovaných pacientů na chelatační léčbě a u 14 bez chelatační léčby [14]. Předpokládá se, že chelatační terapie blokuje proliferaci leukemických kmenových buněk [15]. U pacientů léčených chelátory železa bylo dále pozorováno snížené riziko infekcí mikroorganismy závislými na přísunu volného železa z hostitelského organismu [16].
KLINICKY POUŽÍVANÉ CHELÁTORY ŽELEZA
V současnosti jsou v klinické praxi používány tři chelátory železa – deferoxamin mesylát (DFO), deferasirox a deferipron (DFP), které se významně liší mechanismem působení (obr. 2). Prvním chelátorem používaným u pacientů s MDS byl DFO (Desferal®) [17], který váže volné železo v plazmě („non-transferrin bound iron“, netransferinové železo, NTBI) a indukuje degradaci feritinu v lysozomech. U DFO byla prokázána výrazná protinádorová aktivita s inhibičním vlivem na proliferaci nádorových buněk in vivo i in vitro. Studie u pacientů s neuroblastomem ukázala, že DFO efektivně potlačuje infiltraci kostní dřeně nádorovými buňkami a způsobuje zmenšení nádoru [18]. U leukemických buněčných linií K562 a Daudi byla po ošetření chelátory železa pozorována akumulace buněk v G1 nebo G2 fázi – v závislosti na použité koncentraci chelátoru [19]. Z důvodu krátkého poločasu rozpadu DFO a nutnosti jeho injekčního podání byl postupně v klinické praxi nahrazen orálními chelátory deferasiroxem (Exjade®) [20] a deferipronem (Ferriprox®) [21]. Deferasirox a deferipron jsou malé lipofilní molekuly, které snadno procházejí plazmatickou membránou, a proto chelatují ionty železa přítomné nejen v plazmě, ale i v buňce, včetně železa uvolňovaného z feritinu před jeho lysozomální degradací [3, 22, 23].

VLIV CHELATACE ŽELEZA NA BUNĚČNÝ CYKLUS
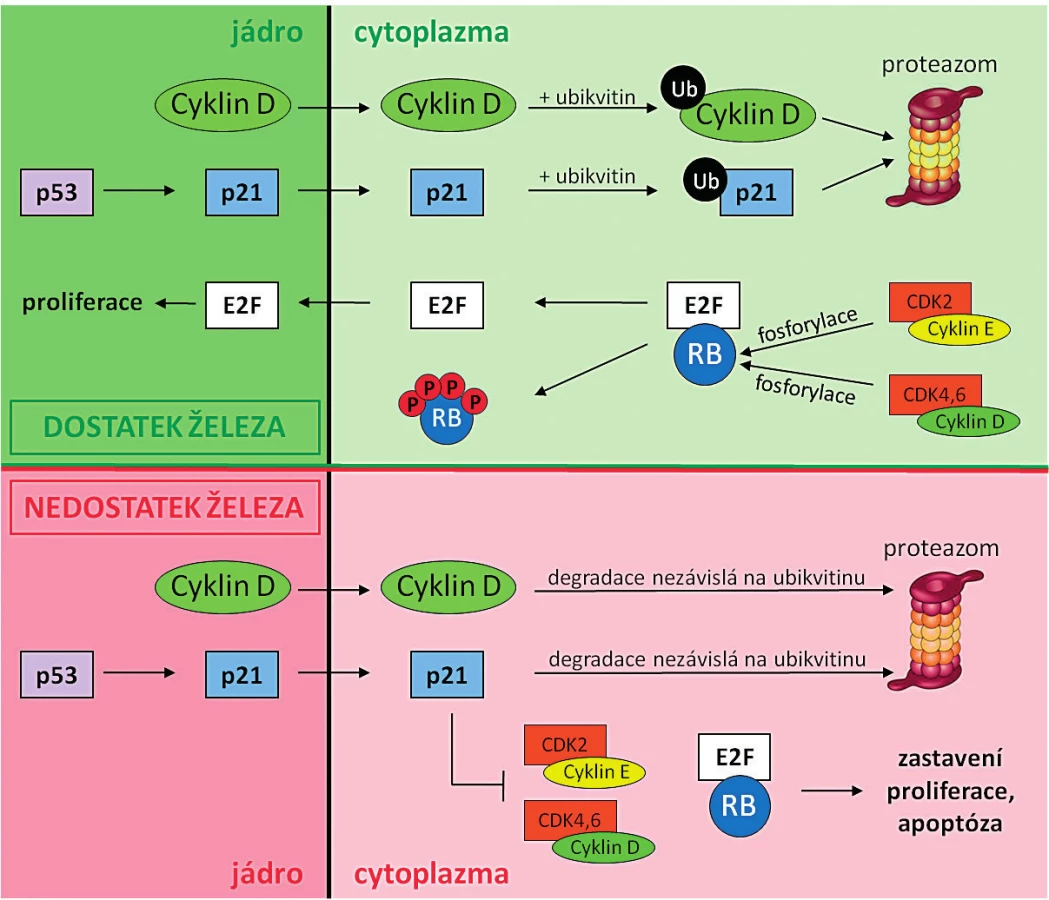
Železo je zcela nezbytné pro buněčnou proliferaci a syntézu DNA a díky tomu jsou rychle proliferující nádorové buňky více citlivé na přítomnost železa než zdravé somatické buňky. Aby nádorové buňky pokryly zvýšené nároky na množství železa, dochází u nich ke zvýšené expresi transferinového receptoru (TFR1), který zabezpečuje import železa do buňky [24]. Nádorové buňky mají také zvýšenou expresi RR (viz kapitola Protinádorové účinky chelátorů železa), což ještě zvyšuje jejich vnímavost na chelataci železa ve srovnání se zdravými somatickými buňkami [25, 26]. RR je tetramerní enzym skládající se ze 2 homodimerních podjednotek R1 a R2. R1 obsahuje aktivní místo pro navázání a redukci ribonukleotidů, R2 pak pro navázání Fe3+ iontů. Železité ionty mají rozhodující úlohu při redukci substrátu. Deplecí železa dochází k inhibici RR aktivity, ke snížení syntézy všech čtyř deoxyribonukleotidů z ribonukleotidů a tím k poruše syntézy a reparace DNA. Odstranění iontů železa tedy vede ke snížení aktivity RR, tím dochází ke snížení hladiny deoxyribonukleotidtrifosfátů v buňce, ke zpomalení a místy k zastavení replikace DNA, což v konečném důsledku vede k zastavení buněčného cyklu a případně k apoptóze [27, 28, 29]. Chelátory železa inhibují nejen RR, ale nepřímo i mnoho molekul zodpovědných za regulaci průchodu buňky buněčným cyklem [9, 30] – obrázek 3.
VLIV CHELATACE ŽELEZA NA INDUKCI APOPTÓZY VYVOLANÉ STRESEM ENDOPLAZMATICKÉHO RETIKULA
V nedávné době byly publikovány práce popisující vznik stresu endoplazmatického retikula (ER) v nádorových buňkách vystavených chelatačním látkám [12]. ER je organela podílející se na celé řadě metabolických pochodů, mezi které patří syntéza, modifikace, popř. degradace mnoha sekretovaných a membránových proteinů. Probíhá zde část syntézy cholesterolu, dále fosfolipidů, acylkoenzymu A, oxidace a redukce steroidních hormonů, desaturace mastných kyselin, některé rozkladné kroky hemu, detoxifikační mechanismy (obsahuje skupinu cytochromů P450) přirozených nebo cizorodých látek, v neposlední řadě regulace uvolňování Ca2+ iontů (většinou ve vázáné formě na proteiny – kalretikulin, BiP („binding immunoglobulin protein“) umožňující správnou funkci jiných organel (mitochondrie aj.). Bylo zjištěno, že vlivem ER stresu dochází k zastavení buněčného cyklu v G1 fázi prostřednictvím indukce p53, p21 a snížení hladiny cyklinu D1 i k zastavení v G2 fázi. Toto zastavení je zprostředkováno kinázou CHK1, která je důležitou součástí signálních drah aktivovaných poškozenou DNA [32, 33]. Obecně je také známo, že po zaregistrování stresu senzory v ER je indukována signální dráha vedoucí ke znovunastolení rovnováhy a snaze buňky vymanit se z ER stresu. V případě dlouhotrvajícího ER stresu může dojít až k apoptóze [34]. Jednou z příčin indukce apoptózy je porušení vnitřní metabolické rovnováhy ER z důvodu hypoxie, abnormální koncentrace Ca2+ iontů, narušení redoxního potenciálu, acidózy a hypoglykemie [35, 36]. Narušení rovnováhy ER se projevuje zejména hromaděním aberantně sbalených proteinů v buňce. Odpovědí buňky na hromadění těchto proteinů je spuštění signálních drah souhrnně označovaných jako „unfolded protein response“ (UPR), které mají obnovit homeostázu a předejít buněčné smrti. Výsledkem je zpomalení translace mRNA, degradace aberantně sbalených proteinů („ER associated degradation“, degradace asociovaná s ER, ERAD dráha), zvýšená exprese chaperonových genů a udržení oxidativního prostředí ER. Současně ER stres zvyšuje expresi GADD34 („growth arrest and DNA damage-inducible protein-34“, protein indukovatelný zastavením růstu a poškozením DNA-34) fosfatázy, která defosforylací eIF2α (eukaryotický iniciační faktor 2α) obnovuje translaci proteinů, a snaží se tak buňku vymanit z ER stresu [34] – obrázek 4. Chelátory železa patří mezi látky, které vyvolávají stres ER vedoucí, stejně jako inhibice RR, k apoptóze [34, 35, 36].

Chelátory železa modulují u lidských nádorových buněk i dráhu řídící autofagii. Autofagie může mít protinádorové i pronádorové účinky v závislosti na stadiu nádoru a tkáňovém mikroprostředí [37]. Úloha železa v regulaci autofagie za fyziologického i patofyziologického stavu buněk je zřejmá, avšak mechanismus propojení mezi ionty železa (respektive chelatací iontů železa) a autofagií prozatím není plně prostudován. Mnoho fagocytovaných proteinů obsahuje železo a autofagie proto hraje důležitou roli v udržení fyziologické hladiny železa v buňce a v regulaci redoxního potenciálu [38]. Byl popsán mechanismus stimulace autofagie chelátory železa prostřednictvím indukce signální dráhy transkripčního faktoru HIF-1α („hypoxia inducible factor-1α“, hypoxií inducibilní faktor-1α) [39] a inhibice proliferační signální dráhy mTOR („mammalian target of rapamycin“; savčí cíl rapamycinu) [40]. Výsledky na buněčných liniích naznačují, že chelatace železa zvyšuje tvorbu autofagozomů [41, 42].
VLIV CHELÁTORŮ ŽELEZA NA AKTIVACI ZÁNĚTOVÉ ODPOVĚDI
Železo má důležitý význam i v regulaci zánětové odpovědi. Koncentrace železa ovlivňuje proliferaci, aktivaci a diferenciaci lymfocytů, NK („natural killer“, přirození zabíječi) buněk a monocytů/makrofágů. Bylo prokázáno, že DFO významně zvyšuje expresi genů zapojených v regulaci zánětové odpovědi včetně prozánětových cytokinů [43]. Popsaným působením chelátorů železa (konkrétně DFO) je aktivace zánětové odpovědi prostřednictvím signální dráhy p38/ERK1,2/NF-κB („extracellular-signal-regulated kinases-1,2/nuclear factor‑κB“; extracelulárním signálem regulované kinázy-1,2/jaderný faktor kappa B) [44]. Prostřednictvím NF-κB dochází k indukci exprese chemokinu MIP-3α („macrophage inflammatory protein-3α“, zánětový protein makrofágů-3α). Chelátory železa ovlivňují i expresi prozánětového IL-1 (interleukin-1) a TNF-α („tumor necrosis factor-α“, faktor nádorové nekrózy-α), které se pak také podílejí na regulaci exprese MIP-3α [44, 45]. DFO-aktivovaná p38 a ERK1,2 pak u buněk stimuluje expresi IL-8 (interleukin-8), který je klíčovým chemokinem regulujícím zánětovou odpověď [43,46].
VLIV CHELATACE ŽELEZA NA PRELEUKEMICKÉ HEMATOPOETICKÉ PROGENITORY U MYŠÍHO MODELU A NA CD34-POZITIVNÍ BUŇKY NEMOCNÝCH S MDS – VLASTNÍ ZKUŠENOSTI
V našich laboratořích se dlouhodobě zabýváme studiem metabolismu iontů železa na buněčné i systémové úrovni [47], studiem odpovědi nádorových a kmenových buněk na poškozenou DNA [48] a máme i rozsáhlé klinické zkušenosti týkající se používání a efektu chelatační terapie u MDS [49]. V posledních několika letech se zabýváme i vlivem chelatace železa na proliferaci, apoptózu a aktivaci vybraných signálních drah u somatických a pluripotentních kmenových buněk in vitro. Ověřili jsme řadu výše zmíněných mechanismů publikovaných jinými autory a zjistili jsme, že klíčovým faktorem ovlivňujícím osud buněk v kultuře s chelatační látkou je koncentrace chelátoru. Dokonce zdánlivě malé rozdíly v koncentraci (jako 5 µM nebo 10 µM DFO v buněčné kultuře) ovlivňují zcela rozdílně funkci některých enzymů, které zajišťují správnou časovou posloupnost jednotlivých fází buněčného cyklu, koordinaci průchodu buněčným cyklem nebo případně spouštějí apoptózu. V in vivo studii prováděné na myším modelu jsme se zaměřili na sledování vlivu chelátoru v koncentraci, která je dosahována v krevní plazmě pacientů podrobovaných chelatační terapii.
V klinicky orientované studii jsme pomocí porovnávání expresních profilů a za použití kvantitativní RT-PCR analyzovali CD34+ buňky kostní dřeně 15 nemocných s nízce rizikovým MDS (podle IPSS), z nichž 6 bylo na chelatační léčbě (Ferriprox, popř. Exjade; na chelatační léčbě 4 měsíce až 3 roky, medián 15 měsíců) a 9 bylo bez chelatační léčby, a srovnali jsme expresní profily těchto nemocných se skupinou 5 zdravých kontrol. Zajímalo nás, jestli mezi diferenciálně exprimovanými geny těchto dvou skupin nemocných a skupinou zdravých dárců budou genové markery, které naznačují aktivaci výše zmíněných předpokládaných mechanismů protinádorových účinků chelátorů železa, kterými jsou inhibice progrese buněčného cyklu, vyvolání stresu ER, akumulace poškozené DNA specificky u nádorových buněk a modulace protinádorové imunitní odpovědi. Vzhledem k řadě omezení, která jsou spojena s analýzami vzorků nemocných (heterogenita buněk, existence subklonů atd.) jsme navíc provedli kontrolní experimentální studii i na myším modelu preleukemie. U tohoto modelu aberantně proliferující myeloidní buňky, které exprimují definovaný onkogen, aktivují signální dráhy odpovědi na poškozenou DNA, což má za následek postupnou aktivaci kontrolních uzlů a pozastavení progrese buněčného cyklu [50]. Myším jsme podávali chelatační látky (DFO, DFP) po dobu 6 týdnů, v dávkách odpovídajících dosaženým plazmatickým koncentracím u chelatovaných nemocných (pro DFO jsme použili dávkování 88,8 mg/kg intraperitoneálně 2krát denně, pro deferipron 166 mg/kg na den orálním podáním).
Zjistili jsme, že chelatace iontů železa ovlivňuje průchod buněčným cyklem u hematopoetických progenitorů (obr. 5A). U chelatovaných nemocných i u myšího modelu na chelatační léčbě jsme detekovali signifikantně zvýšené markery úniku (vyrovnání se) ze stresu ER, ale nikoliv markery vlastního permanentního stresu ER (jako je aktivace genů UPR). Zjistili jsme také ovlivnění metabolismu nukleotidů, aktivaci některých markerů antioxidačního systému a některých prozánětových molekul, o kterých je známo, že u nádorových buněk přispívají ke spouštění signálních drah odpovědi na poškozenou DNA. Nejdůležitějším poznatkem z našich in vivo experimentů je pozorování zvýšené akumulace poškozené DNA (která není kompatibilní s normálním přežitím buňky) specificky v nádorových buňkách při chelatační léčbě (obr. 5B). Tyto výsledky naznačují, že buňky, které jsou vystaveny onkogennímu stresu, jsou zvláště citlivé na deprivaci iontů železa chelatačními látkami, a mohou být specificky eliminovány dlouhodobým působením terapeutických dávek chelátorů.

ZÁVĚR
Z prozatímních výsledků a zkušeností (našich i jiných skupin) vyplývá, že klinicky používané chelátory železa kromě své hlavní funkce modulují i mnoho dalších signálních drah v buňce zapojených v regulaci buněčného cyklu, apoptóze a imunitní odpovědi. Antiproliferační a proapoptotické působení chelátorů železa na nádorové buňky je zřejmé, bude však potřeba ještě mnoho práce k jejich úplnému prostudování. Podrobná charakterizace signálních drah indukovaných chelátory železa bude nezbytná pro správnou a efektivní implikaci těchto nově získaných poznatků do klinické praxe.
Seznam použitých zkratek
AML – akutní myeloidní leukemie
ATF4 – aktivující transkripční faktor 4
ATF6 – aktivující transkripční faktor 6
BiP – binding immunoglobulin protein
BrDU – bromodeoxyuridin
CDK – cyklin-dependentní kináza
CDK2 – cyklin-dependentní kináza 2
CDK4,6 – cyklin-dependentní kináza 4,6
DFO – deferoxamin mesylát
DFP – deferipron
DMT1 – divalent metal transporter-1, transportér dvojmocného železa-1
eIF2α – eukaryotický iniciační faktor 2α
ER – endoplazmatické retikulum
ERAD – ER associated degradation, degradace asociovaná s ER
ERK1,2 – extracellular-signal-regulated kinases-1,2, extracelulárním signálem regulované kinázy-1,2
GADD34 – growth arrest and DNA damage-inducible protein 34, protein indukovatelný zastavením růstu a poškozením DNA-34
GRP78 – 78 kDa glucose-regulated protein, 78 kDa glukózou regulovaný protein
HIF-1α – hypoxia inducible factor-1α, hypoxií inducibilní faktor-1α
IL-1 – interleukin 1
IL-8 – interleukin 8
IPSS – International Prognostic Scoring System
IPSS-R – International Prognostic Scoring System Revised
IRE1 – inositol-requiring 1
LCI – labile cell iron, labilní buněčné železo
LFS – leukemia-free survival, doba do leukemické transformace
LIP – labile iron pool, labilní hotovost železa
LPI – labile plasma iron, nestabilní železo v plazmě
MDS – myelodysplastický syndrom
MIP-3α – macrophage inflammatory protein-3α, zánětlivý protein makrofágů-3α
mTOR – mammalian target of rapamycin, savčí cíl rapamycinu
NDRG1 – N-myc downstream regulated gene-1, N-myc regulovaný gen-1
NF-κB – nuclear factor-κB, jaderný faktor kappa B
NK – natural killers, přirození zabíječi
NTBI – non-transferrin bound iron, netransferinové železo
OS – overall survival, celkové přežití
PERK – protein kinase RNA-like ER kinase
PI – propidium iodide, propidium jodid
pRB – protein retinoblastomu
ROS – reactive oxygen species, reaktivní formy kyslíku
RR – ribonukleotidreduktáza
TFR1 – transferinový receptor-1
TGF-β – transformující růstový faktor-β
TNF-α – tumor necrosis factor-α, faktor nádorové nekrózy-α
Ub – ubikvitin, ubikvitinace
UPR – unfolded protein response
XBP1 – X-box-binding protein-1
Poděkování
Tato práce byla podpořena granty IGA MZ ČR NT/12218, NT/14377, NT/13899, GAČR GP14‑10687P, IGA‑LF‑2015‑015 a IGA‑LF‑2015‑001.
Podíl autorů na rukopisu
LK – napsání rukopisu, provedení části experimentů, příprava finální verze rukopisu
LRK – revize rukopisu, provedení části experimentů, interpretace výsledků experimentů
ZS – provedení části experimentů
MB – analýzy pacientských vzorků, připomínkování rukopisu
PL – provedení části experimentů, revize rukopisu
JČ – autor klinické studie, připomínkování rukopisu
VD – plánování laboratorní studie, příprava finální verze rukopisu, interpretace výsledků experimentů
Čestné prohlášení autora
Autor práce prohlašuje, že v souvislosti s tématem, vznikem a publikací tohoto článku není ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou. Toto prohlášení se týká i všech spoluautorů.
Doručeno do redakce: 12. 3. 2015
Přijato po recenzi: 27. 4. 2015
Mgr. Lenka Křupková, Ph.D.
Ústav biologie,Lékařská fakulta Univerzity Palackého v Olomouci
Hemato-onkologická klinika Lékařská fakulta Univerzity Palackého a FN Olomouc
Hněvotínská 3
775 15 Olomouc
e-mail: lenka.krupkova@fnol.cz
Zdroje
1. Greenberg PL, Tuechler H, Schanz J, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood, 2012;120(12):2454–2465.
2. Bejar R, Levine R, Ebert BL. Unraveling the molecular pathophysiology of myelodysplastic syndromes. J Clin Oncol, 2011; 29(5):504–515.
3. Temraz S, Santini V, Musallam K, Taher A. Iron overload and chela-tion therapy in myelodysplastic syndromes. Crit Rev Oncol Hematol, 2014;91(1):64–73.
4. Richardson DR. Iron chelators as therapeutic agents for the treatment of cancer. Crit Rev Oncol Hematol, 2002; 42(3):267–281.
5. Buss JL, Torti FM, Torti SV. The role of iron chelation in cancer therapy. Curr Med Chem, 2003; 10(12): 1021–1034.
6. Kalinowski DS, Richardson DR. The evolution of iron chelators for the treatment of iron overload disease and cancer. Pharmacol Rev, 2005;57(4):547–583.
7. Richardson DR, Kalinowski DS, Lau S, Jansson PJ, Lovejoy DB. Cancer cell iron metabolism and the development of potent iron chelators as anti-tumour agents. Biochim Biophys Acta, 2009;1790(7):702–717.
8. Thelander L, Graslund A, Thelander M. Continual presence of oxygen and iron required for mammalian ribonucleotide reduction: Possible regulation mechanism. Biochem Biophys Res Commun, 1983;110(3):859–865.
9. Nyholm S, Mann GJ, Johansson AG, Bergeron RJ, Graslund A, Thelander L. Role of ribo-nucleotide reductase in inhibition of mammalian cell growth by potent iron chelators. J Biol Chem, 1993;268(35):26200–26205.
10. Lane DJ, Mills TM, Shafie NH, et al. Expanding horizons in iron chelation and the treatment of cancer: role of iron in the regulation of ER stress and the epithelial-mesenchymal transition. Biochim Biophys Acta, 2014;1845(2):166–181.
11. Chen Z, Zhang D,Yue F, Zheng M, Kovacevic Z, Richardson DR. The iron chelators Dp44mT and DFO inhibit TGF‑β‑induced epithelial-mesenchymal transition via up‑regulation of N‑Myc downstream-regulated gene 1 (NDRG1). J Biol Chem, 2012;287(21):17016–17028.
12. Trondl R, Flocke LS, Kowol CR, et al. Triapine and a more potent dimethyl derivative induce endoplasmic reticulum stress in cancer cells. Mol Pharmacol, 2014;85(3):451–459.
13. Neukirchen J, Fox F, Kündgen A, et al. Improved survival in MDS patients receiving iron chelation therapy – a matched pair analysis of 188 patients from the Düsseldorf MDS registry. Leuk Res, 2012;36(8):1067–1070.
14. Remacha ÁF, Arrizabalaga B, Villegas A, et al. Evolution of iron overload in patients with low-risk myelodysplastic syndrome: iron chelation therapy and organ complications. Ann Hematol, 2015;94(5):779–787.
15. Eberhard Y, McDermott SP, Wang X, et al. Chelation of intracellular iron with the antifungal agent ciclopirox olamine induces cell death in leukemia and myeloma cells. Blood, 2009;114(14):3064–3073.
16. Boelaert JR, de Locht M, Van Cutsem J, et al. Mucormycosis during deferoxamine therapy is a siderophore-mediated infection. In vitro and in vivo animal studies. J Clin Invest, 1993;91(5):1979–1986.
17. Jensen PD, Jensen IM, Ellegaard J. Desferrioxamine treatment reduces blood transfusion requirements in patients with myelodysplastic syndrome. Br J Haematol, 1992;80(1):121–124.
18. Donfrancesco A, Deb G, Dominici C, Pileggi D, Castello MA, Helson L. Effects of a single course of deferoxamine in neuroblastoma patients. Cancer Res, 1990;50(16):4929–4930.
19. Hoyes KP, Hider RC, Porter JB. Cell cycle synchronization and growth inhibition by 3-hydroxypyridin-4-one iron chelators in leukemia cell lines. Cancer Res, 1992;52(17):4591–4599.
20. Yang LP, Keam SJ, Keating GM. Deferasirox: a review of its use in the management of transfusional chronic iron overload. Drugs, 2007;67(15):2211–2230.
21. Cohen A, Galanello R, Piga A, Vullo C, Tricta F. A multi-center safety trial of the oral iron chelator deferiprone. Ann N Y Acad Sci, 1998;850 : 223–226.
22. Richardson DR, Ponka P, Baker E. The effect of the iron(III) chelator, desferrioxamine, on iron and transferrin uptake by the human malignant melanoma cell. Cancer Res, 1994;54(3):685–689.
23. De Domenico I, Ward DM, Kaplan J. Specific iron chelators determine the route of ferritin degradation. Blood, 2009;114(20):4546–4551.
24. Larrick JW, Cresswell P. Modulation of cell surface iron transferrin receptors by cellular density and state of activation. J Supramol Struct, 1979;11(4):579–586.
25. Elford HL, Freese M, Passamani E, Morris HP. Ribonucleotide reductase and cell proliferation. I. Variations of ribonucleotide reductase activity with tumor growth rate in a series of rat hepatomas. J Biol Chem, 1970;245 : 5228–5233.
26. Takeda E, Weber G. Role of ribonucleotide reductase in expression in the neoplastic program. Life Sci, 1981;28(9):1007–1014.
27. Brodie C, Siriwardana G, Lucas J, et al. Neuroblastoma sensitivity to growth inhibition by deferrioxamine: Evidence for a block in G1 phase of the cell cycle. Cancer Res, 1993;53(17):3968–3975.
28. Hileti DPP, Hoffbrand AV. Iron chelators induce apoptosis in proliferating cells. Br J Haematol, 1995;89(1):181–187.
29. Renton FJ, Jeitner TM. Cell cycle‑dependent inhibition of the proliferation of human neural tumor cell lines by iron chelators. Biochem Pharmacol, 1996;51(11):1553–1561.
30. Gao J, Richardson DR. The potential of iron chelators of the pyridoxal isonicotinoyl hydrazone class as effective antiproliferative agents, IV: The mechanisms involved in inhibiting cell‑cycle progression. Blood, 2001;98(3):842–850.
31. Yu Y, Kovacevic Z, Richardson DR. Tuning cell cycle regulation with an iron key. Cell Cycle, 2007;6(16):1982–1994.
32. Bourougaa K, Naski N, Boularan C, et al. Endoplasmic reticulum stress induces G2 cell-cycle arrest via mRNA translation of the p53 isoform p53/47. Mol Cell, 2010;38(1):78–88.
33. Thomas SE, Malzer E, Ordóñez A, et al. p53 and translation attenuation regulate distinct cell cycle checkpoints during endoplasmic reticulum (ER) stress. J Biol Chem, 2013;288(11):7606–7617.
34. Clarke HJ, Chambers JE, Liniker E, Marciniak SJ. Endoplasmic reticulum stress in malignancy. Cancer Cell, 2014;25(5):563–573.
35. Schonthal AH. Endoplasmic reticulum stress: its role in disease and novel prospects for therapy. Scientifica (Cairo), 2012;2012 : 857516.
36. Schonthal AH. Pharmacological targeting of endoplasmic reticulum stress signaling in cancer. Biochem Pharmacol, 2013;85(5):653–666.
37. Kenific CM, Thorburn A, Debnath J. Autophagy and metastasis: another double-edged sword. Curr Opin Cell Biol, 2010;22(2):241–245.
38. Kurz T, Gustafsson B, Brunk UT. Cell sensitivity to oxidative stress is influenced by ferritin autophagy. Free Radic Biol Med, 2011;50(11):1647–1658.
39. Wu Y, Li X, Xie W, Jankovic J, Le W, Pan T. Neuroprotection of deferoxamine on rotenone-induced injury via accumulation of HIF-1 alpha and induction of autophagy in SH-SY5Y cells. Neurochem Int, 2010;57(3):198–205.
40. Pullarkat V, Meng Z, Donohue C, et al. Iron chelators induce autophagic cell death in multiple myeloma cells. Leuk Res, 2014;38(8):988–996.
41. Gutierrez E, Richardson DR, Jansson PJ. The anticancer agent di-2-pyridylketone 4,4-dimethyl-3-thiosemicarbazone (Dp44mT) overcomes prosurvival autophagy by two mechanisms: persistent induction of autophagosome synthesis and impairment of lysosomal integrity. J Biol Chem, 2014;289(48):33568–33589.
42. Sahni S, Bae DH, Lane DJR, et al. Molecular bases of disease: the metastasis supressor, N-myc downstream-regulated gene 1 (NDRG1) inhibits stress-induced autophagy in cancer cells. J Biol Chem, 2014;289(14):9692–9709.
43. Choi EY, Kim EC, Oh HM, et al. Iron chelator triggers inflammatory signals in human intestinal epithelial cells: involvement of p38 and extracellular signal-regulated kinase signaling pathways. J Immunol, 2004;172(11):7069–7077.
44. Lee SK, Lee J, Min SK, et al. Iron chelator differentially activates macrophage inflammatory protein-3alpha/CCL20 in immortalized and malignant human oral keratinocytes. Arch Oral Biol, 2008;53(9):801–809.
45. Fan Y, Wang J, Wei L, He B, Wang C, Wang B. Iron deficiency activates pro-inflammatory signaling in macrophages and foam cells via the p38 MAPK-NF-κB pathway. Int J Cardiol, 2011;152(1):49–55.
46. Lee SK, Jang HJ, Lee HJ, et al. p38 and ERK MAP kinase mediates iron chelator‑induced apoptosis and ‑suppressed differentiation in immortalized and malignant human oral keratinocytes. Life Sci, 2006;79 : 1419–1427.
47. Horvathova M, Ponka P, Divoky V. Molecular basis of hereditary iron homeostasis defects. Hematology, 2010;15(2):96–111.
48. Koledova Z, Kafkova LR, Krämer A, Divoky V. DNA damage-induced degradation of Cdc25A does not lead to inhibition of Cdk2 activity in mouse embryonic stem cells. Stem Cells, 2010;28(3):450–461.
49. Cermak J, Jonasova A, Vondrakova J, et al. Efficacy and safety of administration of oral iron chelator deferiprone in patients with early myelodysplastic syndrome. Hemoglobin, 2011;35(3):217–227.
50. Takacova S, Slany R, Bartkova J, et al. DNA damage response and inflammatory signaling limit the MLL-ENL-induced leukemogenesis in vivo. Cancer Cell, 2012;21(4):517–531.
Štítky
Hematologie a transfuzní lékařství Interní lékařství OnkologieČlánek vyšel v časopise
Transfuze a hematologie dnes

2015 Číslo 3
- Diosmin a hesperidin: Co ukazuje farmakokinetika?
- Evaluace skóre m7-FLIPI u pacientů s folikulárním lymfomem odhalila slibný prediktivní biomarker k určení vhodné chemoterapie
- Neuropatie výrazně zpomaluje proces hojení u povrchových ran
- Diagnostika von Willebrandovy choroby krok za krokem
- Vybíráme z Červené knihy ČHS: Co nového v letošních doporučeních pro diagnostiku a léčbu AML?
Nejčtenější v tomto čísle
- Nové testy pro screening syfilis u dárců krve
- Protinádorové účinky klinicky používaných chelátorů železa – přehled literatury a vlastní zkušenosti
- Získaná uniparentální disomie v buňkách kostní dřeně nemocných s myelodysplastickými syndromy a komplexním karyotypem
- Kazuistika (ne)chelatovaného polytransfundovaného pacienta s 5q minus syndromem
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy