
The Transcription and Translation Landscapes during Human Cytomegalovirus Infection Reveal Novel Host-Pathogen Interactions
Viruses are fully dependent on the cellular translation machinery, and develop diverse mechanisms to co-opt it for their own benefit. However, fundamental questions such as: what is the effect that infection has on the spectrum of host mRNAs that are being translated, and whether, and to what extent, a virus possesses mechanisms to commandeer the translation machinery are still hard to address. Here we show that by simultaneously examining the changes in transcription and translation along Human cytomegalovirus (HCMV) infection, we can uncover extensive transcriptional regulation, but also diverse and dynamic translational control. We were also able to show that, at late time points in infection, translation of viral mRNAs is higher than that of cellular mRNAs. Lastly, we take advantage of our measurements of translation (protein synthesis rate) and integrate these with mass spectrometry measurements (protein abundance). This integration allowed us to unbiasedly reveal dozens of cellular proteins that are being degraded during HCMV infection. Since targeted degradation indicates a strong biological importance, this approach should be applicable for discovering central host functions during viral infection. Our work provides a framework for studying the contribution of transcription, translation and degradation during infection with any virus.
Published in the journal:
. PLoS Pathog 11(11): e32767. doi:10.1371/journal.ppat.1005288
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1005288
Summary
Viruses are fully dependent on the cellular translation machinery, and develop diverse mechanisms to co-opt it for their own benefit. However, fundamental questions such as: what is the effect that infection has on the spectrum of host mRNAs that are being translated, and whether, and to what extent, a virus possesses mechanisms to commandeer the translation machinery are still hard to address. Here we show that by simultaneously examining the changes in transcription and translation along Human cytomegalovirus (HCMV) infection, we can uncover extensive transcriptional regulation, but also diverse and dynamic translational control. We were also able to show that, at late time points in infection, translation of viral mRNAs is higher than that of cellular mRNAs. Lastly, we take advantage of our measurements of translation (protein synthesis rate) and integrate these with mass spectrometry measurements (protein abundance). This integration allowed us to unbiasedly reveal dozens of cellular proteins that are being degraded during HCMV infection. Since targeted degradation indicates a strong biological importance, this approach should be applicable for discovering central host functions during viral infection. Our work provides a framework for studying the contribution of transcription, translation and degradation during infection with any virus.
Introduction
Human cytomegalovirus (HCMV) is a ubiquitous pathogen, infecting the majority of the human population worldwide, leading to severe diseases in newborns and immunocompromised adults. The HCMV genome contains almost 240kb, making it the largest known human virus. Its genome was traditionally estimated to code for approximately 200 open reading frames [1,2], but our recent study showed that many additional, mostly short open reading frames are also translated during infection [3].
During viral infection, cellular gene expression is subjected to rapid alterations induced by both viral and antiviral mechanisms. The differential regulation of cellular transcription and translation distinguishes host pathways that the virus either relies on or actively subverts and can open new therapeutic opportunities and reveal novel principles in cell biology. Over the years, a large body of work was conducted to decipher these changes in a global manner by examining the temporal changes in RNA levels [4–11]. The use of microarrays revealed many biological pathways that are significantly altered during infection and established an important progress in our understanding of how HCMV exploits cellular pathways during infection [6–11]. These studies helped to reveal numerous pathways that are elevated during infection and are important for viral propagation such as cell cycle, DNA damage, transcription and translation factors and energy production. In addition pathways that were reduced during infection were also mapped, such as cell adhesion, cytoskeletal regulators and apoptosis and extracellular matrix. Recently, advancement in mass-spectrometry methods had been used to quantify the cell proteome along HCMV infection giving a wider view on the pathways that are altered during infection [12]. More specifically, this method was used to predict natural killer (NK) and T cells ligands by identifying cell surface molecules which are downregulated during HCMV infection [12]. However, these approaches could not delineate transcriptional, translational and post-translational layers of regulation. It had been shown that unlike many other viruses (including several Herpes viruses, e.g. Herpes simplex virus 1 and 2), the overall impact of HCMV is stimulation of host RNA and protein synthesis [13–15]. Still, fundamental questions such as how and to what extent HCMV changes the spectrum of host mRNAs translation, and whether the virus possess mechanisms to ensure more effective translation of its own mRNAs, have just begun to be addressed [16,17]. In a recent study, changes in host genes translation at 48 hours post HCMV infection were examined by analyzing the fraction of mRNAs associated with polysomes. This study revealed that a significant fraction of cellular mRNAs are translationally activated or repressed by HCMV [17]. In addition, expression of pUL38, a virally encoded mTORC1-activator sufficed to partially recapitulate these translational alterations in uninfected cells, demonstrating that some of the effect is mediated by mTORC1 activation [17].
Here, we have used RNA sequencing (RNA-seq) and ribosome profiling (deep sequencing of ribosomes-protected fragments) to globally map the changes in host genes transcription and translation during HCMV replication. These comprehensive and simultaneous measurements revealed how HCMV orchestrates cellular gene expression at both the transcription and translation levels. We identified several novel pathways that are upregulated during infection and are central for viral propagation. We show that most of the regulation of cellular genes along infection occurs on the level of transcription but our experiments also uncover extensive and dynamic translational regulation of subsets of cellular genes. In addition, our measurements enabled the comparison between translation of viral and host genes, revealing that late in infection viral genes are translated more efficiently than their host counterparts. Finally, by integrating our measurements of protein synthesis rate with recent measurements of protein levels [12] we were able to globally and unbiasedly identify host proteins that are actively targeted for degradation during HCMV infection. We show that BTN2A1 and IGSF8, two cell surface proteins that belong to the immunoglobulin (Ig) superfamily are degraded during HCMV infection. We also reveal that few cytosolic proteins including ROCK1, a key regulator of actin cytoskeleton, are degraded during HCMV infection.
Results
Simultaneous monitoring of RNA levels and translation during HCMV infection
To gain a detailed view of the changes that occur in host genes transcription and translation over the course of HCMV infection, we infected human foreskin fibroblasts (HFF) with the Merlin HCMV strain and harvested cells at 5, 12, 24 and 72 hours post infection (hpi). We also used cells treated with type I interferon (IFN) or cells infected with an irradiated virus, in which viral DNA is inactivated [18], preventing viral gene transcription. We designed our experiment to simultaneously monitor both RNA levels and translation (Fig 1A). Deep sequencing of mRNA (RNA-seq) allows a detailed mapping of transcript levels during infection and these were paired with ribosome footprints (deep sequencing of ribosome-protected mRNA fragments), which allow accurate measurement of protein synthesis by capturing the overall in vivo distribution of ribosomes on a given message [19]. In order to assess the reproducibility of our experiments we have prepared two independent biological replicates for the 5hpi and 72hpi time points. Both the mRNA and footprints read density measurements were highly reproducible (correlation coefficient [R2] = 0.97 and 0.92; SD in ratio between biological replicates corresponded to a 0.18 - and 0.3-fold change, respectively) demonstrating consistency in our experimental methods (Figs 1B and S1). We quantitatively assessed the expression pattern of 10,354 genes. Interestingly, 73% of the transcripts changed by more than 3-fold in their footprints densities along infection, reflecting the drastic changes that occur in cells during HCMV infection (S1 Table). In order to identify patterns of specific cellular pathways that were influenced during infection, we compared the expression of infected samples to mock sample and clustered the mRNA and footprints ratios using partitioning clustering. This approach allowed clustering of the cellular transcripts into ten distinct classes based on similarities in temporal expression profiles in the RNA-seq and ribosome profiling data. Overall we found that changes in ribosome footprints tracked the changes in transcripts abundance (Figs 2A and S2A), which indicates that most of the regulation of host gene expression occurs at the level of transcription. To identify biological processes that are altered during HCMV infection we applied the DAVID software [20]. Most of the time-related clusters showed enrichment for specific biological functions supporting their biological relevance (Fig 2A). Many of these biological functions were captured by previous transcriptomic studies [4,14,15,8,9] and were reported to be affected by HCMV infection such as cell cycle, DNA damage, sterol biosynthesis, ribosome biogenesis and the proteasome [21–25]. The increased sensitivity of deep sequencing approaches allowed us to also identify significant changes in novel biological functions such as RNA processing, protein intracellular transport and transcription regulation. The temporal profile of candidate genes from each of these clusters was confirmed by real-time PCR or western blot analysis (S2B Fig). The full DAVID enrichment analysis is presented in S2 Table.


Identification of up - or down-regulated pathways during HCMV infection
It is likely that many of the genes in the same temporal profile are co-regulated by common transcriptional activators or repressors. Therefore, to find potential regulators that drive the expression of these clusters and to better focus on well-characterized canonical pathways, we turned to Ingenuity Pathway Analysis (IPA) (Ingenuity Systems, www.ingenuity.com). This analysis helped to reveal functional pathways that are significantly changing during infection and identifying upstream molecules that may control the expression of the genes in each cluster.
HCMV is known to induce the cellular DNA damage response (DDR), that includes activation of the ATM, H2AX, NBS1, CHK2, CHK1 and p53 genes [21]. Indeed, we found multiple pathways related to DNA damage that were significantly enriched in cluster 1 (S3 Table). Interestingly, we identified significant upregulation of genes that promote homologous recombination repair (HR), such as Fanconi anemia complementation group D2 protein (FANCD2), BRCA1 and BARD1 (S3 Fig, P-val = 1.25E-16). Recently, it had been suggested that FANCD2 upregulation during HSV1 infection is important for promoting HR on the expanse of non - homologous end-joining [26]. Our results, therefore, suggest that similar strategy may be used in HCMV-infected cells. We also identified significant upregulation of DNA mismatch repair complex that is required for the repair of DNA replication errors (S4 Fig, P-val = 6.3E-12). Mismatch Repair Proteins were shown to be elevated and required for efficient HSV1 replication [27]. The significant elevation we mapped during HCMV infection suggests that the mismatch repair may also be important during HCMV infection and it will be of interest to determine whether mismatch repair proteins contribute to the fidelity of herpesvirus DNA replication. An important upstream regulator of genes in cluster 1 was E2F1 (P-val = 3.14E-16) which was previously shown to be important for HCMV replication [28].
Cholesterol levels are significantly elevated during HCMV infection and cholesterol import was shown to play an important role in this process [23]. Our results also reveal direct upregulation of cholesterol biosynthesis enzymes implying an increase in cholesterol de novo synthesis (P-val = 1.7E-6, cluster1, S3 Table).
In agreement with the overall upregulation in translation in HCMV infected cells [17], we found translation initiation factors to be significantly elevated (S5 Fig, P-val = 7.94E-11). Cluster 2 is also enriched in genes related to assembly of RNA polymerase II (S6 Fig, P-val = 2.4E-5), suggesting a simultaneous upregulation of both the transcription and translation machineries. In addition, we identified upregulation of genes related to tRNA charging and tRNA modifications (P-val = 3.3E10-5, cluster 4, S3 Table) that probably support the higher demand for tRNAs due to the increase in translation.
We have also mapped a significant enrichment in genes involved in proteins quality control (P-val = 5.03E-13, S7 Fig), including upregulation of many of the cell’s chaperones, the proteasome components and deubiquitinating enzymes (DUBs). DUBs form a large family of proteases that cleave ubiquitin chains from target proteins and their up regulation can effect the stability, localization and function of the proteome [29]. Interestingly, DUBs are also encoded by herpesviruses and HCMV encoded DUB (UL48) was shown to influence viral replication [30]. It is possible that the increased cellular DUB activity can increase the stability of target proteins during the progression of infection by inhibiting polyubiquitination.
NRF2 is a transcription factor that plays a key role in cellular defense against oxidative stress. We observed upregulation in NRF2 targets, that included the induction of few detoxifying enzymes and stress response proteins (P-val = 6.6E-5, cluster 5, S3 Table). These results extend previous observations showing NRF2 was elevated during HCMV infection[31].
Downregulated genes (Clusters 3 and 8) were significantly enriched (P-values 1.4E-8 and 1.25E-14 respectively) for genes related to the synthesis of the extra cellular matrix (ECM) (S8 Fig) and for metalloproteinase inhibition (P-values 1.2E-3 and 8.3E-6, S9 Fig), in agreement with previous microarray measurements [9]. Interestingly, collagen was found to restrict HSV-1 infectivity in healthy tissues [32], suggesting a potential motivation to downregulate ECM production. Reduction in metalloproteinase inhibition may have additional effect on reducing ECM protein levels but can also affect proteolytic degradation of cell surface molecules. Indeed, increased proteolytic degradation was demonstrated for the cell surface associated low density lipoprotein related receptor 1 (LRP1) at late phase of infection[23]. In addition proteolytic degradation of cell surface molecules could be employed by the virus to increase shedding of immune stimulatory molecules [33]. Important upstream regulators of genes in these downregulated clusters are β-catenin (Pval = 3.96E-11) and TP63 (Pval = 3.14E-11), in agreement with recent studies showing dysregulation of Wnt/β-catenin signaling pathway during HCMV infection[34]. The enrichment of all pathways and upstream regulators in the different clusters are presented in S3 Table and S4 Table.
In order to test the functional importance of genes we identified as elevated during infection, we tested the effect of knockdown (KD) of few candidate genes on viral replication. We chose genes that fall in different functional categories that were not connected before to HCMV infection. Since we observed elevation in genes responsible for protein transport and localization we targeted two genes that are involved in these processes; SEC11C - a component of the microsomal signal peptidase complex that removes signal peptides from nascent proteins and PEX3 - which is involved in peroxisome biosynthesis and integrity; We also chose two genes that are involved in RNA processing; TRMT1-an enzyme that dimethylates a guanine residue on most tRNAs but its importance is poorly defined and METTL3 - N6-methyltransferase that methylates adenosine residues in mRNAs and was recently shown to be important for RNA stability and translation [35]. Lastly we chose HPRT1 - which is a central enzyme in the purine salvage pathway. Since the expression of these genes could be essential for cell survival, we confirmed that the various KDs did not cause significant cell death and that siRNA-mediated ablation reestablished mRNA levels to these observed in uninfected cells (S10A and S10B Fig). Importantly, KD of SEC11C, PEX3 and HPRT1 significantly reduced viral titers (Fig 2B). In order to preclude off target effects we confirmed that similar effects were obtained using distinct siRNAs that target the same gene (S10C Fig). These results strongly indicate that the elevation in the secretory pathway proteins, the integrity of peroxisomes and the purine salvage pathway are all contributing to viral propagation. Importantly, both PEX3 and HPRT1 KD blocked HCMV at early stages of infection as the expression of early-proteins IE2 and UL44 was reduced (Figs 2C and S10D). In contrast, SEC11C blocked only the late stage of infection as only the expression of the late-protein, pp28 was reduced (Figs 2C and S10D). Although the TRMT1 KD did not significantly affect viral titers we observed a reproducible reduction in pp28 expression (Figs 2C and S10D) suggesting that the TRMT1 dimethylation activity might be playing a subtler role during HCMV infection. Overall, our results increase the breadth of knowledge about the changes that occur in various cellular processes during HCMV infection.
Evaluating the role of translation regulation during HCMV infection
To quantitatively evaluate the role of translational control along HCMV infection, we calculated relative translation efficiency (TE) across our time course. TE is defined as the ratio of ribosome-associated RNA (footprints) to total mRNAs for a given gene (Fig 3A). Replicates indicated high reproducibility (SD of ratio between biological replicates corresponded to a 0.29 fold change), which allowed sensitive measurement of dynamic translational control (Figs 3A and S11A). In order to focus on cases in which translational regulation might play a substantial role in viral replication, we centered on genes that showed more than a 3-fold difference in their TE between any two-time points. In addition, we required that the change in TE would be accompanied by a change in a similar direction in the footprints densities. Based on these criteria we obtained 731 transcripts that showed significant changes in their TE during infection (S5 Table). For each of these genes we calculated log2 TE versus the mock sample and clustered them into five classes based on similarity in their temporal TE profiles. The heat-map of the footprints and mRNA temporal profiles of these genes exemplifies the translation regulation; the changes in footprints are much more pronounced than the changes in mRNA levels (Fig 3B). To characterize the parameters involved in HCMV-dependent translational regulation, we examined specific characteristics of the 5’ untranslated regions (5’UTRs) of the corresponding transcripts, including their length and percent of G+C content. We observed few unique features that define genes from each of these clusters (S11B and S11C Fig), suggesting that several features of 5’UTR may contribute to the TE along infection. In addition, we examined whether the TE clusters show strong enrichment for specific biological processes. Interestingly, cluster 4 (in which translation is induced at 5hpi) is enriched in genes related to the translation machinery, including many of the ribosomal proteins and translation initiation factors. This enrichment suggested that elevated translation of these genes is mediated by mTORC1 activation, as many of mTORC1 targets are related to protein synthesis [36,37]. Supporting this notion, there is the significant overlap (Pval = 5.432e-21) between mRNAs whose translation is repressed by mTOR inhibitors [38,39] and genes found in cluster 4. Genes in cluster 5 (in which translation is induced at 24hpi) were significantly enriched in functions related to cell cycle regulation. Since HCMV is known to arrest the cell cycle at a “pre-S” phase we speculated that translation regulation of this cluster could be attributed to the cell cycle arrest. Indeed, mRNAs whose translation was upregulated in this cluster significantly overlapped with genes that showed enhanced translation at G1 and S phases of the cell cycle (Pval = 9.03e-07 and Pval = 0.0007, respectively) [40]. Further clusters showed interesting patterns of translation regulation but were only weakly enriched for particular biological process (S6 Table). We confirmed the translational regulation for HSP90AB1, one of the genes identified in cluster 5, by showing that protein amounts increase throughout infection while no significant changes in mRNA as measured by real-time PCR are observed (Fig 3C). In order to further validate our translation measurements and to generate a platform that will enable identification of cis-regulatory elements that control translation during HCMV infection, we used a fluorescence-based reporter system to analyze translation of individual transcripts in single, living cells (Figs 3D, 3E, and S11D and [41,42]). The 5’UTRs of translationally regulated genes from clusters 4 and 5 were inserted into the fluorescent reporter and compared with 5’ UTRs of controls mRNAs that did not show HCMV related translational regulation. While the 5’ UTRs of control transcripts did not show significant changes in translation after HCMV infection, the transcripts with 5’UTRs from translationally regulated genes (RPS19, SMC2 and RAD50) showed increased translation after HCMV infection (Fig 3F). These results provide validation of our TE calculations and demonstrate that the regulatory elements required for translational activation of these genes are present in their 5’UTRs. Overall, our data demonstrates that multifaceted translational control of gene expression is carried out during HCMV infection.

Translation efficiency of viral genes outcompetes cellular genes late in HCMV infection
Replication of viruses is completely dependent on the host translational apparatus and many viruses commandeer this machinery to translate their own mRNAs on the expense of cellular mRNAs. In order to evaluate if HCMV evolved mechanisms to co-opt the cells' ribosomes we compared the TE of human genes to that of viral genes at each of the time points along infection. Interestingly, at 5hpi, on average, viral genes are translated less efficiently than human genes (Fig 4A, Pval = 0.0027), this suggests a successful host defense mechanism that lessens the translation of viral mRNAs compared to host mRNAs at the beginning of HCMV infection. However, this effect is diminished later during infection, as at 12hpi and 24hpi human and viral genes do not show any significant difference in their TE. Interestingly, at 72hpi viral mRNAs are, on average, translated more efficiently than human mRNAs (Fig 4A, Pval = 1.29E-05). These same effects were observed in independent biological replicates (S12 Fig). Thus, translation rates of viral mRNAs late in infection are higher than expected from their mRNA prevalence. This effect can also be seen when the fraction of mRNA and footprints reads that map to the virus are plotted along infection (Fig 4B). The molecular mechanism underlying this phenomenon is yet to be studied.

Uncovering immune ligands that are degraded during HCMV infection
HCMV is a paradigm for viral immune evasion and several immune ligands were shown to be targeted for proteosomal degradation by specific viral proteins [43]. One prediction is that molecules that play a role in marking infected cells for immune recognition will be induced at the transcriptional and translational levels (rate of protein synthesis) as a cellular antiviral response, while the virus will stimulate their degradation, causing their protein levels to drop. A recently published quantitative proteomic analysis of host proteins levels during HCMV infection [12] identified many immune molecules that are down regulated during infection. However, down regulation of a given protein could be merely a mirror of transcriptional changes and therefore, does not necessarily indicate degradation. We reasoned that comparing our footprints measurements with these quantitative proteomic measurements, would allow us to systematically identify immune molecules that are targeted for degradation. Indeed, when we examined the profiles of proteins that were shown to be targeted for degradation by HCMV, such as; HLA-A (targeted by US2 and US11 proteins [44,45]) and PVRL2 (a ligand for the activating NK receptor DNAM-1 which is targeted by UL141 [46]), we observed the expected profiles (Fig 5A). We next examined the temporal profiles of the known NK and T cells activating or co-stimulatory ligands for which we had quantitative measurements, including protocadherins that were recently suggested to act as novel activating ligands for NK cells [12]. However, unlike HLA-A and PVRL2 we observed that the regulation of these immune ligands occurs mostly at the transcriptional level as both footprints and mRNA levels were downregulated along infection (Figs 5B and S13). Since active degradation during HCMV infection may indicate biological importance, we looked for inverse correlation between the footprints and protein temporal profiles in a list of potential immune ligands composed of proteins that belong to a few immune-related protein families [12]. This search led us to identify two proteins, BTN2A1 and IGSF8, which belong to the immunoglobulin superfamily and present a profile that suggests degradation (Fig 5C and 5E). Interestingly a recent study that performed plasma membrane profiling showed that the relative abundance of both BTN2A1 and IGSF8 was higher in cells infected with HCMV ΔUS2 compared to those infected with WT virus, further supporting the possibility that these proteins are actively degraded [47]. Real-time PCR measurements from cell lysates along HCMV infection supported elevation in BTN2A1 and IGSF8 mRNA levels (Fig 5D and 5F, upper panels). Since we were not able to obtain specific detection of BTN2A1 and IGSF8 using commercially available antibodies, we expressed these proteins fused to an HA tag in fibroblasts using lentiviral vectors. The vectors also contained green fluorescent protein (GFP) that was expressed from the same transcript using an internal ribosome entry site (IRES) and was used as an internal control. These cells were then infected with HCMV and the kinetics of BTN2A1 and IGSF8 expression along infection was evaluated by western blot analysis. These experiments demonstrated that BTN2A1 and IGSF8 are targeted for degradation since their protein levels were downregulated during HCMV infection (Fig 5D and 5F, middle panels), whereas the levels of the GFP which was expressed from the same transcript was elevated (Fig 5D and 5F, lower panels). Since these proteins were suggested to be effected by the viral US2 protein [47], we tested if US2 affect BTN2A1 and IGSF8 expression by ectopically expressing US2 in fibroblasts expressing either tagged BTN2A1 or tagged IGSF8. Importantly, US2 ectopic expression was sufficient to downregulate BTN2A1 and to lesser extent IGSF8 in uninfected fibroblasts (Fig 6A). We next tested the expression of BTN2A1 and IGSF8 during infection with the AD169VarL virus and a BAC derived AD169VarL virus, in which the US2-US6 region was deleted [48] (the region in which the BAC cassette was inserted). Similar to the results obtained with Merlin strain, both BTN2A1 and IGSF8 were degraded when cells were infected with the AD169VarL parental virus (Figs 6B, S14A, and S14B). In accordance with the plasma profiling results [47], in cells that were infected with the AD169VarL-BAC virus (that is US2-deleted) the expression of BTN2A1 was elevated and resembled the expression pattern of the GFP that was expressed from the same plasmid, strongly suggesting that US2 is essential and sufficient for BTN2A1 degradation (Figs 6B and S14A). Interestingly, however, although in the absence of US2 some of IGSF8 expression was restored, IGSF8 levels are still reduced during infection with the AD169VarL-BAC virus (Figs 6B (second panel) and S14B), indicating that additional viral proteins mediate IGSF8 degradation. In order to identify the viral protein/s that are involved in IGSF8 degradation we performed a pull-down on cells expressing HA tagged-IGSF8 and that were infected with HCMV for 48hr. Isolated viral and host proteins were resolved by electrophoresis and identified by mass spectrometry. Two HCMV proteins were identified in this capture experiment. The first was US9, which was recently shown to selectively target MICA*008 to proteasomal degradation [49] and the second was UL40, which possesses a signal peptide that mimics cellular signal peptides from HLA molecules and regulates the cell surface expression of the NK cell ligand HLA-E [50]. In order to test if these proteins are involved in IGSF8 degradation, we tested the expression of IGSF8 during infection with AD169VarL-BAC derived virus in which US9 or UL40 were deleted. In cells that were infected with the AD169VarL-BAC delta US9 virus, the expression of IGSF8 was elevated and better resembled the GFP levels that was expressed from the same plasmid, suggesting that US9 is contributing to IGSF8 degradation (Figs 6B and S14B). The effect of US9 seemed specific for IGSF8 since US9 deletion had no additional effect on BTN2A1 expression (Figs 6B and S14A, and S14B). In contrast to US9, the deletion of UL40 had no obvious effect on BTN2A1 or IGSF8 expression (Figs 6B, fourth panel, S14A, and S14B). Finally we tested if US9 affect IGSF8 by ectopically expressing US9 in fibroblasts expressing epitope tagged IGSF8. In accordance with the results obtained with US2, ectopic expression of US9 had mild but reproducible effect on IGSF8 expression in uninfected fibroblasts (Fig 6A, middle and right panel). Overall, these results illustrate how our ribosome profiling data in conjunction with recent mass spectrometry measurements [12] allowed us to identify two Ig superfamily proteins that are degraded during HCMV infection.
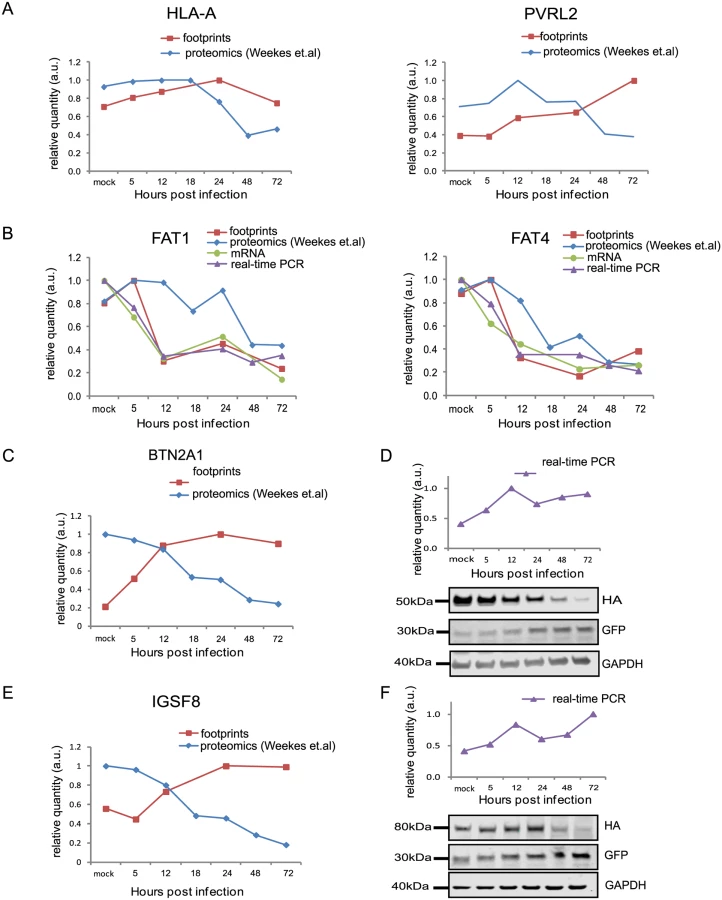

Uncovering novel host proteins that are degraded during HCMV infection
Since targeted degradation of central host proteins by the virus is not limited to immune ligands, we expanded our search and looked for anti-correlations between synthesis rate and steady state protein levels for all human genes for which we had both footprints and proteomic measurements [12]. We limited our search to the simplest profile in which both the footprints measurements and protein levels temporal profiles fit a linear regression (R2 > 0.8) and we focused on cases in which the footprints were upregulated (positive slope), whereas steady state protein levels were downregulated (negative slope). These criteria generated a list of 65 proteins that presented profiles that suggest active degradation (S7 Table, Figs 7A and S15A–S15C, left panels). When the same search was conducted with opposite criteria (requiring that the footprints will be downregulated and protein levels will be upregulated) we found no proteins that passed these conditions, supporting the notion that the list we obtained is biologically meaningful. We focused on five proteins from this list that reflect diverse biological functions; 1. ROCK1—a Rho-associated kinase that is a central regulator of actin cytoskeleton [51]. 2. ERC1—a regulatory subunit of the IκB kinase (IKK) complex [52]. 3. CDC37 - a co-chaperone that binds to numerous kinases and promotes their interaction with the HSP90 complex [53]. 4. WDR61 - a subunit of the human polymerase associated factor (PAF) and SKI complexes that regulate transcription[54]. 5. TIPRL - an inhibitory regulator of protein phosphatase-2A (PP2A). We conducted simultaneous real-time PCR and western blot analysis on cell extracts along HCMV infection and could confirm profiles that suggest active degradation of ROCK1 and ERC1 (Figs 7B and S16A). CDC37, WDR61 and TIPRL also presented profiles that support the premise they might be degraded, but their downregulation was less prominent (S15A–S15C Fig, right panels). To further establish the increased degradation of these proteins during HCMV infection, we examined their half-lives by cycloheximide (CHX) chase assays. As shown in Fig 7C, both ROCK1 and ERC1 show a long half-life in uninfected cells, whereas, in HCMV infected cells their half-lives are considerably decreased (Figs 7C and S16B). Similar results were obtained for CDC37, WDR61 and TIPRL (S15D Fig).
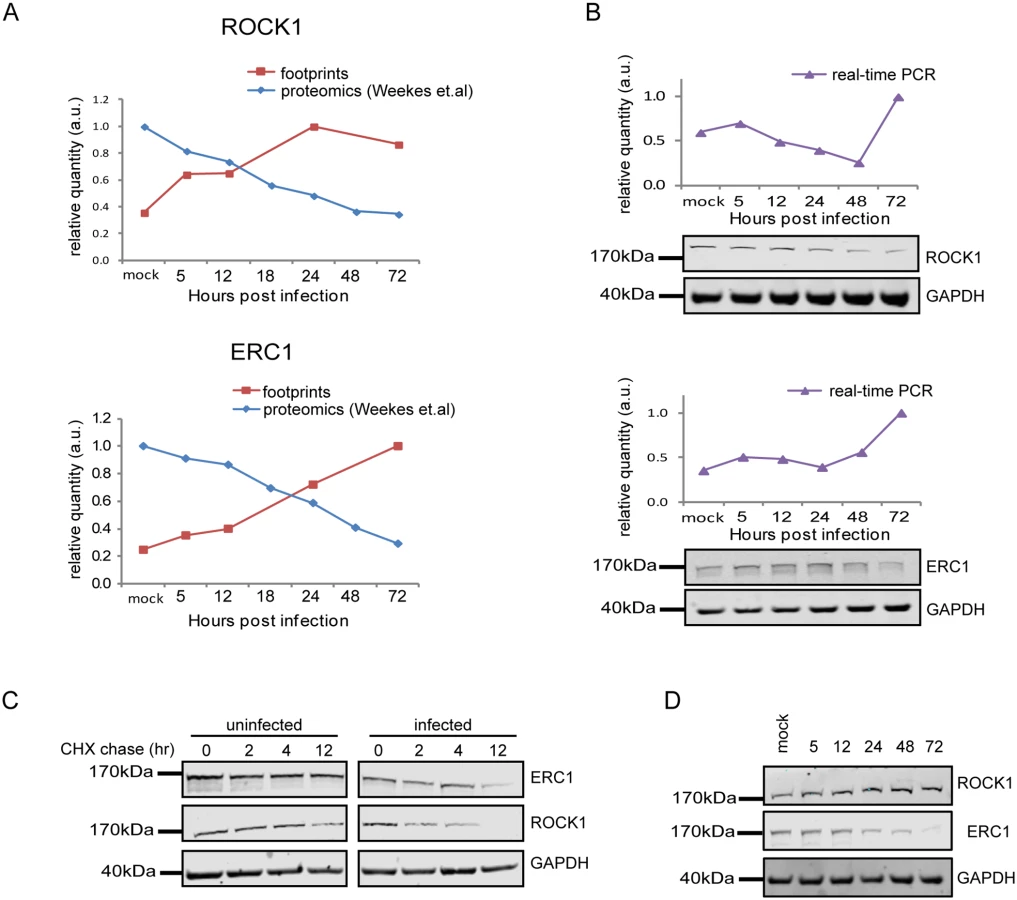
We next tested if the proteins we identified are still degraded when cells are infected with a HCMV laboratory-adapted strain, AD169, in which a 15 kb composing the ULb’ region (genes UL133–UL150) is deleted. Importantly, ERC1, CDC37, WDR61 and TIPRL showed similar reduction in protein levels during infection with the AD169 virus (Figs 7D and S15E). However, ROCK1 levels were significantly elevated during AD169 infection (Fig 7D), suggesting that its degradation might depend on a protein/s that are encoded in the ULb’ region. ROCK1 plays a central role in actin regulation and its KD resulted in rounding up of cells (S17 Fig). pUL135 which is encoded in the ULb’ region was recently shown to remodel the actin cytoskeleton [55]. We therefore tested the effect of pUL135 on ROCK1 expression. We expressed pUL135 in fibroblasts using a lentiviral vector. The vector also contained GFP that was expressed from the same transcript using IRES and was used to identify cells that express the pUL135 protein. As a control, we expressed another viral protein pUL26 in the same manner. As was previously reported [55], expression of pUL135 in fibroblasts induced dramatic changes in cell morphology and cells became rounded up (Fig 8A, GFP expressing cells). We examined ROCK1 expression by immunofluorescence and observed a significant reduction in ROCK1 levels only in cells that express pUL135 (Figs 8A and S18). Similar reduction in ROCK1 levels following pUL135 expression was observed by immunoblotting (Fig 8B) and we confirmed that pUL135 expression did not affect ROCK1 mRNA levels (Fig 8C). Although pUL135 clearly affects ROCK1 expression the magnitude of the observed reduction strongly suggests that additional proteins contribute to ROCK1 degradation.

Overall, our results demonstrate that integration between dynamic translation and proteome measurements enables systematic identification of proteins that are targeted for degradation during infection. Since targeted degradation indicates a strong biological importance this approach could facilitate the discovery of central host functions during viral infection.
Discussion
In this study, we present a comprehensive resource describing temporal changes in cellular gene transcription and translation along HCMV infection. During infection, HCMV extensively manipulates cellular gene expression to maintain conditions favorable for efficient viral propagation.
Analysis of transcription profiles has been the focus of systematic characterization of gene expression during infection [6,8,9,5,56]. However, control of protein production reflects both regulation of mRNA levels and the efficiency with which these messages are translated into proteins. Systematically measuring translation and mRNA levels allowed us to quantitatively evaluate their relative contributions and to reveal novel insights into the viral life cycle.
Our results show that the majority of cellular transcripts, changed by more than 3-fold along infection, exemplifying the radical changes that occur in cells during lytic HCMV infection. Our measurements show that this extensive gene regulation is dominated by changes in mRNA levels, but translational control also regulated the magnitude and timing of protein production during HCMV infection (discussed below).
Although the majority of the pathways that significantly changed during infection were previously mapped, our deep measurements and siRNA experiments allowed us to reveal that peroxisomes might be playing an unrecognized role during HCMV infection. Peroxisomes participate in central pathways of cellular metabolism such as β-oxidation of fatty acids (especially fatty acid chains that are too long to be handled by the mitochondria), amino acid catabolism and detoxification of reactive oxygen species [57]. It will be important to test which of these peroxisome functions are important for HCMV progression.
A more global look at the pathways that are elevated during infection reveals upregulation of modules that are important for cell proliferation (cell cycle, translation and transcription), whereas downregulated modules are related to development and cell-to-cell communication. Since an important dichotomy in the life of a cell is between proliferation and differentiation, these gene expression profiles illustrate that the cell program in infected cells is shifted, and although infected cells are not dividing, their cellular program resembles the cell program of a proliferating transformed cell.
Our simultaneous measurements of mRNA levels and translation rates along infection allowed us to quantitatively evaluate the role of translation regulation in controlling cellular genes expression and to expand recent findings [17]. Using stringent criteria we identified significant changes in TE for almost 10% of cellular genes. By clustering translationally regulated genes based on their TE values, we revealed several distinct temporal profiles suggesting that several independent molecular mechanisms are responsible for the observed translational changes. We were able to connect two of these clusters to known cellular modules; one cluster of genes was significantly enriched in mTORC1 targets implying that the increased translation of mRNAs from this cluster is attributed to mTORC1 activation by the virus [58]. This observation is also supported by the finding that ectopic expression of UL38 (mTORC1-activator) recapitulates a large fraction of the genes whose translation was stimulated by HCMV [17]. A second cluster we identified was enriched in genes involved in cell cycle progression and in mRNAs that showed enhanced translation at G1 and S phases of the cell cycle [40]. Since HCMV infection arrests the cells in a unique G1/S phase it is likely that the induced translation of these mRNAs is the consequence of cell cycle arrest. We observed additional translation regulation profiles, but these were not significantly enriched for any annotated biological process and additional work is needed to delineate the cellular mechanism that drives these changes and their relation to HCMV infection. Overall, our results suggest that functionally related groups of genes are translationally co-regulated and this provides an additional mean to control the expression of a particular subset of mRNAs. Transcription studies have enabled the identification of cis - and trans-transcriptional elements that control diverse cellular processes, whereas a similarly broad understanding of the importance and mechanisms of translational control remains much more elusive. Our data set and clustering approach provides a valuable basis for identifying such cis - and trans-translational regulators.
Unlike many viruses (including several Herpesviruses), HCMV does not completely suppress the synthesis of host proteins in infected cells [1,2]. Our approach allowed, for the first time, to test whether the virus manages to manipulate the cellular translation machinery to preferably translate its own mRNA. By globally comparing the TE of host genes to that of viral genes we made two novel observations; 1. When infection starts, viral mRNAs are translated less efficiently than host mRNAs. This observation intriguingly suggests that the cell possess intrinsic means to distinguish between cellular and viral mRNAs. 2. At 72 hpi a subtle but significant advantage for viral mRNAs translation is observed, suggesting that late in infection the virus does deploy the translation machinery to biasedly translate its mRNAs. Even though this virus does not fully co-opt the translation machinery like other viruses, elucidating the regulatory mechanisms underlying translational reprogramming of both the virus and the host can reveal novel modules the virus relies on, which could ultimately lead to the development of novel therapeutic strategies.
Our work also demonstrates that mass spectrometry and ribosome profiling represent highly complementary approaches; our comparison between changes in rate of protein synthesis, measured by ribosome profiling, and protein abundance, measured by mass spectrometry, revealed novel examples of regulated degradation of cellular proteins during HCMV infection. This integration of ribosome profiling with mass spectrometry measurements along a dynamic process, presents a novel unbiased approach to map protein degradation.
HCMV has evolved a variety of mechanisms to evade the immune response to survive in infected hosts, including elimination of cellular immune ligands from being presented on the cell surface. We reveal here that BTN2A1 and IGSF8, members of the Ig superfamily, are degraded during HCMV infection. BTN2A1 is a cell surface glycoprotein related to the extended family of B7 costimulatory molecules. It was shown to act as a ligand for DC-SIGN [59], a specific dendritic cell receptor, but its role in immune response was never investigated. IGSF8 was identified as a major tetraspanin (CD9 and CD81)-associated protein [60] and was shown to regulate the formation and maintenance of immune synapses [61]. It is therefore possible that degradation of IGSF8 allows protection against cytotoxic immune effector cells by interfering with immune synapse formation. A recent study revealed that US2, a viral protein that was originally defined by its capacity to target MHC molecules for degradation [44], is a pleotropic modulator of cell surface receptors [47]. Hsu et al. [47] unbiased measurements suggested that the US2 protein downregulates BTN2A1 and IGSF8 cell surface expression. Indeed, we show that BTN2A1 degradation is dependent on US2 expression but our analysis indicate that IGSF8 is still degraded even when US2 is not expressed. Our results suggest that US9 probably also plays a role in mediating IGSF8 degradation. Therefore, presenting an additional example for the redundancy in mechanisms HCMV is using to escape immune recognition. Interestingly, US9 was recently shown to selectively target MICA*008, a highly prevalent stress-induced ligand, to proteasomal degradation [49]. Our results suggest that like US2 [47], US9 probably targets a broader set of proteins and future work will have to address the specificity of this viral protein. More broadly, our ability to unbiasedly identify two immune ligands that are degraded during infection strongly argues for the validity of our approach.
Our analysis revealed additional cellular proteins that are degraded during infection. ROCK1 plays a critical role in mediating the effects of small GTPase RhoA on stress fiber formation, focal adhesion and cell motility [62]. Interestingly, these structures were shown to be modulated during HCMV infection [63,64] and the possible role of ROCK1 in modulation of these processes could be studied. We also show that ERC1 is degraded during HCMV infection. ERC1 was shown to be critical for NF-κB activation following ATM induction by genotoxic stress [65]. Since it is known that ATM is activated during HCMV infection [28,66], targeted degradation of ERC1 could serve as an elegant way to antagonize innate immunity response through attenuation of NF-κB signaling.
Finally, our approach is applicable to other viruses or any other pathogen and is useful to gain mechanistic insights into pathogen interference with regulation of mRNA expression, translation and protein degradation.
Materials and Methods
Cells and viruses
Human fibroblasts (CRL-1634) and the HCMV Merlin strain (VR-1590) were obtained from American Type Culture Collection (ATCC). The virus was propagated twice on HFF cells before the preparations of samples for sequencing. Cells were grown on 15cm plates and were infected at a multiplicity of infection (MOI) of 5.
The AD169, AD169VarL, AD169VarL-BAC, AD169VarL-BAC deltaUS9 and the AD169VarL-BAC deltaUL40 were previously described [49,67,68].
Lentiviral constructs and stable cell lines
US2, US9 and UL135 were amplified from cDNA derived from HCMV-infected cells and cloned into the lentiviral vector pHAGE-DsRED(−)-ZsGreen(+). UL135 was cloned under the doxycycline inducible promoter.
Lentivirus was packaged by co-transfection of constructs with the 2nd generation packaging plasmids pMD2.G and PsPax using jetPEI (Polyplus-transfection) into 6-well plates with 293T cells according to protocol. 60 hours post transfection supernatants were collected and centrifuged at 1500 rpm for 5 minutes and filtered through a 0.45 μm filter. HFF expressing BTN2A1, IGSF8 or UL135 cell lines were generated by lentiviral transduction. 12 hours after infection fresh media was added, for UL135 cell line, the medium was supplemented with tet-free serum (Biological industries). Cells were analyzed for expression of proteins using fluorescent microscopy for GFP positive cells.
Ribosome profiling and mRNA-Seq samples preparation
Cylcoheximide treatment was carried out as previously described [3]. Cells were lysed in lysis buffer (20mM Tris 7.5, 150mM NaCl, 5mM MgCl2, 1mM dithiothreitol, 8% glycerol) supplemented with 0.5% triton, 30 U/ml Turbo DNase (Ambion) and 100μg/ml cycloheximide, ribosome protected fragments were then generated as previously described [3]. Total RNA was isolated from infected cells using Tri-Reagent (Sigma).
Polyadenylated RNA was purified from total RNA sample using Oligotex mRNA mini kit (Qiagen). The resulting mRNA was modestly fragmented by partial hydrolysis in bicarbonate buffer so that the average size of fragments would be ~ 80bp. The fragmented mRNA was separated by denaturating PAGE and fragments 50–80 nt were selected as previously described [3]
Sequence alignments, normalization and clustering
Prior to alignment, linker and polyA sequences were removed from the 3’ ends of reads. Bowtie v0.12.7 (5) (allowing up to 2 mismatches) was used to perform the alignments. First, reads that aligned to human rRNA sequences were discarded. All remaining reads were aligned to the concatenated viral (NC_006273.2) and human (hg19) genomes. Finally, still-unaligned reads were aligned to 200bp sequences that spanned splice junctions. Reads with unique alignments were used to compute the total number of reads at each position. Footprints and mRNA densities were calculated in units of reads per kilobase per million (RPKM) in order to normalize for gene length and total reads per sequencing run.
TE was calculated for genes that had more than uniquely aligned 150 reads of mRNA and footprints. For the comparison of between the virus and the host TE only genes with TE >1 were included. For clustering only genes with calculated expression > 3 RPKM in at least one of the condition and a change greater than 3-fold were used. Partitioning clustering was performed using Partek Genomic suits across mRNA, footprints, and TE data. Where indicated, gene lists were analyzed by Ingenuity Pathway Analysis (Ingenuity Systems, Redwood City, CA, USA) using default settings.
5’UTR characterizations
The 5′UTRs were obtained using the known gene ID from the UCSC Genome Browser (GRCh37/hg19). For Each Cluster the 5′UTR length, %G+C content and Gibbs free energy was calculated and compared to background list using Wilcoxon two-sided test.
The translationally regulated clusters we identified (Fig 3B) were compared to genes that were identified as transitionally upregulated during different phases of the cell cycle [40] and to genes that were translationally repressed after mTOR inhibition (log2 ratio <-1, Thoreen et al. [38]) and the enrichment was calculated using hyper Geometric test.
Western blot analysis
Cells were lysed using RIPA buffer. Lysates were nutated at 4°C for 10 min, then centrifuged at 20,000 × g for 15 min at 4°C. Samples were then separated by 4–12% polyacrylamide Bis-tris gel electrophoesis (Invitrogen), blotted onto nitrocellulose membranes and immunoblotted with primary antibodies (αROCK1 ab134181; αERC1 ab180507; αCDC37 ab108305; αWDR61 ab57840; αB7H6 ab138588; αTIPRL ab70795; αGFP (ZsGreen) 632474 (Takara-Clontech); αIE1/IE2 (CH160) ab53495 (Abcam); αHA 3F10 (Roche); αGAPDH 2118S (Cell signaling); αATG3 A3231 (Sigma); αUL44 (CMV ICP36) CA006 (Virusys); αpp28 CA004 (Eastcoast). Secondary antibodies used were Goat anti-rabbit, Goat anti-mouse (IRDye 800CW or IRDye 680RD, Licor), or Goat anti Rat (Alexa Fluor 680, ab175778, Abcam). Reactive bands were detected by Odyssey CLx infrared imaging system (Licor). Protein concentration was measured by Bradford assay (Sigma cat no. B6916). Protein quantification was performed on Licor software.
Real-time PCR
Total RNA was extracted using Tri-Reagent (Sigma) according to protocol. cDNA was prepared using High-Capacity cDNA Reverse Transcription Kit (ABI) according to protocol.
Real time PCR was performed using the SYBR Green PCR master-mix (ABI) on a real-time PCR system StepOnePlus (life technologies) with the following primers (forward, reverse):
RAB12; GCCGTCATGGAAGGTTATTT, CCCTTAGGAAGCCATGAGAG
IRAK1; CAGACAGGGAAGGGAAACAT, AATCACTGTGAAGCCTGTGC
HSP90AB1; GCAGACATCTCCATGATTGG, AAGGAACCTCCAGCAGAAGA
SIX5; CAGTCACCACATCCTTCTGC, GGGAGGGCTGTAACAGAGAG
FAT1; CATCATTGTTGCCAAACCTC, GAGGACGATGGTCATTTGTG
FAT4; AGTGGTGGAACCTGTCACAA, CTCTGCAGGCACTCATTGAT
LAMA2; GGCCTGACTGGGAAATTAAA, CTCGGAAATTCCACAAACCT
ATP5J: GGTGTTACAGCAGTGGCATT, CCTCTCCAGCTCTTGCTGAT
NCOA6; GTCCTGGGTCCAGTAGGAAA, GAGGAGTGGGACTGACCAAT
TSEN54; CCAAGACCTGCCACTGTCTA, GGACAGAGCTTGGTTGGAAT
POLR2L; AGGAGAGCCTTCCATCTCG, ATCTGGCTCTTCAGATTCCG
CDC37; GGTAAATACCAAGCCCGAGA, ATGCCAAAGTGCTTGATCTG
B7H6; ACCCTGGGACTGTCTACCAG, TGAAATAGGCCACCAATGAA
ERC1; TGCAAATCAGAAAGCTGACC, TGGTGGTAGAGGTGGTC
TIPRL; TCCCTGAAATGATGTTTGGA, CTTCAGCACAGGCCACTTTA
ROCK1; TTGGTAGAGGTGCATTTGGA, AAAGCCATGATGTCCCTTTC
WDR61; TGCTCATATTCGTCTTTGGG, ACTTTCCCGACATGAGTTCC
MFGE8; CACTCTGCGCTTTGAGCTAC, TCCAGCTGAAGAGATGCAAG
IGSF8; ACCCTATTTGTGCCTCTGCT, ACAGTCGACACCTGCAAGAC
BTN2A1; AGAGGAATCCACAGGACCAC, GGGACTTAGCCACCCTTACA
RAB12; GCCGTCATGGAAGGTTATTT, CCCTTAGGAAGCCATGAGAG
BTN2A1; AGAGGAATCCACAGGACCAC, GGGACTTAGCCACCCTTACA
Knockdowns
Cells were transfected with siRNA validated for each of the target genes or negative control (TriFECTa Kit DsiRNA Duplex, IDT) in the presence of Lipofectamine RNAiMAX reagent (Life Technologies), according to manufacturer's standard protocol. 24 hours after transfection, cells were infected with HCMV (Merlin strain, MOI 3). All experiments were performed in triplicate, and representative results are reported.
TCID50 assay
104 HFF were plated in 96-well plates and cells were infected with 10-fold serial dilutions of supernatant from knocked-down infected cells, collected 5 days post infection. At 10 days post infection the dilutions showing cytopathic effect were evaluated by light microscopy. The TCID50/ml was calculated using the Spearman-Kaerber method [69]. Experiments were performed at least 3 times and representative figure is presented.
Degron based live cell translation reporter
The fluorescence-based translation reporter was cloned using fusion pcr of three parts; 1. DHFR(Y100I) 2. sfGFP-NLS-P2A 3. NLS-mCherry and cloned into the pHR lentiviral expression vector using BstXI-NotI. 5’ UTRs were inserted using BstX1-BsiWI. Primers to amplify the UTRs used in this study were based on the RNA-seq data to represent the most common UTR splice variant in HFF cells.
Time-lapse microscopy and quantification of fluorescence
All live cell-imaging experiments were performed at 37°C on a AxioObserver Z1 widefield microscope using a 20x air objective and Axiocam 506 mono camera. Cells were grown and imaged in 24-well glass bottom plates and 1 hr before imaging normal growth medium was replaced with DMEM without phenol red, supplemented with 10% FCS and antibiotics. Image analysis was done in Imaris software. For image quantification, images were first corrected for background subtraction using default settings. Segmentation and tracking of each field was performed on the mCherry channel and the GFP mean intensity over time for each segment was measured. The average GFP slope for all segments for each sample was calculated (10<n).
Immunofluorescence
Cells were plated on ibidi slides and fixed in 4% paraformaldehyde for 30 min, washed in PBS (pH 7.4) and permeabilized with 0.2% Triton X-100 in PBS for 10 min, then blocked with 3% BSA in PBS for 30 min. Detection of ROCK1 was performed by immunostaining with anti-ROCK1 antibodies (abcam 156284, 1 : 200 in PBS) 1hr, RT. Cells were washed 3 times with PBS and labeled with anti-rabbit Rhodamine Red-X-conjugated secondary antibody (Jackson ImmunoResearch 711-295-152, 1 : 200 in PBS) 1 hr, RT. Imaging was performed on a AxioObserver Z1 widefield microscope using a 63x oil objective and Axiocam 506 mono camera.
Mass spectrometry analysis of IGSF8-interacting proteins
Samples were digested by trypsin, analyzed by LC-MS/MS on Q Exactive (Thermo). The data was analyzed with Protein Discoverer 1.4 versus Human and HCMV Uniprot database and against decoy databases (in order to determine the false discovery rate -FDR), using the Sequest search engine. The data was also analyzed vs the specific sequences of HCMV Merlin strain. Identifications were filtered with high identification confidence refers to 0.01 FDR, top rank, mass accuracy, and a minimum of 2 identified peptides in the human proteins. Semi-quantitation was done by calculating the peak area of each peptide. The area of the protein is the average of the three most intense peptides from each protein.
Pull down
HFF stably expressing IGSF8-HA or empty vector were collected from 2x15 cm tissue culture plates 48 hours after mock infection or infection with HCMV Merlin strain at MOI ~ 5. 12 hours before harvesting, the proteosomal inhibitor MG-132 was added in final concentration of 10μM. Cells were washed twice in PBS and lysed in 1 ml lysis buffer (150mM NaCl, 2mM CaCl2, 2mM MgCl2, 1% NP-40 in PBS, supplemented with Roche complete protease inhibitor cocktail). Lysis was facilitated by nutating the cells 1 hr at 4°C. Cells were then centrifuged for 15 min, 20,000 rpm 4°C and supernatant was separated and incubated with pre-equilibrated anti-HA magnetic beads (Pierce). Tagged protein binding to beads was performed by nutating cells-beads mixture at 4°C for 1 hr.
Tagged protein bound to anti-HA magnetic beads was separated using magnetic stand, beads were washed 3 times in wash buffer (same composition as lysis buffer with 0.1% NP-40). Elution was performed by incubating the beads with 0.1M glycine pH 2.5 with gentle mixing. Eluate was neutralized with 0.1M Tris pH 8.5. Protein sample buffer was added to eluates and samples were resolved on Bis-Tris-SDS gel 4–12% and stained with Instant Blue staining. The gel was sent to Mass-spectrometry analysis for identification of interacting proteins.
Supporting Information
Zdroje
1. Davison AJ, Dolan A, Akter P, Addison C, Dargan DJ, Alcendor DJ, et al. The human cytomegalovirus genome revisited: comparison with the chimpanzee cytomegalovirus genome. J Gen Virol. 2003/01/21 ed. 2003;84 : 17–28. 12533697
2. Murphy E, Rigoutsos I, Shibuya T, Shenk TE. Reevaluation of human cytomegalovirus coding potential. Proc Natl Acad Sci U S A. 2003/11/01 ed. 2003;100 : 13585–13590. doi: 10.1073/pnas.1735466100 1735466100 [pii] 14593199
3. Stern-Ginossar N, Weisburd B, Michalski A, Le VT, Hein MY, Huang SX, et al. Decoding human cytomegalovirus. Science (80-). 2012/11/28 ed. 2012;338 : 1088–1093. doi: 10.1126/science.1227919
4. Zhu H, Cong JP, Shenk T. Use of differential display analysis to assess the effect of human cytomegalovirus infection on the accumulation of cellular RNAs: induction of interferon-responsive RNAs. Proc Natl Acad Sci U S A. 1998/02/12 ed. 1997;94 : 13985–13990. 9391139
5. Zhu H, Cong JP, Mamtora G, Gingeras T, Shenk T. Cellular gene expression altered by human cytomegalovirus: global monitoring with oligonucleotide arrays. Proc Natl Acad Sci U S A. 1998/11/25 ed. 1998;95 : 14470–14475. 9826724
6. Kenzelmann M, Muhlemann K. Transcriptome analysis of fibroblast cells immediate-early after human cytomegalovirus infection. J Mol Biol. 2000/12/22 ed. 2000;304 : 741–751. doi: 10.1006/jmbi.2000.4271 11124023
7. Browne EP, Wing B, Coleman D, Shenk T. Altered cellular mRNA levels in human cytomegalovirus-infected fibroblasts: viral block to the accumulation of antiviral mRNAs. J Virol. 2001/11/17 ed. 2001;75 : 12319–12330. 11711622
8. Simmen KA, Singh J, Luukkonen BG, Lopper M, Bittner A, Miller NE, et al. Global modulation of cellular transcription by human cytomegalovirus is initiated by viral glycoprotein B. Proc Natl Acad Sci U S A. 2001/06/08 ed. 2001;98 : 7140–7145. doi: 10.1073/pnas.121177598 11390970
9. Hertel L, Mocarski ES. Global analysis of host cell gene expression late during cytomegalovirus infection reveals extensive dysregulation of cell cycle gene expression and induction of Pseudomitosis independent of US28 function. J Virol. 2004/10/14 ed. 2004;78 : 11988–12011. 15479839
10. Challacombe JF, Rechtsteiner A, Gottardo R, Rocha LM, Browne EP, Shenk T, et al. Evaluation of the host transcriptional response to human cytomegalovirus infection. Physiol Genomics. 2004;18 : 51–62. doi: 10.1152/physiolgenomics.00155.2003 15069167
11. Song YJ, Stinski MF. Effect of the human cytomegalovirus IE86 protein on expression of E2F-responsive genes: a DNA microarray analysis. Proc Natl Acad Sci U S A. 2002/02/28 ed. 2002;99 : 2836–2841. doi: 10.1073/pnas.052010099 11867723
12. Weekes MP, Tomasec P, Huttlin EL, Fielding CA, Nusinow D, Stanton RJ, et al. Quantitative temporal viromics: an approach to investigate host-pathogen interaction. Cell. 2014/06/07 ed. 2014;157 : 1460–1472. doi: 10.1016/j.cell.2014.04.028 24906157
13. Tanaka S, Furukawa T, Plotkin SA. Human cytomegalovirus stimulates host cell RNA synthesis. J Virol. 1975;15 : 297–304. 163357
14. Stinski MF. Synthesis of proteins and glycoproteins in cells infected with human cytomegalovirus. J Virol. 1977/09/01 ed. 1977;23 : 751–767. 197270
15. Walsh D, Perez C, Notary J, Mohr I. Regulation of the translation initiation factor eIF4F by multiple mechanisms in human cytomegalovirus-infected cells. J Virol. 2005/06/16 ed. 2005;79 : 8057–8064. 15956551
16. Ziehr B, Lenarcic E, Vincent HA, Cecil C, Garcia B, Shenk T, et al. Human cytomegalovirus TRS1 protein associates with the 7-methylguanosine mRNA cap and facilitates translation. Proteomics. 2015; doi: 10.1002/pmic.201400616
17. McKinney C, Zavadil J, Bianco C, Shiflett L, Brown S, Mohr I. Global reprogramming of the cellular translational landscape facilitates cytomegalovirus replication. Cell Rep. 2014/01/01 ed. 2014;6 : 9–17. doi: 10.1016/j.celrep.2013.11.045 24373965
18. Sullivan R, Fassolitis AC, Larkin EP, Read RB, Peeler JT. Inactivation of thirty viruses by gamma radiation. Appl Microbiol. 1971;22 : 61–5. 4999976
19. Ingolia NT, Brar GA, Stern-Ginossar N, Harris MS, Talhouarne GJS, Jackson SE, et al. Ribosome Profiling Reveals Pervasive Translation Outside of Annotated Protein-Coding Genes. Cell Reports. 2014.
20. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4 : 44–57. doi: 10.1038/nprot.2008.211 19131956
21. Xiaofei E, Kowalik TF. The DNA damage response induced by infection with human cytomegalovirus and other viruses. Viruses. 2014;6 : 2155–85. doi: 10.3390/v6052155 24859341
22. Sanchez V, Spector DH. Subversion of cell cycle regulatory pathways. Curr Top Microbiol Immunol. 2008;325 : 243–62. 18637510
23. Gudleski-O’Regan N, Greco TM, Cristea IM, Shenk T. Increased expression of LDL receptor-related protein 1 during human cytomegalovirus infection reduces virion cholesterol and infectivity. Cell Host Microbe. 2012/07/24 ed. 2012;12 : 86–96. doi: 10.1016/j.chom.2012.05.012 22817990
24. Jarskaja OO, Medzhidova AA, Fedorova NE, Kusch AA, Zatsepina O V. Immunocytochemical reorganization of the nucleolus in human embryo fibroblasts infected with cytomegalovirus in vitro. Dokl Biol Sci. 387 : 589–92. 12577646
25. Tran K, Mahr JA, Spector DH. Proteasome subunits relocalize during human cytomegalovirus infection, and proteasome activity is necessary for efficient viral gene transcription. J Virol. 2010;84 : 3079–93. doi: 10.1128/JVI.02236-09 20042513
26. Karttunen H, Savas JN, McKinney C, Chen Y-H, Yates JR, Hukkanen V, et al. Co-opting the Fanconi anemia genomic stability pathway enables herpesvirus DNA synthesis and productive growth. Mol Cell. 2014;55 : 111–22. doi: 10.1016/j.molcel.2014.05.020 24954902
27. Mohni KN, Mastrocola AS, Bai P, Weller SK, Heinen CD. DNA mismatch repair proteins are required for efficient herpes simplex virus 1 replication. J Virol. 2011;85 : 12241–53. doi: 10.1128/JVI.05487-11 21957315
28. E X, Pickering MT, Debatis M, Castillo J, Lagadinos A, Wang S, et al. An E2F1-mediated DNA damage response contributes to the replication of human cytomegalovirus. PLoS Pathog. 2011;7: e1001342. doi: 10.1371/journal.ppat.1001342 21589897
29. Love KR, Catic A, Schlieker C, Ploegh HL. Mechanisms, biology and inhibitors of deubiquitinating enzymes. Nat Chem Biol. Nature Publishing Group; 2007;3 : 697–705.
30. Kim ET, Oh SE, Lee Y-O, Gibson W, Ahn J-H. Cleavage specificity of the UL48 deubiquitinating protease activity of human cytomegalovirus and the growth of an active-site mutant virus in cultured cells. J Virol. 2009;83 : 12046–56. doi: 10.1128/JVI.00411-09 19759126
31. Lee J, Koh K, Kim Y-E, Ahn J-H, Kim S. Upregulation of Nrf2 expression by human cytomegalovirus infection protects host cells from oxidative stress. J Gen Virol. 2013;94 : 1658–68. doi: 10.1099/vir.0.052142-0 23580430
32. Kolodkin-Gal D, Zamir G, Edden Y, Pikarsky E, Pikarsky A, Haim H, et al. Herpes simplex virus type 1 preferentially targets human colon carcinoma: role of extracellular matrix. J Virol. 2008;82 : 999–1010. doi: 10.1128/JVI.01769-07 17977977
33. Esteso G, Luzón E, Sarmiento E, Gómez-Caro R, Steinle A, Murphy G, et al. Altered microRNA expression after infection with human cytomegalovirus leads to TIMP3 downregulation and increased shedding of metalloprotease substrates, including MICA. J Immunol. 2014;193 : 1344–52. doi: 10.4049/jimmunol.1303441 24973455
34. Angelova M, Zwezdaryk K, Ferris M, Shan B, Morris CA, Sullivan DE. Human cytomegalovirus infection dysregulates the canonical Wnt/beta-catenin signaling pathway. PLoS Pathog. 2012/10/17 ed. 2012;8: e1002959. doi: 10.1371/journal.ppat.1002959 23071438
35. Lee M, Kim B, Kim VN. Emerging Roles of RNA Modification: m6A and U-Tail. Cell. 2014;158 : 980–987. doi: 10.1016/j.cell.2014.08.005 25171402
36. Iadevaia V, Caldarola S, Tino E, Amaldi F, Loreni F. All translation elongation factors and the e, f, and h subunits of translation initiation factor 3 are encoded by 5’-terminal oligopyrimidine (TOP) mRNAs. RNA. 2008;14 : 1730–6. doi: 10.1261/rna.1037108 18658124
37. Meyuhas O. Synthesis of the translational apparatus is regulated at the translational level. Eur J Biochem. 2000;267 : 6321–30. 11029573
38. Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature. 2012;485 : 109–13. doi: 10.1038/nature11083 22552098
39. Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485 : 55–61. doi: 10.1038/nature10912 22367541
40. Stumpf CR, Moreno M V, Olshen AB, Taylor BS, Ruggero D. The translational landscape of the mammalian cell cycle. Mol Cell. 2013;52 : 574–82. doi: 10.1016/j.molcel.2013.09.018 24120665
41. Han K, Jaimovich A, Dey G, Ruggero D, Meyuhas O, Sonenberg N, et al. Parallel measurement of dynamic changes in translation rates in single cells. Nat Methods. 2014;11 : 86–93. doi: 10.1038/nmeth.2729 24213167
42. Tanenbaum ME, Stern-Ginossar N, Weissman JS, Vale RD. Regulation of mRNA translation during mitosis. Elife. 2015;4. doi: 10.7554/eLife.07957
43. Powers C, DeFilippis V, Malouli D, Früh K. Cytomegalovirus immune evasion. Curr Top Microbiol Immunol. 2008;325 : 333–59. 18637515
44. Wiertz EJ, Tortorella D, Bogyo M, Yu J, Mothes W, Jones TR, et al. Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature. 1996;384 : 432–8. 8945469
45. Wiertz EJ, Jones TR, Sun L, Bogyo M, Geuze HJ, Ploegh HL. The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell. 1996;84 : 769–79. 8625414
46. Tomasec P, Wang ECY, Davison AJ, Vojtesek B, Armstrong M, Griffin C, et al. Downregulation of natural killer cell-activating ligand CD155 by human cytomegalovirus UL141. Nat Immunol. 2005;6 : 181–8. 15640804
47. Hsu J-L, van den Boomen DJH, Tomasec P, Weekes MP, Antrobus R, Stanton RJ, et al. Plasma Membrane Profiling Defines an Expanded Class of Cell Surface Proteins Selectively Targeted for Degradation by HCMV US2 in Cooperation with UL141. PLoS Pathog. 2015;11: e1004811. doi: 10.1371/journal.ppat.1004811 25875600
48. Le VT, Trilling M, Hengel H. The cytomegaloviral protein pUL138 acts as potentiator of tumor necrosis factor (TNF) receptor 1 surface density to enhance ULb’-encoded modulation of TNF-alpha signaling. J Virol. 2011/10/07 ed. 2011;85 : 13260–13270. doi: 10.1128/JVI.06005-11 21976655
49. Seidel E, Le VTK, Bar-On Y, Tsukerman P, Enk J, Yamin R, et al. Dynamic Co-evolution of Host and Pathogen: HCMV Downregulates the Prevalent Allele MICA*008 to Escape Elimination by NK Cells. Cell Rep. 2015; doi: 10.1016/j.celrep.2015.01.029
50. Tomasec P, Braud VM, Rickards C, Powell MB, McSharry BP, Gadola S, et al. Surface expression of HLA-E, an inhibitor of natural killer cells, enhanced by human cytomegalovirus gpUL40. Science. 2000;287 : 1031. 10669413
51. Schofield A V., Bernard O. Rho-associated coiled-coil kinase (ROCK) signaling and disease. Crit Rev Biochem Mol Biol. 2013;48 : 301–316. doi: 10.3109/10409238.2013.786671 23601011
52. Ducut Sigala JL, Bottero V, Young DB, Shevchenko A, Mercurio F, Verma IM. Activation of transcription factor NF-kappaB requires ELKS, an IkappaB kinase regulatory subunit. Science. 2004;304 : 1963–7. 15218148
53. Calderwood SK. Cdc37 as a Co-chaperone to Hsp90. Subcell Biochem. 2015;78 : 103–12. doi: 10.1007/978-3-319-11731-7_5 25487018
54. Jaehning JA. The Paf1 complex: platform or player in RNA polymerase II transcription? Biochim Biophys Acta. 2010;1799 : 379–88. doi: 10.1016/j.bbagrm.2010.01.001 20060942
55. Stanton RJ, Prod’homme V, Purbhoo MA, Moore M, Aicheler RJ, Heinzmann M, et al. HCMV pUL135 remodels the actin cytoskeleton to impair immune recognition of infected cells. Cell Host Microbe. 2014;16 : 201–14. doi: 10.1016/j.chom.2014.07.005 25121749
56. Challacombe JF, Rechtsteiner A, Gottardo R, Rocha LM, Browne EP, Shenk T, et al. Evaluation of the host transcriptional response to human cytomegalovirus infection. Physiol Genomics. 2004/04/08 ed. 2004;18 : 51–62. doi: 10.1152/physiolgenomics.00155.2003 15069167
57. Cooper GM. Peroxisomes. Sinauer Associates; 2000.
58. Moorman NJ, Cristea IM, Terhune SS, Rout MP, Chait BT, Shenk T. Human cytomegalovirus protein UL38 inhibits host cell stress responses by antagonizing the tuberous sclerosis protein complex. Cell Host Microbe. 2008/04/15 ed. 2008;3 : 253–262. S1931-3128(08)00087-5 [pii] doi: 10.1016/j.chom.2008.03.002 18407068
59. Malcherek G, Mayr L, Roda-Navarro P, Rhodes D, Miller N, Trowsdale J. The B7 Homolog Butyrophilin BTN2A1 Is a Novel Ligand for DC-SIGN. J Immunol. American Association of Immunologists; 2007;179 : 3804–3811. doi: 10.4049/jimmunol.179.6.3804
60. Stipp CS, Kolesnikova T V, Hemler ME. EWI-2 is a major CD9 and CD81 partner and member of a novel Ig protein subfamily. J Biol Chem. 2001;276 : 40545–54. doi: 10.1074/jbc.M107338200 11504738
61. Gordón-Alonso M, Sala-Valdés M, Rocha-Perugini V, Pérez-Hernández D, López-Martín S, Ursa A, et al. EWI-2 association with α-actinin regulates T cell immune synapses and HIV viral infection. J Immunol. American Association of Immunologists; 2012;189 : 689–700. doi: 10.4049/jimmunol.1103708
62. Riento K, Ridley AJ. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol. 2003;4 : 446–56. 12778124
63. Jones NL, Lewis JC, Kilpatrick BA. Cytoskeletal disruption during human cytomegalovirus infection of human lung fibroblasts. Eur J Cell Biol. 1986;41 : 304–12. 3019700
64. Stanton RJ, McSharry BP, Rickards CR, Wang EC, Tomasec P, Wilkinson GW. Cytomegalovirus destruction of focal adhesions revealed in a high-throughput Western blot analysis of cellular protein expression. J Virol. 2007/05/25 ed. 2007;81 : 7860–7872. doi: 10.1128/JVI.02247-06 17522202
65. Wu Z-H, Wong ET, Shi Y, Niu J, Chen Z, Miyamoto S, et al. ATM - and NEMO-dependent ELKS ubiquitination coordinates TAK1-mediated IKK activation in response to genotoxic stress. Mol Cell. 2010;40 : 75–86. doi: 10.1016/j.molcel.2010.09.010 20932476
66. E X, Pickering MT, Debatis M, Castillo J, Lagadinos A, Wang S, et al. An E2F1-mediated DNA damage response contributes to the replication of human cytomegalovirus. PLoS Pathog. 2011/05/19 ed. 2011;7: e1001342. doi: 10.1371/journal.ppat.1001342 21589897
67. Le VTK, Trilling M, Hengel H. The Cytomegaloviral Protein pUL138 Acts as Potentiator of Tumor Necrosis Factor (TNF) Receptor 1 Surface Density To Enhance ULb’-Encoded Modulation of TNF - Signaling. J Virol. 2011;85 : 13260–13270. doi: 10.1128/JVI.06005-11 21976655
68. Rölle A, Pollmann J, Ewen E-M, Le VTK, Halenius A, Hengel H, et al. IL-12-producing monocytes and HLA-E control HCMV-driven NKG2C+ NK cell expansion. J Clin Invest. 2014;124 : 5305–16. doi: 10.1172/JCI77440 25384219
69. Darling AJ, Boose JA, Spaltro J. Virus Assay Methods: Accuracy and Validation. Biologicals. 1998;26 : 105–110. doi: 10.1006/biol.1998.0134 9811514
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 11
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Dengue Virus Non-structural Protein 1 Modulates Infectious Particle Production via Interaction with the Structural Proteins
- On the Discovery of TOR As the Target of Rapamycin
- Parasite Glycobiology: A Bittersweet Symphony
- Broadening of Virus-Specific CD8 T-Cell Responses Is Indicative of Residual Viral Replication in Aviremic SIV Controllers
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy