
Srdeční AL-amyloidóza s relativně benigním průběhem
Heart amyloidosis with a relatively benign course
The authors present the case of an 81-year old patient with dyspnoea on exertion (NYHA II) and suspicion of coronary artery disease (CAD) due to graphically positive ergometry and detection of wide-complex tachycardia on Holter ECG. Apart from signs of chronic venous insufficiency on lower extremities, the physical examination was normal. The ECG recording showed sinus rhythm, 1st degree AV block and low voltage in limb leads. Signs of pulmonary congestion were found on a chest X-ray. Complex cardiology examination, including coronarography and echocardiography, disproved CAD. However, in regard to echocardiographic finding of possible restrictive cardiomyopathy, endomyocardial biopsy was performed and confirmed our suspicion of cardiac AL-amyloidosis. No amyloid was found in rectal biops. Serum and urine immunofixation were negative, trepanobiopsy disproved diagnosis of multiple myeloma and any other malignant plasma cell dyscrasia. Patient did not have signs of amyloidogenic involvement of other organs. On the grounds the examinations performed we concluded the case as isolated cardiac AL-amyloidosis. Isolated cardiac amyloidogenic involvement is rare (
Key words:
Heart amyloidosis, heart failure, echocardiography, endomyocardial biopsy.
Autoři:
J. Čapek 1; M. Pěnička 1; M. Kment 2
Působiště autorů:
III. interní – kardiologická klinika 3. Lékařské fakulty a FN Královské Vinohrady, Praha
Přednosta: prof. MUDr. Petr Widimský, DrSc., FESC.
1; Pracoviště klinické a transplantační patologie, IKEM, Praha
Přednosta: MUDr. Eva Honsová, Ph. D.
2
Vyšlo v časopise:
Prakt. Lék. 2009; 89(6): 320-323
Kategorie:
Kazuistika
Souhrn
Autoři prezentují případ 81letého pacienta s anamnézou cca 2 roky trvající námahové dušnosti NYHA II a suspekcí na ICHS pro graficky pozitivní ergometrii a záchyt širokokomplexové tachykardie dle Holter EKG. Při fyzikálním vyšetření nebyly popsány patologické odchylky až na známky chronické žilní insuficience. Na EKG byl přítomen sinusový rytmus, AV blok I. stupně a snížená voltáž v končetinových svodech. Na rentgenu srdce a plic byl popsán obraz centrálního městnání. Bylo provedeno komplexní kardiologické vyšetření, včetně koronarografie a echokardiografie, při kterém byla vyloučena diagnóza ICHS. Avšak vzhledem k echokardiografickému nálezu suspektnímu z restriktivní kardiomyopatie byla doplněna endomyokardiální biopsie, jejíž výsledky potvrdily podezření na srdeční AL-amyloidózu. V rektální biopsii amyloid nalezen nebyl. Imunofixace séra a moči byly negativní, trepanobiopsie vyloučila diagnózu mnohočetného myelomu či jinou maligní lymfoproliferaci a pacient neměl známky postižení jiných orgánů. Na základě provedených vyšetření uzavíráme daný případ jako izolovanou srdeční AL-amyloidózu. Izolované srdeční postižení je velmi vzácné (
Klíčová slova:
amyloidóza srdce, srdeční selhání, echokardiografie, endomyokardiální biopsie.
Úvod
Amyloidóza je heterogenní onemocnění či skupina onemocnění, pro které je charakteristické ukládání bílkoviny amyloidu do extracelulární matrix různých tkání a orgánů a s tím spojenou alterací jejich funkce (14). Depozice může být lokalizována pouze na jeden orgán, častěji však bývá postiženo více orgánů současně.
Rozlišujeme celkem 5 typů amyloidózy, lišících se typem i původem amyloidu. Jedná se jednak o vzácné formy jako je sekundární (AA) amyloidóza, která vzniká depozicí proteinu akutní fáze A u některých chronických zánětů, dále amyloidózu na podkladě depozice divokého transthyretinu (prealbuminu), ke které dochází nejčastěji v pokročilém věku, a proto se také nazývá senilní amyloidóza, na rozdíl od autozomálně dominantně dědičné familiární amyloidózy, která se vyskytuje v nižších věkových kategoriích a kde je prekurzorem mutovaný transthyretin. Jedna z nejméně častých a málo významných je izolovaná atriální amyloidóza s depozicí atriálního natriuretického peptidu (15).
Z kardiologického hlediska však představuje největší hrozbu primární AL-amyloidóza, kde jsou depozita amyloidu tvořena lehkými řetězci imunoglobulinu (kappa nebo lambda), které jsou produkovány monoklonální populací plazmatických buněk (1).
Tento typ je nejčastější – tvoří asi 85% všech případů amyloidózy. K srdečnímu postižení dochází až v 50 % případů, přičemž srdeční selhání se vykytuje u 25 % případů. Vedle srdečního selhání se z kardiálních projevů vyskytují dysrytmie (2, 3).
Vlastní pozorování
Dosud relativně zdravý 81-letý pacient, od 78 let léčený pro lehčí hypertenzi, byl odeslán na naše pracoviště k vyloučení ischemické choroby srdeční. Asi 2 roky udával neprogredující ponámahovou dušnost NYHA II, bez dalších symptomů typu bolestí na hrudi či synkopálních stavů. V anamnéze však byla graficky pozitivní ergometrie a dle Holter EKG záchyt širokokomplexové tachykardie spolu s opakovanými úseky akcelerovaného junkčního rytmu s pauzami do 2 sec při přechodu na sinusový rytmus, při kterém měl AV blok I. stupně.
Při fyzikálním vyšetření byl pacient eupnoický, bez cyanózy, normotenzní (TK 110/60 mm Hg) s přiměřenou tepovou frekvencí (61/min). Hlava a krk byly bez významnější patologie, dýchání nad oběma plícemi bylo čisté, sklípkové, akce srdeční byla pravidelná, 2 ozvy ohraničené. Při pohmatu břicha lehce zvětšená játra + 1 cm pod oblouk v inspiriu. Na dolních končetinách byly známky chronické žilní insuficience (hemosiderinové pigmentace a klidné varixy), bez otoků. Na EKG byl zaznamenán sinusový rytmus, osa doprava, AV blokáda I. stupně, bez známek hypertrofie levé komory srdeční, naopak nízká voltáž v končetinových svodech (max. výchylka 5mm).
Echokardiograficky byla zjištěna nezvětšená koncentricky hypertrofická levá komora srdeční (septum 16 mm, zadní stěna 15 mm), myokard měl jiskřivý vzhled (obr. 1). Globální systolická funkce levé komory byla hraniční (ejekční frakce 50 %) při difuzní poruše kinetiky. Byl popsán restriktivní typ plnění levé komory srdeční, a to jak pomocí transmitrálního průtoku (obr. 2), tak tkáňovým dopplerovským vyšetřením (obr. 3). Dále byla zjištěna dilatace obou síní (levá 46 mm, plocha 26 cm2, plocha pravé síně 30 cm2) a jen lehká degenerativní mitrální a aortální insuficience. Pravá komora nebyla zvětšena, tenze v plicnici nebyla zvýšena (odhadovaný systolický tlak v plicnici byl 30–35 mm Hg).



Již na základě tohoto vyšetření (restriktivní plnění u koncentricky hypertrofické levé komory srdeční a zároveň EKG bez obrazu hypertrofie levé komory srdeční s redukcí voltáže v končetinových svodech) bylo v diferenciální diagnostice pomýšleno na suspektní restriktivní, nejspíše infiltrativní kardiomyopatii.
K vyloučení ischemické choroby srdeční (důvod plánovaného přijetí pacienta) byla provedena selektivní koronarografie s nálezem jen nezužující koronární aterosklerózy. Ischemická choroba srdeční tedy byla vyloučena.
Základní laboratorní vyšetření – mineralogram, kreatinin, urea, jaterní testy, CRP, sedimentace, krevní obraz s diferenciálním rozpočtem, koagulační parametry, močový sediment a chemické vyšetření moči byly v normě. Na rentgenu srdce a plic byl popsán obraz centrálního městnání, vinutá hrudní aorta, plicní křídla bez ložiskových změn, ateromatóza aorty.
Vzhledem k podezření na infiltrativní kardiomyopatii připadala v úvahu jedna z nejčastějších příčin – amyloidóza. Další vyšetřování bylo tedy zaměřeno na její vyloučení či potvrzení. Byla indikována endomyokardiální a rektální biopsie, ke které byl pacient pozván v druhé době elektivně. Výsledek rektální biopsie byl při barvení na amyloid (konžská červeň) negativní. Avšak v endomyokardiální biopsii (EMB) byla přítomna ve všech pěti vzorcích výrazná infiltrace intersticia hyalinními hmotami s tinkčními vlastnostmi amyloidu (konžská červeň, saturnová červeň) (obr. 4, 5).


Dle imunohistochemického vyšetření vzorků byly pozitivní kappa řetězce, slabě pozitivní lambda řetězce (obr. 6), A-amyloid i transtyretin byly negativní. Morfologie i imunohistochemie tedy odpovídala AL-amyloidóze.
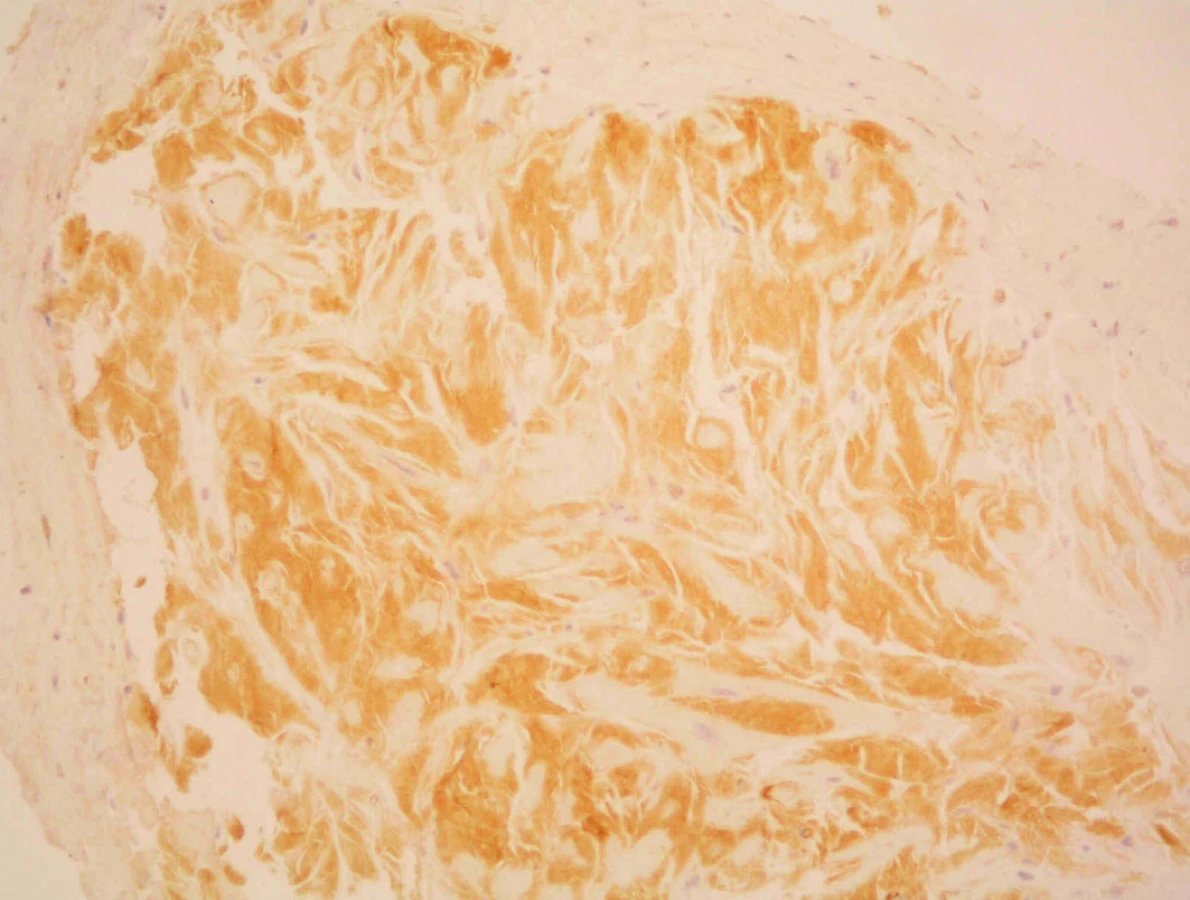
Vzhledem k tomu, že AL amyloidóza bývá velmi často systémové onemocnění (95 %) a bývá často spjata s mnohočetným myelomem, byla další diagnostika zaměřena tímto směrem. Byla doplněna elektroforéza bílkovin séra, imunofixace séra i moči, vyšetření na beta-2-mikroglobulin, znovu jaterní soubor, renální funkce, moč chemicky a sediment – vše v normě. Laktátdehydrogenáza byla hraničně snížena (4,12 μkat/l). Ultrasonograficky byla zjištěna hepatomegalie s obrazem venostázy, obě ledviny byly normálně velké, bez patologických změn jejich morfologie. RTG vyšetření skeletu neprokázalo osteolytická ložiska.
Ve spolupráci s hematology byla provedena ještě trepanobiopsie kostní dřeně, která spolu s dalšími vyšetřeními diagnózu mnohočetného myelomu, potažmo jinou maligní lymfoproliferaci, vyloučila. Rovněž tak systémové postižení ostatních orgánů mimo srdce bylo velmi nepravděpodobné (absence amyloidu v kostní dřeni, intaktní ledvinné funkce, normální ultrasonografie břicha až na lehčí městnavou hepatomegalii, negativní rektální biopsie).
Na základě provedených vyšetření uzavíráme daný případ jako izolovanou srdeční amyloidózu.
Pacient je v péči kardiologů i hematologů. Od počátku léčen léky srdečního selhání – ACE-inhibitory a diuretiky v nevelké dávce. Betablokátory pro sklon k bradykardii a pauzy na Holter EKG nebyly nasazeny. Amiodarone byl nasazen do terapie již před přijetím na naši kliniku, dle později provedených Holter EKG s dobrým efektem na nesetrvalé komorové tachykardie.
V současné době je léčen také vysokodávkovanou kortikoterapií v ambulantní péči hematologů. Doba od diagnózy do nasazení této hematologické terapie, mající za úkol snížit produkci amyoidu, byla v našem případě 9 měsíců pro zdrženlivý postoj pacienta k léčbě. Za tuto dobu nedošlo k výraznější progresi srdečního selhání. Pacient stále toleruje stejnou zátěž, bez nutnosti navýšení diuretické terapie. Vzhledem k tomu, že v literatuře se uvádí průměrný medián přežití pouze 6 měsíců od prvních projevů srdečního selhání (Falk), hodnotíme tento průběh zatím jako relativně benigní.
Diskuse
AL amyloidóza má incidenci 8–12/milion, většinou se manifestuje jako systémové postižení řady orgánů a systémů (5). Na toto nepříliš časté onemocnění se ne vždy myslí, a proto může být přehlédnuto. Diagnostika může být obtížná vzhledem k pestrosti klinické symptomatologie při systémovém postižení.
Ať už je původ amyloidu jakýkoliv, dochází v případě srdečního postižení k extracelulárnímu ukládání amyloidu do myokardu, v síních i komorách, perivaskulárně, v chlopních i v převodním aparátu. Infiltrativní proces má za následek ztlušťování srdečních komor bez jejich dilatace. Dochází k rigiditě pohybu komor jak v systole, tak v diastole, zvýšují se plnící tlaky komor, což zvýší tlaky v síních, které dilatují, přestože se do nich také ukládá amyloid (3).
Uvedené změny se projeví i při echokardiografickém vyšetření jako obraz symetrické hypertrofie myokardu, někdy s jiskřivým vzhledem myokardu, připomínajícím „zrnité sklo“, dále dilatací síní při normální velikosti komor a restriktivním typem plnění levé komory srdeční. To ukazuje na rozhodující význam pečlivého echokardiografického vyšetření spolu s komplexním hodnocením nálezů ostatních vyšetření, včetně EKG, kde nacházíme díky infiltraci srdeční svaloviny a relativnímu i absolutnímu úbytku funkčních kardiomyocytů redukci voltáže QRS komplexů (7).
V našem případě by nález zbytnění komor při absenci těžší hypertenze či tlakového přetížení v podobě aortální stenózy mohl vést k úvaze o hypertrofické kardiomyopatii. Ta však vzhledem k absenci EKG známek hypertrofie levé komory byla nepravděpodobná. Na EKG byla patrna nízká voltáž zejména v končetinových svodech. Restriktivní plnění u koncentricky hypertrofické levé komory srdeční při daném EKG nálezu nás vedlo k úvaze o infiltrativní kardiomyopatii.
Srdeční postižení amyloidózou se klinicky manifestuje nejčastěji jako městnavé srdeční selhání (11), u našeho pacienta prozatím v lehčí formě s dušností NYHA II.
Při postižení převodního aparátu se mohou vyskytnout bradyarytmie s různým postižením sinoatriálního i atrioventrikulárního uzlu. Vzhledem k infiltraci i síňového myokardu a dilataci síní často dochází ke vzniku fibrilace síní. Jindy je amyloidóza substrátem pro vznik monomorfních komorových arytmií.
U našeho pacienta jsou arytmie rovněž přítomny. Kromě výše popsaných širokokoplexových tachykardií, které však dobře reagovaly na terapii amiodaronem, byly přítomny i úseky akcelerovaného junkčního rytmu s pauzami do 2 sec při přechodu na sinusový rytmus, při kterém měl AV blok I. stupně.
Pacient je stran arytmií asymptomatický a zatím bez nutnosti implantace kardiostimulátoru.
Kromě srdce bývají nejčastěji bývají postiženy:
- ledviny (nefrotický syndrom, renální selhání),
- játra (12),
- periferní nervy (senzitivní a autonomní neuropatie),
- měkké tkáně – jazyk (makroglosie),
- gastrointestinální trakt (4).
V našem případě se jednalo o vzácnou primární izolovanou srdeční AL-amyloidózu, což je <5 % všech případů AL amyloidózy (3). Všechna laboratorní a pomocná vyšetření k ověření produkce amyloidu byla negativní.
Diagnózu bez provedení endomyokardiální biopsie tedy nebylo možno stanovit. Navíc endomyokardiální biopsie odliší AL-amyloidózu od senilní amyloidózy, u které nedochází k extrakardiálnímu ukládání amyloidu a která by v našem případě mohla vzhledem k věku pacienta přicházet v úvahu. Lokalizovaná AL-amyloidóza je výsledkem in-situ produkce lehkých řetězců, které nemusí být zachytitelné běžnými metodami, jako je např. imunofixace séra a moči (6).
Ve většině případů je postižení systémové, a k diagnóze srdeční amyloidózy pak může stačit typický echokardiografický a EKG nález spolu s pozitivní imunofixací séra a moči, pozitvní biopsií na amyloid v jiném přístupnějším orgánu či tkáni (podkožní tuk, kostní dřeň) (7). Současně je však nutné vždy vyloučit přítomnost mnohočetného myelomu či jiné maligní lymfoproliferace (RTG skeletu, sedimentace, trepanobiopsie) vzhledem k jinému terapeutickému přístupu.
V poslední době se v diagnostice srdečních onemocnění uplatňuje čím dál více také magnetická rezonance a ta může významně pomoci i v diagnostice srdeční amyloidózy (13, 16).
Léčbu AL-amyloidózy můžeme dělit na
- specifickou, zaměřenou na ovlivnění produkce amyloidu, a
- podpůrnou, symptomatickou terapii srdečního selhání.
Ve spefické terapii AL-amyloidózy se za standardní považuje kombinace melphalanu s kortikoidy v několika cyklech.
Radikálnějším postupem je vysokodávkovaná chemoterapie s následnou autologní transplantací kmenových buněk (ASCT) (4 leté přežití 71 % oproti 41 % u konvenční terapie) (8). Tento agresivní způsob léčby je bohužel spojen s dosti velkou periprocedurální mortalitou (9). Ta se pohybuje od 13 % (7), přes 38 % (5) až po 90 % u pokročilého stadia onemocnění (8).
Spornou, ale v některých případech možnou léčbou pokročilé srdeční amyloidózy je ortotopická transplantace srdce následovaná ASCT do 6 měsíců od srdeční transplantace (10). Z uvedeného vyplývá, že terapie AL, amyloidózy není uspokojivá a její volba není snadná.
My jsme u 81-letého nemocného volili ve spolupráci s hematology stabilizaci nemoci pomocí vyskodávkované kortikoterapie dexamethazonem, ale až s odstupem vzhledem k zdrženlivému přístupu pacienta k léčbě.
Náš případ je pozoruhodný i z hlediska relativně benigního průběhu jinak dosti závažného onemocnění s udávaným mediánem přežití 6 měsíců od prvních projevů srdečního selhání. Zajímavý je 9 měsíců trvající stacionární klinický stav i bez speciální hematologické léčby, která byla nasazena až s odstupem.
Závěr
Amyloidóza je sice vzácné onemocnění, avšak v určitých případech je na něj nutno při diferenciální diagnostice myslet, protože představuje pro pacienta zvláště při postižení srdce velmi závažnou hrozbu, kterou lze ovlivnit terapií. Z našeho případu vyplývá, že při závažném podezření na amyloidózu srdce je endomyokardiální biopsie zcela indikována k definitivnímu vyloučení/potvrzení této choroby, i když základní vyšetření zaměřená na tvorbu amyloidu jsou negativní.
Z hlediska srdečního postižení hraje v diagnostice hlavní roli echokardiografické vyšetření, EKG vyšetření a samozřejmě anamnéza, která však nemusí být vzhledem k různorodosti při systémovém postižení vždy jednoznačná.
Při podezření či průkazu AL-amyloidózy je nutno zjistit, zda je lokalizovaná, či systémová (biopsie rektální sliznice, podkožního břišního tuku, biopsie kostní dřeně) a vyloučit myelom (sternální punkce – lépe biopsie kostní dřeně s průkazem na amyloid, elektroforéza bílkovin séra, imunofixace séra i moči) či potažmo jinou maligní lymfoproliferaci. Od toho se pak odvíjí i terapie.
MUDr. Jan Čapek
III. – interní kardiologická klinika FNKV
Šrobárova 50
100 34 Praha 10
E-mail: jancapek@centrum.cz
Zdroje
1. Abraham, R.S., Geyer, S.M., Price-Troska, T.L., et al. Immunoglobulin light chain variable (V) region genes influence clinical presentation and outcome in light chain-associated amyloidosis (AL). Blood 2003, 101, p. 3801–3808.
2. Dubrey, S.W., Cha, K., Anderson, J. et al. The clinical features of immunoglobulin light-chain (AL) amyloidosis with heart involvement. QJM 1998, 91, p. 141-157.
3. Falk, R.H. Diagnosis and management of the cardiac amyloidoses. Circulation 2005, 112, p. 2047-2060.
4. Gertz, M.A., Comenzo, R., Falk, R.H. et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): A consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis. Am. J. Hematol. 2005, 79, p. 319–328.
5. Gertz, M.A., Lacy, M.Q., Dispenzieri, A. Amyloidosis: Recognition, confirmation, prognosis and therapy. Mayo Clin Proc. 1999, 74, p. 490-494.
6. Hamidi, A.K., Liepnieks, J.J., Nakamura, M. et al. Organspecific (localized) synthesis of Ig ligh chain amyloid. J. Immunol. 1999, 162, p. 5556-5560.
7. Hassan, W., Al-Sergani, H., Mourad, W. et al. Amyloid heart disease. New frontiers and insights in pathophysiology, diagnosis, and management. Tex. Heart Inst. J. 2005, 32, p. 178-184.
8. Kothari, S.S., Ramakrishnan, S., Bahl V.K. et al. Cardiac Amyloidosis –an update. Indian Heart J. 2004, 56, p. 197-203.
9. Krejčí, J. Srdeční amyloidóza. In: Veselka, J. a kol. Hypertrofická kardiomyopatie a příbuzná témata. Praha: Galén 2006, s. 141-148.
10. Maurer, M.S., Raina, A., Hesdorffer, Ch. et al. Cardiac transplantation using extended-donor criteria organs for systemic amyloidosis complicated by heart failure. Transplantation 2007, 83, p. 539-545.
11. Mikulová, A., Toušek, F., Šindelářová, Š. a kol. Progresivní srdeční selhání na podkladě srdeční amyloidózy. Prakt. Lék. 2007, 87, 10, s. 628-631.
12. Park, M.A., Mueller, P.S., Kyle, R.A. Primary (AL) hepatic amyloidosis: clinical features and natural history in 98 patients. Medicine 2003, 82, p. 291–298.
13. Ruberg, F.L., Appelbaum, E., Davidoff, R. et al. Diagnostic and prognostic utility of cardiovascular magnetic resonance imaging in light-chain cardiac amyloidosis. Am. J. Cardiol. 2009, 103, 4, p. 544-549.
14. Selvanayagam, J.B., Hawkins, P.N., Biju, P. et al. Evaluation and managment of the cardiac amyloidosis. J. Am. Coll. Cardiol 2007, 50, p. 2101-2110.
15. Shah, K.B., Inoue, Y., Mehra, M.R. Amyloidosis and the heart: a comprehensive review. Arch. Intern. Med. 2006, 166, p. 1805-1813.
16. Vogelsberg, H., Mahrholdt, H., Deluigi, C.C. et al. Cardiovascular magnetic resonance in clinically suspected cardiac amyloidosis: noninvasive imaging compared to endomyocardial biopsy. J. Am. Coll. Cardiol. 2008, 51, p. 1022-1030.
Štítky
Praktické lékařství pro děti a dorost Praktické lékařství pro dospěléČlánek vyšel v časopise
Praktický lékař

2009 Číslo 6
- Inkontinence jako důsledek operačního zákroku na prostatě
- Efektivita kartáčku Sonicare For Kids u dětí předškolního věku
- Při preskripci inkontinenčních pomůcek je nezbytné hlídat limity
- Na výběru inkontinenčních pomůcek záleží − ale jak se mezi nimi neztratit?
- Diosmin a hesperidin: Co ukazuje farmakokinetika?
Nejčtenější v tomto čísle
- Zdravotní komplikace zneužívání návykových látek a možnosti prevence v primární péči
- Chyby při léčbě deliria tremens
- Syndrom kombinované fibrózy a emfyzému – CPFE syndrom
- Bolestivá noha
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy